Characterization of glycosylation sites for a recombinant IgG1 monoclonal antibody
蓝色花观赏植物花青苷修饰研究进展

蓝色花观赏植物花青苷修饰研究进展黎敏婕,郝文慧,曹晓云,唐佳仪,王未,张玉瑞,杨鼎铉,杜灵娟(西北农林科技大学风景园林艺术学院,陕西咸阳712100)摘要:花青苷是使植物花朵呈色的重要色素之一,能让植物呈现出红、紫和蓝等多种花色。
蓝色花中色素物质主要由花青素糖基化和酰基化高度修饰后积累形成,因此,花青苷修饰是提高花青素稳定性和形成蓝色花色苷的必须步骤。
归纳了花青苷修饰方式对蓝色花呈色调控的相关文献,总结了蓝色花观赏植物花青苷修饰的相关研究现状。
目前蓝色花青苷相关修饰基因的作用方式仍需进一步解析,从而更深一步探究花青苷修饰基因对蓝色花瓣呈色的影响,为培育蓝色花观赏植物新品种提供分子理论基础。
关键词:花青苷修饰;蓝色花;糖基转移酶;酰基转移酶迄今鉴定出的花青素糖基转移酶有2类,分别属于碳水化合物活性酶糖基转移酶1(carbohydrate-ac-tive enzyme glycosyltransferase 1,GT1)家族和糖苷水解酶1(glycoside hydrolase family 1,GH1)家族。
第一类由UDP 依赖的糖基转移酶进行,在细胞质中发挥作用。
第二类是依赖酰基-葡萄糖的糖苷水解酶,在液泡中发挥作用[10-11]。
这些酶将糖分子转移至受体花青素的特定位置。
花青素3-位糖基化是花青素合成途径中第一个稳定的花色苷,而之后的5,7及3’位糖基化将进一步促进蓝色花的形成。
有研究报道,在菊花()中引入蝶豆花()Ct3’5’GT 基因发生3’-和5’-位的糖基化,从而获得了蓝色菊花[12]。
蓝色翠雀()中DgAA7BG-GT1与DgAA7BG-GT2的缺失会导致无法在7-位的酰基上添加葡萄糖,最终形成粉色的花色表型[13-14]。
在百子莲()和风铃草()中也发现了相似功能的AaAA7GT 和CmAA7GT [15-16]。
在GT1家族中,GT 类糖基转移酶主要通过PSPG 基序对糖供体表现出高度特异性。
上海交通大学医学院举办第八届中国医院发展与管理学术大会

1467上海交通大学学报(医学版),2020,40 (11)presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia [J ]. J Clin Endocrinol Metab, 2005, 90(7): 4371-4375.[5] K ranz C, Sun LW, Eklund EA, et al. CDG-Id in two siblings with partiallydifferent phenotypes [J ]. Am J Med Genet, 2007, 143A(13): 1414-1420.[6] R imella-Le-huu A, Henry H, Kern I, et al. Congenital disorder of glycosylationtype Ⅰ d (CDG Ⅰd): phenotypic, biochemical and molecular characterization of a new patient [J ]. J Inherit Metab Dis, 2008, 31(Suppl 2): S381-S386.[7] S tibler H, Stephani U, Kutsch U. Carbohydrate-deficient glycoproteinsyndrome: a fourth subtype [J ]. Neuropediatrics, 1995, 26(5): 235-237.[8] R iess S, Reddihough DS, Howell KB, et al. ALG3-CDG (CDG-Ⅰ d ): clinical,biochemical and molecular findings in two siblings [J ]. Mol Genet Metab, 2013, 110(1/2): 170-175.[9] L epais L, Cheillan D, Frachon SC, et al. ALG3-CDG: Report of two siblingswith antenatal features carrying homozygous p.Gly96Arg mutation [J ]. Am J Med Genet A, 2015, 167(11): 2748-2754.[10] H aeuptle MA, Hennet T. Congenital disorders of glycosylation: an update ondefects affecting the biosynthesis of dolichol-linked oligosaccharides [J ]. Hum Mutat, 2009, 30(12): 1628-1641.[11] N g BG, Shiryaev SA, Rymen D, et al. ALG1-CDG: clinical and molecularcharacterization of 39 unreported patients [J ]. Hum Mutat, 2016, 37(7): 653-660.[12] F rank CG, Eyaid W, Berger EG, et al. Identification and functional analysisof a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL [J ]. Am J Hum Genet, 2004, 75(1): 146-150.[13] Z ufferey R, Knauer R, Burda P, et al. STT3, a highly conserved protein requiredfor yeast oligosaccharyl transferase activity in vivo [J ]. EMBO J, 1995, 14(20): 4949-4960.[14] A ebi M, Gassenhuber J, Domdey H, et al. Cloning and characterization of the ALG3 gene of Saccharomyces cerevisiae [J ]. Glycobiology, 1996, 6(4): 439-444.[15] K orner C. Carbohydrate deficient glycoprotein syndrome type Ⅳ : deficiency ofdolichyl-P-Man: Man5GlcNAc 2-PP-dolichyl mannosyltransferase [J ]. EMBO J, 1999, 18(23): 6816-6822.[16] K ranz C, Basinger AA, Güçsava ş-Çalıko ğlu M, et al. Expanding spectrum ofcongenital disorder of glycosylation Ⅰ g (CDG- Ⅰg): sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality [J ]. Am J Med Genet A, 2007, 143(12): 1371-1378.[17] L i ST, Wang N, Xu S, et al. Quantitative study of yeast Alg1 β-1,4mannosyltransferase activity, a key enzyme involved in protein N-glycosylation [J ]. Biochim Biophys Acta Gen Subj, 2017, 1861(1): 2934-2941.[18] K efala G, Kwiatkowski W, Esquivies L, et al. Application of Mistic toimproving the expression and membrane integration of histidine kinase receptors from Escherichia coli [J ]. J Struct Funct Genomics, 2007, 8(4): 167-172.[19] 李盛陶. 体外重构N-糖基化途径中多萜醇寡糖前体的合成[D ]. 无锡: 江南大学, 2018.[20] L i ST, Lu TT, Xu XX, et al. Reconstitution of the lipid-linked oligosaccharidepathway for assembly of high-mannose N-glycans [J ]. Nat Commun, 2019, 10: 1813.[21] 李庆猛, 李盛陶, 王宁, 等. 酵母来源α-1,2甘露糖转移酶Alg11的异源表达、纯化和活性分析[J ]. 中国生物工程杂志, 2018, 38(6): 26-33.[22] H o SN, Hunt HD, Horton RM, et al. Site-directed mutagenesis by overlapextension using the polymerase chain reaction [J ]. Gene, 1989, 77(1): 51-59.[23] B urgos J, Hemming FW, Pennock JF, et al. Dolichol: a naturally-occurring c100isoprenoid alcohol [J ]. Biochem J, 1963, 88(3): 470-482.[收稿日期][本文编辑]2019-08-30瞿麟平罗冰洁,等 ALG3异常的先天性糖基化疾病相关突变蛋白活性的检测。
蛋白质糖基化修饰的非变性构象分辨质谱研究进展

学 报Journal of China Pharmaceutical University 2023,54(6):674 - 681674蛋白质糖基化修饰的非变性构象分辨质谱研究进展贾翼菲1,王雅梅1,李功玉1,2*(1南开大学化学学院,分析科学研究中心,天津市生物传感与分子识别重点实验室,天津 300071;2物质绿色创造与制造海河实验室,天津 300192)摘 要 糖基化是蛋白质最重要的翻译后修饰之一,能够调控蛋白质的电荷态、结构及分子间相互作用等,进而影响其功能。
糖基化的高度异质性导致传统的结构解析方法很难对糖蛋白进行全面表征。
随着分析技术的发展,质谱在糖蛋白结构解析中发挥了重要作用。
蛋白质组学质谱技术可在多肽水平上对复杂、低丰度蛋白质糖基化修饰的化学组成与位点信息进行鉴定。
非变性质谱技术(native mass spectrometry,nMS)则直接在完整蛋白水平上揭示聚糖异质性及其对蛋白高级结构和相互作用的调控效应。
作为结构质谱的代表性技术,基于离子淌度的nMS受益于离子淌度仪的构象分辨能力和构象去折叠功能,能够在非变性质谱的基础上提供离子的三维动态结构信息,为异构体结构快速鉴定提供不可替代的解决方案。
本文重点介绍了两种新兴离子淌度质谱技术,即非变性动态构象分辨质谱技术和糖型分辨结构质谱技术,并以3种常见蛋白体系为例,介绍其在糖蛋白构象研究领域的最新进展。
关键词蛋白糖基化;非变性质谱技术;构象分辨质谱;离子淌度质谱;碰撞诱导去折叠中图分类号R363 文献标志码 A 文章编号1000 -5048(2023)06 -0674 -08doi:10.11665/j.issn.1000 -5048.2023060901引用本文贾翼菲,王雅梅,李功玉.蛋白质糖基化修饰的非变性构象分辨质谱研究进展[J].中国药科大学学报,2023,54(6):674–681. Cite this article as:JIA Yifei,WANG Yamei,LI Gongyu. Recent progress of protein glycosylation characterization utilizing native conformer-resolved mass spectrometry[J].J China Pharm Univ,2023,54(6):674–681.Recent progress of protein glycosylation characterization utilizing native conformer-resolved mass spectrometryJIA Yifei1, WANG Yamei1, LI Gongyu1,2*1Tianjin Key Laboratory of Biosensing and Molecular Recognition, Research Center for Analytical Science, College of Chemistry, Nankai University, Tianjin 300071; 2Haihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China Abstract Glycosylation of proteins, one of the most prevalent and complex post-translational modifications occurring in nature, plays a crucial role in regulating protein net charge, conformation, binding properties and, ultimately, biological function.Traditional structural techniques are not amenable for glycoproteins due to the inherent heterogeneity of oligosaccharides.With the advances in analytical technique, mass spectrometry dis⁃plays an increasingly crucial role in elucidating the structure of glycoproteins.Mass spectrometry-based pro⁃teomic technique can dissect the chemical composition and site information of low-abundance glycosylation at the peptide level.Instead, native mass spectrometry (nMS) can analyze intact glycoproteins while maintaining the information for glycan heterogeneity, and the insights into the regulatory effects of glycosylation on protein higher order structures and interactions with other proteins or ligands.As a representative structural mass spectrometry tool, ion mobility-based nMS strategy is powered by its conformer-resolving capability and by the feasibility of conformer manipulation through collision-induced unfolding.Consequently, native IM-MS analysis can provide rich information of dynamic protein conformations, allowing for the rapid identification and differ⁃收稿日期2023-06-09 *通信作者Tel:159****1043E-mail:ligongyu@基金项目国家自然科学基金资助项目(No.22104064,No.22293030,No.22293032);国家重点研发计划青年科学家项目资助(No.2022YFA1305200)第 54 卷第 6 期贾翼菲,等:蛋白质糖基化修饰的非变性构象分辨质谱研究进展entiation of protein isoforms in an unprecedented manner.In this review, we briefly introduced two emergingnative IM -MS analytical modes, dynamic conformer -resolving mode and glycoform -resolving mode.Besides, we also discussed the recent progress of conformational and topological characterization of intact glycoproteins with three typical model systems based on two above -mentioned emerging modes of native IM -MS.Key words protein glycosylation; native mass spectrometry; conformer -resolving mass spectrometry; ion mobili⁃ty -mass spectrometry; collision -induced unfoldingThis study was supported by the National Natural Science Foundation of China (No.22104064, No.22293030, No.22293032) and the National Key R&D Program of China (No.2022YFA1305200)糖基化(glycosylation )是一种普遍且重要的翻译后修饰类型,在调控蛋白结构、信号传导、免疫应答、胚胎发育等过程中发挥重要作用[1]。
自由巯基的检测

Free Sulfhydryl in Recombinant Monoclonal AntibodiesWei Zhang*and Marta J.CzuprynWyeth BioPharma,Genetics Institute Campus,One Burtt Road,Andover,Massachusetts01810 Monoclonal antibody(mAb)therapy applications have been growing rapidly in recentyears.Like other recombinant protein drugs,therapeutic mAb’s need to be wellcharacterized to ensure their structural and functional integrity.IgG mAb’s arecomposed of two heavy and two light chains covalently linked by interchain disulfidebonds.Each domain of the heavy or light chain contains one additional disulfide bond.Native IgG mAb’s,with completely formed disulfide bonds,should not bear any freesulfhydryl.This report describes detection and quantification of free sulfhydryl inrecombinant mAb’s produced in Chinese hamster ovary(CHO)cells using a fluorescenttechnique.The method utilizes the fluorescent probe N-(1-pyrenyl)maleimide(NPM).The purified mAb’s appear to be homogeneous under native conditions with ap-proximately0.02mol of free sulfhydryl per mole of protein.Upon denaturation,minorspecies related to the mAb’s are observed on sodium dodecyl sulfate polyacrylamidegel electrophoresis(SDS-PAGE),and the free sulfhydryl level is determined to beapproximately0.1mol/mol of protein.These results suggest that a small portion ofthese recombinant mAb’s lack in intermolecular disulfide bonds but remain nonco-valently associated under native conditions.The formation of the free sulfhydrylcontaining mAb species is likely to occur during the culture process and/or proteinfolding process in the endoplasmic reticulum(ER).IntroductionThe rapid advancement of recombinant monoclonal antibody(mAb)therapy has necessitated detailed char-acterization and analysis of this protein family.Thera-peutic mAb’s derived by recombinant DNA technology, like other genetically engineered protein pharmaceuti-cals,require extensive and stringent characterization of purity,structural integrity,and stability,as molecular modifications may result in undesirable adverse biologi-cal effects,such as increased immunogenicity.Though the general structural features of antibodies have been well documented in literature(1,2),a number of studies have revealed structural variations of recombinant mAb therapeutics.These include isoaspartate formation re-sulting from asparagine deamidation and/or aspartate isomerization(3-5),processing of C-terminal lysine resides(6),methionine and cysteine(Cys)oxidation(7), variability in glycosylation(8-10),mutation introduced during transfection(11),and crossover event between variable genes of heavy and light chains during tran-scription(12).These modifications,due to host-dependent and/or in vitro process-dependent factors,are epigenetic and thus not predicable from the genetic structure of construct alone.They may alter the biological activities or pharmacokinetic properties of therapeutic mAb’s and consequently require substantive characterization to ensure product quality and to provide understanding of structure/function relationships.IgG antibodies have a four-chain structure composed of two heavy chains(HCs)and two light chains(LCs) covalently linked by interchain disulfide bridges(Figure 1).This four-chain structure is also maintained by strong,noncovalent interactions between the N-terminal half of HC-LC pairs and between the C-terminal regions of HC. In addition to interchain disulfide bonds,one intrachain disulfide bond is present and is shielded within each -barrel domain of HC and LC(1,2).While the disulfide linkage between HC and LC always involves the last Cys residue,Cys213according to the Kabat numbering system(13)of the LC,the Cys residue from the HC varies,depending on the IgG subtype(13).In IgG1mAb, the fifth Cys residue(Cys217)of HC forms an inter HC and LC disulfide bond.In IgG2and IgG4mAb’s,it is the third Cys amino acid of HC(Cys123)that links HC and LC.In the hinge region,different IgG subtypes have different numbers of disulfide bond connecting the HCs. In IgG1and IgG4,the two HCs are covalently linked by two disulfide bonds,while in IgG2,there are four disulfide bonds in its hinge region(1,2).In a correctly folded IgG antibody,regardless of the subtype,all Cys residues are involved in disulfide bond formation,there-fore,no free sulfhydryl should be present.During our work on recombinant mAb’s,we detected the presence of some free sulfhydryl as well as nonco-valently associated mAb fragments.We investigated the noncovalently associated species in mAb produced in Chinese hamster ovary(CHO)cells.To this end,we analyzed the structural integrity of these molecules under native and denaturing conditions.Free sulfhydryl content in the mAb’s was determined following deriva-tization with a fluorescent probe.Our results suggest that these noncovalently associated mAb fragments may result from incomplete formation of interchain disulfide bonds within the mAb molecules.*To whom correspondence should be addressed.Email: wxzhang@.Tel:(978)247-2997.Fax:(978)247-2834.509Biotechnol.Prog.2002,18,509−51310.1021/bp025511z CCC:$22.00©2002American Chemical Society and American Institute of Chemical EngineersPublished on Web04/11/2002Materials and MethodsMaterial.The recombinant mAb’s analyzed in the present work were produced in CHO cells and purified at Wyeth BioPharma,Genetics Institute Campus in Andover,MA.Samples included one IgG1,two IgG2(IgG2-I and IgG2-II herein),and one IgG4mAb.N -Acetyl-L -cysteine (Ac-Cys),N -(1-pyrenyl)maleimide (NPM),and N ,N -dimethylformamide (DMF)were obtained from Sigma.Size Exclusion Chromatography (SEC -HPLC).SEC -HPLC analysis of was performed on a Waters Alliance system with a Waters model 2487dual wave-length absorbance detector and a refrigerated autosam-pler controlled at 4°C.A TosoHaas G3000SW XL (7.8mm ×300mm,5µm,250Å)column was equilibrated with a mobile phase consisting of 10mM sodium phosphate,250mM NaCl,pH 7.2at a flow rate of 0.75mL/min.A 20-µl sample of mAb solution at a concentration of approxi-mately 1mg/mL was injected.The elution was monitored at 214nm.The analysis time was 20min.Sodium Dodecyl Sulfate Polyacrylamide Gel Elec-trophoresis (SDS -PAGE).The mAb samples were analyzed using precast Novex 4-20%Tris-Glycine gels.Nonreducing sample buffer consisted of 125mM Tris-HCl,pH 6.8,30%(v/v)glycerol,4%(w/v)SDS,and 0.05%(w/v)bromphenol blue.Reducing sample buffer was made by mixing 5vol of 1M dithiothreitol and 95vol of nonreducing buffer.Next,10µL of the mAb solutions at a concentration of 1mg/mL was mixed with equal volume of sample buffer.The mixture was heated at 100°C for 5min for nonreducing SDS -PAGE and 1min for the reducing gel.The gels were run at 150V (constant voltage)for 1.5h.After electrophoresis,the gels were stained in 0.05%(w/v)Coomassie blue and destained using 10%(v/v)acetic acid,15%(v/v)methanol.Measurement of Sulfhydryl.Sulfhydryl content of the mAb’s was measured by a fluorescent assay.Free sulfhydryl was derivatized by adding 10µL of 2mM NPM in DMF to the following three mAb sample prepara-tions:100µL of mAb solution diluted with 500µL of phosphate-buffered saline (PBS),pH 7.3;100µL of mAb solution diluted with 500µL of 6M guanidine hydro-chloride (GuHCl)in PBS buffer,pH 7.3;100µL of mAb solution diluted with 500µL of 6M GuHCl in PBS,pH 7.3,incubated under argon at 37°C for 2h.After theNPM solution was added,the mixtures were incubated at room temperature for 5min and quenched with 10µL of 50%acetic acid.A set of Ac-Cys standards in PBS,pH 7.3with the concentration ranging from 0.25to 25µM,and a PBS,pH 7.3blank were also prepared by the above procedure.Fluorescence measurement was performed on a Waters model 474scanning fluorescence detector equipped with a cuvette holder.This setting allows measurement of the fluorescence of a sample without using a chromatographic system and therefore simplifies sample handling.A quartz cuvette with 1.0-cm path length was used for fluorescence measurement.The excitation wavelength was set at 330nm,and the emission wavelength was set at 380nm.All samples were analyzed in triplicate.The measured sulfhydryl content was expressed as mole of sulfhydryl per mole of mAb.ResultsSEC -HPLC.Under the native chromatographic con-ditions in this work,all four mAb’s eluted between 10and 11min as a single peak (Figure 2),consistent with a molecular weight of ca.150kDa.The observed level of high molecular weight species (earlier eluting material)was below 1%for these mAb’s.No low molecular weight protein species were detected (the peak at ∼15min was related to salts in the buffer).These results confirmed the identities of these recombinant mAb’s,and they appeared to be homogeneous under the native SEC chromatographic conditions.SDS -PAGE.The four mAb’s had similar banding patterns in both reducing and nonreducing SDS -PAGE (Figure 3).Under reducing conditions,predominantly a single HC band migrating at approximately 50kDa and a LC band migrating at approximately 25kDa were observed (Figure 3A).The band below the LC of the IgG4mAb (Figure 3A,lane 6)was the result of deamidation-related peptide bond cleavage,as identified by electro-blotting and Edman sequencing (data not shown).Under nonreducing conditions,all four mAb’s migrated pre-dominantly as a single band of approximately 150kDa,corresponding to a full mAb molecule consisted of 2HC and 2LC (Figure 3B).However,an additional array of minor bands was also present with apparent molecular weights lower than 150kDa (Figure 3B).Peptidemap-Figure 1.Schematic drawing of IgG antibody.Each mAb consists of two HCs and two LCs covalently linked by interchain disulfide bonds.In IgG1and IgG4antibodies,two disulfide bonds are in the hinge region connecting the two HCs,whereas in IgG2antibody four disulfide bonds are in the hinge region.In addition,Cys213of the LC forms inter-HC and LC disulfide bond with Cys217in IgG1,or Cys123in IgG2and IgG4.In all IgG antibodies,each -barrel domain of the HC and LC contains one intrachain disulfide linkage.ping of these mAb’s detected very little peptide bond cleavage (data not shown).Electroblotting and subse-quent Edman sequencing of these lower bands suggested that they may be mAb missing one LC,HC dimer,a half molecule (one HC and one LC),single HC and LC (data not shown).The bands migrating around 25kDa were all identified to be LC-related (Figure 3B).It has been reported that proteins containing disulfide bonds bind less SDS than their reduced forms (14).Hence,the formation of the multiple LC bands may be due to incomplete reduction of the -barrel disulfide bonds in the LC (15).Under the denaturing SDS -PAGE condi-tions,mAb minor species were resolved,while under the native SEC -HPLC conditions,the mAb’s eluted as one peak.Therefore,these minor species are most likely present in solution as noncovalently associated mAb molecules,or partially linked via disulfide bond.Measurement of Sulfhydryl.To elucidate the origin of these mAb minor species under denaturing conditions,free sulfhydryl content was measured.Though Ellman’s assay has been used for quantification of protein sulfhy-dryl,it lacks adequate sensitivity for sulfhydryl content below the low micromolar range (16).Therefore,the sulfhydryl level in the mAb’s was assessed by a fluores-cent assay.NPM was used as the fluorescent probe because of its specificity,sensitivity,and ease of use (17).In particular,NPM is essentially nonfluorescent until sulfhydryl conjugated,which allows measurement of the fluorescence of the sample without prior reagent separa-tion (17).The fluorescence emission intensities of a series of Ac-Cys standards derivatized by NPM were used to establish a standard curve of fluorescence vs sulfhydryl concentration.The standard curve was linear from 0.25to 25µM with a R 2value of 0.9999(Figure 4)and was then used to determine the sulfhydryl content in the mAb samples.The results were summarized in Figure 5.When measured under nondenaturing conditions in PBS,less than 0.02mol of free sulfhydryl was detected per mole of the IgG2or IgG4mAb,while the IgG1mAb was found to contain approximately 0.03mol of sulfhydryl.To access the potentially buried sulfhydryl,the mAb’s were dena-tured with 5M GuHCl and then modified with NPM.In all cases,sulfhydryl level increased to nearly 0.08mol/mol of protein,3-4times higher than that in the native state.In a third set of experiments,the mAb samples in 5M GuHCl were incubated at 37°C for 2h.The two IgG2mAb’s were found to contain approximately 0.1mol of sulfhydryl per mol of protein,while the IgG1and IgG4mAb’s contained approximately 0.09sulfhydryl per pro-tein molecule.These results indicated that all mAb’s analyzed have similar levels of sulhydryl with the major-ity not accessible in the native state of protein.The increased sulfhydryl levels under denaturingconditionsFigure 2.SEC -HPLC profiles of recombinant mAb’s.(A)IgG1mAb.(B)IgG2-I mAb.(C)IgG2-II mAb.(D)IgG4mAb.Figure 3.SDS -PAGE profiles of recombinant mAb’s under (A)reducing and (B)nonreducing ne 1,molecular weight marker;lane 2,blank;lane 3,IgG1mAb;lane 4,IgG2-I mAb;lane 5,IgG2-II mAb;lane 6,IgG4mAb.also implied the presence of noncovalently associated mAb molecules in solution.DiscussionThe sulfhydryl group of Cys is potentially the most reactive nucleophile of all the functional side chains in proteins.It can exist in the reduced free state or the oxidized state to form disulfide bond.The state of sulfhydryl is often important to the structure and func-tion of a protein (18).Therefore,an assessment of the presence and state of sulfhydryl groups in recombinant protein pharmaceuticals allows an evaluation of their structural and functional integrity.In IgG antibodies,all Cys residues form inter-or intramolecular disulfide bonds,and therefore no free sulfhydryl should exist (Figure 1).However,in the present study,all four recombinant IgG mAb’s produced in CHO cells were found to contain free sulfhydryl groups,and the measured level was elevated upon denaturation.This implied that some Cys residues in these mAb’s stay in the reduced state and do not form either inter-or intrachain disulfide bond.The elevated accessibility of free sulfhydryl under denaturing condi-tions suggested that the majority of these sulfhydryls may reside within a solvent-inaccessible region.This result,combined with those from SEC -HPLC and SDS -PAGE,indicated the presence of noncovalently associated mAb molecules under native conditions.The noncovalent interchain interactions are known to be strong enough to maintain the four-chain structure of IgG antibodies,as reduced and alkylated IgG1did not measurably dissociate under native conditions (1,2).It has been shown that a chimeric IgG1mAb in which the hinge Cys residues were mutated to Ser was able to maintain thefour-chain structure in solution but was found to be predominantly of the half molecule form on nonreducing SDS -PAGE gels (19).Therefore,in the present study,the noncovalently associated species were undetectable under native SEC -HPLC conditions and were only revealed under denaturing conditions,such as SDS -PAGE and in GuHCl solution.The presence of free sulfhydryl groups in these mAb’s implied incomplete formation of disulfide bonds among the detected mAb fragments,which is required for covalent assembly of the four-chain folding of IgG anti-bodies.In CHO and other eukaryotic cells,disulfide bond formation and protein folding proceed within the lumen of the endoplasmic reticulum (ER),where protein oxida-tion takes place upon translocation of nascent peptides into the ER lumen (20).For antibodies,disulfide bond formation is additionally complicated with the balanced transcription of two different genes and the assembly of four polypeptide chains.It has been found that assembly of LC and HC to a complete antibody molecule is the primary rate-limiting step in antibody production and can be speeded up by a high expression rate of LC (21).Therefore,it is likely that the incomplete disulfide bond formation of these mAb’s occurs in vivo in the ER.It is worth pointing out that during the production of the mAb’s analyzed in the present study,low concentrations of both cystine and cysteine were used in the cell culture media,so it is also possible that formation of these hypodisulfide-bonded species occurs in vitro after the mAb’s are secreted into the culture media.However,during the production of recombinant murine interleukin 12in CHO cells using culture media containing cystine and cysteine,a glutathione adduct was found,indicating the incomplete disulfide bond formation in vivo (22).Though the origin of the incomplete disulfide bond formation of these mAb’s is unknown at this time,the presence of free sulfhydryl and noncovalently associated species may be undesirable.Also,accumulation of mis/unfolded proteins has been reported to cause ER stress (23,24)and,subsequently,apoptosis and loss of cell viability (25-27).Moreover,the long-term effect of these undisulfide-bonded mAb species on the stability and biological activity remains to be determined.ConclusionsWe have measured the free sulfhydryl content in recombinant mAb’s by a fluorescent assay.The detected sulfhydryl level was approximately 0.02and 0.1mol/mol of protein under native and denaturing conditions,respectively.The mAb’s appeared to be homogeneous as assessed by SEC -HPLC and reducing SDS -PAGE.However,in addition to the bands corresponding to the intact mAb’s,an array of minor binds was observed on nonreducing SDS -PAGE gels.These results suggest that noncovalently associated species are present in these mAb’s and that these mAb fragments may result from incomplete disulfide bond formation between the HC and LC.The formation of these undisulfide-bonded mAb fragments may occur during the protein folding process in the ER and/or the cell culture process.The long-term effects of these free sulfhydryl-containing minor species on the stability and potency of these mAb’s require further investigation.The fluorescent assay for assess-ment of free sulfhydryl is sensitive and quantitative and is applicable to production monitoring and structural analysis of other recombinant protein and mAb pharma-ceuticals.Figure 4.Ac-Cys fluorescence standard curve.The fluorescence was measured after derivatization with NPM.The excitation and emission wavelengths were set at 330and 380nm,respectively.The curve was linear in the range of 0.25-25µM.Figure 5.Free sulfhydryl content in recombinant mAb’s.Experimental conditions are described in Materials and Meth-ods.AcknowledgmentWe would like to thank Dr.Hubert Scoble for his support on this project and Drs.Mark Hardy,Yin Luo, and Lisa Marzilli for their critical review of the manu-script.We are grateful to Philip Boyle and Jeff Banas for their technical assistance.References and Notes(1)Dorrington,K.J.The structural basis for the functionalversativility of immunoglobulin G.Can.J.Biochem.1974, 56,1087-1101.(2)Burton,D.R.Immunoglobulin G:functional sites.Mol.Immunol.1985,22,161-206.(3)Cacia,J.;Keck,R.;Presta,L.G.;Frenz,J.Isomerization ofan aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE:identifica-tion and effect on binding affinity.Biochemistry1996,35, 1897-1903.(4)Perkins,M.;Theiler,R.;Lunte,S.;Jeschke,M.Determina-tion of the origin of charge heterogeneity in a murine monoclonal antibody.Pharm.Res.2000,17,1110-1117. (5)Harris,R.J.;Kabakoff,B.;Macchi,F.D.;Shen,F.J.;Kwong,M.J.;Andya,D.S.;Shire,J.;Bjork,N.;Totpal,K.;Chen,A.B.Identification of multiple sources of charge heterogeneityin a recombinant antibody.J.Chromatogr.B2001,752,233-245.(6)Harris,R.J.Processing of C-terminal lysine and arginineresidues of proteins isolated from mammalian cell culture.J.Chromatogr.A1995,705,233-245.(7)Kroon,D.J.;Baldwin-Ferro,A.;Lalan,P.Identification ofsites of degradation in a therapeutic monoclonal antibody by peptide mapping.Pharm.Res.1992,9,1386-1393.(8)Weitzhandler,M.;Hardy,M.;Co,M.S.;Avdalovic,N.Analysis of carbohydrates on IgG preparations.J.Pharm.Sci.1994,83,1670-1675.(9)Sheeley,D.M.;Merrill,B.M.;Taylor,L.C.Characterizationof monoclonal antibody glycosylation:comparison of expres-sion systems and identification of terminal R-linked galactose.Anal.Biochem.1997,247,102-110.(10)Bihoreau,N.;Ramon,C.;Lazard,M.;Schmitter,J.M.Combination of capillary electrophoresis and matrix-assisted laser desorption ionization mass spectrometry for glycosyl-ation analysis of a human monoclonal anti-Rhesus(D)anti-body.J.Chromatogr.B1997,697,123-33.(11)Harris,R.J.;Murnane,A.A.;Utter,S.L.;Wagner,K.L.;Cox,E.T.;Polastri,G.D.;Helder,J.C.;Sliwkowski,M.B.Assessing genetic heterogeneity in production cell lines: detection by peptide mapping of a low level Tyr to Gln sequence variant in a recombinant antibody.Bio/Technology 1993,11,1293-1297.(12)Wan,M.;Shiau,F.Y.;Gordon,W.;Wang,G.Y.Variantantibody identification by peptide mapping.Biotechnol.Bioeng.1999,62,485-488.(13)Kabat,D.J.;Wu,T.T.;Reid-Miller,M.;Perry,H.M.;Gottesman,K.S.Sequences of Proteins of ImmunologicalInterest,4th ed;U.S.Department of Health and Human Services:Washington,DC,1987.(14)Pitt-Rivers,R.;Impiombato,F.S.A.The binding of sodiumdodecyl sulphate to various proteins.Biochem.J.1968,109, 825-830.(15)Kikuchi,H.;Goto,Y.;Hamguchi,K.Reduction of the buriedintrachain disulfide bond of the constant fragment of the immunoglobulin light chain:global unfolding under physi-ological conditions.Biochemistry1986,25,2009-2013. (16)Wright,S.K.;Viola,R.E.Evaluation of methods for thequantitation of cysteines in proteins.Anal.Biochem.1998, 265,8-14.(17)Winters,R.A.;Zukowski,J.;Ercal,N.;Matthews,R.H.;Spitz,D.R.Analysis of glutathione,glutathione disulfide, cysteine,homocysteine,and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl)maleimide.Anal.Biochem.1995,227,14-21.(18)Liu,T.-Y.The role of sulfur in proteins.In The Proteins,3rd ed;Neuvath,H.,Hill,R.L.,Eds.;Academic Press: London,1977;pp329-402.(19)Gillies,S. D.;Wesolowski,J.S.Antigen binding andbiological activities of engineered mutant chimeric antibodies with human tumor specificities.Hum.Antibod.Hybridomas 1991,1,47-54.(20)Frand,A.R.;Cuozzo,J.W.;Kaiser,C.A.Pathways forprotein disulfide bond formation.Trends Cell Biol.2000,10, 203-210.(21)Strutzenberger,K.;Borth,N.;Kunert,R.;Steinfellner,W.;Katinger,H.Changes during subclone development and aging of human antibody-producing recombinant CHO cells.J.Biotechnol.1999,69,215-226.(22)Nickbarg,E.B.;Vath,J.;Pittman,D.D.;Leonard,J.E.;Waldburger,K.E.;Bond,M.Structural characterization of the recombinant P40heavy chain subunit monomer and homodimer of murine IL-12.Bioorg.Chem.1995,23,380-396.(23)Dorner,A.J.;Wasley,l.C.;Raney,P.;Haugejorden,S.;Green,M.;Kaufman,R.J.The stress response in Chinese hamster ovary cells.J.Biol.Chem.1990,265,22029-22034.(24)Kaufman,R.J.Stress singaling from the lumen of theendoplasmic reticulum:coordination of gene transscriptional and translational controls.Genes Dev.1999,13,1211-1233.(25)Al-Rubeai,M.;Singh,R.P.Apoptosis in cell culture.Curr.Opin.Biotechnol.1998,9,152-156.(26)Mastrangelo,A.J.;Betenbaugh,M.J.Overcoming apop-tosis:new methods for improving protein-expression systems.Trends Biotechnol.1998,16,88-95.(27)Murphy,T.C.;Woods,N.R.;Dickson,A.J.Expression ofthe transcription factor GADD153is an indicator of apoptosis for recombinant Chinese hamster ovary(CHO)cells.Biotech-nol.Bioeng.2001,75,621-629.Accepted for publication March5,2002.BP025511Z。
LC-MS方法鉴定黄酮类化合物

黄酮类化合物 (flavonoids)
一、 概 述
广泛分布于植物中的天然有氧杂环化合物,主要游离态和与糖结合成苷的形式存在。 藻类、细菌等微生物中没有发现黄酮类化合物的存在。
以前,黄酮类化合物主要是指基本母核2-苯基色原酮类化合物,现在则是泛指两个 苯环(A-与B-环)通过中央三碳链相互联结而成的一系列化合物。
四、黄酮化合物的生物功能 1、使植物免除紫外线对植物的辐射损伤; 2、抵抗昆虫、病原菌; 3、络合有毒金属离子; 4、参与光敏过程与能量转移; 5、调节植物生长等生理活动等。
同种不同属植物的化学何成分有一定相似性,故在植物 化学分类学上作为Chemotaxonomic markers。
五、黄酮的生物活性
对黄酮苷首先对样品进行甲基化、乙基化、三甲基硅烷化等衍生化操作。
化学电离(Chemical ionization,CI)质谱法
对黄酮苷的鉴定效果差。
场解析(Field desorption,FD)质谱法
快原子轰击(Fast atom bombardment,FAB)质谱法
LC-MS法鉴定黄酮
热喷雾(Thermospray ionization,TSP) 质谱
黄酮骨架上结合位点的不同可能与其酶抑制活性、抗氧化活性有关。在Manach 的研 究中,大剂量 Quercetin 给以大鼠后,其甲基化代谢产物 Isorhamnetin 含量很高。而 已知Isorhamnetin对黄嘌呤氧化酶 的抑制能力强于Quercetin 。
黄酮类化合物的MS鉴定
电轰击(Electron impact,EI)质谱法
Stobiecki, M., et al., Profiling of phenolic glycosidic conjugates in leaves of Arabidopsis thaliana using LC/MS. Metabolomics, 2006. 2(4): p. 197-219.
抗体药物N-糖表征及质量研究策略探讨

Pharmacy Information 药物资讯, 2021, 10(3), 85-91Published Online May 2021 in Hans. /journal/pihttps:///10.12677/pi.2021.103012抗体药物N-糖表征及质量研究策略探讨居雪玲1,2,夏晗雪2,周于人2,季昌明2,邢莹莹1*,徐意人2*1中国药科大学生命科学与技术学院,江苏南京2上海药明生物技术有限公司,上海收稿日期:2021年4月11日;录用日期:2021年5月6日;发布日期:2021年5月13日摘要糖基化修饰是抗体药物的关键质量属性(Critical Quality Attributes, CQAs)之一。
在现代抗体工业中,调节和控制糖基化修饰对抗体的药代动力学(PK),活性及免疫原性都有着重要影响。
作为蛋白质众多翻译后修饰中最为复杂且重要的修饰之一,N-糖的表征和质量控制往往充满挑战。
早期研发时,为创新型药物制定高效的N-糖分析策略和为生物类似药制定与原研药的N-糖相似性判定标准是关键的;工业生产时,证明工艺条件变更前后的N-糖质量可比是必要的。
本文综述了抗体药物N-糖基化修饰在不同水平上的表征方法,并结合相关法规,对创新型药物和生物类似药在研发阶段和工业生产阶段的N-糖质量研究策略进行了讨论。
关键词抗体药物,糖基化修饰,分析策略,质量研究Discussion on Characterization andQuality Control Strategies of AntibodyN-GlycosylationXueling Ju1,2, Hanxue Xia2, Yuren Zhou2, Changming Ji2, Yingying Xing1*, Yiren Xu2*1School of Life Science and Technology, China Pharmaceutical University, Nanjing Jiangsu2WuXi Biologics (Shanghai) Co., Ltd., ShanghaiReceived: Apr. 11th, 2021; accepted: May 6th, 2021; published: May 13th, 2021*通讯作者。
Protein Glycosylation蛋白质糖基化

Protein GlycosylationGlycosylation, the attachment of sugar moieties to proteins, is a post-translational modification (PTM) that provides greater proteomic diversity than other PTMs. Glycosylation is critical for a wide range of biological processes, including cell attachment to the extracellular matrix and protein–ligand interactions in the cell. This PTM is characterized by various glycosidic linkages, including N-, O- and C-linked glycosylation, glypiation (GPI anchor attachment), and phosphoglycosylation. Glycoproteins can be detected, purified and analyzed by different strategies, including glycan staining and visualization, glycan crosslinking to agarose or magnetic resin for labeling or purification, or proteomic analysis by mass spectrometry, respectively.IntroductionGlycosylation is a critical function of the biosynthetic-secretory pathway in the endoplasmic reticulum (ER) and Golgi apparatus. Approximately half of all proteins typically expressed in a cell undergo this modification, which entails the covalent addition of sugar moieties to specific amino acids. Most soluble and membrane-bound proteins expressed in the endoplasmic reticulum are glycosylated to some extent, including secreted proteins, surface receptors and ligands, and organelle-resident proteins. Additionally, some proteins that are trafficked from the Golgi to the cytoplasm are also glycosylated. Lipids and proteoglycans can also be glycosylated, significantly increasing the number of substrates for this type of modification.ScopeProtein glycosylation has multiple functions in the cell. In the ER, glycosylation is used to monitor the status of protein folding, acting as a quality control mechanism to ensure that only properly folded proteins are trafficked to the Golgi. Sugar moieties on soluble proteins can be bound by specific receptors in the trans Golgi network to facilitate their delivery to the correct destination. These sugars can also act as ligands for receptors on the cell surface to mediate cell attachment or stimulate signal transduction pathways (1). Because they can be very large and bulky, oligosaccharides can affectprotein–protein interactions by either facilitating or preventing proteins from binding to cognate interaction domains. Because they are hydrophilic, they can also alter the solubility of a protein (2).DistributionGlycosylated proteins (glycoproteins) are found in almost all living organisms that have been studied, including eukaryotes, eubacteria and archae (3,4). Eukaryotes have the greatest range of organisms that express glycoproteins, fromsingle-celled to complex multicellular organisms.Glycoprotein diversityGlycosylation increases the diversity of the proteome to a level unmatched by any other post-translational modification. The cell is able to facilitate this diversity, because almost every aspect of glycosylation can be modified, including:∙Glycosidic linkage—the site of glycan (oligosaccharide) binding∙Glycan composition—the types of sugars that are linked to a particular protein∙Glycan structure—branched or unbranched chains∙Glycan length—short- or long-chain oligosaccharidesGlycosylation is thought to be the most complex post-translational modification because of the large number of enzymatic steps involved (5). The molecular events of glycosylation include linking monosaccharides together, transferring sugars from one substrate to another and trimming sugars from the glycan structure. Unlike other cell processes such as transcription or translation, glycosylation is non-templated, and thus, all of these steps do not necessarily occur during every glycosylation event. Instead of using templates, cells rely on a host of enzymes that add or remove sugars from one molecule to another to generate the diverse glycoproteins seen in a given cell. While it may seem chaotic because of all of the enzymes involved, the different mechanisms of glycosylation are highly-ordered, step-wise reactions in which individual enzyme activity is dependent upon the completion of the previous enzymatic reaction. Because enzyme activity varies by cell type and intracellular compartment, cells can synthesize glycoproteins that differ from other cells in glycan structure (5).Enzymes that transfer mono- or oligosaccharides from donor molecules to growing oligosaccharide chains or proteins are called glycosyltransferases (Gtfs). Each Gtf has specificity for linking a particular sugar from a donor (sugar nucleotide or dolichol) to a substrate and acts independent of other Gtfs. These enzymes are broad in scope, as glycosidic bonds have been detected on almost every protein functional group, and glycosylation has been shown to incorporate most of the commonly occurring monosaccharides to some extent (6).Glycosidases catalyze the hydrolysis of glycosidic bonds to remove sugars from proteins. These enzymes are critical for glycan processing in the ER and Golgi, and each enzyme shows specificity for removing a particular sugar (e.g., mannosidase).Types of glycosylationGlycopeptide bonds can be categorized into specific groups based on the nature of the sugar–peptide bond and the oligosaccharide attached, including N-, O- and C-linked glycosylation, glypiation and phosphoglycosylation. Because N- and O-glycosylation and glypiation are the most commonly detected types of glycosylation, more emphasis in this article will be placed on these modifications.Glycosylation is controlled by moving proteins to areas with different enzyme concentrations; the cell sequesters enzymes into specific compartments to regulate their activity. For example, after a protein is N-glycosylated in the ER, glycan processing occurs in a step-wise fashion by trafficking proteins to distinct Golgi cisternae that contain high concentrations of specific Gtfs and glycosidases.2. Amino acid sequenceBesides the requirement for the right amino acid (e.g., Asn for N-linked; Ser/Thr for O-linked), many enzymes have consensus sequences or motifs that enable formation of the glycosidic bond (6).3. Protein conformation (availability)As proteins are synthesized, they begin to fold into their nascent secondary structure, which can make specific amino acids inaccessible for glycosidic binding. Thus, the target amino acids must be conformationally accessible for glycosylation to occur.Methods to detect and analyze glycoproteinsThe influence that glycoproteins have on biological processes and disease states continues to expand, spurring the development of detection and analytical strategies with increasing sensitivity and throughput to better understanding their diverse structures and biochemistry. Because glycoproteins are a combination of a protein and oligosaccharides, they are more complex to analyze than non-glycosylated proteins. Additionally, the vast diversity in glycan structure and composition add an additional level of complexity to glycoprotein analysis.Glycan staining or labelingBecause of their structure, glycan sugar moieties are not reactive to staining or labeling molecules. To overcome this problem, sugar groups can be chemically restructured with periodic acid, which oxidizes vicinal hydroxyls on sugars (especially sialic acid) to aldehydes or ketones that are then reactive to multiple dyes. The periodic acid-Schiff (PAS) stain uses this reaction to detect and quantify glycoproteins in various biological samples. Periodic acid can also be used to make sugars reactive towards crosslinkers, which can be covalently bound to labeling molecules (e.g., biotin) or immobilized support (e.g., streptavidin) for detection or purification. The two representative protein gels below compare the use of glycan to Coomassie protein staining.Glycoprotein purification or enrichmentLectins can be used to detect and analyze glycoprotein function (52). These glycan-binding proteins have high specificity for distinct sugar moieties. As described previously, lectins facilitate protein folding in the ER, but they are also critical for cell–cell and pathogen–cell attachment. While anti-glycan antibodies can also bind sugar moieties, lectins are used more often because they are less expensive, better characterized and more stable than antibodies (52). Like antibodies, lectins can be conjugated to probes such as horseradish peroxidase, fluorophores and biotin and immobilized to solid support including streptavidin and Thermo Scientific NeutrAvidin Protein. Some of the common uses of lectins include:∙Glycoprotein identification∙Glycoprotein purification/enrichment∙Characterization of cell surface glycoconjugates (e.g., glycoproteins, glycolipids, GPI-anchored molecules) o Detect relative abundanceo Identify tissue and cellular localization∙Glycosylation mutant generation∙Gtf and glycosidase activity analysisMany lectins are commercially available for use in these applications. The most popular lectin is concanavalin A (ConA) from the Jackbean, but other lectins, including Jacalin, wheat germ agglutinin (WGA) and lentil lectin (LCA), are also widely available in different commercial kits. These representative silver stained gels compare results generated using different glycoprotein isolation kits.Glycoproteome and glycome analysis by mass spectrometryGlycoproteins are unique from other post-translationally modified proteins because they are a combination of a protein portion and a glycan portion, both of which can comprise a significant proportion of the molecular weight of the molecule. Thus, glycoproteins can be analyzed as a whole or as individual components. Glycoproteomics is the global analysis of glycosylated proteins and integrates glycoprotein enrichment and proteomic analysis for the systematic identification and quantitation of glycoproteins in complex systems. This subset of proteomics differs from glycomics, which is restricted to all glycans in a system (i.e., the glycome) (53).As with other proteomic analyses, glycoprotein identification and quantitation is performed using mass spectrometry. The basic pipeline for glycoproteomic analysis includes (53):∙Glycoprotein or glycopeptides enrichment∙Multidimensional separation by liquid chromatography (LC)∙Tandem mass spectrometry∙Data analysis via bioinformaticsThis approach can be performed before or after enzymatic cleavage of glycans via endoglycanase H (endo H) orpeptide-N4-(N-acetyl-beta-glucosaminyl)asparagine amidase (PNGase), depending on the type of experiment. Quantitative comparative glycoproteome analysis can be performed by differential labeling with stable isotope labeling by amino acids in cell culture (SILAC) reagents. Additionally, absolute quantitation by selected reaction monitoring (SRM) can be performed on targeted glycoproteins using isotopically labeled, "heavy" reference peptides.Glycoproteomic analysis. Glycoproteins are first digested into glycopeptides and either analyzed directly by liquid chromatography and tandem mass spectrometry (LC-MS/MS) or first deglycosylated and then enriched for glycans or peptides prior to analysis.N-glycosylationThere are many types and mechanisms of glycosylation, and the most common type is N-glycosylation. Glycosylation is often characterized as a post-translational modification. While this is true with other types of glycosylation, N-glycosylation often occurs co-translationally, in that the glycan is attached to the nascent protein as it is being translated and transported into the ER. The "N" in the name of this type of glycosylation denotes that the glycans are covalently bound to the carboxamido nitrogen on asparagine (Asn or N) residues.Because the ER is the site of translation and processing of most membrane-bound and secreted proteins, it is not surprising that most of these are N-linked glycoproteins. Besides being the most common type of glycosylation (90% of glycoproteins are N-glycosylated), N-linked glycoproteins also have large and often extensively branched glycans that undergo multiple processing steps after being bound to proteins.∙Precursor glycan assembly∙Attachment∙Trimming∙MaturationAs described previously, different enzymes are required for each step in during glycosylation, which facilitate diversity in the glycans that are generated (8,9). But N-glycosylation initially occurs identically for all proteins, and the diversity does not manifest until the subsequent trimming and glycan maturation (7).Precursor glycan assemblyOligosaccharides attached via N-glycosidic linkages are derived from a 14-sugar precursor molecule comprised ofN-acetylglucosamine (GlcNAc), mannose (Man) and glucose (Glc). These sugars are added consecutively onto dolichol, a polyisoprenoid lipid carrier embedded in the ER membrane (8,10). The first 7 sugars are donated from sugar nucleotides (UDP- and GDP-sugars) in the cytoplasm and bound to dolichol via a pyrophosphate linkage (-PP-). After theMan5GlcNAc2-PP-dolichol intermediate is completed, the entire complex is flipped into the lumen of the ER, after which thefinal 7 sugars are donated from Man- and Glc-P-dolichol molecules to make the Gcl3Man9GlcNAc2-PP-dolichol precursor glycan.Glycan attachmentGlycosylation is often characterized as a post-translational modification. While this is true with other types of glycosylation, N-glycosylation often occurs co-translationally, in that the glycan is attached to the nascent protein as it is being translated and transported into the ER. The "N" in the name of this type of glycosylation denotes that the glycans are covalently bound to the carboxamido nitrogen on asparagine (Asn or N) residues.Because the ER is the site of translation and processing of most membrane-bound and secreted proteins, it is not surprising that most of these are N-linked glycoproteins. Besides being the most common type of glycosylation (90% of glycoproteins are N-glycosylated), N-linked glycoproteins also have large and often extensively branched glycans that undergo multiple processing steps after being bound to proteins.Glycan assembly and attachment. Precursor glycan synthesis begins on the cytosolic face of the endoplasmic reticulum (ER) and is completed after the structure is flipped into the ER lumen. Oligosaccharide transferase (OSTase) then transfers the precursor glycan to the Asn residue on the nascent protein.One aspect to note is that not all Asn residues with the predicted consensus sequence are glycosylated. N- to C-terminal protein synthesis results in transport of the growing polypeptide into the ER in the same orientation, and protein folding occurs soon after the polypeptide enters the ER. Therefore, as protein folding increases, OSTase is less able to access the consensus sequence for glycan transfer. Indeed, more N-terminal Asn residues are glycosylated than C-terminal Asn residues.Glycan trimming in the EROligosaccharides are trimmed in both the ER and Golgi by glycosidases via hydrolysis. Glycan trimming in the ER, though, serves a different purpose than trimming in the Golgi.In the ER, sugar hydrolysis is used to both monitor protein folding and indicate when proteins should be degraded. Glucosidases I and II remove 2 terminal Glc from the precursor glycan, after which calnexin and calreticulin, which are membrane-bound and soluble (respectively) sugar-binding lectins, bind to the nascent glycoprotein via the remaining Glc and act as chaperones to help the protein fold properly. The final Glc is soon hydrolyzed by glucosidase II, releasing theglycoprotein from the chaperone. Non-native–folded proteins are recognized by UDP-glucose glycoprotein glucosyltransferase, which transfers a Glc to the glycoprotein, and the protein again is bound to the lectin chaperones to facilitate proper protein folding (12). This cycle of Glc addition and removal continues until the protein is correctly folded, at which time it is not reglycosylated, and the glycoprotein is trafficked to the Golgi for further processing (13). The glycan structure for all properly folded glycoproteins that proceed to the Golgi is Man9GlcNAc2 in higher eukaryotes.An ER-resident mannosidase (ERManI) plays a key role in identifying proteins that are unable to fold properly. Proteins that lose 3–4 mannose residues in the ER via ERManI activity are transported out of the ER and deglycosylated by glycanase N (removes the entire glycan en bloc) and delivered to ER-associated degradation (ERAD) (14,15,16,17). It is thought that ERManI acts as a timer of sorts, because it has a slow rate of mannose hydrolysis that allows nascent proteins multiple rounds of reglycosylation to attempt to fold properly before mannose residues are removed and the protein is targeted for degradation (18).Glycan maturation in the GolgiTo this point during glycosylation, all N-linked glycoproteins have the same precursor glycan structure. Glycan processing in the Golgi apparatus combines both trimming and adding sugars to diversify the glycans on individual glycoproteins. As with precursor glycan biosynthesis, this maturation pathway to generate diverse oligosaccharides is highly ordered, such that each step is dependent upon the previous step. To this end, the Golgi segregates specific enzymes into different cisternae to facilitate this step-wise process.Golgi enzyme compartmentalization. Enzymes that mediate glycan processing in the Golgi apparatus are segregated into distinct cisternae to ensure that glycosylation occurs in a step-wise fashion.The final glycan structures can be broadly separated into two groups:∙Complex oligosaccharides—contain multiple sugar types∙High-mannose oligosaccharides—multiple mannose residues∙Hybrid—branches of both high mannose and complex oligosaccharidesGlycans destined to be complex oligosaccharides are trimmed by Golgi mannosidase I and II and glycosylated by GlcNAc transferase, resulting in a common core region (7,12). The core then becomes the substrate for multiple Gtfs that consecutively transfer sugar moieties from sugar nucleotides to build variable-length and -branched oligosaccharide chains of GlcNAc, galactose (Gal), N-acetylneuraminic acid (NANA or sialic acid) and fucose. Any glycoproteins that progress through this processing from the common core stage become resistant to glycan removal by endoglycosidase H (endo H), which is used experimentally to determine if glycoproteins contain high-mannose or complex oligosaccharides.Unlike complex oligosaccharides, high-mannose oligosaccharides do not carry other sugar moieties, although some of the Man residues are often trimmed by Golgi mannosidase I. Whether a glycan is processed into a complex oligosaccharide rather than remaining a high-mannose oligosaccharide is dependent upon the accessibility of the processing enzymes to the glycan, which can be hindered by the glycoprotein conformation. Some glycoproteins have hybrid oligosaccharides, comprising a combination of complex and high-mannose glycans.Glycan maturation. After initial trimming in the ER, the glycoprotein is trafficked to the Golgi, where Golgi mannosidase I removes multiple mannose sugars. Glycans that do not undergo further glycosylation are called high-mannose oligosaccharides. Further sugar addition and removal yields a common core oligosaccharide onto which multiple Gtfs add different sugars to generate the highly variable complex oligosaccharides. Glycan maturation beyond the common core provides endo H insensitivity. Glycans can be high-mannose, complex or a combination of both (i.e., hybrid oligosaccharide).O-glycosylationWhile N-glycosylation is the most common glycosidic linkage, O-glycoproteins also play a key role in cell biology (19). This type of glycosylation is essential in the biosynthesis of mucins, a family of heavily O-glycosylated, high-molecular weight proteins that form mucus secretions. O-glycosylation is also critical for the formation of proteoglycan core proteins that are used to make extracellular matrix components. Additionally, antibodies are often heavily O-glycosylated.O-glycosylation occurs post-translationally on serine and threonine side chains in the Golgi apparatus. N-glycosylation does not preclude the other from occurring, as O-glycosylation commonly occurs on glycoproteins that were N-glycosylatedin the ER. Besides the different linkage, O-glycosylation also differs in the method of glycosylation. While a precursor glycan is transferred en bloc to Asn via N-glycosylation, sugars are added one-at-a-time to serine or threonine residues.O-glycosylation can also occur on hydroxylysine and hydroxyproline, oxidized forms of lysine and proline, respectively, which are found in collagen (19). Additionally, O-linked glycans usually have much simpler oligosaccharide structures than N-linked glycans.MechanismThe O-glycosidic mechanism is not as complex as that of N-glycosylation. Proteins trafficked into the Golgi are most often O-glycosylated by N-acetylgalactosamine (GalNAc) transferase, wh ich transfers a single GalNAc residue to the β-OH group of serine or threonine. To date, there is no known consensus sequence for this enzyme, although structural motifs have been characterized. Some proteins are O-glycosylated with GlcNAc, fucose, xylose, galactose or mannose, depending on the cell and species (6,20). As with N-glycosylation, sugar nucleotides are used as monosaccharide donors for O-glycosylation. Following this first sugar, a highly variable number of sugars (from only a few to greater than 10) are consecutively added to the growing glycan chain. O-glycosylation can also occur in the cytosol and nucleus to regulate gene expression or signal transduction through other Gtfs (21).GlypiationThe covalent attachment of a glycosylphosphatidylinositol (GPI) anchor is a common post-translational modification that localizes proteins to cell membranes. This special kind of glycosylation is widely detected on surface glycoproteins in eukaryotes and some archae (22).GPI anchors consist of a:∙Phosphoethanolamine linker that binds to the C-terminus of target proteins∙Glycan core structure∙Phospholipid tail that anchors the structure in membraneThe structure of glypiation.Both the lipid moiety of the tail and the sugar residues in the glycan core have considerable variation (23,24,25,26,27,28), demonstrating vast functional diversity that includes signal transduction, cell adhesion and immune recognition (29). GPI anchors can also be cleaved by enzymes such as phospholipase C to regulate the localization of proteins that are anchored at the plasma membrane.MechanismSimilar to the precursor glycan used for N-glycosylation, GPI anchor biosynthesis begins on the cytoplasmic leaflet of the ER and is completed on the luminal side. During this process, 3–4 Man and various other sugars (e.g., GlcNAc, Gal) are built onto a phosphatidylinositol (PI) molecule embedded in the membrane using sugars donated from sugar nucleotides and dolichol-P-mannose outside and inside the ER, respectively. Additionally, 2–3 phosphoethanolamine (EtN-P) linker residues are donated from phosphatidylethanolamine in the ER lumen to facilitate binding of the anchor to proteins(30,31,32,33,34).Proteins destined to be glypiated have 2 signal sequences:∙An N-terminal signal sequence that directs co-translational transport into the ER∙ A C-terminal signal sequence that is recognized by a GPI transamidase (GPIT) (29)GPIT does not have a consensus sequence but instead recognizes a C-terminal sequence motif that enables it to covalently attach a GPI anchor to an amino acid in the sequence. This C-terminal sequence is embedded in the ER membrane immediately after translation, and the protein is then cleaved from the sequence and attached to a preformed GPI anchor (35,36,37).C-glycosylationC-mannosylation represents a different approach to glycosylation, because the reaction forms carbon–carbon bonds rather than carbon-nitrogen or carbon-oxygen bonds. C-mannosyltransferase (c-Mtf) links C1 of mannose to C2 of the indole ring of tryptophan (38). The enzyme recognizes the specific sequence Trp-X-X-Trp and transfers a mannose residue from dolichol-P-Man to the first Trp in the sequence (39,40,41).C-mannosylation has been detected in multiple cell lines (42) and rat liver microsomes (40). Specific proteins that areC-glycosylated include Trp2 in RNAse (43), the erythropoietin receptor and IL-12B (5). The biological function ofC-glycosylation is unknown, but current research focuses on the synthesis of C-glycosylated molecules by plants, insects and bacteria for drug discovery, because they are resistant to metabolic hydrolysis.PhosphoglycosylationThis type of post-translational modification is limited to parasites (e.g., Leishmania and Trypanosoma) and slime molds (e.g., Dictyostelium) and is characterized by the linking of glycans to serine or threonine via phosphodiester bonds (44). In some parasitic species, such as Leishmania, phosphoglycosylation is the most abundant post-translational modification (45,46) and is used to make proteophosphoglycans (PPGs), which are critical for protection against host complement (47) and promote parasite aggregation in the host (48). Similar to N-glycosylation, phosphoglycosylation occurs by transfer of a prefabricated phosphoglycan from a membrane-bound molecule via a phosphoglycosyltransferase (PTase), although the exact structure and enzyme varies by species (44).Post-glycosylation modificationsBesides multiple types of glycosylation occurring on the same protein, glycans can be further modified to increase the diversity of glycoproteins in a given proteome. These modifications include:∙Sulfation at Man and GlcNAc residues in the production of glycosaminoglycans (GAGs), which are components of proteoglycans in the extracellular matrix∙Acetylation of sialic acid to facilitate protein-protein interactions (49,50,51)∙Phosphorylation, such as with Man residues on precursor lysosomal proteins (mannose 6-phosphate) to ensure trafficking to lysosomes by binding to mannose 6-phosphate receptor (M6PR) in the GolgiRecommended reading1.Wormald MR et al. (2002) Conformational studies of oligosaccharides and glycopeptides: Complementarity of NMR,X-ray crystallography, and molecular modelling. Chem Rev 102:371–86.2.Rudd PM, Dwek RA (1997) Glycosylation: Heterogeneity and the 3D structure of proteins. Crit Rev Biochem MolBiol 32:1–100.3.Lechner J, Wieland F (1989) Structure and biosynthesis of prokaryotic glycoproteins. Annu Rev Biochem 58:173–94.4.Messner P (1997) Bacterial glycoproteins. Glycoconj J 14:3–11.5.Walsh C (2006) Posttranslational modification of proteins: Expanding nature's inventory. Englewood (CO): Roberts andCo. Publishers. xxi, p 490.6.Spiro RG (2002) Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications ofglycopeptide bonds. Glycobiology 12:43R–56R.7.Trombetta ES (2003) The contribution of N-glycans and their processing in the endoplasmic reticulum to glycoproteinbiosynthesis. Glycobiology 13:77R–91R.8.Burda P, Aebi M (1999) The dolichol pathway of N-linked glycosylation. Biochim Biophys Acta 1426:239–57.9.Dempski RE, Jr., Imperiali B (2002) Oligosaccharyl transferase: Gatekeeper to the secretory pathway. Curr Opin ChemBiol 6:844–50.10.Dean N (1999) Asparagine-linked glycosylation in the yeast Golgi. Biochim Biophys Acta 1426:309–22.11.Bause E, Legler G (1981) The role of the hydroxy amino acid in the triplet sequence Asn-Xaa-Thr(Ser) for theN-glycosylation step during glycoprotein biosynthesis. Biochem J 195:639–44.12.Roth J (2002) Protein N-glycosylation along the secretory pathway: Relationship to organelle topography and function,protein quality control, and cell interactions. Chem Rev 102:285–303.13.Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181–91.14.Ermonval M et al. (2001) N-glycan structure of a short-lived variant of ribophorin I expressed in the Madia214glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology 11:565–76.15.Frenkel Z et al. (2003) Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugarchain trimming to Man6-5GlcNAc2. J Biol Chem 278:34119–24.16.Hosokawa N et al. (2003) Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kongalpha1-antitrypsin by human ER mannosidase I. J Biol Chem 278:26287–94.17.Kitzmuller C et al. (2003) Processing of N-linked glycans during endoplasmic-reticulum-associated degradation of ashort-lived variant of ribophorin I. Biochem J 376:687–96.18.Lederkremer GZ, Glickman MH (2005) A window of opportunity: Timing protein degradation by trimming of sugars andubiquitins. Trends Biochem Sci 30:297–303.19.Gemmill TR, Trimble RB (1999) Overview of N- and O-linked oligosaccharide structures found in various yeastspecies. Biochim Biophys Acta 1426:227–37.20.Dell A, Morris HR (2001) Glycoprotein structure determination by mass spectrometry. Science 291:2351–6.。
2N-linked glycosylation(N-糖基化)
2.4 En bloc transfer of LLO to Asn
Although glycosylation is often referred to as a post-translational modification, transfer usually takes place as the nascent polypeptide emerges in the lumen of the ER and is a co-translational event. Q2 Glycosylation usually takes place on an unfolded polypeptide.
The structure has been conserved among all
eukaryotes.
焦磷酸 多萜醇 dolichol
Glc3Man9GlcNAc2
Q2
Pathway for generation of LLO for protein N-glycosylation
位置/过程/供体
• UDP-Glc, UDP-Gal • UDP-GlcNAc, UDP-GalNAc • UDP-GlcA, UDP-Xyl • GDP-Man, GDP-Fuc
• CMP-Sia (CMP-Neu5Ac)
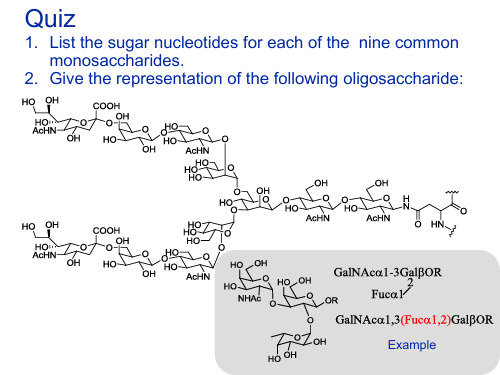
2. Give the representation of the following oligosaccharide:
2.3 Biosynthesis of the N-Glycan Precursor on the Cytosolic Leaflet of the Endoplasmic Reticulum (ER)
“Lipid-Linked Oligosaccharide” (LLO) 脂连接寡糖前体
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Journal of Chromatography A,1218 (2011) 8140–8149Contents lists available at SciVerse ScienceDirectJournal of ChromatographyAj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /c h r o maCharacterization of glycosylation sites for a recombinant IgG1monoclonal antibody and a CTLA4-Ig fusion protein by liquid chromatography–mass spectrometry peptide mappingJacob Bongers a ,∗,John Devincentis a ,Jinmei Fu a ,Peiqing Huang b ,David H.Kirkley a ,Kirk Leister a ,Peiran Liu b ,Richard Ludwig b ,Kathleen Rumney a ,Li Tao b ,Wei Wu b ,Reb J.Russell ba Department of Biologics Process and Product Development,Bristol-Myers Squibb Company,6000Thompson Road,East Syracuse,NY 13057,USA bP.O.Box 5400,Princeton,NJ 08543,USAa r t i c l ei n f oArticle history:Received 15April 2011Received in revised form 27August 2011Accepted 30August 2011Available online 3 September 2011Key words:Antibody CTLA-4Fc-fusionGlycosylation LC–MSPeptide mapa b s t r a c tLiquid chromatography mass spectrometry (LC–MS)peptide mapping can be a versatile technique for characterizing protein glycosylation sites without the need to remove the attached glycans as in conven-tional oligosaccharide mapping methods.In this way,both N-linked and O-linked sites of glycosylation can each be directly identified,characterized,and quantified by LC–MS as intact glycopeptides in a single experiment.LC–MS peptide mapping of the individual glycosylation sites avoids many of the limitations of preparing and analyzing an entire pool of released N-linked oligosaccharides from all sites mixed together.In this study,LC interfaced to a linear ion trap mass spectrometer (ESI-LIT-MS)were used to characterize the glycosylation of a recombinant IgG1monoclonal antibody and a CTLA4-Ig fusion pro-tein with multiple sites of N-and O-glycosylation.Samples were reduced,S-carboxyamidomethylated,and cleaved with either trypsin or endoproteinase Asp-N.Enhanced detection for minor IgG1glycoforms (∼0.1to 1.0mol%level)was obtained by LC–MS of the longer 32-residue Asp-N glycopeptide (4+pro-tonated ion)compared to the 9-residue tryptic glycopeptide (2+ion).LC–MS peptide mapping was run according to a general procedure:(1)Locate N-linked and/or O-linked sites of glycosylation by selected-ion-monitoring of carbohydrate oxonium fragment ions generated by ESI in-source collision-induced dissociation (CID),i.e.204,366,and 292Da marker ions for HexNAc,HexNAc-Hex,and NeuAc,respec-tively;(2)Characterize oligosaccharides at each site via MS and e selected ion currents (SIC)to estimate relative amounts of each glycoform;and (3)Measure the percentage of site-occupancy by searching for any corresponding nonglycosylated peptide.© 2011 Elsevier B.V. All rights reserved.1.IntroductionThe increasing importance of recombinant DNA produced mon-oclonal antibodies and other glycoproteins as therapeutic agents in recent years has placed increasing emphasis on the exten-sive characterization of their chemical structures.Glycosylation is a posttranslational structural modification of secreted proteins manufactured by mammalian cell culture that can have a sig-nificant impact on their solubility,stability,pharmacokinetics,potency,and biological activities [1–16].The N-linked glycosyla-tion at the carboxamide side-chain of a particular asparagine (Asn)∗Corresponding author at:Department of Biologics Process and Product Devel-opment,Bristol-Myers Squibb Company,6000Thompson Road,East Syracuse,NY 13057,United States.Tel.:+13154319370,fax:+13154324821.E-mail address:jacob.bongers@ (J.Bongers).residue or O-linked glycosylation at the alcohol side chain of a particular serine (Ser)or threonine (Thr)residue is only partly determined by the gene DNA sequence and corresponding pro-tein amino acid sequence.Attachment of a carbohydrate precursor to a particular Asn by the oligosaccharide transferase enzyme requires the consensus sequence,Asn-Xaa-Ser/Thr.This consensus sequence is a necessary condition but not a sufficient condition for N-glycosylation,i.e.the enzyme may or may not glycosylate a par-ticular Asn,depending on other factors.The amino acid sequence features and other factors regulating O-linked glycosylation of Ser and Thr residues are even less well understood and less predictable than for those for N-glycosylation.Moreover,after translation of the gene on the ribosome,each population of branched oligosaccharide structures or N-glycans occupying a particular site diverges by a complex series of modifications mediated by multiple glycosidase and glycotransferase enzymes.This enzymatic processing gives rise to structural microheterogeneity in the mature glycoprotein,i.e.a0021-9673/$–see front matter © 2011 Elsevier B.V. All rights reserved.doi:10.1016/j.chroma.2011.08.089J.Bongers et al./J.Chromatogr.A1218 (2011) 8140–81498141heterogeneous population of multiple oligosaccharide structures at each site of glycosylation.To further complicate matters,the occu-pancy of a particular N-linked or O-linked site may or may not be complete[16,17].Clearly,these site-specific attributes of partial occupancy and glycan microheterogeneity place high demands on analytical methods for the structural characterization of glycopro-teins.Oligosaccharide mapping,in which an entire pool of oligosac-charides is released from the protein by hydrazinolysis or,more commonly,enzymatic hydrolysis with peptidyl N-glycosidase (PNGase)or other glycosidase treatment(PNGase F,PNGase A, EndoH,etc.),is widely used for the characterization of pro-tein glycosylation[18–25].The various analytical methods for oligosaccharide mapping include high-pH anion-exchange chro-matography with pulsed amperometric detection(HPAEC-PAD) and a number of different chromatography or electrophoresis based separations,matrix-assisted laser desorption mass spec-trometry(MALDI-TOF MS)and various other MS techniques,and combinations of these including LC–MS and other hyphenated methods.In addition,the released pool of oligosaccharides can be analyzed directly or following derivatization with chromogenic,fluorogenic,or other reagents.A notable example is the sensitive analysis of low microgram amounts offluorescent derivatives of the released carbohydrates with2-aminobenzoic acid(2-AA)or2-aminobenzamide(2-AB)by either normal-phase or reversed-phase LC and LC–MS[19,20].These various established oligosaccharide mapping methods have their own relative advantages and disad-vantages as components of comprehensive analytical strategies for glycoproteins.One limitation shared by all the various oligosaccha-ride mapping methods is that the removal and pooling together of glycans results in the loss of site-specific information on oligosac-charide occupancy and structural microheterogeneity.Of course, relinquishing the site-specific microhetrogeneity information is less of a concern for IgG monoclonal antibodies[26–34]which pos-sess only a single site of glycosylation than it is for proteins with multiple sites of glycosylation.Peptide mapping is a general technique for confirming the amino acid sequences of proteins and characterizing their covalent chemical structures,including glycosylation and other posttrans-lational modifications[35–45].These peptide map methods can be used to good advantage as an alternative or in combination with oligosaccharide mapping methods when it is desirable to preserve site-specific information for proteins with multiple sites of glyco-sylation.Sample preparation involves treatment with trypsin or other proteolytic enzymes to cleave the protein into a reproducible set of peptide fragments amenable to analysis by LC–MS and other methods.Peptide mapping can be considered a general method-ology for identifying,characterizing,quantifying,and locating the sequence site for virtually any covalent modification to a protein, including glycosylation[35–45].Ideally,the appropriate protease or proteases are chosen so as to generate a unique well resolved peptide fragment for each glycosylation site.It is worth noting that the conventional peptide mapping and oligosaccharide map-ping approaches can be used in combination either in parallel or sequentially.For instance,it is generally feasible to chromatograph-ically isolate a pooled glycopeptide fraction specific to a particular N-linked or O-linked site and then further analyze this fraction by oligosaccharide mapping,MALDI-TOF MS,and other off-line techniques.In this way,high resolution oligosaccharide mapping data can be acquired for site-specific samples prepared and iso-lated by LC peptide mapping.Another useful experiment is tofirst deglycosylate the sample with PNGase F and then analyze the deg-lycosylated protein by peptide mapping in order to identify sites of N-glycosylation and to evaluate the percent occupancy for each N-linked site[45].Asn residues with attached N-glycans(occupied) are hydrolyzed to aspartic acid(Asp)by the PNGase F treatment and can be readily distinguished from nonglycosylated Asn residues (nonoccupied).In the current study,LC interfaced to a linear ion trap mass spectrometer(ESI-LIT-MS)was used for the site-specific peptide mapping characterization of a single site of N-linked glycosylation on a recombinant human IgG1monoclonal antibody and multiple sites of N-linked and O-linked glycosylation on a CTLA4-Ig fusion protein[46].This CTLA4-Ig construct consists of a modified IgG antibody Fc[47]fused to a high affinity variant of the extracel-lular domain of cytotoxic T lymphocyte antigen-4(CTLA4).CTLA4 (CD152)is a member of the immunoglobulin(Ig)superfamily and expressed as an extracellular receptor on the surface of activated T cells.CTLA4is similar to the T cell costimulatory protein,CD28, and both molecules bind to B7-1(CD80)and B7-2(CD86)ligands on antigen-presenting cells.CD28transmits a stimulatory signal to T-cells whereas CTLA4transmits a competitive inhibitory signal [47–51].2.Experimental2.1.Materials and reagentsRecombinant DNA derived human IgG1monoclonal antibody and CTLA4-Ig fusion protein were produced in CHO cell lines and purified in-house(Bristol-Myers Squibb,East Syracuse,NY).Dithio-threitol(DTT),and iodoacetamide were purchased from Sigma Aldrich(St.Louis,MO).All solvents including water and acetoni-trile were HPLC grade and were purchased from Burdick and Jackson(Muskegon,MI).Trifluoroacetic acid(TFA)and guanidine hydrochloride were from Pierce(Rockford,IL).Trypsin,modified sequencing grade,was purchased from Promega(Madison,WI), Catalog No.V5113.Endoproteinase Asp-N,sequencing grade,was from Roche(Indianapolis,IN),Catalog No.13874622.2.2.Sample preparationIgG1antibody was reduced,S-carboxyamidomethylated, digested with trypsin,and analyzed by RP-LC according to a stan-dard quality control method for tryptic mapping(Figs.2and3). Antibody samples(500g each)and a water blank were denatured by addition of8M guanidine hydrochloride buffer,pH8.0,to a final concentration of6M guanidine hydrochloride in a volume of 600L.Next,10L of175mM DTT was added and the mixture incubated at50◦C for20min to reduce the disulfide bonds.The denatured and reduced sample was then S-alkylated by the addition of10L of350mM iodoacetamide and incubation at 50◦C in the dark for20min.The alkylated protein was buffer-exchanged(desalted)into digestion buffer(2M urea,50mM Tris, 10mM CaCl2,10mM methionine,pH8.0)by gel-permeation on NAP-5gel columns(GE Healthcare,Catalog No.17-0853-02)by loading0.5mL sample and collecting1.0mL eluate.The buffer-exchanged samples(1mL)were digested with20g each of trypsin(1:50wt:wt enzyme to substrate)at38◦C for5h after which the digest was acidified with25L1.0M HCl to halt the enzyme activity.A separate100L portion of the above reduced and S-carboxyamidomethylated antibody was also digested with 6.6g of endoproteinase Asp-N(1:150wt:wt enzyme to substrate) at37◦C for18.5h and then halted with2.5L of1.0M HCl.CTLA4-Ig fusion protein was reduced,S-carboxyamidomethylated,and digested with trypsin exactly according to a standard quality control method for tryptic peptide mapping.Samples consisting of100L of20mg/mL CTLA4-Ig (2mg)were each mixed with550L of denaturing buffer(8M guanidine hydrochloride,50M Tris/HCl,pH8.0),reduced with 35L of200mM DTT for20min at37◦C,and S-alklylated with8142J.Bongers et al./J.Chromatogr.A 1218 (2011) 8140–8149Fig.1.The IgG1model (A)is that of Padlan [52],a composite of Protein Data Dank (PDB)X-ray crystal structures,2ig2and 1fc2[53]with theoretical hinge.The glycosylation consists of a pair of neutral complex-type biantennary N-glycans (A2G1F)enclosed in the central cavity of the Fc domain.The CTLA4-Ig fusion protein model (B)shows PDB crystal structures of human CTLA4homodimer (PDB 1i85)[70]and human Fc (1fc2)[53].Models of four sialylated A2S2G2F complex-type biantennary N-glycans were added to the CTLA4arms using GlyProt [55]with arbitrary orientations and bond angles.N-Glycans are highly flexible and usually exist in a multitude of rapidly converting conformations [54,55].Complex type biantennary N-glycans are named using the abbreviation “AaSsGgF ”where “a ”is the number of antennae linked to the trimannosyl core,“s ”the number of sialic acid residues (NeuAc),“g ”the number of galactose residues (Gal),and “F ”indicates core-fucosylation (Fuc).O-linked glycans in the hinge-region of CTLA4-Ig are not shown in these models.The CTLA4-Ig tryptic peptides containing glycosylation sites are indicated,i.e.T5,T7,T9,and T14.38.5L 400mM iodoacetamide for 20min at room temperature in the dark.Removal of reagents and buffer-exchange into digestion buffer (50mM Tris,10mM CaCl 2,pH 7.6)was performed on NAP-5Sephadex G-25gel columns (GE Healthcare Catalog No.17-0853-01)by loading 0.5mL sample and collecting 1.0mL eluate.Digestions were performed with 50L of 0.5mg/mL trypsin per 1mL of 1.4mg/mL substrate at 37◦C for 120min and halted with 20L 1.0M HCl.2.3.LC–MS peptide mappingLC–MS was run on a Waters 2695Alliance LC or a Waters Acquity UPLC (Milford,MA)interfaced with a ThermoFinnigan LTQ-XL lin-ear ion-trap mass spectrometer (San Jose,CA)with electrospray ionization acquiring sequential MS full-scans with data-dependent acquisition of MSMS and high resolution zoom-scans for abun-dant ions.The LC–MS data were acquired in positive ion mode in a mass to charge ratio (m /z )range of 200–2000,capillary tem-perature of 275◦C,and a source voltage of 5000V.Tandem mass spectra (MSMS)data were acquired using 15–35%normalized col-lision energy,0.25activation Q and 100ms activation time.LC–MS runs for the CTLA4-Ig fusion protein were also made using in-source CID (SID =96V,m /z 140–380Da scans)for monitoring of carbohy-drate oxonium fragment ions [35–38].IgG1antibody tryptic digests were analyzed by C18reversed-phase HPLC–MS run on a Waters 2695Alliance LC equipped with a 2487UV detector,set at 214and 280nm,and interfaced to the above LTQ-XL linear ion-trap mass spectrometer.Injections of 100L each were made onto a Grace Vydac 218TP52column (2.1mm ×250mm),40◦C,at 0.2mL/min,with eluents (A)0.10%TFA/H 2O and (B)0.09%TFA/95%CH 3CN,and gradient:2%B isocratic for 4min,2–16%B from 5to 30min,16–19.5%B from 30to 60min,19.5–23%B from 60to 84min,23–29%B from 84to 102min,29–30%B from 102to 118min,and 30–42%B from 118to 143min,42–100%B from 143to 145min,100%B isocratic from 145to 146.1min,100–2%B from 146.1to 146.2min,and 100%B isocratic from 146.2to 166min.The flow rate was 0.2mL/min from 0to 145min,0.2to 0.3mL/min from 145to 145.1min,and 0.3mL/min from 145.1to 166min.A rotary switching-valve was used to divert the 2M urea in the digest away from the ESI source for the first 5min of the run.IgG1antibody trypsin and endoproteinase Asp-N digests were analyzed by C18reversed-phase UP LC–MS run on Waters Acquity UPLC instrument equipped with a photodiode array UV detec-tor,set at 214and 280nm,and interfaced to the above LTQ-XL linear ion-trap mass spectrometer.Injections of 50L each were made onto a Waters BEH300C18column (Part No.186003687),1.7m,2.1mm ×150mm,45◦C,at 0.2mL/min,with eluents (A)0.02%TFA/H 2O and (B)0.02%TFA/95%CH 3CN.The tryptic map-ping LC gradient was:1%B isocratic for 6min,1–35%B from 6to 80min,35–100%B from 80to 92min,100%B isocratic from 92to 95min,100–1%B from 95to 96min,and 1%B isocratic from 96to 110min,with a flow rate of 0.2mL/min throughout.The endopro-teinase Asp-N mapping LC gradient was:1%B isocratic for 6min,1–50%B from 6to 80min,50–100%B from 80to 92min,100%B isocratic from 92to 95min,100–1%B from 95to 96min,and 1%B isocratic from 96to 110min,with a flow rate of 0.2mL/min throughout.A rotary switching-valve was used to divert the 2M urea in the digest away from the ESI source for the first 3min of the runs.CTLA4-Ig tryptic digests were analyzed by C18reversed-phase HPLC–MS peptide mapping run on a Waters 2695Alliance LC equipped with a 2487UV detector,set at 215and 280nm,and interfaced to the above LTQ-XL linear ion-trap mass spectrom-eter.Injections of 25L were run on a Grace Vydac 218MS52column (2.1mm ×250mm),at 0.2mL/min,45◦C,with eluents (A)0.02%TFA/H2O and (B)0.02%TFA/95%CH 3CN,and gradient:1.7%B isocratic for 4min,1.7–30%B from 4to 64min,30–50%B from 64to 80min,and 80–100%B from 80to 90min,100%B iso-cratic from 90to 94min,100–1.7%B from 94to 95min,and 1.7%B isocratic from 95to 110min,with a flow rate of 0.2mL/min throughout.J.Bongers et al./J.Chromatogr.A 1218 (2011) 8140–81498143Fig.2.Summary of ESI linear ion-trap LC–MS tryptic peptide mapping results for a recombinant IgG1monoclonal antibody (CHO cell culture derived)showing (A)full-scale 214nm UV and MS base-peak chromatograms;(B)selected ion-current (SIC)chromatograms for the doubly protonated ions of the three main Fc domain tryptic glycopeptides (A2G0F,A2G1F,and A2G2F)and nonglycosylated heavy chain (NGHC);(C)mass spectrum summed across the 24.0–25.2min glycopeptide region with main peak assignments,(D)expanded view of 280nm UV trace of glycopeptides region showing peak area quantitation of mole percent NGHC,and (E)MS and MSMS spectra of the NGHC tryptic peptide.3.Results and discussion3.1.General procedures for LC–MS peptide mapping of glycosylation sitesComplete sequence coverage for the antibody was obtained by overlapping trypsin and endoproteinase Asp-N LC–MS pep-tide maps.The respective peptides can be readily identified by their observed monoisotopic molecular weights determined via on-line high resolution mass spectrometry and their sequences confirmed by tandem mass spectra of their collision induced disso-ciation (CID)fragmentation patterns.Samples were fully reduced with DTT,S-carboxyamidomethylated (S-alkylation of all cysteine residues),and cleaved with either trypsin or endoproteinase Asp-N and then analyzed by LC–MS according to the following general scheme:LC–MS peptide mapping was run according to a general procedure:(1)Locate N-linked and/or O-linked sites of glycosy-lation by single-ion-monitoring (SIM)of carbohydrate oxonium fragment ions generated by ESI in-source collision-induced disso-ciation (CID),i.e.204,366,and 292Da marker ions for HexNAc,HexNAc-Hex,and NeuAc,respectively [35–38];(2)Characterize oligosaccharides at each site via MS and e selected ion currents (SIC)to estimate relative amounts of each glycoform;and (3)Measure the percentage of site-occupancy by searching for any corresponding nonglycosylated peptide.3.2.LC–MS tryptic peptide mapping of an IgG1monoclonal antibodyIgG antibodies usually have a single N-linked glycosylation site in the heavy chain sequence yielding a pair of N-linked oligosac-charides enclosed in a central cavity in between the two C H 2constant-region Ig subunits of the Fc domain of the mature homod-imeric antibody structure as shown in Fig.1[4–6,52–55].The Fc N-glycans are predominantly neutral (nonsialylated)bianten-nary oligosaccharides with zero,one,or two terminal galactose units (A2G0F,A2G1F,and A2G2F).Since the Fc N-glycans are sand-wiched in between the two C H 2constant-region Ig subunits,these oligosaccharides obviously play an integral role in shaping the three dimensional structure of the Fc and modulating effector functions such as binding to the high affinity Fc ␥R receptor and mediating antibody dependent cellular cytotoxicity (ADCC)and complement fixation [4–7].For example,glycoengineering to produce afucosyl Fc N-glycans has been used to make monoclonal antibody thera-peutics with greatly enhanced ADCC [56,57].Conversely,it has been shown that the Fc N-glycosylation of IgG antibody drugs has essen-tially no effect on their pharmacokinetics and serum half-lives [11].The low microheterogeneity and lack of effect on PK clearance are reasonable considering the peculiar disposition of these N-glycans which are almost entirely enclosed and inaccessible inside the Fc domain.8144J.Bongers et al./J.Chromatogr.A 1218 (2011) 8140–8149parison of ESI linear ion-trap LC–MS glycopeptide mapping results for a recombinant IgG1monoclonal antibody obtained with (A)trypsin versus (B)endoproteinase Asp-N using the same digest concentrations,injection volumes,and MS instrument conditions.Selected ion currents are shown for the three major Fc N-glycans (A2G0F,A2G1F,and A2G2F),two minor sialylated glycoforms (A2S1G2F and A2S2G2F),and a minor oligomannose type glycoform (M5).A more comprehensive list of N-glycans observed by LC–MS Asp-N peptide mapping is given in Table 1.Peptide mapping is routinely used for confirming expres-sion of the correct amino acid sequence and characterizing the covalent chemical structures of antibodies and other recombi-nant proteins [40–43].We have found that complete amino acid sequence coverage for recombinant antibodies can usually be obtained by overlapping trypsin and endoproteinase Asp-N pep-tide maps.The respective peptides can be readily identified by their observed monoisotopic molecular weights determined via on-line high resolution mass spectrometry and their sequences confirmed by tandem mass spectra of their collision induced dis-sociation (CID)fragmentation patterns.The other well known IgG1posttranslational modifications,characteristic of IgG1antibodies,were observed for the heavy chain including N-glycosylation at the conserved Fc region Asn-Xaa-Ser/Thr consensus site,cyclization of N-terminal glutamine to pyroglutamate,and proteolytic removal of the C-terminal lysine residue.The LC–MS tryptic peptide map of the reduced/S-alkylated IgG1mAb in Fig.2was obtained according to a standard quality con-trol testing assay and the flow from the UV detector directed into a linear ion-trap mass spectrometer.In addition to provid-ing information on the amino acid sequence and covalent chemical structure of the protein,this LC–MS peptide map contains a con-siderable amount of information on the glycosylation.The three main N-glycans (A2G0F,A2G1F,and A2G2F)at the single N-linkedglycosylation site are typical for the Fc domain of an IgG1anti-body.Minor amounts of the sialylated glycans,A2S1G2F (∼0.4%)and A2S2G2F (∼0.3%)were also found as well as an M5oligoman-nose structure (∼2.0%),and nonglycosylated heavy chain (NGHC)of ∼1.0%,i.e.,an occupancy of ∼99.0%at this asparagine.These minor amounts of nonglycosylation and the immature oligomannose gly-coforms are of interest as possible indicators of cell viability during upstream cell culture processes [2].LC–MS peptide mapping of the antibody with trypsin and endo-proteinase Asp-N are compared in Fig.3and very similar amounts were obtained for this set of glycoforms.The Asp-N glycopeptide (32residues)has multiple basic residues for ionization (protona-tion)and thus tends to give higher signal-to-noise for the selected ion currents (4+ions).Enhanced detection for minor IgG1glyco-forms (∼0.1to 1.0mol%level)was obtained by LC–MS of the longer 32-residue endoproteinase Asp-N glycopeptide (4+protonated ion)compared to the 9-residue tryptic glycopeptide (2+ion).Oppo-site effects on reversed-phase retention times were observed for presence of an additional galactose (hydrophilic)versus sialic acid (hydrophobic)for both the tryptic glycopeptides and the Asp-N glycopeptides.In addition to the mass spectral peak assignments shown in Figs.2and 3,the various glycoforms identified by LC–MS Asp-N mapping are also listed in Table 1.The relative amounts of the glycopeptides as estimated by LC–MS selected ion currentJ.Bongers et al./J.Chromatogr.A 1218 (2011) 8140–81498145Fig.4.LC–MS tryptic map of a CHO cell culture derived recombinant CTLA4-Ig fusion protein showing 215nm UV chromatogram,MS base-peak chromatogram,and selected ion-current chromatograms (SIC’s)for the carbohydrate oxonium fragment marker-ions,N-acetyl hexoseamine (HexNAc,m /z =204Da),hexose N-acetyl hexoseamine disaccharide (HexNAc-Hex,m /z =366Da),and sialic acid (NeuAc,m /z =292Da)generated by in-source collision-induced dissociation (CID).The boxed-in regions indicate glycopeptides corresponding to the three specific N-linked sites and one O-linked site of glycosylation on CTLA4-Ig as shown in Fig.1.Table 1Glycopeptides found for recombinant IgG1monoclonal antibody by LC–MS endoproteinase Asp-N mapping.N-glycan a FormulaFormulaMW calc b (Da)m /z calc 4+(Da)m /z obs 4+(Da)Ret time (min)SIC rel heightRel (%)aglyco C161H258N48O52–3697.9019925.4828925.572.80.8nd A1G0C203H327N51O82C42H69N3O304793.29851199.33191199.771.60.950.20A1G0F C209H337N51O86C48H79N3O344939.35651235.84641236.271.4 4.010.86A2G0C211H340N52O87C50H82N4O354996.37781250.10171250.171.4 5.23 1.13A2G0F C217H350N52O91C56H92N4O395142.43571286.61621286.971.232269.29A2G1F C223H360N52O96C62H102N4O445304.48851327.12941327.571.110221.95A2G2F C229H370N52O101C68H112N4O495467.54141367.89261367.871.011.2 2.41A2S1G1F C234H377N53O104C73H119N5O525596.58401400.15331399.971.7 2.230.48A2S1G2F C240H387N53O109C79H129N5O575758.63681440.66651440.471.6 1.570.34A2S2G2F C251H404N54O117C90H146N6O656049.73191513.44031513.272.1 1.230.26M5C207H334N50O87C46H76N2O354914.32471229.58851229.871.38.81 1.90M6C213H344N50O92C52H86N2O405076.37751270.10171270.071.2 1.530.33M7C219H354N50O97C58H96N2O455238.43041310.61491310.771.2 2.880.62M8C225H364N50O102C64H106N2O505400.48321351.12811351.071.20.820.18M9C231H374N50O107C70H116N2O555563.53601391.89131391.871.00.230.05Sum464.69100aComplex type biantennary N-glycans are named using the abbreviation “AaSsGgFB ”where “a ”is the number of antennae linked to the trimannosyl core,“s ”the number of sialic acid residues (NeuAc),“g ”the number of galactose residues (Gal),and “F ”indicates core-fucosylation (Fuc).Oligomannose type N-glycans are named “Mm ”where m is the number of mannose residues.The amino acid sequence of the Asp-N peptide is DGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQ.bCalculated masses are for the most abundant isotope.peak heights in the Asp-N map were in close agreement with rela-tive amounts found for the corresponding glycoforms in the tryptic map.However,the smaller amounts of minor glycoforms in the ∼0.1to 1.0%range that were below the limits of detection for thetryptic map were readily identified by Asp-N mapping.The rela-tive amounts of the various N-glycans estimated by LC–MS Asp-N peptide mapping selected ion currents in Table 1compare rea-sonably closely with those obtained independently by analyses of8146J.Bongers et al./J.Chromatogr.A 1218 (2011) 8140–8149Fig.5.Mass spectra summed across the four boxed-in regions of the LC–MS tryptic map for the recombinant CTLA4-Ig fusion protein in Fig.4corresponding to the three N-linked sites and one O-linked site of glycosylation on the CTLA4-Ig fusion protein molecule in Fig.1.Major peaks in the spectra are labeled with oligosaccharide structure assignments.Amino acid sequences are shown for the glycosylated tryptic peptides T5and T7(N-linked,CTLA4domain),T9(O-linked,hinge region),and T14(N-linked Fc domain)which correspond to residues 39-83,94-128,133-158,and 203-211of the CTLA4-Ig,respectively.oligosaccharides released from this antibody and similar recom-binant DNA derived monoclonal antibodies via treatment with PNGase F.3.3.LC–MS tryptic peptide mapping of a CTLA4-Ig fusion proteinThe CTLA4-Ig fusion protein shown in Fig.1consists of the extracellular portion of a disulfide-linked homodimeric glycopro-tein receptor,human cytotoxic T-lymphocyte antigen-4(CTLA4)[46–51],fused to a modified human Fc domain [47].CTLA4is a member of the CD28family of receptor/ligands involved in T-cell costimulation [48–51].CTLA4-Ig has two N-linked sites per CTLA4domain,in addition to the above Fc site,plus O-linked glycosyla-tion in the hinge region.The CTLA4domain is an immunoglobulin V (IgV)type of fold and the N-glycosylation sites at Asn 76and Asn 108are contained within tryptic peptides,T5and T7,respectively.Asn 76is located in beta-strand E of the ABED beta-sheet and the Asn 108N-glycosylation site is located in the C-terminal G strand of the CC’FG beta-sheet on the opposite face of the IgV sandwich.The “MYPPPYY loop”connecting beta-strands E and F (residues 97–103)is the key receptor-binding interface for the interaction of the CTLA4homodimer with the B7-1and B7-2ligands on antigen presenting cells.Note that,unlike the atypical case of the N-glycans constrained inside the Fc domain cavity,the N-glycans attachedto the CTLA4domain represent the more usual case of oligosac-charides projecting out from the surface of the protein into the surrounding medium (Fig.1).The four glycopeptide regions in the tryptic map can be visu-alized via single-ion monitoring of in-source CID carbohydrate marker ions in Fig.4.For the CTLA4-Ig fusion protein,the sim-plest of the three N-linked sites is that on the Fc domain T14peptide containing the three neutral biantennary complex-type core-fucosylated N-glycans (A2G0F,A2G1F,and A2G2F)which are typical for virtually all recombinant IgG1antibodies produced by mammalian cell culture.In contrast to the rather simple population of Fc N-glycans,the two N-glycosylation sites on the CTLA4domain show highly diverse microheterogeneous populations of sialylated and neutral forms of bi-,tri-,and tetra-antennary complex-type core-fucosylated glycans with variable numbers of terminal galac-tose (Fig.5).The T7N-glycans contain significantly higher levels of galactose and sialic acid,overall,versus the T5site.The T7glycopeptides are partially resolved into distinct regions on this RP-LC system (Fig.6).The first envelope within the range of approximately 67.8–68.8min consists primarily of the T7glycopep-tide with monosialylated biantennary N-glycans,A2S1G2F and A2S1G1F.The next region from approximately 68.0–70.0min con-tains T7with the disialyated biantennary N-glycan,A2S2G2F,and lesser amounts of T7with the disialylated and trisialylated species,。