LAMMPS软件与分子模拟的实现
LAMMPS软件与分子模拟的实现

LAMMPS软件与分子模拟的实现LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) 是一个基于粒子动力学原理的分子模拟软件。
它使用分子动力学模型来模拟原子、分子或其他粒子在不同温度、压力和相互作用条件下的行为。
它是一个高效、可扩展和灵活的软件,可以模拟从数百到数百万个粒子的多种物理和化学现象。
1. 引入粒子和相互作用模型: LAMMPS实现了多种粒子和相互作用模型。
用户可以指定模拟系统中的粒子类型,包括原子、分子和其他粒子类型。
LAMMPS支持多种相互作用力场模型,如Lennard-Jones和Coulomb 相互作用,以及更复杂的模型如多体相互作用。
2. 粒子动力学模拟: LAMMPS使用经典的牛顿力学原理来模拟粒子在时间和空间上的演化。
它迭代破解了每个粒子所受到的力,并计算粒子的速度和位置。
它使用了一些高效的算法和数据结构来提高模拟效率,如Verlet积分算法和空间分解技术。
3. 温度和压力控制: LAMMPS可以在模拟过程中控制系统的温度和压力。
它采用了多种算法来模拟温度和压力,如Nose-Hoover算法、Berendsen热浴、Langevin动力学和Parrinello-Rahman方法。
这些算法可以在模拟过程中维持系统的平衡状态。
4.边界条件和周期性边界条件:LAMMPS支持各种不同的边界条件。
它可以模拟有限尺寸系统,也可以模拟无限尺寸系统。
对于无限尺寸系统,LAMMPS采用了周期性边界条件,以模拟系统中的无限复制。
5.输入和输出:LAMMPS提供了灵活的输入和输出功能。
用户可以通过输入文件来设置模拟系统的参数,如初始位置、速度、力场模型和模拟时间。
LAMMPS会将模拟结果输出到文件中,用户可以对结果进行分析和后处理。
6.并行计算:LAMMPS是一个并行化的软件,可以在多个计算节点上并行计算,以提高计算效率。
lammps分子模拟石墨烯建长键角

LAMMPS是一款用于进行分子动力学模拟的软件,广泛应用于材料科学、化学、生物学等领域。
在模拟石墨烯的生长过程中,键角的控制是至关重要的。
在模拟中,石墨烯的生长通常是从单个碳原子开始的。
随着时间的推移,这些碳原子会通过化学键连接在一起,形成石墨烯的二维结构。
在这个过程中,控制键角是关键。
键角的大小决定了石墨烯的最终结构和性质。
使用LAMMPS进行模拟时,可以通过调整模拟参数来控制键角。
例如,可以调整碳原子之间的相互作用力,或者改变模拟的温度和压力条件。
这些参数会影响碳原子之间的相对位置,从而影响键角的大小。
通过精细调整这些参数,可以尝试生成具有特定键角大小的石墨烯结构。
这种模拟方法有助于深入了解石墨烯的生长机制,并为实验提供指导。
同时,模拟结果也可以用于预测石墨烯在不同条件下的性质和行为,为实际应用提供理论支持。
总之,使用LAMMPS进行分子模拟是一种有效的方法,可以用来研究石墨烯的生长过程中键角的控制。
通过调整模拟参数,可以深入了解石墨烯的生长机制,并为实验和应用提供有价值的指导。
全原子分子动力学模型 lammps

全原子分子动力学模型 lammps全原子分子动力学模型LAMMPS,是一款非常优秀的分子模拟软件。
它是一款免费的并依托开源社区共同开发的分子模拟软件,在学术界和工业界都具有广泛的应用。
LAMMPS包含许多强大的功能和工具,能够模拟分子、多体相互作用、材料能量和温度等方面,是材料科学、化学、生物学等领域研究的重要工具之一。
下面我们来具体了解一下如何使用LAMMPS进行分子模拟。
第一步:软件安装与配置首先,我们需要前往LAMMPS的官方网站进行下载和安装。
下载的版本可以根据自己的需要选择,一般来说最新的版本越稳定也越实用。
安装之后,我们需要配置环境变量,以便在终端或命令行中可以直接使用LAMMPS。
第二步:建立分子模型在使用LAMMPS进行分子模拟之前,我们需要首先建立分子模型。
这可以通过算法或者数据实验等方式实现。
具体来说,我们需要确定分子的数目、类型、位置等信息。
对于这些信息,可通过多种科学方法获取。
我们建立好分子模型之后,需要将其写入到LAMMPS的输入文件中。
输入文件包含了我们的模型、模拟参数、计算方式和输出等信息,是LAMMPS模拟的核心。
第三步:设置模拟参数LAMMPS除了支持模型参数输入外,还提供了一个非常强大的用户交互机制,以便更灵活地控制模型。
在这里,我们可以设置温度、压力、能量、力场、约束等不同的模拟参数。
不同的模型需要根据具体应用需求进行不同参数的调整,比如需要考虑不同的温度、压力等等。
第四步:运行模拟当我们设置好了LAMMPS的输入文件和模拟参数之后,就可以开始利用LAMMPS进行模拟了。
一般来说,我们可以采用命令行操作,以便更精确地控制模拟进程。
模拟完成之后,我们可以根据之前设置的输出选项进行相应的结果分析。
LAMMPS支持多种输出格式,方便进行分析和后续处理。
总结:通过以上步骤,我们可以看到使用LAMMPS进行分子模拟的过程非常清晰和简单。
LAMMPS强大的功能和灵活性,可以帮助我们快速、准确地获取分子的性质和行为,是当今分子模拟研究领域的重要工具之一。
lammps建水分子

lammps建水分子使用LAMMPS建模水分子LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一种常用的分子动力学模拟软件,可以用于模拟各种复杂的分子体系,包括水分子。
水是一种普遍存在于地球上的重要分子,研究水分子的行为对于理解和解释许多自然现象非常重要。
在本文中,我们将介绍如何使用LAMMPS建模和模拟水分子的方法。
我们需要准备水分子的结构和参数。
水分子由两个氢原子和一个氧原子组成,分子间通过共价键连接。
在LAMMPS中,我们需要定义每个原子的质量、电荷和力场参数。
对于水分子,通常使用TIP3P模型,其中氧原子带负电荷,氢原子带正电荷。
此外,还需要定义原子间的键长、键角和势能函数等参数。
接下来,我们需要创建水分子的初始结构。
可以使用LAMMPS提供的工具或其他建模软件如VMD等来生成水分子的初始结构。
一种常用的方法是在一个正交晶胞中放置水分子,并使用周期性边界条件来模拟无限大的水系统。
我们可以根据需要调整晶胞的大小和水分子的数量来控制模拟系统的尺寸。
在创建初始结构后,我们需要定义模拟系统的边界条件、温度和压力等参数。
对于水分子,通常使用周期性边界条件来模拟无限大的水系统。
温度可以通过控制模拟系统中粒子的速度来实现,可以使用NVT(常温常容)或NPT(常温常压)等模拟模式。
压力可以通过控制模拟系统中粒子之间的相互作用力来实现。
在设置模拟参数后,我们可以运行LAMMPS进行水分子模拟。
LAMMPS使用分子动力学方法模拟粒子在时间上的演化,根据定义的势能函数和初始条件计算粒子之间的相互作用力,并根据牛顿第二定律计算粒子的加速度和速度。
通过迭代计算,我们可以得到水分子在模拟过程中的轨迹和动力学性质。
在模拟结束后,我们可以对模拟结果进行分析和可视化。
LAMMPS 提供了丰富的分析工具和输出选项,可以计算和输出水分子的结构参数(如键长、键角等)、动力学性质(如速度、动能等)和热力学性质(如能量、压力等)。
2.LAMMPS分子动力学模拟-in文件编写

二、LAMMPS分子动力学模拟-in文件编写教程1.说明:in文件是LAMMPS软件的运行程序文件,该文件程序描述了模拟需求指令。
所有模拟指令需根据LAMMPS用户手册,即LAMMPS Users Manual ()文件进行编写。
2.以下将根据一个简单案例进行in文件基本结构说明,该案例中的结构并不固定,可根据需要进行调整。
3.in文件案例:-------------------------------------------------模型基本指令设置------------------------------ # Lennard-Jones crystal (#符号表示不执行该条指令)units real (此命令用于设置模拟的单位类型,有lj or real or metal or si orcgs or electron or micro or nano多种类型,每种类型有各自的单位设定,在后续程序编写中要注意所有数据的单位)boundary p p p (该指令用于设置模型每个维度的边界类型,p为periodic边界,三个p代表x,y,z三个方向都是周期边界)atom_style full (定义在模拟中使用的原子类型,样式的选择决定了data文件中分子结构数据所包含的要素)-------------------------------------------------分子结构模型设置------------------------------#read_data X.data (读入包含lammps运行模拟所需信息的数据文件,data文件中包含了原子坐标、种类、键、角和所带电荷等信息;分子结构也可以通过set,box等指令在in文件中进行设定)read_restart poly.restart.100000 (读入前次模拟保存的运行结果文件,从中断的模拟位置重新启动模拟)---------------------------------原子间作用势类型和参数设定------------------------------ pair_style lj/cut/coul/cut 12 12 (设置用来计算原子对相互作用的势能公式)pair_coeff 1 1 10 10 (根据指定的原子对势能函数,设置势能参数)pair_coeff 2 2 100 10pair_coeff 1 2 10 10------------------------------------键、角类型和参数设定------------------------------------- bond_style harmonicbond_coeff 1 450 1.0bond_coeff 2 500 1.45angle_style harmonicangle_coeff 1 55 109.0angle_coeff 2 55 109.28dihedral_style harmonicdihedral_coeff 1 0.062 1 3dihedral_coeff 2 0.062 1 3---------------------------系统能量最小化方法和参数设定-------------------------------- #min_style sd (选择执行最小化命令时要使用的最小化算法)#minimize 1.0e-5 1.0e-5 100 100 (通过迭代调整原子坐标,实现系统的能量最小化,该指令设置迭代终止条件)--------------------------------------其它模拟相关指令设定----------------------------------- #velocity all create 300.0 200000 (设置或改变一组原子的速度)#velocity all scale 300.0fix 1 all nvt temp 873.0 873.0 1 (设置NVT系综)#fix 2 all temp/rescale 50 673 673 10 1.0 (通过重新调整原子群的速度来重置原子群的温度)compute KE all ke/atom (对一组原子执行计算)variable temp atom c_KE/0.0001292355 (此命令将数值或公式计算结果指配给变量名,以便稍后在输入脚本或模拟过程中使用该变量进行计算)fix 6 all ave/time 10 10000 100000 v_temp file tem.profile (输出时间平均计算结果,写入一个命名为tem.profile的文件)-----------------------------------模拟结果输出相关指令设定------------------------------- timestep 1 (设置分子模拟的时间步长大小)thermo_style custom time temp press density pe (设置将热力学数据打印到屏幕和日志文件的样式和内容)thermo_modify lost ignore flush yesthermo 50restart 100000 poly.restart (每隔这么多个时间步写出一个包含当前模拟数据的重新启动文件)dump 1 all atom 100000 mmpstrj (每100000个时间步将Atom数据转储到mmpstrj文件)run 50000000 (指定运行的步数)。
lammps分子模拟石墨烯建长键角

lammps分子模拟石墨烯建长键角LAMMPS,也称为Large-scale Atomic/Molecular Massively Parallel Simulator,是一个分子动力学模拟软件,可用于模拟原子和分子系统的动力学行为。
本文将介绍如何使用LAMMPS进行石墨烯的模拟,并着重探讨石墨烯中的长键角。
石墨烯是由碳原子形成的二维晶格结构,具有优异的力学、电子和光学性质。
其中的键角是石墨烯模拟中一个重要的参数,可以影响石墨烯的结构和性质。
通过调整键角的大小,可以改变石墨烯的机械强度、电子传输和能带结构等特性。
在LAMMPS中,我们可以使用Morse势函数来描述石墨烯中的键角。
Morse势函数是一种用于描述分子振动的经验势函数。
它的形式为:V(r) = D * (1 - exp(-a * (r - r0)))^2其中V(r)是势能与键长r之间的关系,D是势阱深度,a是振动频率,r0是平衡键长。
首先,我们需要准备一个初始的石墨烯晶胞。
可以使用LAMMPS提供的工具生成一个石墨烯晶胞文件。
然后,我们可以通过定义一个原子类型和键角类型,在输入文件中使用"read_data"命令来读取晶胞文件。
在LAMMPS输入文件中,可以使用"pair_style"和"pair_coeff"命令定义键角的势函数和参数。
例如,我们可以将石墨烯晶胞文件命名为"graphene.data",在输入文件中添加以下命令来定义键角:```# Define the potential and parameters for the bond angle pair_style morsepair_coeff * * D a r0# Define the read_data command to read the graphene cell fileread_data graphene.data```在上述命令中,D、a和r0分别是Morse势函数的参数,它们需要根据实际情况进行选择和调整。
lammps教程

lammps教程
LAMMPS是一个常用的分子动力学模拟软件,用于模拟原子、分子的运动和相互作用。
下面是一份关于如何使用LAMMPS
进行模拟的简单教程。
1. 安装LAMMPS:首先,你需要从官方网站上下载LAMMPS 的最新版本。
根据你的操作系统,选择合适的版本进行下载和安装。
2. 准备输入文件:在使用LAMMPS之前,你需要准备一个输
入文件,用于描述你想要模拟的系统和模拟的条件。
这个文件通常使用文本编辑器创建,扩展名为".in"。
在输入文件中,你
需要定义原子的初始位置、速度、力场参数等。
你也可以在输入文件中指定模拟的时间、温度、压力等参数。
3. 运行LAMMPS:在终端中,使用以下命令来运行LAMMPS,同时指定输入文件:
```
lammps < input_file.in
```
然后,LAMMPS将读取输入文件中的信息,并开始模拟。
4. 分析和可视化结果:LAMMPS输出的模拟结果通常以文本
文件的形式保存。
你可以使用文本处理工具(如awk、sed等)来分析并提取模拟结果中的关键信息。
此外,还可以使用可视
化软件(如VMD、OVITO等)来对模拟结果进行可视化和分析。
注意:以上只是一个简单的教程,介绍了LAMMPS的基本使用方法。
LAMMPS具有非常丰富的功能和参数选项,可以进行复杂的分子动力学模拟。
请参考LAMMPS的官方文档和用户手册,以获取更详细和全面的指导。
lammps分子模拟石墨烯建长键角 -回复

lammps分子模拟石墨烯建长键角-回复在LAMMPS中使用分子模拟来构建石墨烯的建长键角是一个常见的任务。
在本篇文章中,我将详细介绍使用LAMMPS进行石墨烯模拟的步骤,并解释如何添加长键角来模拟石墨烯的特性。
首先,让我们先了解一下LAMMPS是什么。
LAMMPS是一种开源的分子动力学模拟软件,用于模拟原子、分子和离子在各种材料中的行为。
它提供了一系列功能丰富的计算工具和模型,可以帮助研究人员理解和预测材料的性质。
要使用LAMMPS进行石墨烯模拟,我们首先需要创建一个输入文件,我们称之为"input.deck"。
在这个文件中,我们需要定义系统的初始状态,包括原子的初始位置和速度,以及模拟的时间步长等参数。
让我们以一个简单的石墨烯片为例。
首先,我们需要定义石墨烯的晶格结构。
石墨烯由碳原子组成,每个原子有三个相邻原子和一个孤对电子。
为了模拟石墨烯的特性,我们需要引入一个力场,如Lennard-Jones势能,来描述原子之间的相互作用。
我们可以使用LAMMPS提供的现有势场文件,如"ffield.reax"来描述碳原子之间的相互作用。
接下来,在输入文件中,我们需要定义一个"units"块来设置模拟过程中的单位。
例如,我们可以选择以LJ单位来定义长度和能量单位。
使用LJ单位可以简化计算,并提供更方便的结果。
然后,我们需要定义一个"atom_style"块来描述原子的类型和属性。
对于石墨烯,我们只需要定义一种原子类型,即碳原子。
我们也可以设置其他原子属性,如电荷和质量。
在输入文件的下一部分,我们需要定义原子的初始位置。
由于石墨烯具有规则的结构,我们可以使用一些自动生成的脚本来生成初始位置。
例如,我们可以使用LAMMPS提供的"topo hconv"命令来创建一个石墨烯片的初始结构。
接下来,我们可以定义模拟的时间步长、总步数和温度。
lammps分子动力学 能量 平衡

lammps分子动力学能量平衡一、介绍在分子动力学模拟中,能量平衡是一个非常重要的步骤。
通过能量平衡,我们可以确保模拟系统的能量在整个模拟过程中保持稳定,并且系统达到平衡态。
本文将介绍如何使用LAMMPS软件进行分子动力学模拟中的能量平衡。
二、LAMMPS简介LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一个用于分子动力学模拟的开源软件。
它可以模拟原子、分子和大分子等系统的运动,以及系统的能量和力学性质。
LAMMPS提供了丰富的功能和灵活的输入选项,使得用户可以根据自己的需求进行模拟。
三、能量平衡的重要性在进行分子动力学模拟时,能量平衡是非常重要的。
一个能量不平衡的系统可能会导致模拟结果不准确,甚至无法收敛到平衡态。
通过能量平衡,我们可以确保系统的能量在整个模拟过程中保持稳定,并且系统达到平衡态。
能量平衡还可以帮助我们检查模拟参数的选择是否合理,以及模拟过程中是否存在错误。
四、能量平衡的方法1. 步长选择在进行分子动力学模拟时,步长的选择非常重要。
步长过大会导致模拟系统的能量不稳定,步长过小则会增加模拟时间。
一般来说,步长的选择应该结合模拟系统的特点和目标,通过试验和调整来确定一个合适的步长。
2. 温度控制温度控制是能量平衡的一个重要方面。
通过控制系统的温度,我们可以使系统达到热平衡态。
常用的温度控制方法包括NVT和NPT等。
在LAMMPS中,可以使用fix命令来实现温度控制。
3. 压力控制除了温度控制,压力控制也是能量平衡的一个重要方面。
通过控制系统的压力,我们可以使系统达到力学平衡态。
常用的压力控制方法包括NPT和NPH等。
在LAMMPS中,可以使用fix命令来实现压力控制。
4. 能量演化在能量平衡过程中,系统的能量会随着时间的推移而演化。
通过观察系统能量的变化,我们可以判断系统是否达到平衡态。
在LAMMPS中,可以使用compute命令来计算系统的能量,并使用dump命令将能量随时间的变化保存到文件中。
第五讲分子动力学模拟的Lammps实现讲课教案

Lammps计算输入文件
• # create geometry创建初始几何构形
• Lattice hex 0.93 • #指定晶格类型(二维hex)和晶格常数 • Region box block 0 100 0 40 -0.25 0.25 • #定义一个区域 • create_box 5 box • #在指定区域建立一个simulation box,5表示原子类型的种类数 • create_atoms 1 box • #在simulation box中创建类型为1的原子(原子位置初始化)
• #定义左上、左下原子组(便于指定裂纹的存在)
• set
group leftupper type 2
• set
group leftlower type 3
• set
group lower type 4
• set
group upper type 5
• #指定原子类型(便于指定裂纹的存在)
Lammps计算输入文件
Lammps计算输入文件
• region
leftupper block INF 20 20 INF INF INF
• region
leftlower block INF 20 INF 20 INF INF
• group
leftupper region leftupper
• group
leftlower region leftlower
z
u
u
y
x
u
常温30K条件下
金属材料模拟中Lammps的单位
Lammps计算输入文件
• # 3d metal 拉伸模拟 • #模拟条件的初始化 • Units metal • #指定模拟中的单位类型 • boundary s s s • #指定模拟的边界条件 • atom_style atomic • #指定原子类型,原子的属性 • Lattice fcc 3.52 • #指定材料的晶格类型和晶格常数 • region box block -5 5 -5 5 -15 15 • #xlo,xhi,ylo,yhi,zlo,zhi =区域box的上下限 • create_box 1 box • #建立只有1中原子类型的simulation box • create_atoms 1 box • #在simulation box中创建类型为1的原子(原子位置初始化)
第五讲_分子动力学模拟的Lammps实现

第五讲_分子动力学模拟的Lammps实现Lammps是一个用于分子动力学模拟的开源软件,它提供了丰富的功能和灵活的插件,能够模拟各种复杂的分子系统。
在本文中,我们将介绍如何使用Lammps进行分子动力学模拟,并简要介绍一些常用的功能和插件。
例如,下面是一个简单的输入文件示例,用于模拟一个包含100个氩原子的系统:```#输入文件示例#初始化units ljdimension 3boundary p p patom_style atomic#原子定义lattice fcc 0.8442region box block 0 10 0 10 0 10create_box 1 boxcreate_atoms 1 boxmass 1 39.948#相互作用pair_style lj/cut 2.5pair_coeff 1 1 1.0 1.0 2.5#速度初始化#模拟参数thermo 100thermo_style custom step temp etotal press#运行模拟run 1000```在输入文件中,我们首先指定了模拟的基本参数,例如使用Lennard-Jones势函数进行计算(`units lj`),模拟系统的维度为三维(`dimension 3`),周期性边界条件(`boundary p p p`),并且定义了原子的类型(`atom_style atomic`)。
然后,我们定义了原子的初始位置和速度。
在上述示例中,我们使用fcc晶格生成了一个10x10x10的盒子,并将100个氩原子放入其中(`create_box`和`create_atoms`)。
我们还指定了原子的质量(`mass`),这里我们使用了氩的质量。
接下来,我们定义了原子之间的相互作用。
在上述示例中,我们使用了Lennard-Jones势函数(`pair_style lj/cut`),并指定了参数(1.0、1.0和2.5)。
hbn六方氮化硼分子模拟构型
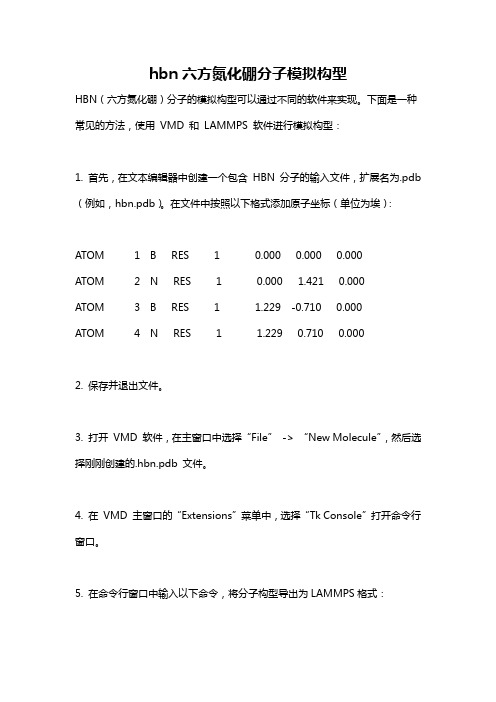
hbn六方氮化硼分子模拟构型HBN(六方氮化硼)分子的模拟构型可以通过不同的软件来实现。
下面是一种常见的方法,使用VMD 和LAMMPS 软件进行模拟构型:1. 首先,在文本编辑器中创建一个包含HBN 分子的输入文件,扩展名为.pdb (例如,hbn.pdb)。
在文件中按照以下格式添加原子坐标(单位为埃):ATOM 1 B RES 1 0.000 0.000 0.000ATOM 2 N RES 1 0.000 1.421 0.000ATOM 3 B RES 1 1.229 -0.710 0.000ATOM 4 N RES 1 1.229 0.710 0.0002. 保存并退出文件。
3. 打开VMD 软件,在主窗口中选择“File”-> “New Molecule”,然后选择刚刚创建的.hbn.pdb 文件。
4. 在VMD 主窗口的“Extensions”菜单中,选择“Tk Console”打开命令行窗口。
5. 在命令行窗口中输入以下命令,将分子构型导出为LAMMPS格式:bashset sel [atomselect top "all"]sel writepdb hbn_lammps.pdb6. 关闭命令行窗口。
7. 打开终端或命令提示符,并转到保存LAMMPS 输入文件的目录。
8. 创建一个名为"hbn.in" 的输入文件,并将以下内容复制到文件中:text# LAMMPS input script for HBN simulation# Initializationunits realatom_style fulldimension 3boundary p p patom_modify map array# Create atomsread_data hbn.data# Potentialpair_style lj/cut 10.0pair_coeff 1 1 0.1500 3.16555787617979pair_coeff 2 2 0.0750 2.56310287433584pair_coeff 1 2 0.1125 2.86433037525732# Simulation settingstimestep 1.0thermo 100thermo_style custom step temp etotal press# Run simulationrun 1000注意:这里的势函数参数(pair_coeff)可能需要根据实际情况进行调整。
lammps非平衡分子动力学

lammps非平衡分子动力学引言:LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款用于分子动力学模拟的软件,它可以模拟各种材料的力学、热力学和动力学性质。
其中,非平衡分子动力学(Non-equilibrium Molecular Dynamics,NEMD)是一种重要的分子动力学模拟方法,它可以模拟材料在非平衡状态下的性质,如热传导、流变性等。
本文将介绍LAMMPS中的非平衡分子动力学方法及其应用。
一、LAMMPS中的非平衡分子动力学方法LAMMPS中实现了多种非平衡分子动力学方法,包括温度梯度法、剪切流法、压力梯度法等。
这些方法都是通过在模拟系统中引入外场来实现的,例如在温度梯度法中,通过在系统中引入温度梯度,使得系统中不同位置的温度不同,从而模拟热传导现象。
在剪切流法中,通过施加剪切力,使得系统中的分子发生流动,从而模拟材料的流变性质。
在压力梯度法中,通过施加压力梯度,使得系统中不同位置的压力不同,从而模拟材料的输运性质。
二、LAMMPS中的非平衡分子动力学应用非平衡分子动力学方法在材料科学中有着广泛的应用,例如在热传导、流变性、输运性质等方面。
其中,热传导是非平衡分子动力学方法的一个重要应用领域。
通过模拟材料中的热传导现象,可以研究材料的热导率、热阻等性质。
此外,非平衡分子动力学方法还可以用于模拟材料的流变性质,例如在高温下,材料的流变性质会发生变化,通过非平衡分子动力学方法可以模拟这种变化。
此外,非平衡分子动力学方法还可以用于模拟材料的输运性质,例如在电池材料中,通过模拟离子输运现象,可以研究电池的性能。
结论:LAMMPS中的非平衡分子动力学方法是一种重要的分子动力学模拟方法,它可以模拟材料在非平衡状态下的性质,如热传导、流变性等。
这些方法在材料科学中有着广泛的应用,可以用于研究材料的性质和优化材料的性能。
lammps中的分子填充功能

lammps中的分子填充功能
LAMMPS(大规模原子/分子并行模拟器)是一款用于模拟原子、
分子和颗粒等系统行为的开源软件包。
其中的分子填充功能是指在模
拟过程中,通过将空间中的分子填充到特定区域来模拟物质的组装和
形态学变化。
分子填充功能在LAMMPS中通过使用相应的命令和参数来实现。
首先,需要创建一个表示空气、溶剂或其他介质的分子体系。
这些分
子体系的大小和形状可以通过调整相关参数来控制。
然后,通过指定
填充算法和填充条件来将分子填充到目标区域。
在LAMMPS中,常用的分子填充算法包括随机填充和扩展填充。
随机填充算法会将分子随机放置到目标区域中,直到达到一定的填充度。
扩展填充算法会先在目标区域中选择一个种子分子,然后以一定
的扩展步长将周围的分子逐渐填充进去,直到达到指定的填充度。
除了填充算法,LAMMPS还提供了一些其他的参数和选项来控制分子填充过程。
例如,可以指定分子的初始速度、旋转角度和初始排列
方式。
还可以设置填充过程中的限制条件,如填充速率、填充压力和
填充温度等。
通过使用LAMMPS的分子填充功能,研究人员可以模拟不同条件
下的分子组装和结构变化,探索物质的性质和行为。
这对于材料科学、化学和生物科学等领域的研究具有重要意义。
lammps化合物建模方法

lammps化合物建模方法LAMMPS(一种大分子模拟软件)是一种基于蒙特卡罗和分子动力学的分子模拟程序。
LAMMPS是一个高度可扩展的分子动力学软件,支持多种计算方法、潜伏势和扩展输入输出等功能,可以通过简单的脚本进行灵活的自定义。
本文介绍了LAMMPS在化合物建模方面的方法。
1. 分子结构构建在使用LAMMPS模拟任何化合物之前,必须构建化合物的分子结构。
在构建化合物分子结构时,最常用的方法是使用化学软件如Gaussian、Chem3D等。
这些软件可以通过在电脑上绘制格子结构或输入化学方程式来构建分子结构。
其他方法包括:(1)在实验室中使用实验技术来确定化合物的分子结构,例如X射线衍射或核磁共振光谱。
(2)通过现有的文献或数据库获取如CCDC等获得分子结构。
2. 分子结构的力场参数设定LAMMPS使用力场参数将分子描述为一系列粒子(原子、分子等)和它们之间相互作用的简单函数形式。
在使用LAMMPS进行分子动力学模拟之前,必须先选择并设定化合物的力场参数。
常用的力场参数有Amber、OPLS-AA和GROMOS。
选择和设定适当的力场参数对于生成可靠结果是至关重要的。
3. 分子结构的几何最优化在分子动力学模拟之前,必须将分子结构进行几何最优化。
几何最优化旨在确定化合物最稳定的可行构象。
最简单的方法是使用化学软件工具如Gaussian、DFT等进行几何优化。
在LAMMPS中也可以使用CGMIN,L-BFGS和FIRE算法等进行几何最优化。
4. 分子动力学模拟的设定在模拟开始之前,必须设定一系列模拟参数,其中最重要的参数是温度、压力和模拟时间步长。
还要确定如何将能量耗散在模拟系统中。
常见的能量消耗方法是把模拟系统与恒温和恒压散热浴或NPT等相互作用。
5. 模拟后的数据处理模拟后,必须对数据进行处理,以确定模拟系统的性质,例如密度、热容等。
数据处理主要包括:(1)使用能量、负荷或其他物理量对模拟系统进行可视化,以评估模拟结果的内容和准确性。
lammps 计算分子数

lammps 计算分子数使用LAMMPS进行分子数计算LAMMPS(大型原子/分子类型多粒子模拟)是一种流行的分子动力学软件,可用于模拟原子和分子的行为。
它被广泛应用于材料科学、化学、生物学等领域,用于研究和解决各种复杂的科学问题。
本文将介绍如何使用LAMMPS进行分子数计算,以及相关的基本原理和步骤。
我们需要准备模拟系统的输入文件。
这个文件包含了系统的初始状态、模拟的时间步长、模拟的总时间等信息。
在LAMMPS中,我们可以使用文本编辑器创建一个输入文件,将所需的信息写入其中。
下面是一个示例输入文件的简化版本:```# LAMMPS input file for molecular dynamics simulation# Initialize simulationdimension 3units realatom_style atomicboundary p p p# Define atom typeslattice fcc 3.52region box block 0 10 0 10 0 10create_box 1 boxcreate_atoms 1 box# Set potentials and simulation parameterspair_style lj/cut 2.5pair_coeff 1 1 1.0 1.0 2.5neighbor 0.3 binneigh_modify delay 5 check no# Run simulationtimestep 0.001thermo 100run 1000```在这个示例中,我们创建了一个简单的立方体模拟系统,其中包含一个原子类型。
我们使用fcc晶格来生成原子,并设置Lennard-Jones势函数作为原子间的相互作用势。
在模拟过程中,我们设定了时间步长为0.001,模拟总时间为1000个时间步。
接下来,我们可以使用LAMMPS来运行模拟并计算分子数。
lammps 蒙特卡洛化学反应

lammps 蒙特卡洛化学反应LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一种用于分子动力学模拟的开源软件,它可以模拟多种化学反应,其中包括蒙特卡洛化学反应。
本文将介绍LAMMPS 中的蒙特卡洛化学反应模拟方法及其应用。
蒙特卡洛方法是一种基于随机抽样的数值计算方法,可以用来模拟和分析复杂的化学反应过程。
在LAMMPS中,蒙特卡洛化学反应模拟是通过在分子动力学模拟中引入蒙特卡洛步骤来实现的。
在蒙特卡洛化学反应模拟中,分子系统中的每个分子都被赋予一个随机的能量状态,并通过随机抽样的方式来选择反应类型和反应路径。
通过在模拟过程中不断更新分子的能量状态和反应路径,可以模拟出分子间的化学反应。
在LAMMPS中,蒙特卡洛化学反应模拟的基本步骤如下:1. 初始化系统:包括定义分子的初始位置和能量状态。
2. 选择反应类型:根据反应的类型和反应路径,通过随机抽样的方式选择要进行的反应。
3. 计算反应概率:根据选择的反应类型和反应路径,计算反应的概率。
4. 进行反应:根据计算得到的反应概率,决定是否进行反应。
如果进行反应,则更新分子的能量状态和反应路径。
5. 更新分子状态:根据反应的结果,更新分子的位置和能量状态。
6. 重复步骤2至步骤5,直到达到模拟的时间或步数。
蒙特卡洛化学反应模拟在化学研究中具有广泛的应用。
它可以用来研究化学反应的速率常数、平衡常数以及反应路径等。
通过模拟不同的反应条件和反应路径,可以得到一系列的化学反应数据,进而对实验中观测到的化学反应进行解释和预测。
蒙特卡洛化学反应模拟还可以用来研究分子间的相互作用和聚集行为。
通过模拟不同的分子结构和反应条件,可以了解分子在不同环境中的行为,并且可以用于设计新的分子材料和催化剂。
蒙特卡洛化学反应模拟是一种强大的工具,可以用来模拟和分析复杂的化学反应过程。
通过LAMMPS软件的应用,可以实现高效的蒙特卡洛化学反应模拟,并为化学研究提供重要的理论指导和预测能力。
lammps计算简单分子

lammps计算简单分子LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一种用于分子动力学模拟的软件工具,它可以模拟和计算分子体系中的原子运动和相互作用。
在本文中,我们将介绍如何使用LAMMPS来计算简单分子系统,并探讨一些计算过程中的关键问题和注意事项。
我们需要准备一个输入文件,该文件包含了分子体系的初始坐标、势能函数以及模拟的时间步长等参数。
在LAMMPS中,分子体系通常以数据文件的形式输入,其中包含了原子的类型、坐标和速度等信息。
我们还需要指定所采用的势能函数,比如经典的力场模型(如Lennard-Jones势能)或量子力学计算所需的波函数等。
接下来,我们需要定义模拟的时间步长和总的模拟时间。
时间步长决定了模拟的精细度,通常选择一个合适的值以保证模拟结果的准确性和稳定性。
总的模拟时间取决于所关注的物理过程,对于简单分子的热力学性质计算通常选择足够长的时间以保证系统达到平衡态。
在模拟过程中,LAMMPS会根据所设定的势能函数和初始条件计算每个原子的运动轨迹,并在每个时间步长更新原子的位置和速度。
根据计算所需的精度和计算资源,我们可以选择不同的计算方法和算法。
例如,对于大规模分子体系,我们可以使用并行计算来加快计算速度。
在模拟结束后,我们可以对模拟结果进行分析和后处理。
LAMMPS 提供了丰富的分析工具和功能,可以计算分子体系的物理性质和结构参数。
比如,我们可以计算分子的能量、温度、压力等热力学性质,还可以分析分子的结构特征,比如键长、键角、配位数等。
需要注意的是,LAMMPS是一个高度可定制和灵活的软件工具,可以根据具体的研究需求进行相应的参数设置和计算方案设计。
不同的分子体系和问题需要不同的模拟策略和算法选择。
因此,在使用LAMMPS进行分子模拟计算时,我们需要根据具体情况进行合理的参数选择和模拟方案设计,以确保计算结果的准确性和可靠性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Lammps常见命令
unit dimension newton 选择单位系统,L-J、real、metal 2D or 3D? 默认是3D
processors
boundary atom style atom modify atom type 告诉lammps在你的模拟中使用何种力场? pair_style, bond_style, angle_style, dihedral_style, improper_style 边界条件:周期性边界or自由边界? 定义你的模拟体系中的原子属性
2. Lammps功能和原理
Lammps初识 Lammps的功能 Lammps的原理 Lammps的特点 Lammps的应用
Lammps初识
Lammps程序是一个经典分子 动力学计算程序。全称 Large-scale Atomic/Molecular Massively Parallel Simulator
Lammps计算输入文件
• • • • • • • • • • • • • • • • # LJ potentials(指定原子作用势) pair_style lj/cut 2.5 #指定lj势,截断半径为2.5 pair_coeff * * 1.0 1.0 2.5 #指定lj势参数 # define groups(便于加载) Region 1 block INF INF INF 1.25 INF INF Group lower region 1 #定义lower组(便于施加外加速度) Region 2 block INF INF 38.75 INF INF INF Group upper region 2 #定义upper组(便于施加外加速度) Group boundary union lower upper #定义总边界组 Group mobile subtract all boundary #定义可动原子组(便于统计温度)
补充内容 Lammps与分子动力学
常用模拟软件
Lammps功能和原理 经典范例
1. 常用的MD模拟软件
NAMD:免费
主要针对与生物和化学软材料体系,程序设计水平高,计 算效率高。有很好的分析辅助软件VMD。
AMBER
主要针对生物体系,也适当兼容一般化学分子。有很好的 内置势能模型,自定义新模型和新分子很方便,有很完善 的维护网站。计算效率不高运算速度慢。
不能(non-features)
- 非图形化界面,不能自动建立分子结构模型和分配力场参 数,不具有复杂的分析的手段,不能可视化输出结果 - 补救:Pizza.py 工具包,用于建模和分析以及可视化,但 是功能不够强大。 - 必须一些其他前后处理软件(几何建模,物理建模,可视 化分析)结合使用,接口方法。
Lammps软件入门
明确自己的问题和方向,选择正确的工具
要做的是什么问题,属于物理,化学,力学,材料,还是都有? 能否具体到希望要作出什么结果?实验和理论上是否有相似的研 究?再看问题是否适合lammps程序?是否有别的程序可以替代选 择或者联合选择?
计算环境搭建可行性分析
现有计算机条件: 硬件水平决定模拟的规模 是否有相关的支持:软件环境 团队学习的重要:交流是非常重要
LAMMPS 免费 一般性分子模拟软件。 兼容当前大多数的势能模型,编程水平高,计算效率高。可以 模拟软材料和固体物理系统。
Materials Explorer 立足于Windows平台的多功能分子动力学软件。拥有强大的分 子动力学计算及Monte Carlo软件包,是结合应用领域来研究 材料工程的有力工具。Materials Explorer可以用来研究有机物 、高聚物、生物大分子、金属、陶瓷材料、半导体等晶体、非 晶体、溶液,流体,液体和气体相变、膨胀、压缩系数、抗张 强度、缺陷等。Materials Explorer软件中包含2Body,3Body ,EAM,AMBER等63个力场可供用户选择。Materials Explorer软件拥有完美的图形界面,方便使用者操作。
Lammps计算输入文件
• • • • • • • # run运行计算 timestep 0.003 #时间间隔步 Thermo 200 #每200步输出热动力学统计量 thermo_modify temp new #计算温度通过new指示的方法计算
3.经典范例
使用L-J势模拟裂纹的扩展 使用EAM势模拟Ni的剪切行为 Cu、Ni等金属的凝固过程模拟 表面能计算
A. 使用L-J势模拟裂纹的扩展
leftupp er 裂纹 uppe r
y
leftlow er x lowe r
Lammps计算输入文件
• # 2d LJ crack simulation(问题的基本初始 化)
养成良好的学习习惯
几个章节必须看(1-1,2,3;2-2,3,5,6,7;3-1,2,3;4-all) 读做例子有感觉(melt,crack,shear) 错误信息自己找(完美的错误提示信息) 随手整理做记录
命令学习(工具体现)
命令名称:基本上告诉你意义 书写格式:脚本语言的特色 格式选项说明:严格遵守,最好理解含义 范例书写:有助于自己写脚本 注意事项:特别的地方 相关命令:命令分类学习,比如输入有那些方式,势函数定 义有哪几类?
CHARMM
主要针对生物体系,也包含部分化学体系。势能模型更新 很快自定义新模型比较方便。计算效率低。
GROMACS 免费 主要针对生物体系,也适当照顾一般化学体系。算法 好,计算效率高。界面友好,维护服务好。 TINKER 免费 一般性分子动力学软件,对生物体系略有偏重。优点 支持多种模型。仍在开发中,某些方面还不完善。 DL-POLY 一般性分子模拟软件,界面友好,计算效率高。维护 服务很好。 Materials Studio
• set • set group leftupper type 2 group leftlower type 3
Lammps计算输入文件
• • • • • • • • • • • • • • • • # initial velocities初始化速度 compute new mobile temp #定义温度的计算(可动区域内统计平均) compute new2 mobile stress/atom #定义原子应力的计算(整个区域) Velocity mobile create 0.01 887723 temp new #按指定的温度(0.01)计算方法,初始化原子的速度 Velocity upper set 0.0 0.3 0.0 #upper原子组y方向的速度为0.3 Velocity mobile ramp vy 0.0 0.3 y 1.25 38.75 sum yes #mobile原子的速初始度从0到0.3线性变化 # fixes施加约束 fix 1 all nve #nve系综的积分算法 fix 2 boundary setforce NULL 0.0 0.0 #边界boundary上力条件,钢化原子,便于加载!!
Lammps的基本原理
编写、输入模拟程序
运行模拟 输出结果
可视化
结果分析
Lammps输入文件的主要组成部分
Initialization Atom definition Settings Run a simulation
后面的两个部分可以按照需要多次重复。
Lammps软件目前的特点
从势场角度看:建模软物质(生物分子,聚合物),固态 材料(金属,半导体),以及粗粒子和介观材料。更一般 的说是lammps程序是用来建模原子/介观/连续尺度物质以 及其在热、力学、化学条件下的性质的模拟软件,因此是 系统化方法。 Lammps程序运行环境:单CPU和多CPU,采用的是消息 响应和模拟扩充 的特性。新版采用C/C++语言书写,周期性发布,以日期 为为准,不断更新一些bug和增加一些功能。脚本语言应 用开发。 美国能源部下属的圣地亚国家实验室发布,主要作者: Steve Plimpton, Aidan Thompson, and Paul Crozier 网上邮件组可以解决和及时交流
官方网址: / 国内交流论坛:
Lammps的功能
能(features)
一般意义(并行化,可扩充,脚本化输入,接口化编译) 专门意义(能建模原子类型,有什么力场,有那些原子操 作,如何设置系综/边界/约束,积分方法,输出控制,前 后图形处理,以及具有一些什么特色功能)
Lammps软件的应用
应用步骤—程序安装
安装平台环境(考虑不同的操作系统,是否并行计算) 简单易行的安装
• Windows下:命令行执行方式 • Linux下:编译选择项 • 几个关键点:编译器的选择;并行库的位置,相关库的位置
应用步骤--实例学习
输入脚本格式书写:3-1节内容,积木式搭建 分块命令学习方法: 几何模型构建:atom_style, boundary, dimension,units create_atoms, create_box, lattice, read_data, read_restart, region, replicate 物理模型构建:angle_coeff, angle_style, bond_coeff, bond_style, dielectric, dihedral_coeff 过程模型构建:Fix:is any operation that is applied to the system during timestepping or minimization. Examples include updating of atom positions and velocities due to time integration, controlling temperature, applying constraint forces to atoms, enforcing boundary conditions, computing diagnostics, etc. 输出模型构建:compute过程计算量,热力学输出量(全局量),局部表征量(单 个原子、组原子)