AutoDock Vina与MGLTools的安装
分子对接

AutoDock和AutoDock Tools 使用教程一、分子对接简介及软件介绍1.分子对接理论基础所谓分子对接就是两个或多个分子之间通过几何匹配和能量匹配而相互识别的过程。
分子对接在酶学研究以及药物设计中具有十分重要的意义。
在酶激活剂、酶抑制剂与酶相互作用以及药物分子产生药理反应的过程中,小分子(通常意义上的Ligand)与靶酶(通常意义上的Receptor)相互互结合,首先就需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,得到一个稳定的复合物构象。
通过分子对接确定复合物中两个分子正确的相对位置和取向,研究两个分子的构象,特别是底物构象在形成复合物过程中的变化,是确定酶激活剂、抑制剂作用机制以及药物作用机制,设计新药的基础。
分子对接计算是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补化学环境互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间最佳的结合模式。
分子对接最初思想起源于Fisher E.的“锁和钥匙模型”(图),认为“锁”和“钥匙”的相识别的首要条件是他们在空间形状上要互相匹配。
然而,配体和受体分子之间的识别要比“锁和钥匙”模型复杂的多。
首先,配体和受体分子的构象是变化的,而不是刚性的,配体和受体在对接过程中互相适应对方,从而达到更完美的匹配。
其次,分子对接不但要满足空间形状的匹配,还要满足能量的匹配。
配体和受体之间的通过底物分子与靶酶分子能否结合以及结合的强度最终是由形成此复合物过程的结合自由能变化ΔG bind所决定的。
互补性(complementarity)和预组坦织(pre-organization)是决定分子对接过程的两个重要原则,前者决定识别过程的选择性,而后者决定识别过理的结合能力。
互补性包括空间结构的互补性和电学性质的互补性。
1958年Koshland提出了分子识别过程中的诱导契合(induced fit)概念,指出配体与受体相互结合时,受体将采取一个能同底物达到最佳结合的构象(图1)。
使用autodock4在linux下进行批量对接(命令行)

使用Autodock4在Linux下进行批量对接(命令行)1.安装1.从官方网站下载MGLTools 和Autodock4压缩包,按说明解压安装。
2.准备MGLTools应用程序:安装完成后,将./mgltools_i86Linux**/MGLToolsPckgs/AutoDockTools/Utilities24目录下的所有应用程序拷贝到个人工作目录。
再将./mgltools_i86Linux**/bin目录下的pythonsh应用程序拷贝到工作目录。
3.将autodock4压缩包中的Autogrid4和Autodock4也放到工作目录。
2.使用1.下载对接的配体和受体文件(*.pdb格式)。
进入工作目录。
2.将PDB格式文件转换成Autodock识别的PDBQT文件:./pythonsh prepare_ligand4.py –l ligand.pdb./pythonsh prepare_receptor4.py –r receptor.pdb –A hydrogens –U waters3.准备autogrid4参数文件:./pythonsh prepare_gpf4.py –l ligand.pdbqt –r receptor.pdbqt4.准备autodock4参数文件:./pythonsh prepare_dpf4.py –l ligand.pdbqt –r receptor.pdbqt5.运行Autogrid4:./autogrid4 –p receptor.gpf6.运行Autodock4:./autodock4 –p ligand_receptor.dpf3.结果分析Autodock 对接结果记录在*.dlg文件中,用Estimated free energy of binding值评价对接结果,值越低表示越容易结合。
从*.dlg文件中提取Estimated free energy of binding值:egrep ‘DOCKD:USER Run|Cluster|Rank|RMSD from reference structure|USER Estimated’ *.dlg后记:本人在使用以上方法进行批量对接过程中,偶尔有报错情况,但大部分对接能跑出结果。
分子对接软件AutoDockVina在太湖之光操作系统上的移植-最新资料

分子对接软件AutoDockVina在太湖之光操作系统上的移植1 “神威?太湖之光”计算系统“神威?太湖之光”计算系统是国家“863计划”重大专项研究成果,是我国第一台全部采用国产处理器构建的超级计算机,由国家并行计算机工程技术研究中心研制[1]。
在2016年6月20日世界TOP500超级计算机排名中,“神威?太湖之光”系统峰值运算性能(125.436PFlops)、持续运算性能(93.015PFlops)、性能功耗比(6.05GFlops/W)三项关键指标均位居世界第一。
“神威?太湖之光”计算系统共包含了40 960个“申威26010”众核处理器。
“申威26010”是由国家“核高基”重大专项支持的我国第一款自主研发的众核处理器,由国家高性能集成电路设计中心研制,性能国际领先,并成功量产,打破了美国对我国的技术封锁。
处理器基于申威(SW-64)指令集,采用片上融合异构众核架构和FCBGA3832封装,单个处理器包含了260个运算核心。
“神威?太湖之光”具有世界领先水平的超大规模系统低功耗控制技术和高密度组装,比目前世界排名第二的系统节能60%以上,单机仓组装密度居世界第一。
同时,基于“神威?太湖之光”系统自主研发软件,建立了基于申威CPU的高性能计算软件生态链。
目前“神威?太湖之光”计算系统开始应用于四个关键领域:先进制造业应用(CFD、CAE)、地球系统建模和天气预报、生物医药领域的计算、大数据分析。
2 分子对接软件AutoDockVinaAutoDock是一款开源的分子模拟软件,最主要应用于执行配体―蛋白分子对接[2~3]。
它由Scripps研究所的Olson实验室开发与维护,目前最新版本为AutoDock 4.2。
AutoDockVina 也是一款由MGL实验室开发的分子对接软件。
与AutoDock 4.0相比,AutoDockVina提高了结合模式预测的平均准确度,通过使用更简单的打分函数加快了搜索速度,并且在处理约20个可旋转键的体系时仍然能提供重现性较好的对接结果。
HCD的Autodock教程

之前自己学着用Autodock,期间各种问题,各种麻烦,各种请教学长,花了整整一个学期才第一次成功的跑完了教程,以后就变得很顺手了。
Autodock最难得是软件的安装和理解软件作者的想法,之后就会很顺手。
我介绍的只是其中一种确定可以成功安装运行的方法,用别的版本的软件和操作系统,会有别的方法,但是大同小异,大家自己看着办吧。
我没有用“科学的语言”来描述这一切操作,只是为了更容易理解。
考究们自己知道就行。
勿喷勿拍。
∙主要软件:算法核心autodock4.2.3_win32,∙图形化界面mgltools_win32_1.5.6rc3,∙编译器python-2.5.2,∙辅助软件(可选):序列剪辑等pymol-1.4.1.win32,编译器python-2.7,制作虚拟小分子chemoffice∙autodock, mgltools, pymol在各自官网上下载,不要用各种软件站的东西。
autodock, mgltools可以用最新版本,但是python版本不能改变(除非最新版的软件要求编译器版本更新)步骤/方法1.软件安装∙我的系统信息:i5处理器,win7,64位专业版(32位无法运行,重装一个系统,一个小时都不用的,而且64位基本完全兼容32的程序,日常用的时候性能也更好),4G内存(2G也能跑),笔记本。
∙注意事项:软件安装前的存放路径最好为全英文,安装路径必须为全英文,最好为默认路径(尤其是编译器必须安在默认路径C盘根目录下,不然以后你会后悔的,程序跑的时候老是报错啊)。
软件安装顺序不能改变。
安装期间杀毒软件、安全软件都关了。
∙安装顺序:1.python-2.5.2(默认路径C:\Python25)2.mgltools_win32_1.5.6rc3(默认路径C:\Program Files(x86)\MGLTools-1.5.6rc3)3.解压autodock4.2.3_win32(我知道你的疑问,没错,是解压,把exe文件解压),提取autodock和autogrid。
AutoDock

AutoDock和AutoDock Tools 使用教程一、分子对接简介及软件介绍1.分子对接理论基础所谓分子对接就是两个或多个分子之间通过几何匹配和能量匹配而相互识别的过程。
分子对接在酶学研究以及药物设计中具有十分重要的意义。
在酶激活剂、酶抑制剂与酶相互作用以及药物分子产生药理反应的过程中,小分子(通常意义上的Ligand)与靶酶(通常意义上的Receptor)相互互结合,首先就需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,得到一个稳定的复合物构象。
通过分子对接确定复合物中两个分子正确的相对位置和取向,研究两个分子的构象,特别是底物构象在形成复合物过程中的变化,是确定酶激活剂、抑制剂作用机制以及药物作用机制,设计新药的基础。
分子对接计算是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补化学环境互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间最佳的结合模式。
分子对接最初思想起源于Fisher E.的“锁和钥匙模型”(图),认为“锁”和“钥匙”的相识别的首要条件是他们在空间形状上要互相匹配。
然而,配体和受体分子之间的识别要比“锁和钥匙”模型复杂的多。
首先,配体和受体分子的构象是变化的,而不是刚性的,配体和受体在对接过程中互相适应对方,从而达到更完美的匹配。
其次,分子对接不但要满足空间形状的匹配,还要满足能量的匹配。
配体和受体之间的通过底物分子与靶酶分子能否结合以及结合的强度最终是由形成此复合物过程的结合自由能变化ΔG bind所决定的。
互补性(complementarity)和预组坦织(pre-organization)是决定分子对接过程的两个重要原则,前者决定识别过程的选择性,而后者决定识别过理的结合能力。
互补性包括空间结构的互补性和电学性质的互补性。
1958年Koshland提出了分子识别过程中的诱导契合(induced fit)概念,指出配体与受体相互结合时,受体将采取一个能同底物达到最佳结合的构象(图1)。
autodock命令行的用法

autodock命令行的用法一、概述autodock是一款功能强大的自动化 docking 工具,用于自动化地进行蛋白质对接和药物分子对接等生物化学研究。
在autodock中,命令行是一种常用的操作方式,可以通过命令行来执行各种操作,如安装软件、运行 docking 计算、分析结果等。
本篇文章将详细介绍autodock命令行的用法,帮助用户更好地使用autodock进行生物化学研究。
二、安装和配置1. 安装autodock软件:首先需要在计算机上安装autodock软件,可以从官方网站下载相应的安装包,按照提示进行安装。
2. 配置环境变量:安装完成后,需要配置相应的环境变量,以便在命令行中能够正确地找到autodock软件的位置。
三、常用命令1. docking计算:使用autodock进行 docking 计算时,需要指定目标蛋白和配体分子的结构文件、 docking 计算的参数文件等信息。
常用的命令如下:a. dock -i input.pdb -o output.txt -p parameters.dat -r reference.pdb -n num_iterations其中,-i参数指定输入文件路径,-o参数指定输出文件路径,-p 参数指定参数文件路径,-r参数指定参考文件路径,-n参数指定最大迭代次数。
b. dock -l:用于列出当前目录下的所有 docking 计算文件,包括输入文件、输出文件和参数文件等。
c. dock -h:用于查看 autodock 命令行的帮助文档,了解更多关于 docking 计算的用法和参数说明。
2. 分析结果:docking计算完成后,可以使用命令行来分析结果。
常用的命令如下:a. analyze -i input.txt -o output.txt -p parameters.dat其中,-i参数指定输入文件路径,-o参数指定输出文件路径,-p 参数指定参数文件路径。
autodock_tutorial_new说明

Autodock Tutorial1Autodock的简介及安装Autodock是一个免费的计算机辅助药物设计软件,它包含autodock、autogrid及一个可视化的操作界面autodock tools(MGL Tools)三个部分。
Autodock及其相关软件可以在网站/上找到相应的下载链接。
由于autodock最初是基于Linux为操作平台开发的软件,若要在Windows系统中安装则会出现一定的系统兼容性问题,尽管新开发的版本安装说明表明了可以直接安装,但是在实际操作中我们发现,新版本还是需要先安装Cygwin及Python 2.5才能正确的安装autodock相关组件。
其中Cygwin可以在Windows系统里创建一个模拟的Linux 环境,它需要在网络上安装。
安装好后将文件名为的文件从C:\cygwin\bin文件夹里的cygwin1.dll拷贝到C:\Windows\System32文件夹里。
安装好Cygwin及Python 2.5后就可以安装autodock组件了,其中autodock及autogrid只需要解压后将之拷贝到C:\Windows\System32文件夹下及C:\cygwin\bin文件夹下即可。
MGL Tools和Windows系统下其它文件的安装方式一样,双击图标即可实现安装,需要注意的是尽量使MGL Tools保存路径名为英文字母以减少不必要的麻烦。
除了autodock系列组件以外,还推荐安装Chemoffice、Chimera或Accelrys作为辅助软件。
2配体文件的准备将需要计算的小分子在Chemoffice中画好后粘贴至Chem3D中,计算最低能量后另存为.pdb文件,关闭Chemoffice。
打开Chimera,打开pdb文件,运行Tools→Structure Editing→AddH进行加氢。
或在Accelrys中选中分子后运行Chemistry→Hydrogens→Add polar进行极性氢的添加,在这里只添加极性氢的原因是autodock软件在计算时会自动去掉非极性氢。
autok-vina详解

生物计算事业部 Department of Computational Biology
一、软件安装
(2)环境变量的设置
将autodock的路径添加到环境变量中,64位系统 默认的安装路径为:C:\Program Files(x86)\The Scripps Research Institute\Autodock\4.2.6, 并在dos中测试程序是否能够正常运行
生物计算事业部 Department of Computational Biology
三、对接准备及操作
(2)准备受体分子
在“File->Read Molecule”中加载1hsg.pdb文件
生物计算事业部 Department of Computational Biology
三、对接准备及操作
生物计算事业部 Department of Computational Biology
三、对接准备及操作
(4)准备配体分子
在"Ligand->Torsion Tree->Choose Torsions"中可 以设置配体中可以旋转的键。同时指定在活动键 时需要移动的最少的原子数目为6, 在"Ligand>Torsion Tree->Set Number of Active Torsions“ 中设置。
二、Vina安装
安装软件选择autodock_vina_1_1_2_win32.exe, 安装过程中软件初始位置及安装路径均不能含有中文
注意安装路径!
生物计算事业部 Department of Computational Biology
二、Vina安装
环境变量的设置:
在“计算机->属性->高级系统设置”中打开“环境 变量”设置:
autodock-vina.

生物计算事业部 Department of Computational Biology
一、AutoDock-Vina介绍
Autodock Vina,它是继Scripps实验室的Autodock4对接 程序之后由Oleg Trott等人推出的使用新的打分函数、预测准 确率更高并支持并行化的更高效易用的对接程序。它的使用 方法和Autodock 4类似,只是少了调用AutoGrid程序(原 Autodock 4 中需要用到)这一步。
二、ADT介绍
生物计算事业部 Department of Computational Biology
三、对接准备及操作
本练习以HIV蛋白1HSG.pdb及其生物活性抑制剂 ind.pdb作为实例进行分子对接演示: (1)将默认的工作路径调整到当前操作的文件夹下 (便于程序的运行)在"File->Preferences->set" 中设置
生物计算事业部 Department of Computational Biology
一、软件安装
(3)mgltools的安装
生物计算事业部 Department of Computational Biology
一、软件安装
(3)mgltools的安装
安装成功后会出现下面的界面
生物计算事业部 Department of Computational Biology
二、Vina安装
安装软件选择autodock_vina_1_1_2_win32.exe, 安装过程中软件初始位置及安装路径均不epartment of Computational Biology
二、Vina安装
环境变量的设置:
在“计算机->属性->高级系统设置”中打开“环境 变量”设置:
autodock-vina

生物计算事业部 Department of Computational Biology
五、分子对接
(2) 运行autodock
确认参数文件及其他文件的文件路径果分析 Department of Computational Biology
生物计算事业部 Department of Computational Biology
一、软件安装
(3)mgltools的安装
生物计算事业部 Department of Computational Biology
一、软件安装
(3)mgltools的安装
安装成功后会出现下面的界面
生物计算事业部
Department of Computational Biology
生物计算事业部 Department of Computational Biology
一、软件安装
(2)环境变量的设置
将autodock的路径添加到环境变量中,64位系统 默认的安装路径为:C:\Program Files(x86)\The Scripps Research Institute\Autodock\4.2.6, 并在dos中测试程序是否能够正常运行
在ADT菜单栏中选择"ligand->input->open",加载配 体文件ind.pdb,弹出的警告点击确定即可。
生物计算事业部 Department of Computational Biology
三、对接准备及操作
(4)准备配体分子
在"Ligand->Torsion Tree->Choose Torsions"中可 以设置配体中可以旋转的键。同时指定在活动键 时需要移动的最少的原子数目为6, 在"Ligand>Torsion Tree->Set Number of Active Torsions“ 中设置。
Autodock_vina_使用方法

Autodock vina 使用方法需要软件为autodocktool、vina、pymol需要文件为蛋白质文件:.pdb 小分子配体文件:.mol21、打开autodocktools软件File > Read Molecule > *.pdbEdit > Charges > Add Kollman Charges > 确定Edit > Hydrogens > Add Hydrogens > Polar Only > OKWithBondOrderYesFloat Dashborad Widget > Atom(pdb name)Grid > Macromolecule > choose > pdb name > Select MoleculeGrid > Grid box > 首先把Spacing (angstrom)设为1然后把number of points in x-dimension值设在一个合理范围number of points in y-dimensionnumber of points in z-dimension使晶格能够囊括所以蛋白质原子最后把x center 值设在一个合理范围y centerz center使晶格中心能够在蛋白质分子中心把所有值记录在*.txt文件中。
2、打开Raccoon 软件(此步骤也可以用autodocktools完成)Add ligands > *.mol2 (现在Files of type中改成*.mol2) > open此步骤可把.mol2文件批量转化为.pdbqt文件(而autodocktools软件只能单个转化)。
3、打开终端,进入目的文件夹首先创建一个*.txt文件(利用步骤1中的*.txt文件的数值),比如:receptor =center_x = 2center_y = 6center_z = -7size_x = 25size_y = 25size_z = 25num_modes = 9然后把所有小分子配体进行虚拟筛选,利用一下批处理脚本:for f in ligand_*.pdbqt; dob=`basename $f .pdbqt`echo Processing ligand $bmkdir -p $bvina --config --ligand $f --out ${b}/ --log ${b}/done之前一直在看别人发的帖子,赶脚自己也应该发一个。
MGLTools-1.5.6分析AutoDock分子对接结果

MGLTools-1.5.6分析AutoDock分⼦对接结果分⼦对接图形化软件MGLTools-1.5.6在Windows7上分析AutoDock分⼦对接结果作者:shims在对接完成之后需要分析分⼦对接结果,也是做分⼦对接最重要的部分。
跑分⼦对接是简单的可是如何分析得到得数据是考验个⼈经验的。
限于知识⽔平的限制这⾥只介绍简单的查看分⼦对接结果中的构象并查看相关的性质。
通过Analyze-Dockings-Open来打开分⼦对接的输出⽂件,该⽂件⼀般是dlg后缀,如这⾥的⽂件为complex.dlg。
加载分⼦对接结果⽂件之后会提⽰加载的构象数⽬,这⾥AutoDock对接了2000个构想。
如果想查看配体分⼦在受体分⼦中的位置,需要单独加载受体分⼦结构,通过Analyze-Macromolecule-Open打开⽂件窗⼝,选择分⼦对接使⽤的受体分⼦结构的pdbqt⽂件加载到MGLTools软件中,并及时的在显⽰窗⼝显⽰出来。
如果不需要查看在受体分⼦上的位置可以跳过这⼀步。
加载了分⼦对接结果之后通过Analyze-Conformations-Play, rankd by energy来打开构象播放窗⼝,在该窗⼝中会将所有的构象按照能量由低到⾼的顺序排列,即能量最低的序号为1。
如果不需要按照能量排序⽽是使⽤默认的对接结果⽂件中的顺序可以选择Play来打开播放窗⼝。
在打开的播放窗⼝中间显⽰的是当前的构象序号,如这⾥显⽰的是1,即能量最低的构象。
左右两边实⼼的单箭头是向左右移动⼀个。
两个实⼼的箭头是向前或向后播放。
两个实⼼箭头加上竖线是直接跳转到第⼀个或最后⼀个构象。
图中⽤红⾊框标识出来的按钮是⽤来打开播放设置窗⼝Set Play Options。
在播放设置窗⼝中可是设置随着显⽰构象的变化显⽰当前构象的信息,选择Show Info,就会弹出构象信息窗⼝Conformaion * Info,其中*是当前构象序号。
用于药物研发的分子对接软件的使用技巧

用于药物研发的分子对接软件的使用技巧随着药物研发领域的发展和进步,计算机辅助药物设计的重要性日益凸显。
分子对接技术被广泛应用于药物研发中,可以预测化合物与受体的结合模式,从而指导新药分子的设计和优化。
而为了更好地进行分子对接研究,研究人员常常会使用一些专业的分子对接软件。
本文将介绍一些常见的用于药物研发的分子对接软件的使用技巧。
1. VinaVina是一种广泛应用的免费分子对接软件,它能够高效地进行分子对接计算。
以下是一些使用Vina的技巧:- 安装和配置:首先,需要从官方网站或源代码库中下载Vina软件,并按照安装指南进行安装。
然后,配置软件的运行环境,包括指定受体和配体分子的文件路径,并设置其他参数如计算网格的大小等。
- 导入受体和配体:使用Vina进行分子对接前,需要将受体和配体的结构文件导入到软件中。
一般情况下,受体和配体的文件格式是pdb或pdbqt。
- 设置搜索空间:Vina需要设置一个搜索空间,用于指定受体结构中比较有可能发生配体结合的区域。
通过定义一个接受区域的坐标范围,可以限制对接计算的搜索空间大小,提高计算效率。
- 运行对接计算:配置完毕后,即可运行Vina进行分子对接计算。
在计算过程中,软件会通过评分函数对每个配体进行评估,并给出一个分数,用于描述配体与受体结合的亲和性。
2. AutodockAutodock是另一种广泛应用于药物研发领域的分子对接软件,它提供了一套完整的分子对接计算流程,并具备良好的可视化功能。
以下是一些使用Autodock的技巧:- 导入受体和配体:在使用Autodock进行分子对接前,需要先将受体和配体的pdb文件导入到软件中。
此外,还可以对受体和配体进行处理,如添加氢原子、给配体加电荷等。
- 设置搜索空间:与Vina类似,Autodock也需要设置一个搜索空间以缩小对接计算的范围。
可以通过手动指定参考原子的坐标范围,或者利用软件提供的自动搜索功能找到合适的搜索空间。
AutoDock中文使用说明

AutoDock和AutoDock Tools 使用教程一、分子对接简介及软件介绍1.分子对接理论基础所谓分子对接就是两个或多个分子之间通过几何匹配和能量匹配而相互识别的过程。
分子对接在酶学研究以及药物设计中具有十分重要的意义。
在酶激活剂、酶抑制剂与酶相互作用以及药物分子产生药理反应的过程中,小分子(通常意义上的Ligand)与靶酶(通常意义上的Receptor)相互互结合,首先就需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,得到一个稳定的复合物构象。
通过分子对接确定复合物中两个分子正确的相对位置和取向,研究两个分子的构象,特别是底物构象在形成复合物过程中的变化,是确定酶激活剂、抑制剂作用机制以及药物作用机制,设计新药的基础。
分子对接计算是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补化学环境互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间最佳的结合模式。
分子对接最初思想起源于Fisher E.的“锁和钥匙模型”(图),认为“锁”和“钥匙”的相识别的首要条件是他们在空间形状上要互相匹配。
然而,配体和受体分子之间的识别要比“锁和钥匙”模型复杂的多。
首先,配体和受体分子的构象是变化的,而不是刚性的,配体和受体在对接过程中互相适应对方,从而达到更完美的匹配。
其次,分子对接不但要满足空间形状的匹配,还要满足能量的匹配。
配体和受体之间的通过底物分子与靶酶分子能否结合以及结合的强度最终是由形成此复合物过程的结合自由能变化ΔG bind 所决定的。
互补性(complementarity)和预组坦织(pre-organization)是决定分子对接过程的两个重要原则,前者决定识别过程的选择性,而后者决定识别过理的结合能力。
互补性包括空间结构的互补性和电学性质的互补性。
1958年Koshland提出了分子识别过程中的诱导契合(induced fit)概念,指出配体与受体相互结合时,受体将采取一个能同底物达到最佳结合的构象(图1)。
使用autodock4在linux下进行批量对接(命令行)

使用autodock4在linux下进行批量对接(命令行)使用Autodock4在Linux下进行批量对接(命令行)1.安装1.从官方网站下载MGLTools 和Autodock4压缩包,按说明解压安装。
2.准备MGLTools应用程序:安装完成后,将./mgltools_i86Linux**/MGLT oolsPckgs/AutoDockT ools/Utilities 24目录下的所有应用程序拷贝到个人工作目录。
再将./mgltools_i86Linux**/bin目录下的pythonsh应用程序拷贝到工作目录。
3.将autodock4压缩包中的Autogrid4和Autodock4也放到工作目录。
2.使用1.下载对接的配体和受体文件(*.pdb格式)。
进入工作目录。
2.将PDB格式文件转换成Autodock识别的PDBQT文件:./pythonsh prepare_ligand4.py –l ligand.pdb./pythonsh prepare_receptor4.py –r receptor.pdb –A hydrogens –U waters3.准备autogrid4参数文件:./pythonsh prepare_gpf4.py –l ligand.pdbqt –r receptor.pdbqt4.准备autodock4参数文件:./pythonsh prepare_dpf4.py –l ligand.pdbqt –r receptor.pdbqt5.运行Autogrid4:./autogrid4 –p receptor.gpf6.运行Autodock4:./autodock4 –p ligand_receptor.dpf3.结果分析Autodock 对接结果记录在*.dlg文件中,用Estimated free energy of binding值评价对接结果,值越低表示越容易结合。
prac1_autodock

蛋白处理过程:
建目录:c:/ADtest 下载1HSG.pdb文件到:c:/ADtest 将其中的: “HOH A”和“HOH B” 替换成“HOH W” 将:MK1 B 替换成: MK1 C
药物分子设计上机练习
打开ADT
药物分子设计上机练习
读入受体分子: 1HSG: HIV II protease与L-735,524复合 两种方式: 两种方式: A: A:File->open B: 在Dashboard中: 中 右键点击PMV Molecules. 右键点击
药物分子设计上机练习
药物分子设计上机练习
SOFTWARE
药物分子设计上机练习
Most common docking programs—trends in the percentage of citations per year for the five most common docking programs, analyzed from ISI Web of Science considering any of the original references asindicated in Table III. 药物分子设计上机练习
下载及安装: 最新版4.2.3
药物分子设计上机练习
下载及安装Vina: 最新版autodock_vina_1_1_2_win32
AutoDock Vina: open-source drug discovery, molecular docking virtual screening multi-core capability, high performance and enhanced accuracy and ease of use designed and implemented by Dr. Oleg Trott in the Molecular Graphics Lab at The Scripps Research Institute. Vina Video Tutorial
迈迪工具及离线包安装教程
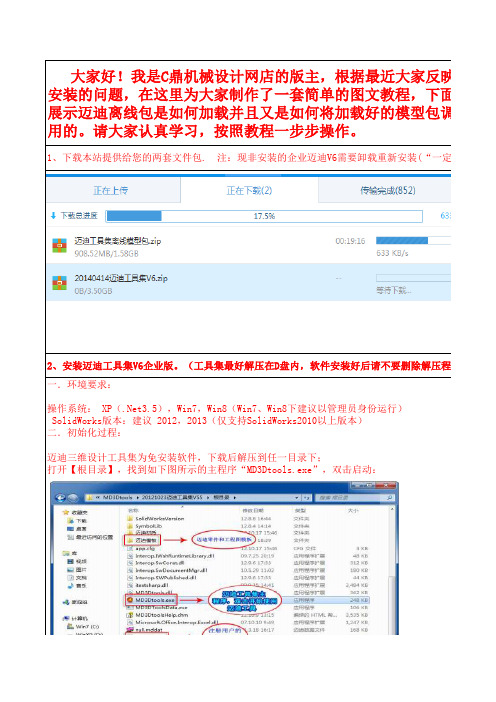
大家好!我是C鼎机械设计网店的版主,根据最近大家反映的安装的问题,在这里为大家制作了一套简单的图文教程,下面我将为展示迈迪离线包是如何加载并且又是如何将加载好的模型包调出来供用的。
请大家认真学习,按照教程一步步操作。
1、下载本站提供给您的两套文件包. 注:现非安装的企业迈迪V6需要卸载重新安装(“一定要2、安装迈迪工具集V6企业版。
(工具集最好解压在D盘内,软件安装好后请不要删除解压程序一.环境要求:操作系统: XP(.Net3.5),Win7,Win8(Win7、Win8下建议以管理员身份运行)SolidWorks版本:建议 2012,2013(仅支持SolidWorks2010以上版本)二.初始化过程:迈迪三维设计工具集为免安装软件,下载后解压到任一目录下;打开【根目录】,找到如下图所示的主程序“MD3Dtools.exe”,双击启动:若首次使用迈迪工具,会自动启动初始化程序:(若程序目录被移动、重命名后也会被认为首次1.选择SolidWorks版本:下拉列表中列出了当前电脑所安装的所有SolidWorks版本(不支持201用的版本。
2.在SolidWorks中注册插件:选中后打开SolidWorks会存在迈迪插件。
3.网络版设置:设置是否在企业内网中共享模型库。
作为单机版使用:不使用局域网的功能,但仍然可以使用在线配件库,以及在线更新。
建议个人用户选作为内网客户端:需要首先在企业内网中配置一台迈迪工具集服务器。
请参考相关帮助。
作为内网服务器:配置当前电脑为内网中的迈迪工具集服务器,详细设置方法请参考相关帮助。
设置完成后,单击【确定】进入下一步。
提示将插件注册文件加入到注册表中:单击【是】注册迈迪插件。
【注意】:如果没有弹出上面的注册提示窗口,说明您的系统打开注册表文件(.reg)的方式不要手工将文件【根目录\迈迪插件注册文件.reg】用注册表编辑器打开,才能在SolidWorks中注册插件首次使用迈迪工具,会提示启动SolidWorks。
AutoDock编译安装
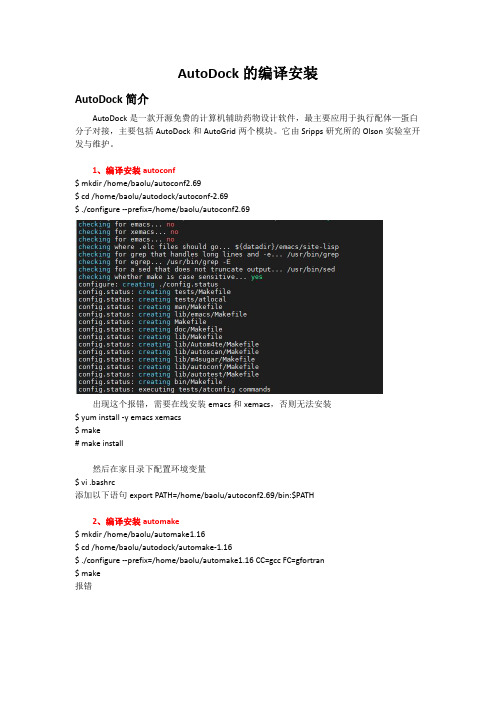
AutoDock的编译安装AutoDock简介AutoDock是一款开源免费的计算机辅助药物设计软件,最主要应用于执行配体—蛋白分子对接,主要包括AutoDock和AutoGrid两个模块。
它由Sripps研究所的Olson实验室开发与维护。
1、编译安装autoconf$mkdir/home/baolu/autoconf2.69$cd/home/baolu/autodock/autoconf-2.69$./configure--prefix=/home/baolu/autoconf2.69出现这个报错,需要在线安装emacs和xemacs,否则无法安装$yum install-y emacs xemacs$make#make install然后在家目录下配置环境变量$vi.bashrc添加以下语句export PATH=/home/baolu/autoconf2.69/bin:$PATH2、编译安装automake$mkdir/home/baolu/automake1.16$cd/home/baolu/autodock/automake-1.16$./configure--prefix=/home/baolu/automake1.16CC=gcc FC=gfortran$make报错这时需要修改Makefile,在3694行的末尾加上--no-discard-stderr$vi Makefile重新make和make install配置环境变量$vi.bashrc添加语句export PATH=/home/baolu/automake1.16/bin:$PATH3、编译安装autodock解压后进入autodock目录$mkdir/home/baolu/AutoDock$cd/home/baolu/autodock/src/autodock$./configure--prefix=/home/baolu/AutoDock$make&&make install编译安装完成,在指定安装目录里生成bin文件夹,其中有autodock4和autodock4.omp 两个可执行文件。
vina教程

需要安装包:MGLTools,Vina
假设我们做蛋白质与小分子对接,protein.pdb + ligand.pdb
1. 打开Autogridtools(包含在MGL里):读入蛋白质pdb, 加氢原子(一般PDB下载得到的PDB文件可能
保存为Macromolecular,命名为protein.pdbqt
设置该蛋白受体的对接位置Grid Box,包括:七类参数(盒子中心
读入配体ligand.pdb,这里可以选择默认的可扭转键,也可以人为将
保存配体为ligand.pdbqt
2. vina --help 查看具体说明
将这两个pdbqt文件放入vina工作路径下
运行vina脚本(推荐):
vina --config conf.txt --log log.txt
#####conf.txt####
receptor = protein.pdbqt
ligand = ligand.pdbqt
center_x = 11 # Grid box 设定的参数
center_y = 90.5
center_z = 57.5
size_x = 22
size_y = 24
size_z = 28
##################
运行后得到两个文件:ligand_out.pdbqt----包含最优的前N个构想,按从优到差排序,可直接用pymol
log.txt----包含最优的前N个构想的具体信息
3. 用户可用pymol打开ligand_out.pdbqt和protein.pdb,查看对接的位置
或是打开实验所得的配体构像与对接最优构象做对比。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
AutoDock Vina(简称Vina)是一款免费开源的分子对接软件,由美国斯克利普斯研究所(The Scripps Research Institute)Oleg Trott博士开发。
Vina在AutoDock 4的基础上做了极大的改进,主要是算法上的改进,促使它在结合模式预测的平均准确率和计算速度方面都远超AutoDock 4。
更多介绍请访问:
Vina有Windows、Linux、MacOSX等不同系统版本以及源代码。
可从这里免费:
Vina只负责对接计算,它并不提供蛋白大分子和配体小分子的准备功能。
该部分功能则由MGLTools来完成。
因
此,通常也需要安装MGLTools
下面简单讲解Vina和MGLTools的安装。
Windows系统中安装Vina
1、双击点击二进制文件autodock_vina_1_1_2_win32.msi,按提示点击安装即可。
2、由于Scripps Array下面。
3、为
Linux
1
2
3
4
Windows
Linux
1
2
3、敲以下
chmod
4
5
——仅供参考。