LY2228820_862507-23-1_DataSheet_MedChemExpress
2015-2021年FDA批准的505b2药物

APPLICATIONNUMBERPROPRIETARY NAME ESTABLISHED NAME通用名APPLICANTNDA208746(1)PEMETREXED培美曲塞HOSPIRA INC NDA 206610ACETAMINOPHEN对乙酰氨基酚RISINGNDA 214313NOREPINEPHRINEBITARTRATE IN 5%DEXTROSENOREPINEPHRINEBITARTRATE酒石酸去甲肾上腺素BAXTER HEALTHCARECORPNDA 204803POSIMIR BUPIVACAINE布比卡因DURECT CORPNDA 204957ACETAMINOPHEN对乙酰氨基酚B BRAUN MEDICAL INCNDA 212994AZSTARYS SERDEXMETHYLPHENIDATE ANDDEXMETHYLPHENIDATE对甲苯磺酸盐和对甲苯磺酸盐COMMAVETHERAPEUTICS SANDA 211844MIDAZOLAM咪达唑仑INFORLIFE SANDA 213072ROSZET ROSUVASTATIN ANDEZETIMIBE瑞舒伐他汀和依折麦布ALTHERAPHARMACEUTICALSLLCNDA 214154NEXTSTELLIS DROSPIRENONE ANDESTETROL TABLETS屈螺酮和雌醇片MAYNE PHARMA LLCNDA 212045KLOXXADO NALOXONE HCL盐酸纳洛酮HIKMA PHARMACEUTICALS USA INCNDA214657(1)PEMETREXED SANDOZ INCNDA 211988ZYNRELEF BUPIVACAINE ANDMELOXICAM布比卡因和美洛昔康HERON THERAPEUTICSINCNDA 214253LEVOTHYROXINESODIUMCUSTOPHARM INCNDA 211488CAMCEVI LEUPROLIDE亮丙瑞林FORESEE PHARMACEUTICALS CO LTDNDA 214846MYFEMBREE RELUGOLIX 40 MG,ESTRADIOL 1 MG,AND NORETHINDRONEACETATE 0.5MGRELUGOLIX 40 mg、雌二醇 1 mg 和醋酸炔诺酮0.5 mgMYOVANT SCIENCESGMBHNDA 213378LYBALVI OLANZAPINE ANDSAMIDORPHAN奥氮平和沙美芬ALKERMES INCNDA 215025SODIUMPHENYLACETATE ANDSODIUM BENZOATE苯乙酸钠和苯甲酸钠MAIAPHARMACEUTICALSINCNDA 213218SOAANZ TORSEMIDE托拉塞米SARFEZ PHARMACEUTICALS INCNDA 213536REZIPRES EPHEDRINEHYDROCHLORIDE盐酸麻黄碱ETONPHARMACEUTICALSINCNDA 212156MICAFUNGIN米卡芬净PAR STERILE PRODUCTS LLCNDA 210282DAPTOMYCIN达托霉素HOSPIRA INC NDA 214965VERKAZIA CYCLOSPORINE环孢素SANTEN INCNDA 212303(1)DULUTEGRAVIR,LAMIVUDINE, ANDTENOFOVIRDISOPROXIL度替拉韦、拉米夫定和富马酸替诺福韦酯LUPIN LTDNDA 214902TWYNEO TRETINOIN ANDBENZOYL PEROXIDE维 A 酸和过氧化苯甲酰SOL-GELTECHNOLOGIES LTDNDA 215143SUCCINYLCHOLINECHLORIDE琥珀胆碱氯化物HIKMAPHARMACEUTICALSUSA INCNDA 210735CYCLOPHOSPHAMIDE环磷酰胺EUGIA PHARMA SPECIALITIES LTDNDA 213895VANCOMYCIN万古霉素XELLIA PHARMACEUTICALS APSNDA 214826LOREEV XR LORAZEPAM劳拉西泮ALMATICA PHARMA LLCNDA 211566(1)SITAGLIPTIN西他列汀ZYDUS WORLDWIDEDMCCNDA 213436TRUDHESA DIHYDROERGOTAMINEMESYLATE甲磺酸二氢麦角胺IMPEL NEUROPHARMANDA 215133SERTRALINEHYDROCHLORIDE盐酸舍曲林ALMATICA PHARMALLCNDA 212854ZIMHI NALOXONEHYDROCHLORIDE盐酸纳洛酮ADAMISPHARMACEUTICALSCORPNDA 213426SEGLENTIS CELECOXIB ANDTRAMADOLHYDROCHLORIDE塞来昔布和盐酸曲马多KOWAPHARMACEUTICALSAMERICA INCNDA 213978TYRVAYA VARENICLINESOLUTION伐尼克兰溶液OYSTER POINTPHARMA INCNDA 211950XIPERE TRIAMCINOLONEACETONIDE曲安奈德BAUSCH AND LOMBINCNDA 214028VUITY PILOCARPINEHYDROCHLORIDE盐酸匹洛卡品ABBVIE INCNDA 210526DYANAVEL XR AMPHETAMINE安非他明TRIS PHARMA INCNDA 213005(1)YUTREPIA TREPROSTINIL曲前列尼尔LIQUIDIATECHNOLOGIESNDA 214679EPRONTIA TOPIRAMATE托吡酯AZURITY PHARMACEUTICALS INCNDA 214869DHIVY CARBIDOPA ANDLEVODOPACARBIDOPA 和左旋多巴AVIONPHARMACEUTICALSLLCNDA 215668(1)BENDAMUSTINEHYDROCHLORIDE盐酸苯达莫司汀DR REDDYSLABORATORIES LTDNDA 213312FYARRO SIROLIMUS PROTEIN-BOUND PARTICLESSIROLIMUS 蛋白结合颗粒AADI BIOSCIENCEINCNDA 215422LYVISPAH BACLOFEN巴氯芬SAOL THERAPEUTICS RESEARCH LTDNDA 215650XACIATO CLINDAMYCINPHOSPHATE磷酸克林霉素DARE BIOSCIENCEINCNDA 215423ENTADFI TADALAFIL ANDFINASTERIDE他达拉非和非那雄胺VERU INCNDA 215935TARPEYO BUDESONIDE布地奈德CALLIDITAS THERAPEUTICS ABNDA 215019DARTISLA ODT GLYCOPYRROLATE甘氨酰吡咯酸EDENBRIDGE PHARMACEUTICALS LLCNDA 2I4032ILLUCCIX KIT FOR THEPREPARATION OF GA-68 PSMA-11GA-68 PSMA-11 制备试剂盒TELIXPHARMACEUTICALS USINCNDA 215395LANREOTIDE ACETATE醋酸来那度胺INVAGEN PHARMACEUTICALS INCNDA 214133RECORLEV LEVOKETOCONAZOLE左酮康唑STRONGBRIDGE DUBLIN LTDREVIEW CLASSIFI CATION 505(B)(2)APPROVALAPPROVAL DATE批准类型S Y1/8/2021Type 3 - New Dosage FormS Y1/15/2021Type 5 - New Formulation or New ManufacturerS Y1/15/2021Type 5 - New Formulation or New ManufacturerS Y2/1/2021Type 3 - New Dosage FormS Y2/18/2021Type 5 - New Formulation or New ManufacturerS Y3/2/2021Type 1 - New Molecular Entity and Type 4 - New CombinationS Y3/22/2021Type 5 - New Formulation or New ManufacturerS Y3/23/2021Type 4 - New CombinationS Y4/15/2021Type 1 - New Molecular EntityS Y4/29/2021Type 5 - New Formulation or New ManufacturerS Y5/6/2021Type 5 - New Formulation or New ManufacturerP Y5/12/2021Type 4 - New CombinationS Y5/17/2021Type 5 - New Formulation or New ManufacturerS Y5/25/2021Type 2 - New Active IngredientS Y5/26/2021Type 4 - New CombinationS Y5/28/2021Type 1 - New Molecular Entity and Type 4 - New CombinationS Y6/10/2021Type 5 - New Formulation or New ManufacturerS Y6/14/2021Type 5 - New Formulation or New ManufacturerS Y6/14/2021Type 5 - New Formulation or New ManufacturerNew ManufacturerS Y6/21/2021Type 5 - New Formulation or New ManufacturerS,O Y6/23/2021Type 5 - New Formulation or New ManufacturerS Y6/25/2021Type 4 - New Combination S Y7/26/2021Type 4 - New CombinationS Y8/20/2021Type 5 - New Formulation or New ManufacturerS Y8/25/2021Type 5 - New Formulation or New ManufacturerP Y8/26/2021Type 5 - New Formulation or New ManufacturerS Y8/27/2021Type 3 - New Dosage FormS Y9/2/2021Type 2 - New Active IngredientS Y9/2/2021Type 5 - New Formulation or New ManufacturerS Y10/4/2021Type 3 - New Dosage FormS Y10/15/2021Type 5 - New Formulation or New ManufacturerS Y10/15/2021Type 4 - New Combination S Y10/15/2021Type 3 - New Dosage Form S Y10/22/2021Type 3 - New Dosage FormS Y10/28/2021Type 5 - New Formulation or New ManufacturerS Y11/4/2021Type 3 - New Dosage Form S Y11/4/2021Type 3 - New Dosage FormS Y11/5/2021Type 3 - New Dosage FormS Y11/12/2021Type 5 - New Formulation or New ManufacturerNew ManufacturerP,O Y11/22/2021Type 5 - New Formulation or New ManufacturerS Y11/22/2021Type 3 - New Dosage FormP Y12/7/2021Type 5 - New Formulation or New ManufacturerS Y12/9/2021Type 4 - New CombinationP,O Y12/15/2021Type 5 - New Formulation or New ManufacturerS Y12/16/2021Type 3 - New Dosage FormS Y12/17/2021Type 3 - New Dosage Form and Type 4 - New CombinationS Y12/17/2021Type 5 - New Formulation or New ManufacturerS,O Y12/30/2021Type 2 - New Active Ingredient适应症NSCLC、Mesothelioma间皮瘤(注射剂)止痛、退烧(注射剂)升高低血压(注射剂)局部麻醉(注射剂)止痛、退烧CNS兴奋剂注射麻醉剂降低LDL-C预防妊娠阿片类拮抗剂,用于阿片类过量NSCLC、Mesothelioma间皮瘤术后止痛(注射剂)粘液性水肿昏迷myxedema coma晚期前列腺癌子宫平滑肌瘤月经量过多成人精神分裂症、成人双相 i 型障碍急性高血氨症成人心力衰竭、肾病相关水肿麻醉状态的低血压念珠菌血症、急性播散性念珠菌病、念珠菌腹膜炎和脓肿cSSSI、Bacteremia菌血症春季角结膜炎(Tentative Approval)痤疮局部治疗作为全身麻醉的辅助治疗、便于气管插管、在手术或机械通气期间提供骨骼肌松弛label not available败血症、感染性心内膜炎、皮肤和皮肤结构感染、骨感染、下呼吸道感染、艰难梭菌相关性腹泻、金黄色葡萄球菌引起的小肠结肠炎label not availablelabel not available偏头痛急性治疗MDD、OCD紧急治疗阿片类药物过量治疗需要阿片类镇痛剂且替代治疗效果不佳的成人急性疼痛。
碧云天生物技术 Beyotime Biotechnology 生物素标记EMSA探针说明书

碧云天生物技术/Beyotime Biotechnology订货热线:400-168-3301或800-8283301订货e-mail:******************技术咨询:*****************网址:碧云天网站微信公众号生物素标记EMSA探针-β-Catenin/TCF (0.2μM)产品编号产品名称包装GS018B 生物素标记EMSA探针-β-Catenin/TCF (0.2µM) 200µl产品简介:生物素标记EMSA探针-β-Catenin/TCF是用于EMSA(也称gel shift)研究的生物素(Biotin)标记的β-Catenin/TCF consensus oligonucleotide。
这个生物素标记的双链寡核苷酸含有公认的β-Catenin/TCF结合位点,可以用作EMSA研究时的探针。
β-Catenin/TCF consensus oligo的序列如下:5'-CCC TTT GAT CTT ACC-3'3'-GGG AAA CTA GAA TGG-5'本生物素标记EMSA探针已经过纯化,可以直接用于EMSA结合反应。
本生物素标记EMSA探针可以和碧云天的化学发光法EMSA试剂盒(GS009)配套使用。
一个包装的生物素标记探针可以进行约200-400个样品的EMSA检测。
包装清单:产品编号产品名称包装GS018B 生物素标记EMSA探针-β-Catenin/TCF (0.2µM) 200µl—说明书1份保存条件:-20ºC保存,一年有效。
注意事项:避免加热到40ºC以上,温度过高会导致双链DNA探针解聚成单链。
而单链无法用于EMSA研究。
对于基于生物素标记的EMSA检测的详细操作可以参考碧云天的化学发光法EMSA试剂盒(GS009)的使用说明。
本产品仅限于专业人员的科学研究用,不得用于临床诊断或治疗,不得用于食品或药品,不得存放于普通住宅内。
Trigonox B(滴苷但羊水)产品数据表单说明书

Product Data SheetTrigonox BDi-tert-butyl peroxideTrigonox® B is a pure peroxide in liquid form.CAS number110-05-4EINECS/ELINCS No. 203-733-6TSCA statuslisted on inventory Molecular weight 146.2Active oxygen contentperoxide10.94%SpecificationsAppearance Clear liquidAssay≥ 99.0 %ApplicationsTrigonox® B (Di-tert-butyl peroxide) can be used for the market segments: polymer production, polymer crosslinking and acrylics production with their different applications/functions. For more information please check our website and/or contact us.Half-life dataThe reactivity of an organic peroxide is usually given by its half-life (t½) at various temperatures. For Trigonox® B in chlorobenzene half-life at other temperatures can be calculated by using the equations and constants mentioned below:0.1 hr at 164°C (327°F)1 hr at 141°C (286°F)10 hr at 121°C (250°F)Formula 1kd = A·e-Ea/RTFormula 2t½ = (ln2)/kdEa153.46 kJ/moleA 4.20E+15 s-1R8.3142 J/mole·KT(273.15+°C) KThermal stabilityOrganic peroxides are thermally unstable substances which may undergo self-accelerating decomposition. The lowest temperature at which self-accelerating decomposition may occur with a substance in the packaging as used for transport is the Self-Accelerating Decomposition Temperature (SADT). The SADT is determined on the basis of the Heat Accumulation Storage Test.SADT80°C (176°F)Method The Heat Accumulation Storage Test is a recognized test method for thedetermination of the SADT of organic peroxides (see Recommendations on theTransport of Dangerous Goods, Manual of Tests and Criteria - United Nations, NewYork and Geneva).StorageDue to the relatively unstable nature of organic peroxides, a loss of quality will occur over a period of time. To minimize the loss of quality, Nouryon recommends a maximum storage temperature (Ts max. ) for each organic peroxide product.Ts Max.40°C (104°F) andTs Min.-30°C (-22°F) to prevent crystallizationNote When stored according to these recommended storage conditions, Trigonox® Bwill remain within the Nouryon specifications for a period of at least 6 months afterdelivery.Packaging and transportIn North America Trigonox® B is packed in non-returnable, five gallon polyethylene containers of 30 lb net weight and steel drums of 100 or 340 lb net weight. In other regions the standard packaging is a 30-liter HDPE can (Nourytainer®) for 20 kg peroxide. Delivery in a 200 l steel drum for 150 kg peroxide is also possible in a number of countries. Both packaging and transport meet the international regulations. For the availability of other packed quantities consult your Nouryon representative. Trigonox® B is classified as Organic peroxide type E; liquid, Division 5. 2; UN 3107.Safety and handlingKeep containers tightly closed. Store and handle Trigonox® B in a dry well-ventilated place away from sources of heat or ignition and direct sunlight. Never weigh out in the storage room. Avoid contact with reducing agents (e. g. amines), acids, alkalis and heavy metal compounds (e. g. accelerators, driers and metal soaps). Please refer to the Safety Data Sheet (SDS) for detailed information on the safe storage, use and handling of Trigonox® B. This information should be thoroughly reviewed prior to acceptance of this product. The SDS is available at /sds-search.Major decomposition productsAcetone, Methane, tert-ButanolAll information concerning this product and/or suggestions for handling and use contained herein are offered in good faith and are believed to be reliable.Nouryon, however, makes no warranty as to accuracy and/or sufficiency of such information and/or suggestions, as to the product's merchantability or fitness for any particular purpose, or that any suggested use will not infringe any patent. Nouryon does not accept any liability whatsoever arising out of the use of or reliance on this information, or out of the use or the performance of the product. Nothing contained herein shall be construed as granting or extending any license under any patent. Customer must determine for himself, by preliminary tests or otherwise, the suitability of this product for his purposes.The information contained herein supersedes all previously issued information on the subject matter covered. The customer may forward, distribute, and/or photocopy this document only if unaltered and complete, including all of its headers and footers, and should refrain from any unauthorized use. Don’t copythis document to a website.Trigonox® and Nourytainer are registered trademarks of Nouryon Functional Chemicals B.V. or affiliates in one or more territories.Contact UsPolymer Specialties Americas************************Polymer Specialties Europe, Middle East, India and Africa*************************Polymer Specialties Asia Pacific************************2022-6-30© 2022Polymer crosslinking Trigonox B。
华蟾毒素(CBG)酶联免疫吸附检测试剂盒 (ELK8271) 说明书

华蟾毒素(CBG)酶联免疫吸附检测试剂盒货号:ELK8271规格:96T(本试剂盒仅供体外研究使用,不用于临床诊断)灵敏度:91.3ng/mL检测范围:312.5-20000ng/mL特异性:可检测重组或天然的CBG,且不与其它相关蛋白交叉反应。
试剂盒组成中文名称英文名称规格保存条件48T96T酶标板(可拆)Pre-coated Microplate 6条x8孔12条x8孔4°C/-20°C(6个月)冻干标准品Standard(lyophilized)124°C/-20°C(6个月)标准品&样品稀释液Standard/Sample Diluent Buffer10mL20mL4°C生物素结合物(100×)Biotinylated-Conjugate(100x)30μL60μL4°C/-20°C(6个月)生物素结合物稀释液Biotinylated Conjugate Diluent5mL10mL4°C浓缩HRP酶结合物(100×)Streptavidin-HRP(100×)60μL120μL4°C/-20°C(6个月)酶结合物稀释液HRP Diluent6mL12mL4°C浓缩洗涤液(25×)Wash Buffer(25×)10mL20mL4°C显色底物溶液(TMB)TMB Substrate Solution6mL10mL4°C(避光)反应终止液Stop reagent3mL6mL4°C封板覆膜Plate Covers12常温产品说明书Instruction manual1份1份常温特别说明1.打开包装后请及时检查所有物品是否齐全完整。
所有试剂的批号见标签。
2.试剂盒保存:-20°C(长时间存放,试剂按照标签提示温度保存);2-8°C(一周内使用);避免反复冻融,过期后请勿使用。
富马酸替诺福韦酯--------印度药典

NOTE - Prepare the solutions immediately before use. Test solution. Dissolve 100 mg of the substance under examination in 50 ml of methanol. Reference solution (a). A 0.2 per cent w/v solution of tenofovir disoproxil jitmarate RS in methanol. Reference solution (b). Dilute 1.0 ml ofreference solution (a) to 100.0 ml with methanol. Reference solution (c). Dissolve 10 mg ofthejUmaric acid in 50 ml of methanol.
TELMISARTAN TABLETS
IP 2010
Solvent mixture. 80 volumes of buffer solution prepared by diluting 5.0 ml of triethylamine to 2000ml with water and 20 volumes of methanol. Test solution. Weigh and powder 20 tablets. Disperse a quantity ofpowder containing about 100 mg ofTelmisartan in 100.0 ml ofsolvent mixture, sonicate for 45 minutes and filter. Reference solution. A 0.0005 per cent w/v solution of telmisartan RS in the solvent mixture.
接触碟产品目录

5
产品规格
90 mm 预灌装培养基平皿:用于空气中沉降菌的测定以及实验室 相关微生物指标的测定等。 75 mm 预灌装培养基平皿:用于 MILLIPORE 浮游生物检测仪配 套使用。 55 mm 预灌装培养基接触性平皿(Rodac Plate 接触碟) :用于设 备、车间、人员、包装材料等表面微生物的测定。
海南办事处 海口市南沙路 78 号南沙公寓 320 室 570206 电话: (0898)66703090 传真: (0898)66703090
8
2
产品用途
本品适用于物品表面(洁净厂房、机械设备、洁净服、包装材料等)表 面微生物的检测。广泛应用于制药、食品、医院以及化妆品等企事业单位 的环境微生物检测,比传统的棉签方法更简便、准确。可用于对洁净环境 中的微生物进行动态/静态监测,以及检验消毒效果等。
培养基种类(进口) :
∗ 胰蛋白大豆琼脂(TSA) ∗ 含卵磷脂及吐温 80 胰蛋白大豆琼脂(TSAWLP) ∗ 沙堡氏琼脂(SDA) ∗ 麦康凯琼脂(MCA) ∗ 营养琼脂 NA ∗ 玫瑰红纳琼脂 RBA、R2A 等 可在培养基中添加青霉素酶等催化酶,用于去除生产中产生的抗生素 粉尘的抗菌性。
江苏益玛生物科技有限公司(EM-BIO) ,成立于 2011 年 7 月,注册资
金 500 万元。公司创建于国内唯一的国家级医药产业园区——江苏泰州・中国医 药城,是国内首家与国外著名制药企业合作,按照欧盟 EMEA 标准,开发无菌环 境检测产品的民营企业;公司紧跟新版 GMP 要求,研发出高水平、高质量的预灌 装培养基平皿系列产品,产品的灌装及包装等主要环节,均在 B 级背景下的 A 级 半隔离器中完成,为国内首创。产品经与国际知名企业产品对照试验,性能优良, 反应灵敏,品质稳定,完全达到欧盟相关技术要求。目前,已有多家国内外知名 药企使用本公司产品。
FDA批准的精准医疗诊断体外器械一览表List of Cleared or Approved Companion Diagnostic Devices
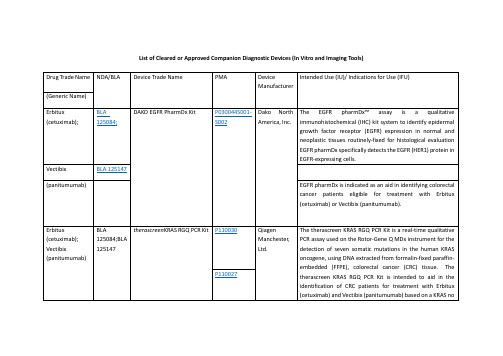
Drug Trade Name
NDA/BLA
Device Trade Name
PMA
Device Manufacturer
Intended Use (IU)/ Indications for Use (IFU)
(imatinibmesylate)
NDA 021588
The c-KitpharmDxis indicated as an aid in the differential diagnosis of gastrointestinal stromal tumors (GIST). After diagnosis of GIST, results from c-KitpharmDxmay be used as an aid in identifying those patients eligible for treatment withGleevec/Glivec(imatinibmesylate).
(deferasirox)
Gilotrif
NDA 201292
therascreenEGFR RGQ PCR Kit
P120022
QiagenManchester, Ltd.
ThetherascreenEGFR RGQ PCR Kit is a real-time PCR test for the qualitative detection of exon 19 deletions and exon 21 (L858R) substitution mutations of the epidermal growth factor receptor (EGFR) gene in DNA derived from formalin-fixed paraffin-embedded (FFPE) non-small cell lung cancer (NSCLC) tumor tissue. The test is intended to be used to select patients with NSCLC for whom GILOTRIF (afatinib), an EGFR tyrosine kinase inhibitor (TKI), is indicated. Safety and efficacy of GILOTRIF (afatinib) have not been established in patients whose tumors have L861Q, G719X, S768I, exon 20 insertions, and T790M mutations, which are also detected by thetherascreenEGFR RGQ PCR Kit.
糖尿病患者诊断应用血清C肽及糖化血红蛋白联合检测的价值分析

DOI:10.16658/ki.1672-4062.2023.14.085糖尿病患者诊断应用血清C肽及糖化血红蛋白联合检测的价值分析倪胜南,陈少,陈一鸣泗阳康达医院检验科,江苏宿迁223700[摘要]目的探讨糖尿病患者诊断应用血清C肽联合糖化血红蛋白检测的价值。
方法将2022年1月—2023年1月泗阳康达医院收治的74例疑似糖尿病患者作为研究对象,检测入组患者糖化血红蛋白(glycosylated hemoglobin, HbA1c)以及血清C肽水平,以口服葡萄糖耐量试验(glucose tolerance test check, OGTT)为金标准,统计血清C肽联合糖化血红蛋白检测与单一项目检测的敏感性、特异度和诊断符合率。
结果74例疑似糖尿病患者根据葡萄糖耐量试验结果,确诊患者67例,确诊率为90.54%(67/74);与血清C肽、HbA1c单一检测相比,血清C肽+HbA1c联合检测敏感度更高,差异有统计学意义(P<0.05);血清C肽+HbA1c联合检测的特异度略高于血清C肽、HbA1c单一检测,但差异无统计学意义(P>0.05);联合检测诊断符合率明显高于血清C 肽、HbA1c单项检测,差异有统计学意义(P<0.05)。
结论血清C肽与糖化血红蛋白是临床诊断糖尿病的重要参考指标,二者表达水平的变化有助于检测患者胰岛素分泌功能,评估疾病严重程度,两者联合检验灵敏性与特异度良好,有助于早期明确诊断,临床参考价值较高。
[关键词] 糖尿病;血清C肽;糖化血红蛋白;诊断价值[中图分类号] R446.1 [文献标识码] A [文章编号] 1672-4062(2023)07(b)-0085-04Analysis of the Value of the Diagnostic Application of Combined Serum C-peptide and Glycosylated Hemoglobin Testing in Patients with Diabetes MellitusNI Shengnan, CHEN Shao, CHEN YimingDepartment of Laboratory Medicine, Siyang Kangda Hospital, Suqian, Jiangsu Province, 223700 China[Abstract] Objective To explore the value of applying serum C-peptide combined with glycated hemoglobin test for the diagnosis of diabetic patients. Methods A total of 74 patients with suspected diabetes admitted to Siyang Kangda Hospital from January 2022 to January 2023 were selected as the research objects. The levels of glycosylated hemoglo‐bin (HbA1c) and serum C-peptide were detected. Oral glucose tolerance test (OGTT) was used as the gold standard. The sensitivity, specificity and diagnostic coincidence rate of serum C-peptide combined with glycosylated hemoglo‐bin detection and single item detection were statistically analyzed. Results According to the results of glucose toler‐ance test, 67 patients were diagnosed in 74 patients with suspected diabetes, and the diagnosis rate was 90.54% (67/ 74). Compared with the single detection of serum C-peptide and HbA1c, the sensitivity of combined detection of se‐rum C peptide and HbA1c was higher, and the difference was statistically significant (P<0.05). The specificity of com‐bined detection of serum C-peptide and HbA1c was slightly higher than that of single detection of serum C-peptide and HbA1c, but the difference was no statistically significant (P>0.05). The diagnostic coincidence rate of combined detection was significantly higher than that of single detection of serum C-peptide and HbA1c, and the difference was statistically significant (P<0.05). Conclusion Serum C-peptide and glycosylated hemoglobin are important reference indexes for clinical diagnosis of diabetes mellitus, and changes in the expression levels of the two can help to detect the insulin secretion function of patients and assess the severity of the disease. The sensitivity and specificity of the [作者简介]倪胜南(1991-),女,本科,主管检验师,研究方向为免疫学、分子生物学检验。
XL228-SDS-MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:Oct.-06-2018Print Date:Oct.-06-20181. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :XL228Catalog No. :HY-15749CAS No. :898280-07-41.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:************************1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureNot a hazardous substance or mixture.2.2 GHS Label elements, including precautionary statementsNot a hazardous substance or mixture.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:XL-228;XL 228Formula:C22H31N9OMolecular Weight:437.54CAS No. :898280-07-44. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation. 7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature:Powder-20°C 3 years4°C 2 yearsIn solvent-80°C 6 months-20°C 1 monthShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Clean spillages in asafe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance White to off-white (Solid)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGThis substance is considered to be non-hazardous for transport.IATAThis substance is considered to be non-hazardous for transport.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2018 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel:609-228-6898Fax:609-228-5909E-mail:***********************Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
盐酸利多卡因注射剂遗传毒性杂质研究_NormalPdf

Journal of China Pharmaceutical University2020,51(4):466-471学报盐酸利多卡因注射剂遗传毒性杂质研究冼芷然1,孙春萌2,骆雪芳1*,钟文英1**(1中国药科大学理学院药物质量研究中心,南京211198;2中国药科大学药学院,南京211198)摘要确定2,6-二甲基苯胺为盐酸利多卡因注射液中遗传毒性杂质,N-氯乙酰-2,6-二甲基苯胺为潜在遗传毒性杂质,建立LC-MS/MS方法,用色谱柱Agilent ZORBAX Eclipse Plus C18(4.6mm×250mm,5μm)对原料、自制制剂及原研制剂进行遗传毒性杂质研究。
研究结果表明自制制剂中杂质2,6-二甲基苯胺与N-氯乙酰-2,6-二甲基苯胺除由原料引入外,可能分别由氧化条件或碱性条件下降解引入,为盐酸利多卡因注射液的遗传毒性风险评估和工艺优化提供参考与指导。
关键词盐酸利多卡因注射液;遗传毒性杂质;LC-MS/MS中图分类号R917文献标志码A文章编号1000-5048(2020)04-0466-06doi:10.11665/j.issn.1000-5048.20200412引用本文冼芷然,孙春萌,骆雪芳,等.盐酸利多卡因注射剂遗传毒性杂质研究[J].中国药科大学学报,2020,51(4):466–471.Cite this article as:XIAN Zhiran,SUN Chunmeng,LUO Xuefang,et al.Profiling of genotoxic impurities in a lidocaine hydrochloride injec‐tion[J].J China Pharm Univ,2020,51(4):466–471.Profiling of genotoxic impurities in a lidocaine hydrochloride injection XIAN Zhiran1,SUN Chunmeng2,LUO Xuefang1*,ZHONG Wenying1**1Drug Quality Research Center,College of Science,China Pharmaceutical University;2School of Pharmacy,China Pharmaceutical University,ChinaAbstract2,6-dimethylbenzenamine was determined as a genotoxic impurity in lidocaine hydrochloride injec‐tion,and2-chloro-N-(2,6-dimethylphenyl)acetamide was determined as potential genotoxic impurity.An LC-MS/ MS method was established to research the profiling of genotoxic impurities in active pharmaceutical ingredients (API),homemade preparation and reference preparation on column Agilent ZORBAX Eclipse Plus C18(4.6mm×250mm,5μm).The results show that in the homemade preparation the2,6-dimethylbenzenamine and the 2-chloro-N-(2,6-dimethylphenyl)acetamide may be degraded under oxidation condition and alkaline condition in addition to the introduction from API preparation process.This study provides guidance for genotoxic risk assess‐ment and prescription process optimization of lidocaine hydrochloride.Key words lidocaine hydrochloride injection;genotoxic impurities;LC-MS/MS盐酸利多卡因(lidocaine hydrochloride)为临床上常制成盐酸利多卡因注射剂应用于局部麻醉药[1]和抗心律失常药物等[2-3]。
白术等中草药对仔猪腹泻病防控和促生长效果研究

专题论述20231254白术等中草药对仔猪腹泻病防控和促生长效果研究马大才(广西鹿寨县寨沙镇农业农村服务中心,广西柳州 545604)摘要:仔猪腹泻是指仔猪因消化系统疾病引起的腹泻现象,在生猪养殖阶段是尤为突出的一类疾病,如果不能合理控制,会使大量仔猪出现死亡情况。
导致仔猪出现腹泻的原因非常多,养殖户需对其不断探索和分析,依照实际情况选用相匹配的方法予以解决。
主要介绍了仔猪腹泻发病原因,探讨白术等中草药对仔猪腹泻病防控的优势及其具体应用,为相关人士提供参考。
关键词:仔猪腹泻病;白术;中草药Prevention and Control of Diarrhea Disease in Piglets with Chinese Herbs such as Atractylodes macrocephalaResearch on growth promoting effectsMa Da-cai(Agricultural and Rural Service Center, Zhaisha Town, Luzhai County,Liuzhou,Guangxi 545604)Abstract: Piglet diarrhea refers to the phenomenon of diarrhea caused by digestive system diseases in piglets, which is particularly prominent in the pig breeding stage. If not properly controlled, it can lead to a large number of piglets dying. There are many causes of diarrhea in piglets, and farmers need to continuously explore and analyze them, and choose matching methods to solve them according to the actual situation. This article mainly introduces the causes of piglet diarrhea, explores the advantages and specific applications of Chinese herbal medicines such as Atractylodes macrocephala in the prevention and control of piglet diarrhea, and provides reference for relevant personnel.Key words: Piglet diarrhea disease;Atractylodes macrocephala;Chinese herbal medicine 中图分类号:S858.28 文献标志码:B 文章编号:1003-8655(2023)01-0094-03近年来,随着养殖业逐渐增多,规模越来越大,仔猪腹泻病发病率也呈上升趋势。
IFCC Aspartate Aminotransferase 检测手册说明书

ASTAspartate Aminotransferase IFCCMANUAL RX MONZAINTENDED USEFor the quantitative in vitro determination of AspartateAminotransferase (AST) in serum and plasma. This product is suitable for manual use and on the Rx Monza analyser.Cat. No. AS 1202 R1a. Buffer/Substrate 1 x 70 ml 20 x 2 ml R1b. Enzyme/Coenzyme/ 20 x 2 ml α-oxoglutarate GTIN: 05055273200416AS 1204 R1a. Buffer/Substrate 1 x 105 ml 10 x 10 ml R1b. Enzyme/Coenzyme/ 10 x 10 ml α-oxoglutarate GTIN: 05055273200423AS 1267 R1a. Buffer/Substrate 1 x 105 ml 5 x 20 ml R1b. Enzyme/Coenzyme/ 5 x 20 ml α-oxoglutarate GTIN: 05055273200430AS 2359 R1a. Buffer/Substrate 5 x 100 ml 5 x 100 ml R1b. Enzyme/Coenzyme/ 5 x 100 ml α-oxoglutarate GTIN: 05055273200454UV METHODThis is an optimised standard method according to the concentrations recommended by the IFCC.CLINICAL SIGNIFICANCE (1,2,3,4)The aminotransferases are a group of enzymes that catalyse the inter conversions of amino acids and α-oxoacids by transfer of amino groups. AST (aspartate aminotransferase or glutamate oxaloacetatetransaminase) has been found in the cytoplasm and the mitochondria of cells that have been studied. In cases of mild tissue damage, e.g. liver, the predominant form of serum AST is that from the cytoplasm, with a smaller amount coming from the mitochondria. Severe tissue damage will result in more mitochondrial enzyme being released. Elevated levels of AST can signal myocardial infarction, hepatic disease, muscular dystrophy and organ damage.Although heart muscle is found to have the most activity of the enzyme, significant activity has also been seen in the brain, liver, gastric mucosa, adipose tissue and kidneys of humans.The IFCC has now recommended (1980) standardised procedures for AST determinations including:-1. optimization of substrate concentrations.2. Employment of Tris buffers (instead of phosphate, which has beenshown to inhibit recombination of the apoenzyme with pyridoxal phosphate).3. Pre-incubation of combined buffer and serum to allow sidereactions with NADH to occur. 4. Substrate start (α-oxoglutarate)5. Optional pyridoxal phosphate activation.This is an optimised standard method according to the recommendations of the IFCC.PRINCIPLEα-oxoglutarate reacts with L-aspartate in the presence of AST to form L-glutamate plus oxaloacetate. The indicator reaction utilises the oxaloacetate for a kinetic determination of NADH consumption. AST -oxoglutarate + L-aspartate L-glutamate + oxaloacetate MDH oxaloacetate + NADH + H + L-malate + NAD +SPECIMEN COLLECTION AND PREPARATION (5) Serum:- Use serum free from haemolysis.Plasma:- EDTA or heparin can be used as the anticoagulant.Plasma should be separated from cells within one hour after collection.Specimens should be refrigerated if not used immediately:-Specimens stored longer than 3 days should be frozen at -20︒C.REAGENT COMPOSITIONContents Concentrations in the TestR1a. Buffer/Substrate Tris buffer 80 mmol/l, pH 7.5 L-aspartate 240 mmol/l R1b. Enzyme/Coenzyme/α-oxoglutarate α-oxoglutarate 12 mmol/l MDH ≥420 U/l LD ≥600 U/l NADH 0.18 mmol/lSAFETY PRECAUTIONS AND WARNINGS For in vitro diagnostic use only. Do not pipette by mouth.Exercise the normal precautions required for handling laboratory reagents.Solution R1a contains Sodium Azide. Avoid ingestion or contact with skin or mucous membranes. In case of skin contact, flush affected area with copious amounts of water. In case of contact with eyes or if ingested, seek immediate medical attention.Sodium Azide reacts with lead and copper plumbing, to form potentially explosive azides. When disposing of such reagents flush with large volumes of water to prevent azide build up. Exposed metal surfaces should be cleaned with 10% sodium hydroxide.Health and Safety data sheets available on request.The reagents must be used only for the purpose intended by suitably qualified laboratory personnel, under appropriate laboratory conditions.STABILITY AND PREPARATION OF REAGENTS R1a. Buffer/SubstrateContents ready for use. Stable up to the expiry date when stored at +2 to +8︒C.R1b. Enzyme/Coenzyme/α-oxoglutarate Reconstitute one vial of Enzyme/Coenzyme/α-oxoglutarate R1b with the appropriate volume of Buffer/Substrate R1a: 2 ml for the 20 x 2 ml kit (AS 1202) 10 ml for the 10 x 10 ml kit (AS 1204) 20 ml for the 5 x 20 ml kit (AS 1267) Stable for 14 days at +2 to +8︒C or 24 hours at +15 to +25︒C. Cat. AS 2359 5 x 100 mlReconstitute one vial of Enzyme/Coenzyme/α-oxoglutarate R1b with a portion of Buffer/Substrate R1a and then transfer the entire contents to bottle R1a rinsing bottle R1b several times. Stable for 14 days at +2 to +8︒C or 24 hours at +15 to +25︒C.MATERIALS PROVIDED Buffer/SubstrateEnzyme/Coenzyme/ -oxoglutarateMATERIALS REQUIRED BUT NOT PROVIDEDRandox Assayed Multisera Level 2 (Cat. No. HN 1530) and Level 3 (Cat. No. HE 1532)Randox Calibration Serum Level 3 (Cat. No. CAL 2351) RX series Saline (Cat. No. SA 3854)PROCEDUREAspirate fresh ddH 2O and perform a new Gain Calibration in flow cell mode. Select AST in the Run Test screen and carry out a water blank as instructed.Pipette into a test tube:Sample 0.05 ml Reagent 0.5 mlMix and aspirate into the Rx Monza.CALIBRATION FOR RX MONZAThe use of Saline and Randox Calibration Serum Level 3 isrecommended for calibration. Calibration is recommended with change of reagent lot or as indicated by quality control procedures.FOR MANUAL USEWavelength: 340 nm (Hg 334 nm or Hg 365 nm) Cuvette: 1 cm light path Temperature: 25/30/37︒C Measurement: against airPipette into cuvette: Macro MicroSample 0.2 ml 0.1 ml Enzyme/Coenzyme/ α-oxoglutarate R1 2.0 ml 1.0 mlMix, read initial absorbance after 1 minute. Read again after 1, 2 and 3 minutes. Note: If the absorbance change per minute is between 0.11 and 0.16 at 340/Hg 334 nm 0.06 and 0.08 at Hg 365 nmuse only the values for the first 2 minutes for the calculation.MANUAL CALCULATIONTo calculate the AST activity, use the following formulae:U/l = 1746 x A 340 nm/min U/l = 1780 x A Hg 334 nm/min U/l = 3235 x A Hg 365 nm/minSTANDARDISATIONRandox Calibration Serum Level 3 is traceable to AST reference material JSCC TS01.QUALITY CONTROLRandox Assayed Multisera, Level 2 and Level 3 are recommended for daily quality control. Two levels of controls should be assayed at least once a day. Values obtained should fall within a specified range. If these values fall outside the range and repetition excludes error the following steps should be taken:1. Check instrument settings and light source.2. Check cleanliness of all equipment in use.3. Check water. Contaminants, i.e. bacterial growth, maycontribute to inaccurate results. 4. Check reaction temperature.5. Check expiry date of kit and contents.6. Contact Randox Laboratories Customer Technical Services, Northern Ireland +44 (0) 28 9445 1070.SPECIFICITY/INTERFERENCE (6,7)Gross haemolysis will produce falsely elevated test results. The effects of various drugs on AST activity should be taken intoconsideration in the case of patients receiving large doses of drugs.The analytes below were tested up to the following levels and were found not to interfere: Haemoglobin 250 mg/dl Free Bilirubin 25 mg/dl Conjugate Bilirubin 25 mg/dl Triglycerides 1000 mg/dlIntralipid ® 200 mg/dlA list of substances and conditions known to effect AST activity in vivo is given by both Young et al and Friedman et al. Norepresentation is made by Randox Laboratories Ltd regarding the completeness of these lists and the accuracy of the information contained therein.NORMAL VALUES IN SERUM (8,9) +25︒C +30︒C +37︒C Men up to 18 U/l up to 25 U/l up to 37 U/l Women up to 15 U/l up to 21 U/l up to 31 U/lIt is recommended that each laboratory establish its own reference range to reflect the age, sex, diet and geographical location of the population.SPECIFIC PERFORMANCE CHARACTERISTICS The following performance data were obtained using an Rx Monza analyser running at +37o C.LINEARITYThis method is linear up to 562 U/l. If the sample concentration exceeds this value, dilute the sample 1+9 with 0.9% NaCl solution and re-assay. Multiply the result by 10.SENSITIVITYThe minimum detectable concentration of AST with an acceptable level of precision was determined as 9.3 U/l.PRECISIONIntra AssayLevel 2 Level 3Mean (U/l) 35.6 153SD 1.66 1.47CV(%) 4.65 0.96n 20 20Inter AssayLevel 2 Level 3Mean (U/l) 35.6 153SD 1.77 7.10CV(%) 4.96 4.63n 20 20CORRELATIONThis method (Y) was compared with another commerciallyavailable method (X) and the following linear regression equationobtained:Y = 1.07X + 4.9and a correlation coefficient of r = 0.997543 patient samples were analysed spanning the range 28 to 559U/l.REFERENCES1. Wroblewski F, La Due J.S: Ann Intern Med. 1956; 45: 801.2. Wroblewski F, La Due J.S: Proc Soc Exp Biol Med 1956;91: 569.3. Bergmeyer HU, Bowers GN Jr, et al: Clin Chem 1977; 23:887.4. Bergmeyer HU, Bowers GN Jr, et al: J.Clin Chem ClinBiochem 1980; 18: 521-534.5. Tietz N W: Fundamentals of Clinical Chemistry ed 3.Philadelphia, WB Saunders Co. 1987, pg 372.6. Young D S, et al: Clin Chem 1975, 21; No5.7. Friedman RB, et al: Clin Chem 1980, 26; No4.8. Wallnofer H, Schmidt.E, Schmidt FW, eds: Synopsis derLeberkrankheiten Stuttgart, Georg Thieme Verlag, 1974.9. Thefeld W, et al: Dtsch Med Wschr 1974; 99: 343.Revised 26 Apr 16 biRev. 003THIS PAGE IS INTENTIONALLY BLANK。
Bioanalytical Method ValidationGuidance for Indust

Guidance for Industry Bioanalytical Method ValidationU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Veterinary Medicine (CVM)May 2001BPGuidance for Industry Bioanalytical Method ValidationAdditional copies are available from:Drug Information Branch (HFD-210)Center for Drug Evaluation and Research (CDER)5600 Fishers Lane, Rockville, MD 20857 (Tel) 301-827-4573Internet at /cder/guidance/index.htmorCommunications Staff (HFV-12)Center for Veterinary Medicine (CVM)7500 Standish Place, Rockville, MD 20855 (Tel) 301–594-1755Internet at /cvmU.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)Center for Veterinary Medicine (CVM)May 2001BPTable of ContentsI.INTRODUCTION (1)II.BACKGROUND (1)A.F ULL V ALIDATION (2)B.P ARTIAL V ALIDATION (2)C.C ROSS-V ALIDATION (3)III.REFERENCE STANDARD (4)IV.METHOD DEVELOPMENT: CHEMICAL ASSAY (4)A.S ELECTIVITY (4)B.A CCURACY, P RECISION, AND R ECOVERY (5)C.C ALIBRATION/S TANDARD C URVE (5)D.S TABILITY (6)E.P RINCIPLES OF B IOANALYTICAL M ETHOD V ALIDATION AND E STABLISHMENT (8)F.S PECIFIC R ECOMMENDATIONS FOR M ETHOD V ALIDATION (10)V.METHOD DEVELOPMENT: MICROBIOLOGICAL AND LIGAND-BINDING ASSAYS (11)A.S ELECTIVITY I SSUES (11)B.Q UANTIFICATION I SSUES (12)VI.APPLICATION OF VALIDATED METHOD TO ROUTINE DRUG ANALYSIS (13)A CCEPTANCE C RITERIA FOR THE R UN (15)VII.DOCUMENTATION (16)A.S UMMARY I NFORMATION (16)B.D OCUMENTATION FOR M ETHOD E STABLISHMENT (17)C.A PPLICATION TO R OUTINE D RUG A NALYSIS (17)D.O THER I NFORMATION (19)GLOSSARY (20)GUIDANCE FOR INDUSTRY1Bioanalytical Method ValidationI.INTRODUCTIONThis guidance provides assistance to sponsors of investigational new drug applications (INDs), new drug applications (NDAs), abbreviated new drug applications (ANDAs), and supplements in developing bioanalytical method validation information used in human clinical pharmacology, bioavailability (BA), and bioequivalence (BE) studies requiring pharmacokinetic (PK) evaluation. This guidance also applies to bioanalytical methods used for non-human pharmacology/toxicology studies and preclinical studies. For studies related to the veterinary drug approval process, this guidance applies only to blood and urine BA, BE, and PK studies.The information in this guidance generally applies to bioanalytical procedures such as gas chromatography (GC), high-pressure liquid chromatography (LC), combined GC and LC mass spectrometric (MS) procedures such as LC-MS, LC-MS-MS, GC-MS, and GC-MS-MS performed for the quantitative determination of drugs and/or metabolites in biological matricessuch as blood, serum, plasma, or urine. This guidance also applies to other bioanalytical methods, such as immunological and microbiological procedures, and to other biological matrices, such as tissue and skin samples.This guidance provides general recommendations for bioanalytical method validation. The recommendations can be adjusted or modified depending on the specific type of analytical method used. II.BACKGROUND1 This guidance has been prepared by the Biopharmaceutics Coordinating Committee in the Center for Drug Evaluation and Research (CDER) in cooperation with the Center for Veterinary Medicine (CVM) at the Food and Drug Administration.This guidance has been developed based on the deliberations of two workshops: (1) Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies (held on December 3B5, 19902 ) and (2) Bioanalytical Methods Validation C A Revisit With a Decade of Progress (held on January 12B14, 20003).Selective and sensitive analytical methods for the quantitative evaluation of drugs and their metabolites (analytes) are critical for the successful conduct of preclinical and/or biopharmaceutics and clinical pharmacology studies. Bioanalytical method validation includes all of the procedures that demonstrate that a particular method used for quantitative measurement of analytes in a given biological matrix, such as blood, plasma, serum, or urine, is reliable and reproducible for the intended use. The fundamental parameters for this validation include (1) accuracy, (2) precision, (3) selectivity, (4) sensitivity, (5) reproducibility, and (6) stability. Validation involves documenting, through the use of specific laboratory investigations, that the performance characteristics of the method are suitable and reliable for the intended analytical applications. The acceptability of analytical data corresponds directly to the criteria used to validate the method.Published methods of analysis are often modified to suit the requirements of the laboratory performing the assay. These modifications should be validated to ensure suitable performance of the analytical method. When changes are made to a previously validated method, the analyst should exercise judgment as to how much additional validation is needed. During the course of a typical drug development program, a defined bioanalytical method undergoes many modifications. The evolutionary changes to support specific studies and different levels of validation demonstrate the validity of an assay’s performance. Different types and levels of validation are defined and characterized as follows:A.Full Validation•Full validation is important when developing and implementing a bioanalytical method for the first time.•Full validation is important for a new drug entity.• A full validation of the revised assay is important if metabolites are added to an existing assay for quantification.B.Partial ValidationPartial validations are modifications of already validated bioanalytical methods. Partial validation can range from as little as one intra-assay accuracy and precision determination to a nearly full2 Workshop Report: Shah, V.P. et al., Pharmaceutical Research: 1992; 9:588-592.3 Workshop Report: Shah, V.P. et al., Pharmaceutical Research: 2000; 17:in press.validation. Typical bioanalytical method changes that fall into this category include, but are not limited to:•Bioanalytical method transfers between laboratories or analysts•Change in analytical methodology (e.g., change in detection systems)•Change in anticoagulant in harvesting biological fluid•Change in matrix within species (e.g., human plasma to human urine)•Change in sample processing procedures•Change in species within matrix (e.g., rat plasma to mouse plasma)•Change in relevant concentration range•Changes in instruments and/or software platforms•Limited sample volume (e.g., pediatric study)•Rare matrices•Selectivity demonstration of an analyte in the presence of concomitant medications•Selectivity demonstration of an analyte in the presence of specific metabolitesC.Cross-ValidationCross-validation is a comparison of validation parameters when two or more bioanalytical methods are used to generate data within the same study or across different studies. An example of cross-validation would be a situation where an original validated bioanalytical method serves as thereference and the revised bioanalytical method is the comparator. The comparisons should be done both ways.When sample analyses within a single study are conducted at more than one site or more than one laboratory, cross-validation with spiked matrix standards and subject samples should be conducted at each site or laboratory to establish interlaboratory reliability. Cross-validation should also be considered when data generated using different analytical techniques (e.g., LC-MS-MS vs.ELISA4) in different studies are included in a regulatory submission.All modifications should be assessed to determine the recommended degree of validation. The analytical laboratory conducting pharmacology/toxicology and other preclinical studies for regulatory submissions should adhere to FDA=s Good Laboratory Practices (GLPs)5 (21 CFR part 58) and to sound principles of quality assurance throughout the testing process. The bioanalytical method for human BA, BE, PK, and drug interaction studies must meet the criteria in 21 CFR 320.29. The analytical laboratory should have a written set of standard operating procedures (SOPs) to ensure a complete system of quality control and assurance. The SOPs should cover all aspects of analysis from the time the sample is collected and reaches the laboratory until the results of the analysis are reported. The SOPs also should include record keeping, security and chain of sample custody4 Enzyme linked immune sorbent assay5 For the Center for Veterinary Medicine, all bioequivalence studies are subject to Good Laboratory Practices.(accountability systems that ensure integrity of test articles), sample preparation, and analytical tools such as methods, reagents, equipment, instrumentation, and procedures for quality control and verification of results.The process by which a specific bioanalytical method is developed, validated, and used in routine sample analysis can be divided into (1) reference standard preparation, (2) bioanalytical method development and establishment of assay procedure, and (3) application of validated bioanalytical method to routine drug analysis and acceptance criteria for the analytical run and/or batch. These three processes are described in the following sections of this guidance.III.REFERENCE STANDARDAnalysis of drugs and their metabolites in a biological matrix is carried out using samples spiked with calibration (reference) standards and using quality control (QC) samples. The purity of the reference standard used to prepare spiked samples can affect study data. For this reason, an authenticated analytical reference standard of known identity and purity should be used to prepare solutions of known concentrations. If possible, the reference standard should be identical to the analyte. When this is not possible, an established chemical form (free base or acid, salt or ester) of known purity can be used. Three types of reference standards are usually used: (1) certified reference standards (e.g., USP compendial standards); (2) commercially supplied reference standards obtained from a reputable commercial source; and/or (3) other materials of documented purity custom-synthesized by an analytical laboratory or other noncommercial establishment. The source and lot number, expiration date, certificates of analyses when available, and/or internally or externally generated evidence of identity and purity should be furnished for each reference standard.IV.METHOD DEVELOPMENT: CHEMICAL ASSAYThe method development and establishment phase defines the chemical assay. The fundamental parameters for a bioanalytical method validation are accuracy, precision, selectivity, sensitivity, reproducibility, and stability. Measurements for each analyte in the biological matrix should be validated. In addition, the stability of the analyte in spiked samples should be determined. Typical method development and establishment for a bioanalytical method include determination of (1) selectivity, (2) accuracy, precision, recovery, (3) calibration curve, and (4) stability of analyte in spiked samples.A.SelectivitySelectivity is the ability of an analytical method to differentiate and quantify the analyte in thepresence of other components in the sample. For selectivity, analyses of blank samples of theappropriate biological matrix (plasma, urine, or other matrix) should be obtained from at leastsix sources. Each blank sample should be tested for interference, and selectivity should be ensured at the lower limit of quantification (LLOQ).Potential interfering substances in a biological matrix include endogenous matrix components, metabolites, decomposition products, and in the actual study, concomitant medication and other exogenous xenobiotics. If the method is intended to quantify more than one analyte, each analyte should be tested to ensure that there is no interference.B.Accuracy, Precision, and RecoveryThe accuracy of an analytical method describes the closeness of mean test results obtained by the method to the true value (concentration) of the analyte. Accuracy is determined by replicate analysis of samples containing known amounts of the analyte. Accuracy should be measured using a minimum of five determinations per concentration. A minimum of three concentrations in the range of expected concentrations is recommended. The mean value should be within 15% of the actual value except at LLOQ, where it should not deviate by more than 20%. The deviation of the mean from the true value serves as the measure of accuracy.The precision of an analytical method describes the closeness of individual measures of an analyte when the procedure is applied repeatedly to multiple aliquots of a single homogeneous volume of biological matrix. Precision should be measured using a minimum of five determinations per concentration. A minimum of three concentrations in the range of expected concentrations is recommended. The precision determined at each concentration level should not exceed 15% of the coefficient of variation (CV) except for the LLOQ, where it should not exceed 20% of the CV. Precision is further subdivided into within-run, intra-batch precision or repeatability, which assesses precision during a single analytical run, and between-run, inter-batch precision or repeatability, which measures precision with time, and may involve different analysts, equipment, reagents, and laboratories.The recovery of an analyte in an assay is the detector response obtained from an amount of the analyte added to and extracted from the biological matrix, compared to the detector response obtained for the true concentration of the pure authentic standard. Recovery pertains to the extraction efficiency of an analytical method within the limits of variability. Recovery of the analyte need not be 100%, but the extent of recovery of an analyte and of the internal standard should be consistent, precise, and reproducible. Recovery experiments should be performed by comparing the analytical results for extracted samples at three concentrations (low, medium, and high) with unextracted standards that represent 100% recovery.C.Calibration/Standard CurveA calibration (standard) curve is the relationship between instrument response and known concentrations of the analyte. A calibration curve should be generated for each analyte in thesample. A sufficient number of standards should be used to adequately define the relationship between concentration and response. A calibration curve should be prepared in the same biological matrix as the samples in the intended study by spiking the matrix with known concentrations of the analyte. The number of standards used in constructing a calibration curve will be a function of the anticipated range of analytical values and the nature of theanalyte/response relationship. Concentrations of standards should be chosen on the basis of the concentration range expected in a particular study. A calibration curve should consist of a blank sample (matrix sample processed without internal standard), a zero sample (matrix sample processed with internal standard), and six to eight non-zero samples covering the expected range, including LLOQ.1.Lower Limit of Quantification (LLOQ)The lowest standard on the calibration curve should be accepted as the limit ofquantification if the following conditions are met:C The analyte response at the LLOQ should be at least 5 times the responsecompared to blank response.C Analyte peak (response) should be identifiable, discrete, and reproducible witha precision of 20% and accuracy of 80-120%.2.Calibration Curve/Standard Curve/Concentration-ResponseThe simplest model that adequately describes the concentration-response relationshipshould be used. Selection of weighting and use of a complex regression equation should be justified. The following conditions should be met in developing a calibration curve:C#20% deviation of the LLOQ from nominal concentrationC#15% deviation of standards other than LLOQ from nominal concentrationAt least four out of six non-zero standards should meet the above criteria, including the LLOQ and the calibration standard at the highest concentration. Excluding thestandards should not change the model used.D.StabilityDrug stability in a biological fluid is a function of the storage conditions, the chemical properties of the drug, the matrix, and the container system. The stability of an analyte in a particular matrix and container system is relevant only to that matrix and container system and should not be extrapolated to other matrices and container systems. Stability procedures should evaluate the stability of the analytes during sample collection and handling, after long-term (frozen at theintended storage temperature) and short-term (bench top, room temperature) storage, and after going through freeze and thaw cycles and the analytical process. Conditions used in stability experiments should reflect situations likely to be encountered during actual sample handling and analysis. The procedure should also include an evaluation of analyte stability in stock solution.All stability determinations should use a set of samples prepared from a freshly made stock solution of the analyte in the appropriate analyte-free, interference-free biological matrix. Stock solutions of the analyte for stability evaluation should be prepared in an appropriate solvent at known concentrations.1.Freeze and Thaw StabilityAnalyte stability should be determined after three freeze and thaw cycles. At least three aliquots at each of the low and high concentrations should be stored at the intendedstorage temperature for 24 hours and thawed unassisted at room temperature. Whencompletely thawed, the samples should be refrozen for 12 to 24 hours under the sameconditions. The freeze–thaw cycle should be repeated two more times, then analyzedon the third cycle. If an analyte is unstable at the intended storage temperature, thestability sample should be frozen at -700C during the three freeze and thaw cycles.2.Short-Term Temperature StabilityThree aliquots of each of the low and high concentrations should be thawed at roomtemperature and kept at this temperature from 4 to 24 hours (based on the expectedduration that samples will be maintained at room temperature in the intended study) and analyzed.3.Long-Term StabilityThe storage time in a long-term stability evaluation should exceed the time between the date of first sample collection and the date of last sample analysis. Long-term stabilityshould be determined by storing at least three aliquots of each of the low and highconcentrations under the same conditions as the study samples. The volume of samples should be sufficient for analysis on three separate occasions. The concentrations of allthe stability samples should be compared to the mean of back-calculated values for the standards at the appropriate concentrations from the first day of long-term stabilitytesting.4.Stock Solution StabilityThe stability of stock solutions of drug and the internal standard should be evaluated at room temperature for at least 6 hours. If the stock solutions are refrigerated or frozenfor the relevant period, the stability should be documented. After completion of thedesired storage time, the stability should be tested by comparing the instrumentresponse with that of freshly prepared solutions.5.Post-Preparative StabilityThe stability of processed samples, including the resident time in the autosampler, should be determined. The stability of the drug and the internal standard should be assessedover the anticipated run time for the batch size in validation samples by determiningconcentrations on the basis of original calibration standards.Although the traditional approach of comparing analytical results for stored samples with those for freshly prepared samples has been referred to in this guidance, other statistical approaches based on confidence limits for evaluation of an analyte=s stability in abiological matrix can be used. SOPs should clearly describe the statistical method andrules used. Additional validation may include investigation of samples from dosedsubjects.E.Principles of Bioanalytical Method Validation and Establishment•The fundamental parameters to ensure the acceptability of the performance of a bioanalytical method validation are accuracy, precision, selectivity, sensitivity,reproducibility, and stability.• A specific, detailed description of the bioanalytical method should be written. This can be in the form of a protocol, study plan, report, and/or SOP.•Each step in the method should be investigated to determine the extent to which environmental, matrix, material, or procedural variables can affect the estimation of analyte in the matrix from the time of collection of the material up to and including the time ofanalysis.•It may be important to consider the variability of the matrix due to the physiological nature of the sample. In the case of LC-MS-MS-based procedures, appropriate steps should be taken to ensure the lack of matrix effects throughout the application of the method,especially if the nature of the matrix changes from the matrix used during method validation.• A bioanalytical method should be validated for the intended use or application. All experiments used to make claims or draw conclusions about the validity of the methodshould be presented in a report (method validation report).•Whenever possible, the same biological matrix as the matrix in the intended samples should be used for validation purposes. (For tissues of limited availability, such as bone marrow, physiologically appropriate proxy matrices can be substituted.)•The stability of the analyte (drug and/or metabolite) in the matrix during the collection process and the sample storage period should be assessed, preferably prior to sampleanalysis.•For compounds with potentially labile metabolites, the stability of analyte in matrix from dosed subjects (or species) should be confirmed.•The accuracy, precision, reproducibility, response function, and selectivity of the method for endogenous substances, metabolites, and known degradation products should beestablished for the biological matrix. For selectivity, there should be evidence that thesubstance being quantified is the intended analyte.•The concentration range over which the analyte will be determined should be defined in the bioanalytical method, based on evaluation of actual standard samples over the range,including their statistical variation. This defines the standard curve.• A sufficient number of standards should be used to adequately define the relationship between concentration and response. The relationship between response and concentration should be demonstrated to be continuous and reproducible. The number of standards used should be a function of the dynamic range and nature of the concentration-responserelationship. In many cases, six to eight concentrations (excluding blank values) can define the standard curve. More standard concentrations may be recommended for nonlinear than for linear relationships.•The ability to dilute samples originally above the upper limit of the standard curve should be demonstrated by accuracy and precision parameters in the validation.•In consideration of high throughput analyses, including but not limited to multiplexing, multicolumn, and parallel systems, sufficient QC samples should be used to ensure control of the assay. The number of QC samples to ensure proper control of the assay should be determined based on the run size. The placement of QC samples should be judiciously considered in the run.•For a bioanalytical method to be considered valid, specific acceptance criteria should be set in advance and achieved for accuracy and precision for the validation of QC samples over the range of the standards.F.Specific Recommendations for Method Validation•The matrix-based standard curve should consist of a minimum of six standard points, excluding blanks, using single or replicate samples. The standard curve should cover the entire range of expected concentrations.•Standard curve fitting is determined by applying the simplest model that adequately describes the concentration-response relationship using appropriate weighting and statistical tests for goodness of fit.•LLOQ is the lowest concentration of the standard curve that can be measured with acceptable accuracy and precision. The LLOQ should be established using at least five samples independent of standards and determining the coefficient of variation and/orappropriate confidence interval. The LLOQ should serve as the lowest concentration on the standard curve and should not be confused with the limit of detection and/or the low QC sample. The highest standard will define the upper limit of quantification (ULOQ) of an analytical method.•For validation of the bioanalytical method, accuracy and precision should be determined using a minimum of five determinations per concentration level (excluding blank samples).The mean value should be within ±15% of the theoretical value, except at LLOQ, where it should not deviate by more than ±20%. The precision around the mean value should not exceed 15% of the CV, except for LLOQ, where it should not exceed 20% of the CV.Other methods of assessing accuracy and precision that meet these limits may be equally acceptable.•The accuracy and precision with which known concentrations of analyte in biological matrix can be determined should be demonstrated. This can be accomplished by analysis ofreplicate sets of analyte samples of known concentrations C QC samples C from anequivalent biological matrix. At a minimum, three concentrations representing the entire range of the standard curve should be studied: one within 3x the lower limit of quantification (LLOQ) (low QC sample), one near the center (middle QC), and one near the upperboundary of the standard curve (high QC).•Reported method validation data and the determination of accuracy and precision should include all outliers; however, calculations of accuracy and precision excluding values that are statistically determined as outliers can also be reported.•The stability of the analyte in biological matrix at intended storage temperatures should be established. The influence of freeze-thaw cycles (a minimum of three cycles at twoconcentrations in triplicate) should be studied.•The stability of the analyte in matrix at ambient temperature should be evaluated over a time period equal to the typical sample preparation, sample handling, and analytical run times.•Reinjection reproducibility should be evaluated to determine if an analytical run could be reanalyzed in the case of instrument failure.•The specificity of the assay methodology should be established using a minimum of six independent sources of the same matrix. For hyphenated mass spectrometry-basedmethods, however, testing six independent matrices for interference may not be important.In the case of LC-MS and LC-MS-MS-based procedures, matrix effects should beinvestigated to ensure that precision, selectivity, and sensitivity will not be compromised.Method selectivity should be evaluated during method development and throughout methodvalidation and can continue throughout application of the method to actual study samples.•Acceptance/rejection criteria for spiked, matrix-based calibration standards and validation QC samples should be based on the nominal (theoretical) concentration of analytes.Specific criteria can be set up in advance and achieved for accuracy and precision over therange of the standards, if so desired.V.METHOD DEVELOPMENT: MICROBIOLOGICAL AND LIGAND-BINDING ASSAYSMany of the bioanalytical validation parameters and principles discussed above are also applicable to microbiological and ligand-binding assays. However, these assays possess some unique characteristics that should be considered during method validation.A.Selectivity IssuesAs with chromatographic methods, microbiological and ligand-binding assays should be shown to be selective for the analyte. The following recommendations for dealing with two selectivity issues should be considered:1.Interference From Substances Physiochemically Similar to the Analyte•Cross-reactivity of metabolites, concomitant medications, or endogenouscompounds should be evaluated individually and in combination with the analyteof interest.•When possible, the immunoassay should be compared with a validated reference method (such as LC-MS) using incurred samples and predetermined criteria foragreement of accuracy of immunoassay and reference method.。
1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐质量标准

1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐质量标准1. 引言1.1 概述一-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(简称EPH-HCl)是一种重要的有机化合物,具有广泛的应用价值。
EPH-HCl在医药领域被广泛用作药物原料,特别是作为治疗注意力缺陷多动障碍(ADHD)和嗜睡症(narcolepsy)的药物。
此外,EPH-HCl还可用于合成农药、染料、功能材料等领域。
1.2 文章结构本文共分为五个主要部分。
引言部分对文章的研究背景进行了概述,并介绍了文章的结构和目的。
第二部分将详细描述EPH-HCl的性质和用途,包括其化学性质和生物活性以及在不同领域中的应用情况。
第三部分将讨论制备EPH-HCl的方法和工艺流程,包括原料准备、反应条件和控制参数以及工艺流程简介。
第四部分将列出EPH-HCl的质量标准与检验方法,包括外观与纯度要求、含量测定方法以及杂质限量要求与检测方法。
最后,第五部分将总结本文的内容和研究结果,并对EPH-HCl的质量标准进行评价和展望。
1.3 目的本文旨在全面介绍一-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的性质、制备方法及工艺流程,重点讨论其质量标准与检验方法。
通过对EPH-HCl的研究和分析,能够为相关领域的科学研究和工业应用提供参考依据,并促进该化合物在医药、农药、染料等领域的进一步开发和应用。
2. 一-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的性质和用途2.1 化学性质:一-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐是一种白色结晶性固体,具有较好的溶解性。
它是一种非挥发性有机化合物,分子式为C10H24ClN3O,相对分子质量为217.77。
其熔点约为174-176摄氏度。
该化合物在常温下稳定,不易受热分解。
2.2 生物活性:一-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐是一种药物中间体,在药物领域有广泛的应用。
它是β受体阻滞剂的重要原料之一,可用于合成各类心血管系统药物。
德谷门冬双胰岛素注射液治疗2_型糖尿病临床效果及安全性探讨

DOI:10.16658/ki.1672-4062.2023.17.098德谷门冬双胰岛素注射液治疗2型糖尿病临床效果及安全性探讨林生,谢平,陈予福州市长乐区人民医院内分泌科,福建福州350200[摘要]目的研究德谷门冬双胰岛素注射液治疗2型糖尿病的临床效果及安全性。
方法选取于2022年7月—2023年4月福州市长乐区人民医院收治的2型糖尿病患者98例为研究对象,采用随机抓阄法分为两组,每组49例。
两组均联用常规降糖药物治疗,对照组采用甘精胰岛素注射液治疗,观察组采用德谷门冬双胰岛素注射液治疗。
对比两组临床治疗效果、临床症状好转时间和胰岛素用量情况、糖代谢指标、胰岛素功能指标、不良反应发生情况、心血管不良事件发生情况。
结果观察组总有效率高于对照组,差异有统计学意义(P<0.05)。
观察组尿酮体转阴时间、血糖达标时间、胰岛素用量均优于对照组,差异有统计学意义(P< 0.05)。
观察组空腹血糖、餐后2 h血糖、糖化血红蛋白均低于对照组,差异有统计学意义(P<0.05)。
观察组胰岛β细胞功能指数高于对照组,胰岛素抵抗指数、空腹胰岛素低于对照组,差异有统计学意义(P<0.05)。
两组恶心呕吐、倦怠乏力、低血糖总发生率比较,差异无统计学意义(P>0.05)。
两组心绞痛、心力衰竭总发生率比较,差异无统计学意义(P>0.05)。
结论德谷门冬双胰岛素注射液治疗2型糖尿病临床效果显著优于甘精胰岛素注射液,但是治疗安全性无显著变化。
[关键词] 2型糖尿病;德谷门冬双胰岛素注射液;不良反应;心血管不良事件[中图分类号] R59 [文献标识码] A [文章编号] 1672-4062(2023)09(a)-0098-04Discussion on the Clinical Effect and Safety of Insulin Degludec and Insu⁃lin Aspart Injection in the Treatment of Type 2 Diabetes MellitusLIN Sheng, XIE Ping, CHEN YuDepartment of Endocrinology, Changle District People's Hospital, Fuzhou, Fujian Province, 350200 China[Abstract] Objective To study the clinical effect and safety of insulin degludec and insulin aspart injection in the treatment of type 2 diabetes mellitus. Methods A total of 98 patients with type 2 diabetes admitted to Fuzhou Changle District People's Hospital from July 2022 to April 2023 were selected as the study objects and divided into two groups with 49 cases in each group by random lottery method. Both groups were treated with conventional hypoglycemic drugs, the control group was treated with insulin glargine injection, and the observation group was treated with Degu asparton double insulin injection. The clinical therapeutic effect, time of improvement of clinical symptoms, insulin dosage, glucose metabolism index, insulin function index, occurrence of adverse reactions and cardiovascular adverse events were compared between the two groups. Results The total effective rate of the observation group was higher than that of the control group, and the difference was statistically significant (P<0.05). The time of urine ketone body turning negative, blood glucose reaching standard and insulin dosage in observation group were better than those in control group, and the differences were statistically significant (P<0.05). Fasting plasma glucose, 2-hour postprandial blood glucose and glycated hemoglobin in the observation group were lower than those in the control group, and the differences were statistically significant (P<0.05). The function index of islet β cells in observation group was higher than that in control group, the insulin resistance index and fasting insulin was lower than that in control group, the dif⁃ference was statistically significant (P<0.05). There was no statistically significant difference in the total incidence of [作者简介]林生(1981-),男,本科,副主任医师,研究方向为糖尿病及其并发症的相关临床研究。
Y-27632_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:Y–27632 is an ATP–competitive inhibitor of ROCK–I and ROCK–II , with K i of 220 nM and 300 nM for ROCK–I and ROCK–II , respectively.IC50 & Target: Ki: 220/300 nM (ROCK–I/II)[1]In Vitro: Y–27632 inhibits the ROCK family of kinases 100 times more potently than other kinases including protein kinase C,cAMP–dependent kinase and myosin light chain kinase. Y–27632 prolongs the lag time and delays the appearance of BrdU–labeled cells in a concentration–dependent manner, delays of about 1 and 4 h are noticed in the Swiss 3T3 cells treated with 10 and 100 μM Y–27632, respectively [1]. Y–27632 promotes neuronal differentiation of adipose tissue–derived stem cells (ADSCs). Compared to 1.0and 2.5 μM Y–27632 induced groups, percentages of neuroal–like cells achieved a peak in the 5.0 μM Y–27632 induced group [2].In Vivo: Y–27632 (5 and 10 mg/kg) significantly prolongs the onset time of myoclonic jerks when compare with saline group.Y–27632 (5 and 10 mg/kg) significantly prolongs the onset time of clonic convulsions when compare with saline group [3].Treatment with Dimethylnitrosamine (DMN) causes a significant decrease in rat body and liver weight (DMN–S group) compared with control animals (S–S group). Oral Y27632 (30 mg/kg) essentially prevents this DMN–induced rat body and liver weight loss (DMN–Y group)[4].PROTOCOL (Extracted from published papers and Only for reference)Kinase Assay:[1]Recombinant ROCK–I, ROCK–II, PKN, or citron kinase is expressed in HeLa cells as Myc–tagged proteins by transfection using Lipofectamine, and is precipitated from the cell lysates by the use of 9E10 monoclonal anti–Myc antibodycoupled to G protein–Sepharose. Recovered immunocomplexes are incubated with various concentrations of [32P]ATP and 10 mg of histone type 2 as substrates in the absence or presence of various concentrations of either Y–27632 or Y–30141 at 30°C for 30 min in a total volume of 30 μL of the kinase buffer containing 50 mM HEPES–NaOH, pH 7.4, 10 mM MgCl 2, 5 mM MnCl 2, 0.02% Briji 35, and 2 mM dithiothreitol. PKCa is incubated with 5 μM [32P]ATP and 200 μg/mL histone type 2 as substrates in the absence or presence of various concentrations of either Y–27632 or Y–30141 at 30°C for 10 min in a kinase buffer containing 50 mM Tris–HCl,pH 7.5, 0.5 mM CaCl 2, 5 mM magnesium acetate, 25 μg/mL phosphatidyl serine, 50 ng/mL 12–O–tetradecanoylphorbol–13–acetate and 0.001% leupeptin in a total volume of 30 μL. Incubation is terminated by the addition of 10 μL of 43 Laemmli sample buffer.After boiling for 5 min, the mixture is subjected to SDS–polyacrylamide gel electrophoresis on a 16% gel. The gel is stained withCoomassie Brilliant Blue, and then dried. The bands corresponding to histone type 2 are excised, and the radioactivity is measured [1]. Cell Assay: Y–27632 is dissolved in water and stored [1].[1]HeLa cells are plated at a density of 3×104 cells per 3.5–cm dish. The cells are cultured in DMEM containing 10% FBS in the presence of 10 mM Thymidine for 16 h. After the cells are washed with DMEM containing 10% FBS, they are cultured for an additional 8 h, and then 40 ng/mL of Nocodazole is added. After 11.5 h of theNocodazole treatment, various concentrations of Y–27632 (0–300 μM), Y–30141, or vehicle is added and the cells are incubated for another 30 min [1].Animal Administration: Y–27632 is dissolved in 0.9% NaCl (saline) (Mice)[3].Product Name:Y–27632Cat. No.:HY-10071CAS No.:146986-50-7Molecular Formula:C 14H 21N 3O Molecular Weight:247.34Target:ROCK; ROCK; ROCK Pathway:TGF–beta/Smad; Stem Cell/Wnt; Cell Cycle/DNA Damage Solubility:DMSO: ≥ 32 mg/mLY–27632 is dissolved in saline (final concentration 2%) (Rat)[4].[3][4]Mice[3]Male, inbred Swiss albino mice (2–3 months old) weighing 25–30 g are used. Mice are injected with a sub–convulsive dose of PTZ (35 mg/kg, i.p.) (on Mondays, Wednesdays and Fridays) of each week for a total of 11 injections. After each PTZ injection, mice are observed for 30 min and the occurrence of convulsive activity is recorded. After 30 min, the mice are then injected with either Fasudil (25 mg/kg, i.p.) or Y–27632 (5 mg/kg, i.p.) and returned to their home cages until the next injection. Control mice for Fasudil andY–27632 receives saline.Rat[4]Male Wistar Kind A rats (200–250 g) are used. DMN (1 g/mL) is diluted ten times with saline (final concentration 1%) and 10 mg/kg per day of DMN is injected intraperitoneally (i.p.) on the first 3 days of each week for 4 weeks. Y27632 is given orally once per day at a dose of 30 mg/kg for 4 weeks starting on the day of the first injection of DMN. The dose of 30 mg/kg corrects hypertension in several rat models without toxicity. Twenty rats are randomized into four experimental groups (n=5 in each group) as follows: (1) S–S (injection of saline i.p. and oral administration of saline); (2) S–Y (injection of saline i.p. and oral administration of Y27632); (3) DMN–S (DMN i.p. and oral administration of saline); (4) DMN–Y (DMN i.p. and oral administration of Y27632). The rats are weighed every week. They are sacrificed at the end of the fourth week and the liver is excised. In addition, a blood sample is taken immediately before the rats are sacrificed.References:[1]. Ishizaki T, et al. Pharmacological properties of Y–27632, a specific inhibitor of rho–associated kinases. Mol Pharmacol. 2000 May;57(5):976–83.[2]. Xue ZW, et al. Rho–associated coiled kinase inhibitor Y–27632 promotes neuronal–like differentiation of adult human adipose tissue–derived stem cells.Chin Med J (Engl). 2012 Sep;125(18):3332–5.[3]. Inan S, et al. Antiepileptic effects of two Rho–kinase inhibitors, Y–27632 and fasudil, in mice. Br J Pharmacol. 2008 Sep;155(1):44–51.[4]. Tada S, et al. A selective ROCK inhibitor, Y27632, prevents dimethylnitrosamine–induced hepatic fibrosis in rats. J Hepatol. 2001 Apr;34(4):529–36.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
15523666_迷迭香酸对哮喘小鼠氧化性肺损伤的保护作用

$ 材 料 与 方 法
$C$ 材 料 !3!3!试验动物!0""8XDC[ 级雌性 &;B&&S 小鼠购自 广 西 医 科 大 学 实 验 动 物 中 心"动 物 合 格 证
号!D#2*%桂'"8!$?888"# 试 验 前 适 应 性 饲 养:P" 自由饮水 饮 食"饲 养 环 境 温 度 "$ c l! c"相 对 湿 度 $85 "085 # !3!3" 主 要 试 剂 和 药 品 9,D$D,'$1DZ?CQ 检测试 剂 盒 均 购 自 碧 云 天 生 物 技 术 研 究 所*,(; 购自 DEX\@ 公 司*液 态 铝 佐 剂 购 自 >OHM\I 公 司# 肿节风药材购自广 西 玉 林 中 桂 药 材 有 限 公 司"药 材
%!3广西大学动物科学技术学院"南宁 4:8884*"3广西畜牧研究所"南宁 4:888!'
摘要为评价迷迭香酸对哮 喘 小 鼠 模 型 氧 化 性 肺 损 伤 的 保 护 作 用"本 研 究 用 卵 清 蛋 白 %,(;'致 敏$激 发 雌 性 &;B&&S小鼠建立哮喘模型"并用 ,(; 和 Z"," 联合激发小鼠作为氧化肺损伤阳性对照模型#在最后一次滴鼻激 发 "$O 后 "取 支 气 管 肺 泡 灌 洗 液 %&;B['进 行 细 胞 计 数 并 测 定 活 性 氧 %9,D'$超 氧 化 物 歧 化 酶 %D,''和 谷 胱 甘 肽 过 氧化物酶%1DZ?CQ'水平"取左侧肺脏固定做 Z= 染色#结果显示"迷迭香酸可明显减少 &;B[ 中 细 胞 总 数 和 嗜 酸 性粒细胞数目"显著抑制肺组织和 &;B[ 中 9,D的产生"升高 D,' 和 1DZ?CQ水 平"改 善 肺 组 织 病 理 变 化# 本 试 验 结 果 表 明 "迷 迭 香 酸 对 氧 化 肺 损 伤 起 到 明 显 的 保 护 作 用 # 关 键 词 迷 迭 香 酸 *哮 喘 *氧 化 性 肺 损 伤 *抗 氧 化 中 图 分 类 号 9"0434 文 献 标 识 码 ; 文 章 编 号 !+6!?6":+%"8!6'!"?:+48?8+
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Caution: Not fully tested. For research purposes only Medche c . s s e r p x e m e h c d e m . w w w : b e AW Sm Uo ,c 0 4. 5s 8s 0e r p Jx Ne ,m n e oh t c e cd ne i rm P @ ,o y f an Wi : l ni oa sm n iE k l i W 8 1
Product Data Sheet
LY2228820 862507-23-1 HY-13241 612.74 C26H37FN6O6S2 >98%
Product Name: CAS No.: Cat. No.: MWt: Formula: Purity :
Solubility:
DMSO ≥33mg/mL Water ≥ 121mg/mL Ethanol ≥2.6mg/mL
Mechanisms: Pathways:MAPK ; Target:p38 MAPK Biological Activity: LY2228820 is a novel and potent p38MAPK inhibitor (the IC50 for p38αMAPK and p38βMAPK were 7 nM and 3 nM, respectively). IC50 value: 7/3 nM(p38α/p38β MAPK) [1] Target: p38MAPK in vitro: LY2228820 inhibits p38α, as well as the level of phosphoMAPKAPK-2 (pMK2) in RAW 264.7 cells, with IC50 values of 7 nM and 34.3 nM, respectively. Furthermore, LY2228820 inhibits lipopolysaccharide (LPS)-induced TNFα formation in murine peritoneal macrophages, with IC50 of 5.2 nM [1]. n multiple myeloma (MM) cells, including INA6, RPMI-8226, U266, and RPMI-Dox40, LY2228820 (200 nM–800 nM) significantly blocks p38MAPK signaling, as revealed by its inhibition on phosphorylation of HSP27, a downstream target of p38MAPK, without affecting the expression level of HSP27. LY2228820 (200 nM–400 nM) enhances bortezomib-induced cytotoxicity and apoptosis, but LY2228820 alone doesn't inhibit the gr... References: [1]. Mader M, et al. Imidazolyl benzimidazoles and imidazo[4,5-b]pyridines as potent p38alpha MAP kinase inhibitors with excellent in vivo antiinflammatory properties. Bioorg Med Chem Lett, 2008, 18(1), ( ), 179-183. [2]. Ishitsuka K, et al. p38 mitogen-activated protein kinase inhibitor LY2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma; therapeutic implications. Br J Haematol, 2008, 141(5), 598-606.