纳米材料和纳米结构第十一讲-XRD-2
第十一章纳米粒子和粒子团与沸石的组装体系

离子交换法 气相注入法 固相扩散法
在沸石孔隙中组装 半导体团簇的方法
光氧化法 内延MOCVD 内延CVD
(a)离子交换法
离子交换是最常用的一种方法。 适用条件:尤其对制备II-VI族化合物较为方 便,对于I-VII族的卤化银及某些III-V族半导 体团簇也可用。但不适用于单质半导体纳米团 簇的制备。 分为两个步骤:(1)阳离子的引入, (2)阴离子的引入。 此法缺点:容易引入杂质,难以得到高纯的半 导体纳米团簇。
图11.12形象地示出了 WO3团簇的形成过程。 精确的晶格和电子结 构研究表明,在这些 材料中,含W的组份都 很好地控制在沸石的 孔隙中。这些化合物 的存在对于沸石的结 晶习性、完整性没有 影响,对于晶胞的大 小也只产生很微小的 影响。
某 些 典 型 的 沸 石 结 构 示 于 图 11.1
11.1.2 金属纳米粒子和金属离子团簇 与沸石组装体系的合成
沸石是一种三维的阳离子交换器,因此制备 含有金属前驱体的方法有两种,分别为: 1、离子交换法; 2、化合物吸附法。 表11.2列出了含有金属前驱体的沸石转 变成金属粒子/沸石或离子团簇/沸石组装 体系的方法。
每个笼中含有4.9,5.4和5碱 金属原子的Na—,K—和Rb— LTA沸石的磁、光性能进行比较, 试样由气相沉积获得。图11.7示 出这三种沸石的反射谱。很清楚, 在约2.0eV处,表面等离子激发 支配Na和K纳米粒子的光谱,而 Rb纳米粒子中表面等离子带变得 不明显,由1p 1d的跃迁在1.6 和2.0eV处出现两个独立的激发。
11.1.1 沸石结构的描述
沸石是一种硅酸铝,一般表示式为
其中M为单价阳离子,D为二价阳离子,它们中和因四价Si 原子被三价Al原子所替代引入的负电荷。 沸石是一种多孔的介质,它是由一系列不同的规则通道 和孔洞构成,进入这些间隙孔洞是通过不同数量的四面体 构成的窗口。这些通道和孔洞的尺寸对于沸石的特性是很 关键的,笼中空间可容纳金属和非金属纳米粒子。
纳米材料的表征方法

STM的优点
它有原子量级的极高分辨率(横向可达0.1nm,纵向可达 0.01nm),即能直接观察到单原子层表面的局部结构 。 比如表 面缺陷、表面吸附体的形态和位置等. STM能够给出表面的三维图像 STM可在不同的环境条件下工作,包括真空、大气、低温,甚至 样品可浸在水中或电解液中,所以适用于研究环境因素对样品表 面的影响. 可研究纳米薄膜的分子结构.
原子力显微镜AFM
AFM的主要应用
纳米材料的 形貌测定 生物材料研 究 黏弹性材料 的表面加工
X射线衍射法(XRD)
XRD是鉴定物质晶相的有效手段。 利用XRD谱图可以推断出纳米材料的结晶度和 层状结构的有序度。 利用XRD图结合Debye-scherrer公式,又衍射 峰的半高宽计算对应晶面方向上的平均粒径 D=Rλ/βcosθ
பைடு நூலகம்
D为粒子直径,R为Scherrer常数(0.89), λ为入射X光波长 (0.15406 nm),θ为衍射角(°),β为衍射峰的半高峰 宽(rad)。
XRD在纳米材料中的应用
物相结构的分析 介孔材料的分析 纳米薄膜的厚度以及界面结构的测定.
SEM的主要功能
三维形貌的观察和分析
观察分析纳米材料的形貌 直接观察大样品的原始表面
扫描隧道显微镜(STM)
扫描隧道显微 镜工作原理示 意图
STM针尖
STM在纳米材料中的应用
测量单分子、单个纳米颗粒、单根纳米线和纳米管 等的电学、力学以及化学特性.
对表面进行纳米加工,构建新一代的纳米电子器件.
磁原子力显微镜或者专用的仪器纳米材料表征手段透射电子显微镜tem的主要功能研究纳米材料的结晶情况观察纳米材料的形貌分散情况评估纳米粒子的粒径
纳米材料课XRD刘青玲(132302)

XRD的主要用途
1、物相结构分析
不同的晶体有不同的晶体结构→ 不同的点阵和晶胞→ 不同的 衍射谱→ 不同的衍射峰位置和衍射峰强度。理论上没有完全 相同者。 测定各种标准物质、材料的衍射谱,将其衍射峰的位置和强 度作成数据库。实际是收集所有已发表的工作,加以分类、 整理、鉴别和可能的验证,作成数据库。 测定所需鉴定的样品,得到它的衍射峰的位置和强度,与数 据库对比。和数据库中的哪种或哪几种物质的数据相符,即 确定样品为那几种物质所组成。与比对指纹确认人同一原理。
比如:我们实验室用铁的配合物做的XRD,吡啶类配 体,Fe 2+,不同阴离子配位得到的XRD图
20000
16000
Intensity(AU)
12000
8000
4000
0 5 10 15 20 25 30
2(Deg)
由此可以说明,这四种晶体的晶格是一样 的,阴离子只是游离在晶格中的。
Thank
*单晶体衍射获得的是衍射 *粉末衍射为晶体粉末的集X NhomakorabeaD的原理
何谓X射线衍射? □只有结晶体才会造成X射线衍射 □结晶体为具有一定结构的原子团(分子)在三维空 间的周期排列
Cl
XRD的原理
• 布拉格公式: 2dsinθ=nλ 式中: λ,X射线波长; n,衍射级数; d,晶面间距; θ,衍射半角。
3、晶体内的微结构分析
• 微结构类型: 微应变:晶体内的微应力造成的点阵畸变 反相畴:相邻两畴中部分原子排列反向 各种位错:局部原子排列位置发生错误。 会引起微应力、微应变。 • 微结构对衍射谱的影响: 多数使衍射峰加宽
微应力与微应变
微应变(微应力)ε与衍射线增宽β间的关系
/ 4 tan
纳米材料PPT课件

利用微生物作为生物反应器,通过发酵或培养微生物来制备纳米材料。该方法 具有高产量、环保等优点,但需要选择合适的微生物种类和生长条件。
03
纳米材料的应用领域
能源领域
高效电池
01
利用纳米材料提高电池的能量密度和充电速度,延长电池寿命。
太阳能电池
02
通过纳米结构设计提高太阳能电池的光电转换效率,降低成本。
纳米材料分类
01
02
03
按组成分类
根据组成元素的种类,纳 米材料可分为金属、非金 属和复合材料等类型。
按维度分类
根据在纳米尺度上的维度 数,纳米材料可分为零维 (0D)、一维(1D)和 二维(2D)纳米材料。
按应用领域分类
根据应用领域,纳米材料 可分为电子、能源、环境、 生物医学等领域所需的特 定功能材料。
微乳液法
利用两种互不相溶的溶剂在表面活性剂的作用下形成微乳液,然后在微乳液中加入反应物 进行化学反应,最终得到纳米材料。该方法可制备出粒径均匀、形貌可控的纳米材料,但 制备过程较为复杂。
生物法
生物分子自组装
利用生物分子间的相互作用,如氢键、离子键等,将生物分子组装成纳米结构。 该方法具有条件温和、环保等优点,但制备过程较慢且产量较低。
燃料电池
03
利用纳米材料改善燃料电池的氧电极反应性能,提高燃料电池
的效率和稳定性。
医学领域
药物传输
利用纳米材料作为药物载体,实现药物的定向传输和精确释放。
医学成像
利用纳米材料提高医学成像的分辨率和对比度,为疾病诊断提供 更准确的信息。
生物检测
利用纳米材料的高灵敏度特性,实现生物分子的快速、高灵敏度 检测。
化学法
纳米材料的结构与性质PPT幻灯片

(2)表面原子数的增加 随着晶粒尺寸的降低,表面原子所占的比例、比表面积急剧提 高,使处于表面的原子数也急剧增加,平均配位数急剧下降。
表面原子数占全部原子 数的比例和粒径之间的 关系
14
(3)表面能 如果把一个原子或分子从内部移到界面,或者说
增大表面积,就必须克服体系内部分子之间的吸 引力而对体系做功。
11
(1)比表面积的增加 比表面积常用总表面积与质量或总体积的比值表示。
质量比表面积、体积比表面积 当颗粒细化时,粒子逐渐减小,总表面积急剧增大,
比表面积相应的也急剧加大。
12
如:把边长为1cm的立方体逐渐分割减小的立方 体,总表面积将明显增加。
边长
1 cm 10-5 cm (100 nm) 10-6 cm (10 nm) 10-7 cm (1 nm)
第二章 纳米材料的结构与性能
2.1 纳米材料的分类及特性 2.2 纳米微粒的物理特性 2.3 纳米碳材料 2.4 纳米晶体材料 2.5 纳米复合材料
1
2.1 纳米材料的分类及特性
纳米材料:三维空间中至少有一维处于1~100nm尺度
范围内或由纳米基本单元构成的材料。
一、纳米材料的分类 按结构(维度)分为4类: (1)零维纳米材料:空间三个维度上尺寸均为纳米
尺度—纳米颗粒、原子团簇等。 (2)一维纳米材料:在空间二个维度上尺寸为纳米
尺度—纳米丝、纳米棒、纳米管等。 (3)二维纳米材料:只在空间一个维度上尺寸为纳
米尺度—纳米薄膜、多层薄膜等。 (4)三维纳米材料:由纳米材料基本单元组成的块
体 2
按组成分类 纳米金属、纳米晶体、纳米陶瓷、纳米玻璃、纳 米高分子、纳米复合材料 按应用分类 纳米电子材料、纳米光电子材料、纳米生物医用 材料、纳米敏感材料、纳米储能材料 按材料物性分类 纳米半导体材料、纳米磁性材料、纳米非线性光 学材料、纳米铁电体、纳米超导材料、纳米热电 材料
纳米材料的制备与表征方法详解

纳米材料的制备与表征方法详解纳米材料是指具有至少一维尺寸在1-100纳米范围内的材料。
由于其特殊的尺寸效应和表面效应,纳米材料具有许多独特的物理、化学和生物性质,广泛应用于能源、电子、生物医学等领域。
本文将详细介绍纳米材料的制备与表征方法,以帮助读者更好地了解和应用这些材料。
一、纳米材料的制备方法1. 物理法物理法是指利用物理原理和方法制备纳米材料。
常见的物理法包括磁控溅射、蒸发凝聚、惰性气氛法等。
磁控溅射是将靶材置于真空室中,然后通过气体离子轰击靶材表面,使靶材原子冲击脱离并堆积在基底上,从而获得纳米薄膜。
蒸发凝聚是将材料加热到显著高于其熔点的温度,使其蒸发并在冷凝器上再凝结为纳米颗粒。
惰性气氛法是在惰性气氛中利用高温反应或氧化物还原反应生成纳米材料。
2. 化学法化学法是指利用化学反应和溶液合成方法制备纳米材料,常见的化学法包括溶胶-凝胶法、聚合物溶胶法等。
溶胶-凝胶法是将溶胶(纳米颗粒的前体)悬浮在溶液中,通过控制温度、浓度和pH值等条件使其凝胶形成纳米材料。
聚合物溶胶法是将聚合物与金属盐或金属前体形成配合物,然后通过控制溶液组成和pH值等条件制备纳米材料。
3. 生物法生物法是指利用生物体、生物分子和生物反应合成纳米材料。
常见的生物法有生物还原法、生物矿化法等。
生物还原法是利用微生物、酶或植物等生物体将金属离子还原为金属纳米材料。
生物矿化法是利用生物体或生物分子作为催化剂,在无机物晶体表面上沉积金属纳米颗粒。
二、纳米材料的表征方法1. 透射电子显微镜(TEM)透射电子显微镜是用来观察纳米材料形貌和晶体结构的重要工具。
它通过透射电子束穿透样品,产生透射电镜像,并从中获得样品纳米颗粒的尺寸、形状和分布情况以及晶体结构信息。
2. 扫描电子显微镜(SEM)扫描电子显微镜可用于观察纳米材料的表面形貌和拓扑结构。
它通过聚焦电子束扫描样品表面,形成二次电子、反射电子和荧光X射线等信号,并通过探测二次电子图像来获得样品的表面形貌和微观结构。
纳米材料的表征方法和工具介绍

纳米材料的表征方法和工具介绍随着纳米科技的迅速发展,纳米材料的研究和应用越来越重要。
然而,纳米材料的特殊性质决定了常规材料表征方法的局限性,因此需要采用专门的方法和工具来对纳米材料进行表征。
本文将介绍几种常用的纳米材料表征方法和工具,帮助读者更好地了解纳米材料的特性。
在纳米材料的表征中,最常用的方法之一是透射电子显微镜(TEM)。
TEM利用电子束替代了可见光,可以提供比光学显微镜更高的分辨率。
通过将样品置于电子束中,可以观察纳米材料的形貌、尺寸和结构等。
此外,TEM还常常结合能量散射谱(EDS)分析,用于确定纳米材料的元素成分和组成。
TEM是一种非常强大的工具,可以提供关于纳米材料的详细微观结构信息。
扫描电子显微镜(SEM)是另一种常用的纳米材料表征工具。
不同于TEM,SEM可以提供更大的视野,并且可以用于观察表面形貌和表面组成。
SEM使用电子束扫描样品表面,通过测量电子的反射和散射来生成显微图像。
此外,SEM还可以通过探针激发技术(EDS)分析表面的元素成分。
与TEM相比,SEM更适用于纳米材料的表面形貌和排列的研究。
除了电子显微镜,纳米材料的结构表征也可以借助X射线衍射(XRD)来实现。
XRD是一种基于材料对X射线的散射规律进行分析的技术。
通过测量样品对X射线的散射强度和角度,可以确定纳米材料的结晶结构、晶粒大小和晶格参数等信息。
XRD常用于研究纳米材料的晶体结构和相变行为,对于纳米化材料的结构调控非常有价值。
此外,拉曼光谱也是一种常用的纳米材料表征方法。
拉曼光谱通过测量光的散射来获得样品的振动信息,可以得到纳米材料的分子结构、纳米颗粒的大小以及纳米结构的应变等信息。
相较于其他表征方法,拉曼光谱具有非侵入性、无需样品处理等优点,适用于对纳米材料进行原位、非破坏性的表征。
特别是在研究碳纳米管、纳米颗粒和纳米二维材料时,拉曼光谱被广泛应用。
另外,热重分析(TGA)也是表征纳米材料性质的重要方法之一。
纳米材料

纳米材料的简介所谓“纳米”是一种长度的度量单位,一纳米相当于10-9米[1]。
更直观来说,一张纸的厚度大约是十万个纳米,一个金原子的直径大约是三分之一个纳米。
纳米材料又被称为纳米结构材料,是指在三维空间中至少有一维处于纳米尺度范围或由它们作为基本单元构成的具有特殊性能的材料。
目前,国际上将处于1到100纳米尺度范围内的超微颗粒及其致密的聚集体以及由纳米微晶所构成的材料统称为纳米材料。
纳米科技的发展研究起初只是在纳米级别的材料上发现一些新的特殊性质,而纳米科技的最终目标是能够在纳米尺度上实现材料的自由编排、组装原子分子、观察和操作极微观世界,从而加工、制造具有特定功能的材料[2]。
1纳米材料的特性和分类1.1纳米材料的特性1.1.1量子尺寸效应当粒子尺寸下降到某一值时,其费米能级附近的电子能级由准连续变为离散能级的现象和纳米半导体微粒存在不连续的最高被占据分子轨道和最低未被占据的分子轨道能级,能隙变宽现象均称为量子尺寸效应。
量子尺寸效应带来的能级改变、能隙变宽,使微粒的发射能量增加,光学吸收向短波方向移动(蓝移)。
直观上表现为样品颜色的变化,如CdS微粒的颜色随尺寸的减小由黄色逐渐变为浅黄色,当黄金被细分到小于光波波长的尺寸时,即失去了原有的富贵光泽而呈黑色[3]。
1.1.2小尺寸效应处在纳米量级的材料其尺寸和体积很小,所包含的原子、电子数目非常有限,许多现象已经不能用传统的大块物质的性质说明。
当纳米材料的颗粒尺寸与光波波长、徳布罗意波长以及超导态的相干长度或者透射深度等物理特征尺寸相当或者更小时,材料周期性边界条件将被破坏,使得材料表面层附近的原子密度减小,从而引起材料的光、电、热、磁和催化等特性呈现出新的变化。
小尺寸效应的主要影响是使得材料的强度和硬度提高以及金属材料的电阻提高;磁性颗粒的矫顽力提高和磁有序态向磁无序态转变。
例如金属纳米微粒的熔点远低于块状金属,纳米银粉的熔点能降低到100℃[2]。
纳米结构的表征和物理分析

纳米结构的表征和物理分析纳米材料的制备发展至今已经有了一定的成熟性,但是如何对纳米结构进行表征和物理分析却成为了当前研究中的一个重点和难点。
本文将介绍几种常见的纳米结构表征手段和物理分析方法,希望对纳米研究领域的同行们有所启示。
一、透射电镜透射电镜(Transmission Electron Microscopy, TEM)是一种常见的纳米结构表征手段,它能够直接观察样品内部的微观结构。
这种技术是通过电子学原理实现的,将一束电子束通过样品,其中由于电子的波长非常短,能够穿透纳米材料并被屏幕记录下来,这样就可以对样品的微观结构进行观察和分析。
二、扫描电子显微镜扫描电子显微镜(Scanning Electron Microscopy,SEM)具有高分辨率、宽视野、高辐射稳定性等特点,常被用于纳米材料的表面形貌、粒径大小、分布规律的表征。
在SEM中,电子束被聚焦成一个非常小的点,它会在样品表面扫描,并逆反射回来。
通过对反射电子的探测,可以获得样品表面形貌信息。
三、X射线衍射X射线衍射(X-Ray Diffraction,XRD)是一种常见的物理分析手段,它基于光学原理,通过探测样品对X射线的反射或散射来研究纳米材料的晶格结构、晶粒大小、物相组成等。
XRD仪器通常将X射线束定向照射样品,在样品的晶格周期性排布的规律下,经过反射后,能够在检测器上形成一系列强度特异的衍射峰,这些衍射峰可以反映出样品的晶体结构。
四、原子力显微镜原子力显微镜(Atomic Force Microscopy,AFM)是一种常见的纳米结构表征手段,它基于原子力的测量,可以获取材料表面的自然形貌、粗糙度、分子结构、磁性质等信息。
AFM通过在样品表面扫描一个非常尖锐的“探针”,通过测量探针表面受到的原子力变化,可以得到样品表面的形貌和微观结构。
五、拉曼光谱拉曼光谱是一种常见的物理分析手段,它通过分析样品对激光激发后的散射光,来研究纳米材料的晶体结构、结构特征、化学键特征等。
纳米材料的表征

原子力显微镜主要特征是不要求电导的表面, 因为 它测量的是扫描探针和它的样品表面间的相互作用 力, 包括静电的、范德华的、摩擦的、表面张力的 (毛细的)和磁力的, 因此它克服了STM方法不足并 成为它的互补。
由于仪器可以调节到所测量对象特定力有敏感作用 的范围, 故其可测量样品范围扩展到有机、无机、 生物材料及技术样品。
不同于STM, 从AFM探针所获得是每一个表面点力的 图。这力的图可解释为表面结构的反映, 是磁的、 静电的诸种力的几何图。
AFM的主要应用
纳米材料的形貌测定 生物材料研究 黏弹性材料的表面加工
二次电子就是在单电子激发过程中被入射电子轰击出来的核外电子,其信号主要来自样品 表面 5-10μm深度范围,能量较低,一般小于50 eV.
advantage
有较高的放大倍数, 20-100万倍之间连续 可调;
有很大的景深, 视野大, 成像富有立体感, 可直接观察各种试样凹凸不平表面的细 微结构;
利用XRD谱图可以推断出纳米材料的结晶度和层状结构的有序度。
D=Rλ/βcosθ 其中式中, D为粒子直径, R为Scherrer常数(0.89), λ为入射X 光波长(0.15406 nm), θ为衍射角(°), β为衍射峰的半高峰 宽(rad)。
5.1.4 X射线法(XRD
1894年,德国物理学家伦琴发现了具有穿透力 的新型X射线,这是一种波长很短的电磁波,波 长是0.05~0.25nm.
1912年,德国物理学家劳厄发现了X射线通过晶 体时产生衍射现象.
1912年,小布拉格提出著名的布拉格方程. 1913年,老布拉格设计出第一台X射线分光计,
纳米材料实验 XRD测定TiO2纳米粉体的晶型并用谢乐公式计算粒度
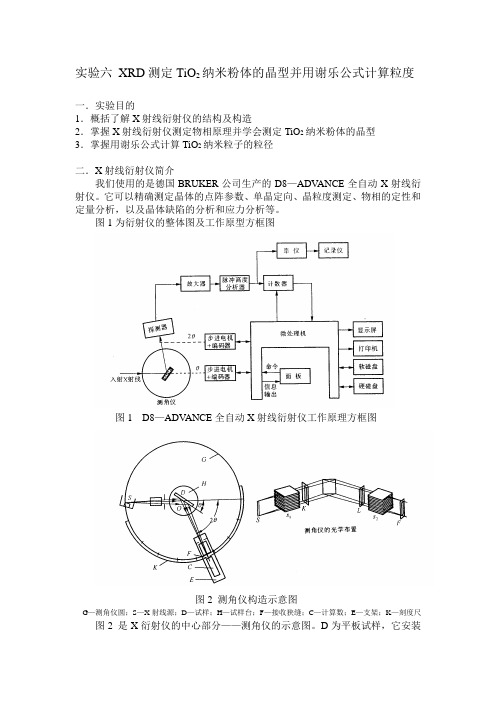
实验六XRD测定TiO2纳米粉体的晶型并用谢乐公式计算粒度一.实验目的1.概括了解X射线衍射仪的结构及构造2.掌握X射线衍射仪测定物相原理并学会测定TiO2纳米粉体的晶型3.掌握用谢乐公式计算TiO2纳米粒子的粒径二.X射线衍射仪简介我们使用的是德国BRUKER公司生产的D8—ADV ANCE全自动X射线衍射仪。
它可以精确测定晶体的点阵参数、单晶定向、晶粒度测定、物相的定性和定量分析,以及晶体缺陷的分析和应力分析等。
图1为衍射仪的整体图及工作原型方框图图1 D8—ADV ANCE全自动X射线衍射仪工作原理方框图图2 测角仪构造示意图G—测角仪圆;S—X射线源;D—试样;H—试样台;F—接收狭缝;C—计算数;E—支架;K—刻度尺图2 是X衍射仪的中心部分——测角仪的示意图。
D为平板试样,它安装在试样台H 上,试样台可围绕垂直于图面的轴O 旋转。
S 为X 射线源,也就是X 射线靶面上的线状焦斑,它与图面相垂直,与衍射仪轴平行。
由射线源射出的发散X 射线,照射试样后即形成一根收敛的衍射光束,它在焦点F 处聚集后射进记数馆C 中。
F 处有一接收狭缝,它与计数管同安装在可围绕O 旋转的支架E 上,其角位置2θ可以从刻度尺K 上读出。
衍射仪的设计使H 和E 的转动保持固定的关系,当H 转过θ度时,E 即转过2θ度。
这种关系保证了X 射线相对于试样的“入射角”与“反射角”始终相等,使得从试样产生的衍射线都正好能聚焦并进入计数管中。
计数管能将X 射线的强弱情况转化为电信号,并通过计数率仪、电位差将信号记录下来。
当试样连续转动时,衍射仪就能自动描绘出衍射强度随2θ角的变化情况。
测角仪的光学布置也在图2中展出。
S 为靶面的线焦点,其长轴方向为竖直。
入射线和衍射线要通过一系列狭缝光阑。
K 为发散狭缝,L 为防发散狭缝,F 为接受狭缝,分别限制入射线及衍射线束在水平方向的发散度。
防散射狭缝还可以排斥非试样的辐射,使峰底比得到改善。
纳米TiO2的XRD谱图分析2

化学化工学院材料化学专业实验报告实验名称:纳米TiO2的XRD谱图分析年级:2010级日期:2012年9月12日姓名:学号:一、预习部分(一)XRD介绍1、关于XRDXRD即X-ray diffraction 的缩写,X射线衍射,通过对材料进行X射线衍射,分析其衍射图谱,获得材料的成分、材料内部原子或分子的结构或形态等信息的研究手段。
X射线是一种波长很短(约为20~0.06埃)的电磁波,能穿透一定厚度的物质,并能使荧光物质发光、照相乳胶感光、气体电离。
在用电子束轰击金属“靶”产生的X射线中,包含与靶中各种元素对应的具有特定波长的X射线,称为特征(或标识)X射线。
X射线是原子内层电子在高速运动电子的轰击下跃迁而产生的光辐射,主要有连续X射线和特征X射线两种。
晶体可被用作X光的光栅,这些很大数目的原子或离子/分子所产生的相干散射将会发生光的干涉作用,从而影响散射的X射线的强度增强或减弱。
由于大量原子散射波的叠加,互相干涉而产生最大强度的光束称为X射线的衍射线。
满足衍射条件,可应用布拉格公式:2dsinθ=nλ。
应用已知波长的X射线来测量θ角,从而计算出晶面间距d,这是用于X射线结构分析;另一个是应用已知d的晶体来测量θ角,从而计算出特征X射线的波长,进而可在已有资料查出试样中所含的元素。
2、X 射线衍射相分析方法定性分析原理根据衍射数据d hkl和值I值,将数据按强度I/I1归一化处理。
根据d hkl测得的和相对强度I/ I1与国际粉末衍射标准联合会(JCPDS) 收集发行的粉末衍射图谱集卡片相对照,符合者可以断定这个未知样品就是卡片上注明了结构和名称的物质。
定量分析(1)单线条法(外标法、直接对比法):测量混合样品中欲测相(A相)某根衍射线的强度并与纯A相同一条强度对比,既可定出A相在混合样品中的相对含量A相的质量分数:W A=I A/(I A)0(2)内标法:分析谱图,确定待测试样中含有的多个物相,根据各相的质量吸收系数不同,先完成工作曲线,确定混合物中的不同组分的含量,根据工作曲线则可以测得其中组分的含量。
纳米材料样品制备和XRD测量

序言随着科技的进步与发展,纳米技术的日益显现出其重要性,尤其在材料方面,取得了很大的成就,例如碳纳米管等纳米材料。
而对其性质的研究也是较为重要的方面,通过相应的技术手段来获得较为成熟的技术以及对所制得的纳米材料的相关性质以及参数的定性和定量的测量,以便更好地了解该纳米材料的同时又可以为其应用打下基础。
于是XRD的相关技术开始应用到纳米材料的鉴定和测量上面,而XRD有包括晶体型和粉末型两种仪器,为纳米材料的测量提供了方便。
所以如何了解其的制备以及相关参数的测量,是之至重要的。
一、基本知识简介1.纳米相关定义纳米:1nm = 10-9m;四倍原子直径。
纳米尺度:尺寸大小处于1~100 nm,相当分子尺寸,分子相互作用的空间。
纳米材料:广义上,在三维空间中至少有一维处于纳米尺度或由它们作为基本单元构成的材料。
大致包括:一维——纳米粉末;二维——纳米纤维(如碳纳米管),直径处于纳米尺度而长度较大的线状材料;三维——纳米膜。
纳米效应颗粒小到纳米级时所表现的特有性质,例如表面效应(当粒径的减小到一定范围时,纳米粒子的表面积、表面能表现为迅速增加以及表面原子数迅速增加。
此外其具有不饱和性质,易于其他原子想结合而稳定下来从而表现出良好的化学和催化活性。
)、小尺寸效应、量子尺寸效应以及宏观量子隧道效(微观粒子具有贯穿势垒的能力)。
2. XRD:X射线衍射分析;其装置包括粉末型、晶体型两种常用的仪器。
测量主要采用标准对照法,即利用XRD仪器测得待测材料的衍射图像,再与已知的数据库进行对比,所得到的极其相似的图像所对应的物质即为所测。
所用软件:Jade(主要的),采取PDF卡片对照的方式求得。
其中,X射线是原子内层电子在高速运动电子的轰击下跃迁而产生的光辐射,主要有连续X射线和特征X射线两种。
二、制备方法1. 化学制备1.1 溶胶-凝胶法主要步骤是先选择制备金属化合物,然后金属化合物在适当的溶剂中溶解,经过溶胶、凝胶化过程而固化,再经低温热处理而得到纳米粒子。
XRD在纳米材料上的应用

XRD在镍包覆纳米二氧化钛实验中的应用应用化学陈阳1282080033摘要本文主要采用化学镀的方法在纳米二氧化钛表面包覆一层镍。
通过溶胶-凝胶的方法制备出二氧化钛纳米粒子,用X射线衍射仪检测分析二氧化钛的锐钛矿型和金红石型含量。
用X射线衍射仪检测分析包覆后的二氧化钛的组成。
在二氧化钛包覆镍的过程中对二氧化钛活化采用不同的方法,用X射线衍射仪分别检测分析包覆效果并进行比较分析。
关键词:二氧化钛;包覆;镍;X射线衍射1 概述X射线衍射分析(XRD)在纳米材料研究中的应用X射线衍射分析较常用于物相的定向和定量分析以及晶粒度、介孔结构等的测定1.1 XRD定性分析原理及应用XRD定性分析是利用XRD衍射角位置以及强度来鉴定未知样品的物相组成。
各衍射峰的角度及其相对强度是由物质本身的内部结构决定的。
每种物质都有其特定的晶体结构和晶胞尺寸,而这些又都与衍射角和衍射强度又着对应对应关系。
因此,可以根据衍射数据来鉴别晶体结构。
通过将未知物相的衍射花样与已知物相衍射花样相比较,可以逐一鉴定出样品中的各种物相。
目前可以利用粉末衍射卡片进行直接比对,也可以通过计算机数据库直接进行检索。
1.2 XRD定量分析原理及应用XRD定量分析是利用衍射线的强度来确定物相含量的。
每一种物相都有各自的特征衍射线,而衍射线的强度与物相的质量分数成正比。
各物相衍射线的强度随该相含量的增加而增加。
目前对于XRD物相定量分析最常用的方法主要有单线条法、直接比较法、内标法、增量法以及无标法。
1.3 XRD测定晶粒度XRD测定晶粒度是基于衍射线的宽度与材料晶粒大小有关这一现象。
对于TiO2纳米粉体,其主要衍射峰2θ为21.5°,可指标化为(101)晶面图(1.1)。
当采用铜靶作为衍射源,波长为0.154nm,衍射角的2θ为25.30°,测量获得的半高宽为0.375°,一般Scherrer常数取0.89.根据Scherrer公式,可以计算获得晶粒的尺寸此外,根据晶粒大小,还可以计算纳米粉体的比表面积。
CuxSy微纳米材料的制备及其XRD表征

CuxSy微纳米材料的制备及其XRD表征摘要:研究纳米材料的方法很多,本课题首先用四种不同的条件用水热法和复合碱媒介法成功合成了CuxSy微纳米材料,用去离子水多次清洗收集好样品,并用XRD工具对其进行分析和比较,着重分析其微观形态与化学成分。
结果发现不同的合成方法合成的微纳米材料微观结构,微观尺寸都不一样,并且都含有少量杂质。
关键词:CuxSy;微纳米材料;XRD1 纳米材料的基本特性纳米材料是指三维空间尺度至少有一维处于纳米量级(1100nm)的材料,它是由尺寸介于原子、分子和宏观体系之间的纳米粒子所组成的新一代材料。
2 纳米材料制备过程本实验进行了四组不同的实验,具体流程如下:①反应物:氢氧化钠、硫粉、氯化铜、乙醇、去離子水;生长温度:180℃;生长时间:24h。
第一步:合成。
用电子称精确量3g 氢氧化钠(NaOH)、0.08g 硫粉(S)后放入Teflon中后混合均匀,作为反应物。
加入10ml水,摇匀,待其冷却以后再打开盖子加入 0171g氯化铜(CuCl2)和2ml乙醇然后盖上盖子并摇匀,最后将罐子放入与其配套的钢制套筒中并且拧紧,并对套筒对应编号为1。
后将其反应体系放入温度为180℃的马弗炉中,加热24h取出。
第二步,收集。
同样方法合成以下几组:②反应物:氢氧化钠、硫粉、氯化铜、去离子水;生长温度:180℃;生长时间:24h。
③反应物:氢氧化钠、硫化钠、氯化铜、去离子水、乙醇;生长温度:180℃;生长时间:24h。
④反应物:氢氧化钠、硫化钠、氯化铜、去离子水;生长温度:180℃;生长时间:24h。
3 XRD表征测试结果4 结论图1到4所示样品分别为1、2、3、4号样品,图谱峰型很好,表明结晶状态良好。
1号样品分别在15.17°和39.41°出现最高峰;2号样品分别在12.57°和39.51°出现最高峰;3号样品分别在36.12°和39.18°出现最高峰;4号样品分别在36.12°和39.16°出现最高峰;四个图谱都在39°附近处出现最高峰,并且峰很尖锐,此处峰正是代表我们所需样品CuxSy微纳米材料,但是在其他位置也出现了其他的峰,含有其他杂质成分,其中包含有CuO和Cu2O。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
61
无 机 相 字 母 索 引
这种索引是按照物相英文名称的第一 个字母为顺序编排条目。每个条目占 一横行。物相的英文名称写在最前面, 其后,依次排列着化学式,三强线的 d值和相对强度,卡片编号,最后是 参比强度(I/Ic)。
30
31
STD模式覆盖了现有的许多非偶合衍射设备, 如薄膜分析用: 1、GAD(glancing angle X-ray diffractometry):这是 一种掠角入射技术,其入射角a处于0.6~10 之间, 所以穿透深度很浅:t=0.13a/m 2、TFD(thin film diffractometry):这是一种平行光 入射技术,其入射角a处于1~100 之间,当a小时, X射线穿透深度也浅 3、S-B(Seemann-Bohlin diffractometry ), 早期的非偶 合薄膜分析设备,3-6度,胶片记录。
X
4
5
衍 射 仪 基 本 结 构 方 框 图
6
7
8
9
10
高分辨衍射仪 (D8-Discovre型,Bruker公司1999年产品)
11
测 角 仪 简 介
测角仪是X射线的核心组成部分 试样台位于测角仪中心,试样台的中 心轴ON与测角仪的中心轴(垂直图 面)O垂直。 试样台既可以绕测角仪中心轴转动, 又可以绕自身中心轴转动。
45
1. PDF卡片简介; 2. PDF试样图; 3. PDF试样结构图; 4. PDF卡片内容。
PDF 卡 片
46
卡 片 简 介
J.D.Hanawalt等人于1938年首先发起,以d-I 数据组代替衍射花样,制备衍射数据卡片 的工作。 1942年“美国材料试验协会(ASTM)” 出版约1300张衍射数据卡片(ASTM卡 片)。 1969年成立了“粉末衍射标准联合委员 会”,由它负责编辑和出版粉末衍射卡片, 称为PDF卡片。 现在由ICDD(International Center for Diffraction Data), PDF-1: d, I PDF-2: card PDF-3: pattern
47
卡 片
48
卡 片
49
卡 片 的 内 容 ( 1 )
(1)1a、1b、1c,三个位置上的数据 是衍射花样中前反射区(2 θ <90°) 中三条最强衍射线对应的面间距,1d 位置上的数据是最大面间距。 (2)2a、2b、2c、2d, 分别为上述各 衍射线的相对强度,其中最强线的强 度为100.
值连同它的误差标写在每页的顶部。每
个条目由第一处面间距d1值决定它应属
于哪一组。每组内按d2值递减顺序编排
条目,对d2值相同的条目,则按d1值递减 顺序编排。不同的d值对应误差:
59
Hanawalt 索 引 说 明 ( 三 )
1980年版本编排规则为: (1)对I3/I2≤0.75的相,以d1d2的编排 顺序出现一次; (2)对I3/I2>0.75和I2/I1<0.75的相,以 d1d2和d1d3的编排顺序出现二次; (3)对I3/I2>0.75和I2/I1≥0.75的相,以 d1d2,d1d3和d2d3的编排顺序出现三次。
35
基于衍射强度测量的应用来自(1)物相的定量分析,结晶度的测定
衍 射 分 析 应 用
(2) 平衡相图的相界的测定;
(3) 第三类应力的测定;
(4) 有序固溶体长程有序度的测定;
(5) 多晶体材料中晶粒择优取向的极图、 反极图和三维取向分布的测定; (6) 薄膜厚度的测定。
36
基于衍射线型分析的应用 (1) 多晶材料中位错密度的测定,层 错能的测定,晶体缺陷的研究; (2) 第二类(微观残余)应力的测定; (3) 晶粒大小和微应变的测定;
12
测 角 仪 说 明 - 1
13
测 角 仪 说 明 - 2
14
15
16
17
18
19
测 角 仪 的 光 路 布 置
测角仪要求与 X射线管的线 焦斑联接使用, 线焦斑的长边 与测角仪中心 轴平行。 采用狭缝光阑 和梭拉光阑组 成的联合光阑。
20
测 角 仪 的 光 路 布 置 说 明 - 1
52
卡 片 的 内 容 ( 4 )
(6)试样来源,制备方法;化学分析, 有时亦注明升华点(S.P.),分解温 度(D.T.),转变点(T.P.),摄照 温度等。
53
卡 片 的 内 容 ( 5 )
(7)物相的化学式和名称。 在化学式 之后常有一个数字和大写英文字母的 组合说明。数字表示单胞中的原子数; 英文字母表示布拉菲点阵类型:C— 简单立方;B—体心立方;F—面心立 方;T—简单正方;U—体心正方; R—简单菱方;H—简单六方;O简单 斜方;Q—底心斜方;S—面心斜方; M—简单单斜;N—底心单斜;Z—简 单三斜。
衍 射 分 析 应 用
37
基于衍射位置和强度的测定
衍 射 分 析 应 用
(1) 物相的定性分析
(2) 相消失法测定相平衡图中的相界; (3) 晶体(相)结构,磁结构,表面结构,界 面结构的研究
38
同时基于衍射位置、强度和线型的 Rietveld多晶结构测定
需输入原子参数(晶胞中各原子的坐标、 占位几率和湿度因子)、点阵参数、波长、 偏正因子、吸收系数、择优取向参数等。
54
卡 片 的 内 容 ( 6 )
(8)矿物学名称。右上角的符号标 记表示:*—数据高度可靠;i—已指 标化和估计强度,但可靠性不如前者; O—可靠性较差;C—衍射数据来自 理论计算。 (9)晶面间距,相对强度和干涉指 数。 (10)卡片的顺序号。
55
卡 片 索 引
PDF卡片索引是一种能帮助实验者从 数万张卡片中迅速查到所需要的PDF 卡片的工具书。由JCPDS编辑出版手 册有: Hanawalt无机物检索手册; 有机相检索手册; 无机相字母索引; Fink无机索引; 矿物检索手册等品种。
21
测 角 仪 的 光 路 布 置 说 明 - 2
22
单 色 器 - 1
23
单 色 器 - 2
24
b 不 对对 称称 反反 射射 准( 聚 焦 几 何 ( 2 ) ) a Bragg Bragg b=aq; q(q)/ q ba
被测晶平面与试样表 面的夹角Y=q-a
25
26
27
28
32
影 响 衍 射 线 强 度 的 各 种 因 素
1 Ik k Fk I A Vk K t R
因 素 可调节的各因子以提高强度
I A 1、仪器特性 k 2、试样性质 3、制样及实验条件 I, A, V, K 4、衍射几何因子
Fk
R
33
小 结
Bragg 方程 方位角方程 强度方程 扫描模式
56
Hanawalt 无 机 相 数 值 索 引
索引的编排方法是:每个相作为一个 条目,在索引中占一横行。每个条目 中的内容包括: 衍射花样中八条强线的面间距和相对 强度,按相对强度递减顺序列在前面, 随后,依次排列着,化学式,卡片编 号,参比强度(I/I)。 索引说明:
57
Hanawalt 索 引 说 明 ( 一 )
III
表面反射 透过反射
Common Bragg Diffraction, Match, Transmission Sample Tilting Diffraction, Angular Dispersion Analysis
29
偶合扫描模式有对称和非对称之分, 这里对称与否是指入射线和反射线相对于 试样表面而言的,而对晶面来说都是对称 的.这是布拉格定律所规定的. 有七种不同的扫描模式,它们之间有 共性也有特性.从表中看出,对称模式只 有一种“CBD”,该模式同时具有对称、偶 合及表面反射三者的特征,因而它出现在I 及 III 中,其它六种都是非对称偶合或非对 称非偶合扫描模式.
44
定 性 相 分 析 的 判 据
通常用d(晶面 间距表征衍射线 位置)和I(衍射 线相对强度)的 数据代表衍射花 样。用d-I数据作 为定性相分析的 基本判据。
定性相分析方法 是将由试样测得 的d-I数据组与已 知结构物质的标 准d-I数据组 (PDF卡片)进 行对比,以鉴定 出试样中存在的 物相。
51
卡 片 的 内 容 ( 3 )
(4)晶体学数据:Sys.—晶系;S.G— 空间群;a0、b0、c0,α、β、γ—晶胞 参数;A= a0/b0 ,C= c0 / b0 ;Z—晶 胞中原子或分子的数目; Ref—参 考资料。 (5)光学性质: εα、nωβ、εγ—折射 率;Sign—光性正负;2V—光轴夹 角;D—密度;mp—熔点;Color— 颜色; Ref—参考资料。
60
Hanawalt 索 引 说 明 ( 四 )
1982年版本又作了进一步改进,它的 编排规则为: (1)所有的相最少都以的编排顺序 出现一次; (2)对I2/I1>0.75和I3/I1 ≤0.75的相, 以d1d2和d1d3的编排顺序出现二次; (3)对I3/I1 >0.75和I4 /I1 ≤0.75的相, 以d1d2, d2d1 和d3 d1的编排顺序出现三 次; (4)对I4/I1 >0.75的相,以d1d2,d2d1 , d3 d1和d4 d1的编排顺序出现四次;
每个条目中,衍射线的相对强度分为10个 等级,最强线为100用×表示,其余者均 以小10倍的数字表示,写在面间距d值的 右下角处。参比强度I/I是被测相与刚玉按 1:1重量配比时,被测相最强线峰高与刚 玉最强线峰高之比。