GRA_Ex-25_LCMS_22504_MedChemExpress
磁力架说明书_Magnetic Stand Manual_MCE
1包装清单产品概述MCE 磁力架是磁珠产品专用配套设备,支持 MCE 全线磁珠类产品,内含强磁磁芯,可实现快速高效的分离。
MCE 磁力架 (200 μL / 2 mL / 15 mL ) 采用独特的三明治槽设计,磁条可抽出,可容纳 200 μL PCR 管,1.5 mL EP 管,2 mL EP 管,15 mL 离心管。
本产品适用于抗体纯化、免疫沉淀 (IP )、免疫共沉淀 (Co-IP )、细胞分选和核酸分离等实验。
2操作说明31. 将装有磁珠悬液的 EP 管/离心管置于磁力架对应的样品孔中,静置数分钟后磁珠被吸附聚集于管壁,溶液恢复澄清。
2. 用移液器或吸管从管底将溶液吸出,或小心倾倒出液体。
3. 抽去磁力条,加入复溶液体,轻缓震荡即可混合均匀,进行下一步操作。
注:磁性分离的时间与磁珠粒径有关,磁珠粒径越小,磁性分离时间越长。
此外,溶液的黏稠程度以及溶液的成分也会对磁性分离时间产生影响。
5注意事项1. 根据实验参数和样品体积不同,可调整磁芯位置或选用不同样式的磁力架。
2. 为减少操作过程中磁珠的损失,请将样品管底端插入磁力架底部的凹槽内。
当磁珠吸附在管壁上后,缓慢倾去上清,或用移液枪吸尽。
3. 由于磁力架有强大的磁场,请远离手机、电脑、手表、起博器、磁铁等易被磁力干扰的物体,尤其是刀具,以免对操作人员造成伤害。
4. 如需同时使用多个磁力架 (≥2 个) ,应分开放置,避免磁场之间产生干扰。
不要把多个磁棒放在一起,以防止夹伤。
5. 请勿与强酸、强碱等腐蚀性溶剂直接接触。
6. 请勿拆卸磁块。
7. 为保护外壳,请勿长时间暴露在阳光和紫外线下。
8. 为保持磁力架磁性,请勿置于高温和强外界磁场环境中。
9. 使用后请及时清洁,妥善放置在干燥环境中。
Magnetic StandMedChemExpress MedChemExpress 400-820-3792 电话: ************ 传真: ************Email: t *********************MCE Hotline: 400-820-3792Contents HY-K0200Magnetic Stand 200 μL-2 mL-15 mL。
薄层色谱指纹图谱在丹参药材质量评价中的应用研究_蒋轶伦

-第44卷 第6期厦门大学学报(自然科学版)V ol.44 N o.6 2005年11月Journal of Xiam en U niversity (Natural Science)No v.2005薄层色谱指纹图谱在丹参药材质量评价中的应用研究收稿日期:2005 05 23基金项目:科技部"九五"攻关计划中药现代化重中之重项目:中药现代化生产中重金属测定的标准化及质量控制(99 929 02 13);香港创新科技署与香港新世界生物科技有限公司联合资助项目作者简介:蒋轶伦(1981-),女,硕士研究生.*通讯作者:mt2elp@蒋轶伦1,李 伟1,庄峙厦1,黎先春2,王小如1,2*(1.厦门大学化学化工学院化学系,现代分析科学教育部重点实验室,福建厦门361005;2.国家海洋局第一海洋研究所,青岛现代分析技术及中药标准化重点实验室,山东青岛266061)摘要:利用薄层色谱(T L C)技术对中药材丹参的有效成分进行了指纹图谱分析.以不同等级的中江丹参和不同采收期的丹参、南丹参为分析对象,采用适宜的水溶性及脂溶性成分提取方法和T LC 分析条件,可获得其相应的T L C 图像及色谱图.通过对不同丹参药材的色谱图像的分析,结合化学计量学方法对其各主要指标成分的信息进行提取,初步构建了丹参的T L C 指纹图谱.结果表明,在254nm 波长下获取脂溶性和水溶性成分共7个共有指纹峰,各峰相对比移值(R f )和峰面积的相对标准偏差分别小于1.7%和4.0%.通过对薄层色谱斑点的R f 值、斑点颜色和大小的比较,并进行主成分分析和聚类分析,实现了对不同等级的丹参进行归属,并确立了最佳采收期.本实验建立的薄层色谱指纹图谱方法具有较高的稳定性、良好的精密度和重现性,可用于丹参药材的质量评价和控制.关键词:丹参药材;T LC 指纹图谱;图像分析;化学计量学;质量评价中图分类号:O 658 文献标识码:A 文章编号:04380479(2005)06 0801 05 丹参系唇形科鼠尾草属植物丹参的干燥根及根茎.丹参药材含有的有效成分类型包括脂溶性的二萜醌和水溶性的丹酚酸二大类,其中脂溶性成分包括丹参酮I 、丹参酮IIA 、隐丹参酮等,广泛应用于治疗化脓性感染及痤疮等;水溶性成分包括丹酚酸B 、迷迭香酸、原儿茶醛、原儿茶酸、丹参素等,主要用于治疗心血管疾病[1,2].因药典所载丹参野生资源渐少,各地纷纷引种栽培.为澄清混乱,寻找优良品种,进行开发利用,需对丹参药材进行系统的品种鉴定和质量评价[3].指纹图谱是现阶段可以全面反映中药材内在质量的一个有效手段,它突破传统的线性思维,根据指纹图谱的模糊属性,着眼于宏观规律性的特征分析[4].本文通过对不同等级及不同采收期中江丹参药材进行T LC 指纹图谱研究,为丹参药材的生产和质量控制提供科学可行的依据.1 实 验1.1 仪器与试药瑞士卡玛薄层色谱自动点样仪;Reprostar 3薄层色谱数码成像系统;w inCAT S 薄层色谱工作站;双槽展开缸;硅胶GF254预制板(德国Merck).甲醇、二氯甲烷、苯、醋酸乙酯、氯仿、甲酸均为分析纯,水为双蒸水;丹参酮IIA 对照品、丹参酮I 对照品、隐丹参酮对照品、丹参素钠对照品、原儿茶酸对照品、原儿茶醛对照品、咖啡酸对照品由中国生物制品检验所提供;丹酚酸B 对照品由中国药科大学周荣汉教授提供.丹参药材购自来源地四川中江药材公司(中江丹参由中国科学院昆明植物研究所李锡文教授鉴定为Salv ia.m iltio rrhiza Bg e.).不同商品等级中江丹参及不同采收期南丹参、丹参样品信息见表1、2.1.2 样品制备随机取10株丹参根部药材洗净,自然风干10d 后60 烘干,粉碎过60目筛.水溶性供试液:取待测药材粉末1.0g ,2%氨水超声提取60min,提取液于沸水浴中加热45m in(冰醋酸调pH 至3~4)后过滤.以乙酸乙酯萃取滤液,萃取液用氮气挥干,所得水溶性成分以1.0mL 甲醇溶解.脂溶性供试液:取待测药材粉末0.5g ,加甲醇/二氯甲烷(8/2,体积比)混合溶液10mL,置具塞试管中超声提取30m in 后过滤[5].对照品溶液:取2.0m g 丹参酮IIA 、丹参酮I 、隐丹参酮、表1 不同商品等级丹参药材T ab.1 Danshen samples w it h7different co mmercial gr ades特级品一级品二级品三级品四级品五级品六级品样品号123456789101112131415161718192021表2 中江南丹参、丹参药材动态样品*T ab.2 Danshen samples collected at differ ent harv est times南丹参丹参样品号23456791011121314151617181920采收期288317331359370388160190220250280282310311334353374386 *1号、8号及21号为混合标准物质(T he sample number of mix ed standard substances was1,8and21)原儿茶醛、原儿茶酸、咖啡酸和4.0mg丹酚酸B对照品,分别加1.0mL甲醇溶解;取1.0mg丹参素钠,加1.0mL25%的甲酸甲醇溶解.1.3 薄层色谱过程固定相:20cm 10cm硅胶60GF254高效薄层板(M erck),110 活化0.5h,干燥器中冷却至室温.水溶性展开剂:氯仿/乙酸乙酯/苯/甲酸(12/10/5/3,体积比);脂溶性展开剂:苯/乙酸乙酯(19/1,体积比)[1].点样:供试品及混合对照品溶液各2 L,底边距10 mm.展开方式:展开剂预平衡30min,上行展开8 0.5cm.检测:置紫外光254nm下检视,在254nm下获取色谱图像并同时生成色谱图和色谱数据.1.4 薄层色谱识别在水溶性供试品色谱中,在与对照品相应的位置上,显相同暗黄色斑点;主斑点自下而上为丹参素、丹酚酸B、原儿茶酸、原儿茶醛和咖啡酸;在脂溶性供试品色谱中,在与对照品色谱相应的位置上,显相同的暗红色斑点.主斑点自下而上为隐丹参酮,丹参酮I和丹参酮IIA.2 结果和讨论2.1 指纹图谱方法学考察2.1.1 稳定性试验取同一样品的供试液,分别在0, 6,12,24和48h点样检测指纹图谱,各主要色谱峰R f 值的RSD低于1.7%,峰面积RSD低于3.3%,表明同一样品在相同色谱条件下所测成分的R f值及色谱峰面积均取得良好的一致性.2.1.2 精密度试验取同一样品的供试液,同板连续点样5次,检测指纹图谱,各主要色谱峰R f值RSD低于1.2%,峰面积RSD低于2.6%,表明同一样品在相同色谱条件下连续点样时,各成分R f值及色谱峰面积精密度良好.方法精密可靠.2.1.3 重现性试验取同一批丹参药材的样品5份,按供试品溶液制备方法平行制备,同一条件下检测指纹图谱,各主要色谱峰R f值RSD低于1.7%,峰面积RSD低于4%,表明实验方法及仪器重现性均良好. 2.2 丹参TLC指纹图谱图像分析2.2.1 中江丹参药材TLC指纹图像等级间差异考察对样品和对照品依次点样、饱和、展开,在紫外光254 nm下获取7个等级、各10个批次的中江丹参药材色图1 不同等级中江丹参药材脂提物(上)及水提物(下)H PT L C图像(检测波长:254nm)F ig.1 H P T L C gr aph o f lipid so luble(up)and waterso luble(dow n)co mponents of Danshen w ithco mmercial gr ade S to g rade V I谱图像(图1).结果显示,7个等级、各10个批次的中江丹参药材脂溶性提取物色谱图像,在与对照品丹参酮IIA和隐丹参酮色谱斑点相对应处,不同等级样品间面积的大小和颜色的深浅均无明显差异,表明就主要指标成分的含量和物质群指纹图像而言,中江丹参脂溶性提取物的内在质量与药材的分级无明显的相802厦门大学学报(自然科学版) 2005年关.水溶性提取物色谱图像在与对照品原儿茶醛和丹酚酸B 色谱斑点相对应处,不同等级样品间面积的大小和颜色的深浅除了特级品与I 级品比较相近外,I 级品、II 级品和III 级品药材水溶性提取物的内在质量与药材的分级存在明显的正相关,而IV 级品、V 级品和VI 级品药材则未表现出明显的相关性.提示传统的依据丹参药材的色泽、粗细及均匀程度等外观性状进行分级的方法,在一定程度上反映了水溶性提取物的内在质量.2.2.2 中江丹参药材TLC 指纹图像动态分布考察中药材一般都有固定的采收时间,但对于采收时间跨度较大的中药材,应考察不同采收时间的指纹图谱,分析采收时间对指纹图谱,乃至对生物活性的影响.对样品和对照品依次点样、饱和、展开,在紫外光254nm 下获取不同生长周期的中江南丹参(时间跨度为100d)、中江丹参(时间跨度为226d)药材薄层色谱图像(图2).图2 不同生长周期的中江南丹参、丹参药材脂提物(上)及水提物(下)H PT LC 图像(检测波长:254nm)F ig.2 H P T L C gr aph o f lipid so luble (up)and waterso luble (dow n)components of Danshen co llected at differ ent har vest times结果显示,在紫外光254nm 下进行检视,脂溶性和水溶性提取物色谱图像与对照品隐丹参酮、丹参酮I 、丹参酮IIA 、原儿茶醛和丹酚酸B 色谱斑点相对应的样品色谱斑点处,不同采收期的南丹参,其斑点面积的大小和颜色的深浅无明显差异;生长期在160~386d 内的丹参药材,其隐丹参酮斑点面积的大小和颜色的深浅与采收期呈负相关;生长期在160~250d 内的丹参药材,其丹参酮I 、丹参酮IIA 、原儿茶醛和丹酚酸B 斑点面积的大小和颜色的深浅与采收期呈正相关,生长期在250~386d 内,上述斑点面积的大小和颜色的深浅无明显差异.图3 不同等级中江丹参药材水溶性成分指纹图谱聚类图Fig.3 Cluster ing plo t of water soluble com ponents inDanshen samples of commercial gr ade S to g rade V I2.3 丹参药材TLC 指纹图谱计量学分析2.3.1 不同等级中江丹参药材TLC 指纹图谱聚类分析比较7个等级,各10个批次中江丹参药材薄层色谱,确定其水溶性成分的共有峰为5个(R f 值在0.2~0.7之间).按照R f 值由小到大的顺序对其进行编号.取2号峰丹酚酸B 作为参照物,计算各峰的相对峰面积.以共有峰的相对峰面积为指标,对不同等级中江丹参药材水溶性成分进行聚类分析,以Pearson 相关系数为测度的聚类结果见图3.2.3.2 不同等级中江丹参药材TLC 指纹图谱主成分分析对7个等级,各10个批次中江丹参药材,脂溶性、水溶性成分共选取7个共有峰,以共有峰的相对峰面积为指标,进行主成分分析,结果见表3、表4.主成表3 主成分分析相关矩阵的特征值T ab.3 Eig env alues o f the PCA co rr elatio n mat rix 主成分特征值差异百分比累计贡献率12345675.583442510.800421690.400358550.141173670.046956430.027527940.000119224.783020820.400063140.259184880.094217240.019428490.027408720.79760.11430.05720.02020.00670.00390.00000.79760.91200.96920.98930.99611.00001.0000803 第6期 蒋轶伦等:薄层色谱指纹图谱在丹参药材质量评价中的应用研究表4 主成分分析特征向量T ab.4 T he PRIN COM P procedur e eigenvector s峰 号PC1PC2P C3P C4PC5PC6PC71 2 3 4 5 6 7-0.35570.3861-0.41460.3996-0.36180.39820.32100.38130.3656-0.14050.32920.18090.2439-0.70730.63300.00590.11110.0128-0.7524-0.10200.10050.21300.5650-0.21090.05050.2830-0.63370.32620.4237-0.5416-0.46510.30980.31170.13940.31300.22970.2327-0.3047-0.77110.09460.42740.11740.22220.22410.66490.19600.28960.40930.4101图4 不同等级中江丹参药材T LC指纹图谱主成分分析线性投影图F ig.4 PCA linear pr ojectiv e plot o f bo th lipid and waterso luble com ponents in Danshen samples of commer cial g rade S to g rade VI分1和主成分2的累计贡献率为0.9120(即91.2%),PC1(主成分1)=-0.3557P1+0.3861P2-0.4146P3+0.3996P4-0.3618P5+0.3982P6+0.3210P7,由此表达式可以看出,各峰对主成分1贡献十分相近,选取的7个共有峰具有指纹代表性.以主成分1和主成分2做线性投影图(图4).由图可见,如表1所示的不同等级21个丹参样品,各等级内在此投影图上可较好的聚为一类.由聚类及主成分分析的结果可知,经10个批次样品的实验,7个等级各自基本可聚为一类,说明各等级内(批次间)丹参药材主要指标成分含量分布情况是十分相似的,此法可用于考察不同等级丹参的归属.2.3.3 中江丹参药材动态分布TLC色谱数据显著性差异考察采收期时间跨度为100d的南丹参,其脂提与水提主要指标成分峰面积经方差分析,P=0.994,表明100d内6个采收时间点,各主要指标成分含量无显著性差异(P>0.05).以积分峰面积为指标,考察不同采收期丹参药材各主要成分含量与采收期关系(图5).从图中可以看出,其反映的趋势与图像考察是一致的.H arv est t ime/day图5 不同采收期中江丹参药材主成分T L C动态分布:隐丹参酮, :丹参酮IIA, :原儿茶醛, :丹参酮I, :丹酚酸BF ig.5 T he g ro wth dynamic trends o f Danshen co llectedat different harvest times上述结果表明,生长期在288~388d范围内,南丹参脂溶性和水溶性提取物的内在质量已基本稳定;就主要指标成分的含量而言,生长期在160~250d范围内,中江丹参药材中丹参酮IIA、原儿茶醛、丹参酮I和丹酚酸B的含量与采收期呈正相关,隐丹参酮作为早期次生代谢物和后期次生代谢物的前体[6],其含量随着植物的生长而呈下降趋势;生长期在250~386d范围内,中江丹参脂溶性和水溶性提取物的内在质量已基本稳定.3 结 论本研究采用薄层色谱方法,同时、快速地获取中江丹参药材的TLC图像,并生成基于光密度的薄层色谱图及量化的数据信息,通过对薄层色谱图像的直观比较,结合指纹图谱技术及化学计量学,对不同等级和不同采收期的丹参药材进行了质量评价.通过稳定性、精密度及重现性考察,显示本方法适用于丹参药材质量的评价.804厦门大学学报(自然科学版) 2005年参考文献:[1] 国家药典委员会.中华人民共和国药典(一部)[M ].北京:化学工业出版社,2000.57-58.[2] 杨保津.丹参的活性成分[J].中成药研究,1986,3:36-37.[3] 徐国均,徐珞珊,主编.常用中药材品种整理和质量研究(第一册)[M ].福州:福建科学技术出版社,1994.140-168.[4] 曹进,饶毅,沈群.中药指纹图谱及其建立原则[J].中药新药与临床药理,2001,12(3):200-203.[5] 胡建林,张荣平,李惠兰.3种滇产鼠尾草属植物丹参酮IIA 的薄层扫描定量[J].中成药,2000,22(10):723-724.[6] 徐任生.丹参生物学及其应用[M ].北京:科学出版社,1990.7-9.Quality Assessment of Radix Salvia Miltiorrhiza (Danshen)by TLC Fingerprinting TechniqueJIA NG Yi lun 1,LI Wei 1,ZH UA NG Zhi x ia 1,Frank S.C.Lee 2,WA NG Xiao ru 1,2*(1.Department of Chemistry ,Colleg e of Chemist ryand Chemica l Eng ineering ,T he K ey L aborat or y of A naly tical Sciences o f the M OE,Xiamen U niver sity,Xiamen 361005,China;2.Q ingdao K ey L ab of A na lyt ical T echnolo gy Development and St andardization o f Chinese M edicines,F ir st Instit ute O ceanog ra phy o f SOA ,Q ing dao 266061,China)Abstract:T he purpose of this st udy is to develo p and apply t he thin lay er chr omato gr aphy (T L C)technique to the finger printingstudies of Danshen (Radix Salv ia M iltio rr hiza)dr ug s.A rang e o f Danshen samples w ith differ ent co mmer cial g rades or har vested af ter diff er ent g ro wing time was analyzed for qualit y assessment and reg ulato ry purposes.A nd the composit ional va riations o f these au thentic Danshen dr ug s wer e inv est igated using an analy tical pr otoco l invo lv ing w ater o r or ganic solvent ex tr act ions,fo llowed by T L C assay of the extr acts,and subsequent T L C fing erprint ing analysis by imag e com par ison combined w ith chemometr ics methods.T he analytical perfor mances of the method wer e ev aluated by ex amining the stability,precision and r epr oducibility ,r espectively.Excellent results w ere observ ed and the RSDs of relat ive R f v alues and peak a reas w ere respectiv ely less than 1.7%and 4.0%.T he analysis re sults o btained sug gested t hat a t otal of seven co mmon peaks wer e identified in the T L C chr omato gr ams under the 254nm detection wav eleng th.Based on the compariso n o f the R f v alues of the common peaks,the depth and size of t he T L C spo ts and in co mbination with cluster analysis and pr incipal co mpo nent analysis (P CA ),the att ribute o f quality g rading and optimal har vest t ime o f the Dansh en material w ere ident ified.T hus,the dev eloped T L C chro matog raphic finger pr inting metho d is consider ed as an useful and effectiv e too l for the qualit y contro l o f the D anshen drug material.Key words:Radix Salvia M iltio rrhiza;T L C fing erpr int ing;image analysis;chemometr ics;quality assessment805 第6期 蒋轶伦等:薄层色谱指纹图谱在丹参药材质量评价中的应用研究。
Gelucire-14-44-SDS-MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:Nov.-23-2018Print Date:Nov.-23-20181. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :Gelucire 14/44Catalog No. :HY-Y1892CAS No. :121548-04-71.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:sales@1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureNot a hazardous substance or mixture.2.2 GHS Label elements, including precautionary statementsNot a hazardous substance or mixture.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:NoneFormula:N/AMolecular Weight:N/ACAS No. :121548-04-74. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation.7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature:Pure form-20°C 3 years4°C 2 yearsIn solvent-80°C 6 months-20°C 1 monthShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Cleanspillages in a safe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance White to off-white (Oil)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGThis substance is considered to be non-hazardous for transport.IATAThis substance is considered to be non-hazardous for transport.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2018 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
高效液相色谱法测定复方刺五加糖浆中紫丁香苷含量

高效液相色谱法测定复方刺五加糖浆中紫丁香苷含量【摘要】目的建立复方刺五加糖浆的含量测定方法。
方法采用高效液相色谱法测定复方刺五加糖浆中紫丁香苷的含量。
以甲醇-水(8∶2)为流动相,以岛津VP-ODSC18柱(5 μm 4.6×150 mm)为固定相,检测波长为265 nm。
结果紫丁香苷在0.080 5~1.61 μg范围内有良好的线性关系,r=0.999 7。
平均回收率为97.4%(n=6),RSD=0.55%。
结论该法操作简便准确,可作刺五加糖浆中紫丁香苷的质量控制方法。
【Abstract】Objective To study determination method of Syringin in Compound Ciwujia Syrup by HPLC.Methods Using VP-ODS C18column(5 μm 4.6×150mm),the mobile phase was methanol-water(8:2)at the rate 1.0 ml/min,the wavelength of detector was 265 nm.Results The linear rang was 0.080 5~1.61 μg(r=0.999 7).The average recovery was 97.4%(RSD=0.55%,n=6).Conclusion The method is simple,rapid,reliable.It can be used for the quality control of Compound Ciwujia Syrup.【Key words】Compound Ciwujia Syrup;Syringin;HPLC复方刺五加糖浆为医院制剂,其处方中含有刺五加浸膏、五味子、蔗糖及苯甲酸钠。
其中有效成分之一的紫丁香苷(即刺五加苷B)具有抗疲劳的作用,对中枢神经的兴奋与抑制均有影响。
玉足海参糖胺聚糖对血液中钙离子影响的研究

玉足海参糖胺聚糖对血液中钙离子影响的研究1朱伟1,张家骊1,蒋毅2,朱劼1,任蓓霞1,龚逸奕11.江南大学医药学院,江苏无锡(214122);2.上海开润生物医药有限公司,上海(200000)E-mail: 541206@摘要:目的:研究玉足海参糖胺聚糖(GAG)对血液中钙离子的影响。
方法:大白兔耳缘静脉取血进行体外复钙凝血实验,考察GAG对复钙凝血时间的影响。
考查GAG浸泡24 h后的毛细管对小鼠眼眶后静脉丛取血过程中血液Ca2+的影响。
将大鼠随机分为5组,正常对照组、模型对照组、肝素钠组、GAG低剂量组、GAG高剂量组,每组6只。
静脉注射一周后,取血备查血液中钙离子(Ca2+)浓度,除正常对照组外,其他各组均用角叉菜胶构建尾部血栓模型,观察不同时间的黑尾长度。
结果GAG对复钙凝血时间的影响可因充足Ca2+的加入而减小。
用GAG浸泡24 h后的毛细管对小鼠眼眶后静脉丛进行取血可显著降低血液中Ca2+的浓度。
GAG高剂量组黑尾长度显著减少,GAG高、低剂量组 Ca2+浓度显著降低。
结论:GAG能有效减轻角叉菜胶所致的大鼠尾部血栓,并能显著影响血液中Ca2+的浓度。
关键词:糖胺聚糖;血栓形成;角叉菜胶;钙离子1. 前言玉足海参糖胺聚糖(holothurian glycosaminoglycan, GAG)是从我国南海一带海域广泛存在的的玉足海参体壁中提取的一种含岩藻糖分支的糖胺聚糖[1],在临床上主要用于血栓性疾病的预防和治疗。
实验和临床研究发现,GAG具有抗凝血、抗血栓、降低血液黏度和降低血脂等作用[2-4],且可通过多种机制发挥其抗凝血和抗血栓作用[3-5]。
Ca2+是血液中极其重要的活性离子,其作为凝血因子Ⅳ在凝血过程中发挥极其重要的作用,凝血因子Ⅸ、Ⅹ等的激活及相关凝血因子复合物/激活物的形成均必需Ca2+的参与,凝血酶(Ⅱ)及纤维蛋白多聚体的形成中Ca2+也是必不可少的,可以说Ca2+几乎参与了血液凝固的整个过程[6]。
槲皮素分子印迹聚邻苯二胺敏感膜电化学传感器

槲皮素分子印迹聚邻苯二胺敏感膜电化学传感器王微;孙红;赫春香【摘要】以槲皮素为模板分子,邻苯二胺为功能单体,采用循环伏安法制备了槲皮素分子印迹薄膜修饰电极,并将其用于槲皮素的检测.在HAc-NH4Ac缓冲溶液(pH 4.0)中,以微分脉冲伏安法为电化学激发信号,槲皮素在0.37 V(VS.SCE)处产生一个灵敏的氧化峰,峰电流与其浓度在8.00×10-8~1.00×10-3 mol/L 范围内呈线性关系,实验检出限为5.00×10-8 mol/L.将新方法应用于2种银杏叶类药物中槲皮素的测定,回收率在99.2%~102%之间.【期刊名称】《高师理科学刊》【年(卷),期】2012(000)006【总页数】3页(P50-52)【关键词】槲皮素;邻苯二胺;分子印迹膜;微分脉冲伏安法【作者】王微;孙红;赫春香【作者单位】辽宁师范大学化学化工学院,辽宁大连 116029;辽宁师范大学化学化工学院,辽宁大连 116029;辽宁师范大学化学化工学院,辽宁大连 116029【正文语种】中文【中图分类】O657.1槲皮素(Quercetin,Qu)具有多种药用价值,因而在黄酮类化合物中占有重要的地位[1],其测定方法的研究在鉴定生产原料品质、评价生物制剂工艺以及药代动力学分析等方面具有重要意义.目前,检测槲皮素的方法主要有毛细管电泳法[2]、高效液相色谱法[3]、荧光法[4]和电化学检测方法[5-7].将分子印迹技术应用于传感器的研制是近年来出现的新技术[8].邻苯二胺(o-pheny-lenediamine,OPD)是制备分子印迹电化学传感器常用的功能单体,采用电化学方法可以直接在基体电极表面聚合成带有模板印迹的聚合物[9].这种分子印迹技术不需要交联剂和致孔剂,具有简便、快速的优点.本文以槲皮素为模板分子,邻苯二胺为功能单体,制备了槲皮素分子印迹电化学传感器,并建立了微分脉冲伏安法测定槲皮素的新方法.本方法具有灵敏度高、选择性好、方法简便等优点.LK9805电化学分析仪(天津市兰力科电子科技发展有限公司);三电极系统:以石墨(SG)、聚邻苯二胺/石墨、槲皮素/邻苯二胺/石墨分子印迹修饰电极为工作电极,饱和甘汞电极为参比电极(文中全部电位均相对于饱和甘汞电极),铂丝电极为辅助电极.槲皮素、芦丁、桑色素、葛根素和山奈酚(由西安天本生物工程有限公司提供,纯度>98%).除指明外其它化学试剂均为分析纯.实验用水为石英亚沸二次蒸馏水. 1.2.1 分子印迹修饰电极的制备将光谱纯石墨棒制成蜡浸石墨电极(SG),并清洗至洁净[10].以SG为工作电极,含有1.00×10-2 mol/L OPD,4.00×10-4mol/L Qu,0.10 mol/L KCl的混合溶液为修饰剂,在+1.50 ~-1.00 V电位范围内,以0.05 V/s的扫速连续循环伏安扫描30圈.取出电极,用水冲洗,再置于pH 7.0 的磷酸盐缓冲溶液(PBS)中,循环扫描30圈,得到具有槲皮素印迹的聚邻苯二胺石墨修饰电极(MIPCME).不加入模板分子(槲皮素)制备非分子印迹修饰电极(NIPCME).1.2.2 槲皮素的测定以MIPCME为工作电极,静置吸附300 s,-0.30 V为起点电位,在-0.30~0.60 V电位范围内记录微分脉冲伏安曲线,根据 0.37 V 处的峰电流进行定量分析.分别以SG,NIPCME,MIPCME为工作电极,测定5 mmol/L K3Fe(CN)6(0.1 mol/L KNO3中)的循环伏安曲线(见图1).结果表明,K3Fe(CN)6在NIPCME电极上没有电化学响应,在MIPCME上出现氧化还原峰,但是峰电流明显小于其在SG电极上的响应.说明NIPCME是电惰性的,而MIPCME上存在空穴.分别以SG,MIPCME,NIPCME为工作电极,按照实验方法测定5.00×10-5 mol/L槲皮素.结果表明,槲皮素在MIPCME上的峰电流远远大于在SG电极上电流响应(见图2).分别试验了槲皮素在HAc-NH4Ac(pH 4.00~6.00 ),NH3-NH4Cl(pH8.00~10.00),PBS(pH 4.00~10.00)中的电化学响应.结果表明,以pH 4.0 的HAc -NH4Ac缓冲溶液作为支持电解质时峰电流最大.实验发现,吸附时间对峰电流有明显的影响,最佳吸附时间为300 s.在8.00×10-8~1.00×10-3mol/L范围内,槲皮素的浓度与峰电流呈线性关系,回归方程ip(μA)= 3.14 c(μmol/L)+ 4.33(r=0.998),实验检出限5.00×10-8 mol/L.一支电极平行测定6组同浓度的槲皮素,响应值的相对标准偏差(RSD)为1.2 %;平行制备4支电极,RSD为3.3 %.在允许存在10 % 误差的前提下,桑色素、葛根素和山奈酚不干扰测定.芦丁对本方法有一定的干扰,最大共存量为槲皮素的60倍.此外,实验还表明,测定体系中允许共存300倍的十二烷基磺酸钠,150倍的抗坏血酸,100倍的聚乙烯醇,50倍的草酸和柠檬酸钠.其它常见金属离子均不干扰测定.测定了舒血宁注射液(1#)和银杏叶分散片(2#)中槲皮素的质量分数.银杏叶中黄酮苷的代表成分是槲皮苷、山萘酚苷、芦丁苷和鼠李糖苷[11].据此对药品进行预处理:将试样溶液与 50 mL 0.5 % 的硫酸和45 mL无水乙醇混合,加热回流1 h,冷却,用无水乙醇定容至100 mL.处理后槲皮苷转化成槲皮素,芦丁水解转化成槲皮素,结果见表1,平均回收率(n=3)分别为99.2%,102%.表明此方法适用于实际样品中槲皮素的测定.【相关文献】[1] Luo Yiping.Advances in Pharmacological Research of Flavonoids[J].Asia-Pacific Traditional Medicine,2010(4):132-134[2] Dadakova E,Kalinova J.Determination of quercetin glycosides and free quercetin in buckwheat by capillary micellar electrokimetic chromatography[J].Journal of Separation Science,2010,33:1633-1638[3] Du FY,Xiao XH.Lonic lipquid aqueous solvent-based microwave-assisted hydrolysis for the extraction and HPLC determination of myricetin and quercetin from Myrica rubra leaves[J].Biomedical Chromatography,2011,25:472-478[4] Chen Daiwu,Xie Qingji.Fluorescent Studies on the Interactions of Quercetin with Aminoacid[J].Chemical Research,2009(4):78- 81[5] 轩卫华,曹红,龚晓武,等.槲皮素在悬汞电极上的伏安行为研究[J].分析试验室,2011,29(11):32-35[6] Ziyatdinova G,Aytuganova I,Nizamova A,et al.Cyclic voltammetry of natural flavonoids mwnt-modified electrode and their determination inpharmaceuticals[J].Collection of Czechoslovak chemical communications,2011,76:1619-1631[7] 王明艳,许兴友,马卫兴,等.4-(3-吡啶基)-2-巯基咪唑修饰金电极一阶微分线性扫描伏安法直接测定复方鱼腥草片中槲皮素[J].理化检验:化学分册,2010,46(10):1125-1128,1131[8] 朱玲艳,王宗花,陈小印,等.聚吡咯/碳纳米管分子印迹修饰电极对槲皮素的选择性测定[J].分析测试学报,2011,30(1):18-23[9] Tanji Y, Wanzhi W,Jinxiang Z. Selective detection of dopamine in the presence of ascorbic acid by use of glassy-carbon electrodes modified with both polyaniline film and multi-walled carbon nanotubes with incorporated β-cyclodextrin[J].Analytical and Bioanalytical Chemistry,2006,386:2087-2094[10] 李晓娟,赫春香.铁氰化镍/银复合修饰电极的制备及对氟康唑的电吸附作用[J].高师理科学刊,2009,29(1):68-71[11] 唐于平,王颖,楼凤昌,等.银杏叶中的黄酮醇苷类成分[J].药学学报,2000,35(5):363-366。
毛细管胶束电动色谱-紫外间接检测环胞素制剂中的有机溶剂

毛细管胶束电动色谱-紫外间接检测环胞素制剂中的有机溶剂刘浩;陈代杰【摘要】用毛细管胶束电动色谱-紫外间接检测环胞素软胶囊及环胞素注射液中的有机溶剂含量.采用非涂层弹性石英毛细管;操作缓冲液为含0.26mol/L十二烷基硫酸钠的0.02mol/L苯巴比妥钠溶液(pH9.0);检测波长为235nm(间接检测);电泳过程中在进样端始终外加适当的压力使基线保持稳定.以甲醇为内标物,乙醇和1,2-丙二醇均在2.6~18mg/ml范围内呈良好的线性关系.连续进样分析所得峰面积和迁移时间的相对标准偏差均不大于1.3%.环胞素软胶囊中乙醇和1,2-丙二醇含量测定方法的回收率分别为99,2%和100.7%;环胞素注射液中乙醇含量测定方法的回收率为99.5%.【期刊名称】《中国抗生素杂志》【年(卷),期】2010(035)002【总页数】4页(P111-114)【关键词】毛细管胶束电动色谱;紫外间接检测;有机溶剂;环胞素软胶囊;环胞素注射液【作者】刘浩;陈代杰【作者单位】上海医药工业研究院,上海,200040;上海市食品药品检验所,上海,201203;上海市食品药品检验所,上海,201203【正文语种】中文【中图分类】R978.1~+1环胞素为常用免疫抑制剂,临床上用于防止器官移植后的移植物排斥作用。
环胞素的脂溶性较强,在水中几乎不溶。
环胞素软胶囊的处方中主要采用乙醇和1,2-丙二醇作助溶剂,而环胞素注射液的处方中则主要采用乙醇和聚氧乙烯蓖麻油作助溶剂。
环胞素软胶囊及环胞素注射液的进口复核标准(JX20000339和JX20040257)规定应对软胶囊中的乙醇和1,2-丙二醇以及注射液中的乙醇加以控制,含量限度均为标示量的80.0%~120.0%,检测方法为以高分子多孔小球为载体的填充柱气相色谱(GC)法。
然而,由于环胞素和高沸点的聚氧乙烯蓖麻油等辅料在色谱柱载体上的不可逆吸附,分析过程中色谱柱会逐渐被污染变色甚至阻塞,具体表现为柱效降低,分离度减小,色谱峰变形。
紫外可见吸收光谱习题集和答案

五、紫外可见分子吸收光谱法(277题)一、选择题( 共85题)1. 2 分(1010)在紫外-可见光度分析中极性溶剂会使被测物吸收峰( )(1) 消失(2) 精细结构更明显(3) 位移(4) 分裂2. 2 分(1019)用比色法测定邻菲罗啉-亚铁配合物时,配合物的吸收曲线如图1所示,今有a、b、c、d、e滤光片可供选用,它们的透光曲线如图2所示,你认为应选的滤光片为( )3. 2 分(1020)欲测某有色物的吸收光谱.下列方法中可以采用的是( )(1) 比色法(2) 示差分光光度法(3) 光度滴定法(4) 分光光度法4. 2 分(1021)按一般光度法用空白溶液作参比溶液.测得某试液的透射比为10%.如果更改参比溶液.用一般分光光度法测得透射比为20% 的标准溶液作参比溶液.则试液的透光率应等于( )(1) 8% (2) 40% (3) 50% (4) 80%5. 1 分(1027)邻二氮菲亚铁配合物.其最大吸收为510 nm.如用光电比色计测定应选用哪一种滤光片?( )(1) 红色(2) 黄色(3) 绿色(4) 蓝色6. 2 分(1074)下列化合物中.同时有n→π*.π→π*.σ→σ*跃迁的化合物是( )(1) 一氯甲烷(2) 丙酮(3) 1,3-丁二烯(4) 甲醇7. 2 分(1081)双波长分光光度计的输出信号是( ) (1) 试样吸收与参比吸收之差(2) 试样在λ1和λ2处吸收之差(3) 试样在λ1和λ2处吸收之和(4) 试样在λ1的吸收与参比在λ2的吸收之差8. 2 分(1082)在吸收光谱曲线中.吸光度的最大值是偶数阶导数光谱曲线的( )(1) 极大值(2) 极小值(3) 零(4) 极大或极小值9. 2 分(1101)双光束分光光度计与单光束分光光度计相比.其突出优点是( )(1) 可以扩大波长的应用范围(2) 可以采用快速响应的检测系统(3) 可以抵消吸收池所带来的误差(4) 可以抵消因光源的变化而产生的误差10. 2 分(1105)在紫外光谱中.λmax最大的化合物是( )11. 2 分 (1106)用实验方法测定某金属配合物的摩尔吸收系数ε.测定值的大小决定于( )(1) 配合物的浓度 (2) 配合物的性质(3) 比色皿的厚度 (4) 入射光强度12. 2 分 (1173)下列结构中哪一种能产生分子荧光? () OHNO 2COOHI(1)(2)(3)(4)13. 2 分 (1198)1198有下列四种化合物已知其结构.其中之一用 UV 光谱测得其λmax 为 302nm.问应是哪种化合物? ( )CH 3CH CHCOCH 3CH 3CH 3(4)(3)(2)BrOHOOCH 33CH 3(1)14. 2 分 (1217) 许多化合物的吸收曲线表明.它们的最大吸收常常位于 200─400nm 之间.对这一光谱区应选用的光源为 ( )(1) 氘灯或氢灯 (2) 能斯特灯(3) 钨灯 (4) 空心阴极灯灯15. 5 分 (1231)下列四种化合物中,在紫外光区出现两个吸收带者是 ( )(1)乙烯 (2)1,4-戊二烯(3)1,3-丁二烯 (4)丙烯醛16. 2 分 (1232)助色团对谱带的影响是使谱带 ( )(1)波长变长 (2)波长变短(3)波长不变 (4)谱带蓝移17. 5 分 (1233)对化合物 CH 3COCH=C(CH 3)2的n — *跃迁,当在下列溶剂中测定,谱带波长最短的是 ( )(1)环己烷 (2)氯仿(3)甲醇 (4)水紫外-可见吸收光谱曲线呈高斯分布的是( )(1)多普勒变宽(2)自吸现象(3)分子吸收特征(4)原子吸收特征19. 2 分(1300)指出下列哪种是紫外-可见分光光度计常用的光源?( )(1) 硅碳棒(2) 激光器(3) 空心阴极灯(4) 卤钨灯20. 2 分(1301)指出下列哪种不是紫外-可见分光光度计使用的检测器?( )(1) 热电偶(2) 光电倍增管(3) 光电池(4) 光电管21. 2 分(1302)指出下列哪种因素对朗伯-比尔定律不产生偏差?( )(1) 溶质的离解作用(2) 杂散光进入检测器(3) 溶液的折射指数增加(4) 改变吸收光程长度22. 1 分(1303)分子荧光过程是( )(1) 光致发光(2) 能量源激光发光(3) 化学发光(4) 电致发光23. 1 分(1305)在分子荧光测量中, 在下列哪一种条件下, 荧光强度与浓度呈正比? ( )(1) 荧光量子产率较大(2) 在稀溶液中(3) 在特定的激发波长下(4) 用高灵敏度的检测器下列哪种方法的测量灵敏度高? ( )(1) 磷光分析法(2) 荧光分析法(3) 紫外-可见分光光度法(4) 目视比色法25. 2 分(1307)已知相对分子质量为320的某化合物在波长350nm处的百分吸收系数(比吸收系数)为5000, 则该化合物的摩尔吸收系数为( )(1)1.6×104L/(moL·cm) (2)3.2×105 L/(moL·cm)(3)1.6×106 L/(moL·cm) (4)1.6×105 L/(moL·cm)26. 2 分(1308)在310nm时, 如果溶液的百分透射比是90%,在这一波长时的吸收值是( )(1) 1 (2) 0.1 (3) 0.9 (4) 0.0527. 1 分(1309)荧光分析法和磷光分析法的灵敏度比吸收光度法的灵敏度( )(1) 高(2) 低(3) 相当(4) 不一定谁高谁低28. 2 分(1324)紫外-可见吸收光谱主要决定于( )(1) 分子的振动、转动能级的跃迁(2) 分子的电子结构(3) 原子的电子结构(4) 原子的外层电子能级间跃迁29. 1 分(1333)指出下列说法中哪个有错误? ( )(1) 荧光和磷光光谱都是发射光谱(2) 磷光发射发生在三重态(3) 磷光强度I p与浓度c的关系与荧光一致(4) 磷光光谱与最低激发三重态的吸收带之间存在着镜像关系30. 2 分(1334)指出下列不正确的说法?( )(1) 分子荧光光谱通常是吸收光谱的镜像(2) 分子荧光光谱与激发波长有关(3) 分子荧光光谱较激发光谱波长长(4) 荧光强度与激发光强度呈正比31. 2 分(1335)下列哪一种分子的去激发过程是荧光过程? ( )(1) 分子从第一激发单重态的最低振动能级返回到基态(2) 分子从第二激发单重态的某个低振动能级过渡到第一激发单重态(3) 分子从第一激发单重态非辐射跃迁至三重态(4) 分子从第一激发三重态的最低振动能级返回到基态32. 2 分(1336)下列哪种说法有错误? ( )(1) 荧光分子的激发光谱与发射波长无关(2) 荧光分子的激发光谱的荧光强度是激发波长的函数(3) 在分子荧光光谱法中吸收与激发光谱常可以互换(4) 得到荧光分子的激发光谱方法与常规吸收光谱方法是两种基本相同的方法33. 2 分(1338)在荧光光谱中, 测量时, 通常检测系统与入射光的夹角呈( )(1) 180°(2) 120°(3) 90°(4) 45°34. 2 分(1339)某荧光物质的摩尔吸收系数为2.0×105L/(mol cm),当用激发光强度为50(随机单位)去激发该荧光物质, 若吸收池为1.0cm, 化合物浓度为5.0 ×10-7mol/L,测得荧光强度为2.3(随机单位), 则该化合物的荧光量子效率约为( )(1) 0.2 (2) 0.46 (3) 23 (4) 2.335. 2 分(1340)某化合物在λmax=356nm处, 在乙烷中的摩尔吸收系数εmax=87 L/(mol⋅cm), 如果用1.0cm吸收池,该化合物在已烷中浓度为1.0 ×10-4mol/L,则在该波长处, 它的百分透射比约为( )(1) 87% (2) 2% (3) 49% (4) 98%36. 2 分(1341)某化合物的浓度为1.0 ×10-5mol/L,在λmax=380nm时, 有透射比为50%, 用1.0cm吸收池, 则在该波长处的摩尔吸收系数εmax /[L/(mol⋅cm)]为( )(1) 5.0 ×104 (2) 2.5 ×104 (3) 1.5 ×104 (4) 3.0 ×10437. 2 分(1342)在分光光度计的检测系统中, 以光电管代替硒光电池, 可以提高测量的( )(1) 灵敏度(2) 准确度(3) 精确度(4) 重现性38. 2 分(1343)基于发射原理的分析方法是( )(1) 光电比色法(2) 荧光光度法(3) 紫外及可见分光光度法(4) 红外光谱法39. 2 分(1344)基于吸收原理的分析方法是( )(1) 原子荧光光谱法(2) 分子荧光光度法(3) 光电直读光谱法(4) 紫外及可见分光光度法40. 2 分(1346)在紫外-可见分光光度计中, 强度大且光谱区域广的光源是( )(1) 钨灯(2) 氢灯(3) 氙灯(4) 汞灯41. 1 分(1355)硒光电池主要用于检测( )(1) X射线(2) 紫外光(3) 可见光(4) 红外光42. 2 分(1357)荧光分光光度计与紫外-可见分光光度计的主要区别在于( )(1) 光路(2) 光源(3) 单色器(4) 光电倍增管43. 2 分(1367)物质的紫外-可见吸收光谱的产生是由于( )(1) 分子的振动(2) 分子的转动(3) 原子核外层电子的跃迁(4) 原子核内层电子的跃迁44. 1 分(1371)工作波长范围较宽的光度计为( )(1) 581-G型滤光光度计(2) 72型分光光度计(3) 721 型分光光度计(4) 751 型分光光度计45. 2 分(1372)在一定波长处, 用2.0 cm比色皿测得某试液的透光度为60%, 若改用3.0 cm比色皿时, 该试液的吸光度为( )(1) 0.11 (2) 0.22 (3) 0.33 (4) 0.4446. 1 分(1374)阶跃线荧光的波长( )(1)大于所吸收的辐射的波长(2)小于所吸收的辐射的波长(3)等于所吸收的辐射的波长(4)正比于所吸收的辐射的波长47. 2 分(1381)双波长分光光度计的输出信号是( )(1) 试样与参比吸收之差(2) 试样与参比吸收之和(3) 试样在λ1和λ2处吸收之差(4) 试样在λ1和λ2处吸收之和48. 1 分(1752)下面哪一种电子能级跃迁需要的能量最高? ( )(1) σ→σ*(2) n→σ *(3) π→π* (4) π→σ*49. 2 分(1753)化合物中CH3--Cl在172nm有吸收带,而CH3--I的吸收带在258nm处,CH3--Br 的吸收带在204nm ,三种化合物的吸收带对应的跃迁类型是( )(1) σ→σ*(2) n→π*(3) n→σ * (4)各不相同50. 2 分(1754)某化合物在乙醇中λmax乙醇=287nm,而在二氧六环中λmax二氧六环=295nm.该吸收峰的跃迁类型是()(1) σ→σ* (2) π→π*(3) π→σ* (4) π→π*51. 2 分(1755)一化合物溶解在己烷中,其λmax己烷=305 nm.而在乙醇中时.λ乙醇=307nm.引起该吸收的电子跃迁类型是( )(1) σ→σ * (2)n→π *(3) π→π* (4) n→σ*52. 2 分(1756)在分子CH3的电子能级跃迁中,下列哪种电子能级跃迁类型在该分子中不发生( )(1) σ→π* (2) π→σ*(3) n→σ* (4) n→π*53. 2 分(1757)一化合物在235nm处有最大吸收值,用1.0 cm的吸收池,化合物的浓度为2.0×10-4mol/L,透射比为20%, 则在该波长处的摩尔吸收系数εmax /[L/(moL·cm)]为( )(1) 5.0×103 (2) 3.5×103 (3) 2.5×103 (4) 1.0×10354. 1 分(1758)在254nm时.如果溶液的百分透射比是10%.其吸光度值为()(1) 1 (2) 0.9 (3) 0.1 (4) 0.0555. 2 分(1759)某化合物在己烷中(λmax=220nm)的摩尔吸收系数εmax=14500L/(moL·cm).若用1.0cm吸收池.1.0×10-4mol/L的该化合物在该波长处的百分透射比为()(1) 5% (2) 3.5% (3)10% (4)50%56. 2 分(1760)对某特定的仪器.其透射比的标准偏差为0.006.对某溶液测得的透射比T=0.015 时那么浓度的相对标准偏差是()(1) +2.5% (2) +5.0% (3) +9.5% (4) +12.5%57. 2 分(1761)对某特定的仪器.其透射比的标准偏差为0.006.当测得溶液的百分透射比T=64.8%时.则浓度的相对标准偏差是()(1) +6.6% (2) +4.2% (3) +3.4% (4) +2.1%58. 2 分(1762)对某特定的仪器.其透射比的标准偏差为0.006.当测得溶液的吸光度A=0.334时. 则浓度的相对标准偏差是 ( )(1) +0.6% (2) +1.7% (3) +3.5% (4) +7.6% 59. 2 分 (1763)比较下列化合物的UV -VIS 光谱λmax 大小( )CH 3CHON(CH 3)2(a)OHOCl CH 3COOC 2H 5(b)COOHCl(CH 3)2N(C)(1)a>b>c (2)c>a>b (3)b>c>a (4)c>b>a 60. 2 分 (1764)比较下列化合物的UV -VIS 吸收波长的位置(λmax )( )(C)CH 3OCH 3OC(b)COOHOCl(a)O(1) a>b>c (2) c>b>a (3)b>a>c (4)c>a>b 61. 2 分 (1765)在紫外-可见吸收光谱中.下列具有最大吸收波长的物质是( )O(1)(2) (3)(4)62. 2 分 (1766)在紫外-可见光谱区有吸收的化合物是( )(1) CH 3-CH=CH-CH 3 (2) CH 3-CH 2OH(3) CH 2=CH-CH 2-CH=CH 2 (4) CH 2=CH-CH=CH-CH 363. 2 分(1767)Fe和Cd 的摩尔质量分别为55.85g/mol和112.4g/mol.各用一种显色反应用分光光度法测定.同样质量的两元素分别被显色成容积相同的溶液.前者用2cm吸收池.后者用1cm吸收池.所得吸光度相等.此两种显色反应产物的摩尔吸收系数为()(1) εFe≈2εCd (2) εCd ≈2εFe(3) εCd≈4εFe (4) εFe≈4εCd64. 2 分(1768)双波长分光光度计和单波长分光光度计的主要区别是()(1)光源的个数(2)单色器的个数(3)吸收池的个数(4)单色器和吸收池的个数65. 1 分(1769)物质的颜色是由于选择性地吸收了白光中的某些波长所致.CuSO4溶液呈蓝色是由于它吸收了白光中的()(1) 蓝色光(2) 绿色光(3) 黄色光(4) 红色光66. 2 分(1770)符合朗伯-比尔定律的有色溶液稀释时.其最大吸收峰的波长位置()(1) 向长波方向移动(2) 向短波方向移动(3) 不移动.但最大吸收峰强度降低(4) 不移动.但最大吸收峰强度增大67. 2 分(1771)某金属离子X和R试剂形成一有色配合物.若溶液中X的浓度为1.0×10-4mol/L.用1cm吸收池在525nm处测得吸光度为0.400.则此配合物在525nm处的摩尔吸收系数为( )(1) 4.0×10-3 (2) 4.0×103(3) 4.0×10-4 (4) 4.0×10468. 2 分(1772)以下三种分析方法:分光光度法(S)、磷光法(P)和荧光法(F).具有各不相同的灵敏度.按次序排列为( )(1) P<F<S (2) S=F<P (3) P<S<F (4) F>P>S69. 2 分(1773)A和B二物质紫外-可见吸收光谱参数如下:物质λ1时的摩尔吸收系数λ2时的摩尔吸收系数/[L/(moL·cm)]A 4,120 0.00B 3,610 300若此二种物质的某溶液在λ1时在1.00cm 吸收池中测得A=0.754.在λ2时于10.0cm 吸收池中测得A =0.240.问B的浓度是多少?()(1) 0.64×10-5mol/L (2) 0.80×10-5 mol/L(3) 0.64×10-4mol/L (4) 0.80×10-4mol/L70. 1 分(1774)分光光度法中.为了减小浓度测量的相对误差.配制的试样溶液的透射比应控制在什么范围?()(1) 小于1% (2) 1%-10%(3) 30%-50% (4) 90%-99%71. 2 分(1775)下列哪种方法可用于测定合金中皮克数量级(10-12)的铋?()(1)分光光度法(2)中子活化(3)极谱法(4)电位滴定法72. 2 分(1776)K I O4法氧化Mn2+到MnO4-.然后用分光光度法测定.选择合适的空白为()(1) 蒸馏水(2) 试剂空白(3) 除K I外的试剂空白(4) 不含K I O4的溶液空白73. 1 分(1777)在分光光度法中.运用朗伯-比尔定律进行定量分析采用的入射光为()(1)白光(2)单色光(3)可见光(4)紫外光74. 2 分(1778)在分光光度法中.运用朗伯-比尔定律进行定量分析采用的入射光为()(1)白光(2)单色光(3)可见光(4)紫外光75. 2 分(1779)邻二氮菲亚铁配合物的最大吸收波长为510nm.如用光电比色计测定时应选哪种滤光片?()(1)红色(2)黄色(3)绿色(4)蓝色76. 2 分(1780)分子运动包括有电子相对原子核的运动(E电子)、核间相对位移的振动(E振动)和转动(E转动)这三种运动的能量大小顺序为()(1) E振动>E转动>E电子(2) E转动>E电子>E振动(3) E电子>E振动>E转动(4) E电子>E转动>E振动77. 2 分(1781)现有紫外-可见吸收光谱相互干扰的A和B两组分.它们的最大波长分别为λA和λB.若用双波长测定A组分的含量.则下面哪一种选择λ1和λ2的方法是正确的?()(1)使λ1和λ2分别等于λA和λB(2)选λ1等于λA.选λ2使B组分在λ2的吸光度和它在λ1处的吸光度相等(3)选λ1等于λA.选λ2为A.B两组分吸收峰相交处的波长(4)选λ1等于λB.选λ2使A组分在λ2的吸光度和它在λ1处的吸光度相等78. 1 分(1782)某化合物在乙醇中的λmax=240nm.εmax=13000L/(moL·cm).则该UV-VIS吸收谱带的跃迁类型是()(1) n→σ* (2) n→π* (3) π→π* (4) σ→σ*79. 2 分(1783)在分子荧光法中.以下说法中正确的是()(1)激发过程中的电子自旋虽不变.但激发态已不是单重态(2)激发态电子的自旋不成对.此状态称为单重态(3)激发三重态能级比相应激发单重态能级要低一些(4)单重态到三重态的激发概率高于单重态到单重态80. 2 分(1784)在分子荧光分析法中.以下说法正确的是()(1)分子中π电子共轭程度越大.荧光越易发生.且向短波方向移动(2)只要物质具有与激发光相同的频率的吸收结构.就会产生荧光(3)分子中π电子共轭程度越大.荧光越易发生.且向长波方向移动(4)非刚性分子的荧光强于刚性分子81. 2 分(1785)在分子荧光分析法中.下面说法正确的是()(1)荧光发射光谱不随激发波长的变化而改变(2)荧光发射光谱要随激发波长的变化而改变(3)荧光激发光谱与它的紫外-可见吸收光谱互为镜像对称关系(4)荧光发射光谱与它的紫外-可见吸收光谱形状相似且波长位置也一样82. 2 分(1786)在分子荧光分析法中.下面说法不正确的是()(1)吸电子基团常使荧光增强(2)将一个高原子序数的原子引入到π体系中.使荧光减弱(3)与π电子体系作用小的取代基引入.对荧光影响不明显(4)给电子基团常使荧光增强83. 5 分(1787)化合物(1)的烯醇式乙酰化产物可能是(2)和(3).它的紫外吸收λmax为238nm(lgεmax=4.2)。
Thiamet_G_LCMS_16482_MedChemExpress
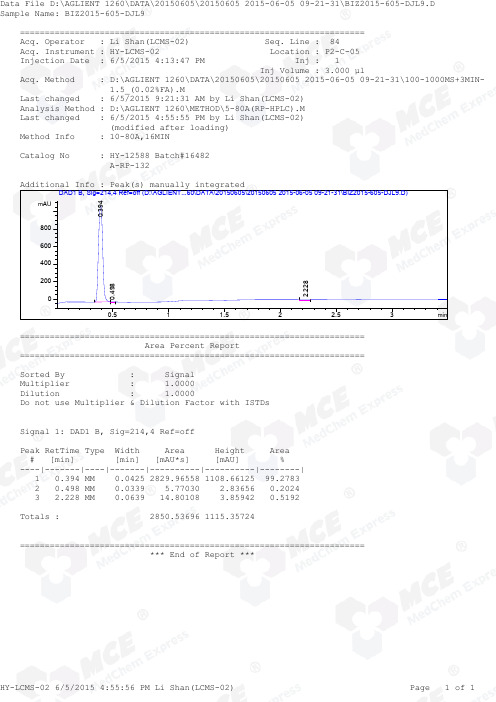
=====================================================================Acq. Operator : Li Shan(LCMS-02) Seq. Line : 84Acq. Instrument : HY-LCMS-02 Location : P2-C-05Injection Date : 6/5/2015 4:13:47 PM Inj : 1 Inj Volume : 3.000 µl Acq. Method : D:\AGLIENT 1260\DATA\20150605\20150605 2015-06-05 09-21-31\100-1000MS+3MIN- 1.5_(0.02%FA).M Last changed : 6/5/2015 9:21:31 AM by Li Shan(LCMS-02)Analysis Method : D:\AGLIENT 1260\METHOD\5-80A(RP-HPLC).M Last changed : 6/5/2015 4:55:55 PM by Li Shan(LCMS-02) (modified after loading)Method Info : 10-80A,16MIN Catalog No : HY-12588 Batch#16482 A-RP-132 Additional Info : Peak(s) manually integrated min0.51 1.52 2.53mAU200400600800DAD1 B, Sig=214,4 Ref=off (D:\AGLIENT...60\DATA\20150605\20150605 2015-06-05 09-21-31\BIZ2015-605-DJL9.D)0.394 0.498 2.228 ===================================================================== Area Percent Report ===================================================================== Sorted By : Signal Multiplier : 1.0000Dilution : 1.0000Do not use Multiplier & Dilution Factor with ISTDs Signal 1: DAD1 B, Sig=214,4 Ref=off Peak RetTime Type Width Area Height Area # [min] [min] [mAU*s] [mAU] %----|-------|----|-------|----------|----------|--------| 1 0.394 MM 0.0425 2829.96558 1108.66125 99.2783 2 0.498 MM 0.0339 5.77030 2.83656 0.2024 3 2.228 MM 0.0639 14.80108 3.85942 0.5192 Totals : 2850.53696 1115.35724 ===================================================================== *** End of Report ***=====================================================================Acq. Operator : Li Shan(LCMS-02) Seq. Line : 84Acq. Instrument : HY-LCMS-02 Location : P2-C-05Injection Date : 6/5/2015 4:13:47 PM Inj : 1 Inj Volume : 3.000 µl Acq. Method : D:\AGLIENT 1260\DATA\20150605\20150605 2015-06-05 09-21-31\100-1000MS+3MIN- 1.5_(0.02%FA).M Last changed : 6/5/2015 9:21:31 AM by Li Shan(LCMS-02)Analysis Method : D:\AGLIENT 1260\METHOD\5-80A(RP-HPLC).M Last changed : 6/5/2015 4:56:55 PM by Li Shan(LCMS-02) (modified after loading)Method Info : 10-80A,16MIN Catalog No : HY-12588 Batch#16482 A-RP-132 Additional Info : Peak(s) manually integrated min0.51 1.52 2.530100000200000300000400000500000MSD1 TIC, MS File (D:\AGLIENT 1260\DATA\20150605\20150605 2015-06-05 09-21-31\BIZ2015-605-DJL9.D) ES-API, Pos, Sca0.392MS Signal: MSD1 TIC, MS File, ES-API, Pos, Scan, Frag: 50 Spectra averaged over upper half of peaks. Noise Cutoff: 1000 counts. Reportable Ion Abundance: > 10%. Retention Mol. Weight Time (MS) MS Area or Ion 0.392 2110102 250.10 I 249.10 Im/z 100200300400500600020406080100*MSD1 SPC, time=0.377:0.413 of D:\AGLIENT 1260\DATA\20150605\20150605 2015-06-05 09-21-31\BIZ2015-605-DJL9.D ES-API,Max: 377429251.1 249.1 *** End of Report ***。
药用镀铝复合膜中铝含量测定方法的研究

临床医药文献杂志Journal of Clinical Medical 2019 年第 6 卷第 4 期2019 Vol.6 No.4174・卫生论坛・药用镀铝复合膜中铝含量测定方法的研究邹茄1,常亮2,朱碧君2*(1.江西中医药大学,江西南昌 330004,2.江西省药品检验检测研究院、江西省药品与医疗器械工程技术研究中心,江西南昌 330029)【摘要】目的 研究聚酯/镀铝聚酯/聚乙烯药用复合膜中铝含量的测定方法,并考察测定方法的可行性。
方法 对样品进行前处理后,采用EDTA返滴定法测定药用复合膜中铝的含量。
结果 采用EDTA返滴定法测得精密度、重复性、稳定性试验的相对标准偏差均小于1.5%,加标回收率为92.90%~97.97%。
采用EDTA返滴定法测定了71个厂家99批次的药用镀铝复合膜样品中的铝含量。
结论 采用EDTA返滴定法准确可靠,重复性高,适用于药用镀铝复合膜中铝的含量测定。
【关键词】EDTA返滴定法;药用复合膜;铝【中图分类号】R917 【文献标识码】A 【文章编号】ISSN.2095-8242.2019.04.174.02目前常量Al的测定采用滴定法,微量及痕量Al的分析采用原子吸收法、原子发射光谱法、色谱法、X射线荧光光谱法及分光光度分析等方法[1]。
镀铝膜厚度一般为0.02~0.06 µm,假定镀铝的密度与铝的密度相当,约2.7 g/cm3,则可算出铝含量约为5~16 mg/1000cm2。
本实验取样量为1000 cm2时,样品中铝含量已达到mg级别,故采用常量法进行测定较合适,即采用EDTA返滴定法进行测定。
本文采用EDTA返滴定法,测定了来源于71个厂家99批次药用复合膜样品。
采用铝标准溶液为对照品溶液,测定铝的含量,探讨了EDTA返滴定法测定镀铝复合膜样品中铝含量的可行性。
1 试药与仪器样品:聚酯/镀铝聚酯/聚乙烯药用复合膜,71个厂家共99批次样品,镀铝聚酯原料膜。
Febuxostat_SDS_MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:Jul.-04-2017Print Date:Jul.-04-20171. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :FebuxostatCatalog No. :HY-14268CAS No. :144060-53-71.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:sales@1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureGHS Classification in accordance with 29 CFR 1910 (OSHA HCS)Acute toxicity, Oral (Category 4), H302Acute aquatic toxicity (Category 1), H400Chronic aquatic toxicity (Category 1), H4102.2 GHS Label elements, including precautionary statementsPictogramSignal word WarningHazard statement(s)H302 Harmful if swallowed.H410 Very toxic to aquatic life with long lasting effects.Precautionary statement(s)P264 Wash skin thoroughly after handling.P270 Do not eat, drink or smoke when using this product.P273 Avoid release to the environment.P301 + P312 IF SWALLOWED: Call a POISON CENTER or doctor/ physician if you feel unwell.P330 Rinse mouth.P391 Collect spillage.P501 Dispose of contents/ container to an approved waste disposal plant.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:TEI 6720; TMX 67Formula:C16H16N2O3SMolecular Weight:316.37CAS No. :144060-53-74. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation.7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature:Powder-20°C 3 years4°C 2 yearsIn solvent-80°C 6 months-20°C 1 monthShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Cleanspillages in a safe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance White to off-white (Solid)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGUN number: 3077Class: 9Packing group: IIIEMS-No: F-A, S-FProper shipping name: ENVIRONMENTALLY HAZARDOUS SUBSTANCE, SOLID, N.O.S.Marine pollutant: Marine pollutantIATAUN number: 3077Class: 9Packing group: IIIProper shipping name: Environmentally hazardous substance, solid, n.o.s.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2017 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
顶空气相色谱-电子捕获检测器法测定明胶空心胶囊中2-氯乙醇残留量

分析测试新成果 (299 ~ 303)顶空气相色谱-电子捕获检测器法测定明胶空心胶囊中2-氯乙醇残留量程 磊1,郭文旭1, 2,王丹丹1,陈 超1(1. 浙江省食品药品检验研究院,浙江 杭州 310052;2. 中国药科大学,江苏 南京 210009)摘要:建立明胶空心胶囊中2-氯乙醇残留量的顶空气相色谱-电子捕获检测器分析测定方法. 明胶空心胶囊溶液经NaCl 溶液盐析后失去明胶的物理性质,采用顶空气相色谱-电子捕获检测器对明胶空心胶囊溶液中2-氯乙醇含量进行分析. 试验结果表明,2-氯乙醇在10~35 µg/g 范围内呈良好线性关系,r 为0.999 4,定量限和检出限分别为10和5 µg/g ,回收率为92.1%~102.2%,相对标准偏差(RSD )均小于10%,使用所建立的方法对样品进行了检验.方法简单准确,灵敏度高,有效解决了明胶空心胶囊基质效应,可用于市售明胶空心胶囊中2-氯乙醇的定量分析.关键词:明胶空心胶囊;2-氯乙醇;盐析;气相色谱-电子捕获检测器;顶空气相色谱中图分类号:O657. 7+1 文献标志码:B 文章编号:1006-3757(2023)03-0299-05DOI :10.16495/j.1006-3757.2023.03.008Determination of 2-Chloroethanol Residues in Gelatin Hollow Capsules by Headspace Gas Chromatography-Electron Capture Detector MethodCHENG Lei 1, GUO Wenxu 1, 2, WANG Dandan 1, CHEN Chao1(1. Zhejiang Institute for Food and Drug Control , Hangzhou 310052, China ;2. China Pharmaceutical University , Nanjing 210009, China )Abstract :A method was developed to determine the residues of 2-chloroethanol in gelatin hollow capsules using headspace gas chromatography-electron capture detector method. The physical properties of gelatin disappeared when the gelatin hollow capsule solution was salting out with NaCl solution. The content of 2-chloroethanol in gelatin hollow capsule solution was analyzed by headspace gas chromatography-electron capture detector method. The results showed that 2-chloroethanol had a good linear relationship in the range of 10~35 µg/g, with an r value of 0.999 4. The limits of quantitation and limits of detection were 10 and 5 µg/g, respectively. The recoveries ranged from 92.1%~102.2%, the relative standard deviation (RSD) were all less than 10%. The samples were tested by the method. The method is simple,accurate and sensitive and effective in solving the matrix effect of gelatin hollow capsules, and can be used for the quantitative analysis of 2-chloroethanol in commercially available gelatin hollow capsules.Key words :gelatin hollow capsules ;2-chloroethanol ;salting out ;gas chromatography-electron capture detector ;headspace gas chromatography由于明胶空心胶囊具有掩盖药物气味、提高生物利用度和制作缓释试剂等特性,已经成为应用最广泛的药用剂型之一. 明胶空心胶囊是采用动物(主要为猪、牛)皮、骨、筋腱中的胶原质,通过部分碱和/或酸解、酶解、热解提纯制得的蛋白质制品[1]. 明胶空心胶囊的质量安全一直是世界用药安全问题收稿日期:2023−07−06; 修订日期:2023−08−15.作者简介:程磊(1988−),男,助理工程师,主要从事药用辅料及药品包装材料的研究,E-mail :通信作者:王丹丹(1982−),女,副主任药师,主要从事药用辅料及药品包装材料的研究,E-mail :.第 29 卷第 3 期分析测试技术与仪器Volume 29 Number 32023年9月ANALYSIS AND TESTING TECHNOLOGY AND INSTRUMENTS Sep. 2023的焦点,作为蛋白质品,其中细菌的控制尤为重要,因此,灭菌成为整个胶囊生产工艺中必不可少的环节之一. 在某些生产工艺中,廉价的环氧乙烷被用作明胶空心胶囊的灭菌剂[2],环氧乙烷的消毒特性来源于其高挥发性和反应性,这导致环氧乙烷消毒后产生潜在的衍生物,包括1, 4-二氧六环、乙醛和卤代乙醇[3]. 因此,环氧乙烷在明胶空心胶囊中主要以其衍生物形式存在. 2-氯乙醇(2-CE)是环氧乙烷的主要衍生物,也是明胶空心胶囊中最易残留的杂质之一.欧盟已明确规定禁止使用环氧乙烷对胶囊产品进行熏蒸消毒,并将环氧乙烷和2-CE的最大残留量同时进行限定,但在许多食品检验中,仅观察到2-CE的残留物,不存在环氧乙烷. 厂家表明,食品中可能存在2-CE,与非法使用环氧乙烷作为熏蒸剂/消毒剂无关[4]. 近期通过结构活性关系和体外测试方法验证了2-CE不具备遗传毒性,也不是啮齿动物致癌物[5]. 因此有必要将2-CE进行单独质量限定以提高检测效率,而不是向环氧乙烷的限度标准看齐. 欧洲与中国均对2-CE最大残留量进行严格限定,《中华人民共和国药典》2020版规定:明胶空心胶囊中2-CE限度为20 mg/kg[6],采用直接进样气相色谱-火焰离子化检测器(GC-FID)法对2-CE含量进行测定. 但该方法前处理较麻烦,仪器信号不够灵敏,不仅大幅降低了检验效率,还增加了有机试剂的损耗. 欧洲对2-CE的最大残留限量尤为严格,采用德国开发的测定2-CE的方法[7],其将2-CE 与环氧乙烷(EO)共同衍生化为2-碘乙醇,再通过气相色谱-电子捕获检测器(GC-ECD)方法进行检测[8]. 2020年,欧盟收到越来越多关于芝麻(特别是印度芝麻)中存在环氧乙烷的通知,要求对所有批次中的50%进行检测. 与此同时,基于分散固相萃取样品制备技术(QuEChERS)、固相萃取(SPME)纯化和气相-质谱/质谱(GC-MS/MS)开发了一种更精确、高通量的方法[9],该方法虽然可以实现2-CE的痕量测定,但是该方法主要用于进口香料、芝麻等食品的测定,操作步骤繁琐,回收率低,不适用于具有胶凝特性的胶囊产品,并且其需要使用MS/MS进行痕量分析,可能在许多实验室中不容易实现.因此,开发一种适合形势发展的2-CE检测手段尤为重要. 本文以2-CE中的氯原子为抓手,以顶空进样方式,建立了气相色谱-离子检测器测定明胶空心胶囊中2-CE含量的方法. 将顶空GC-ECD与GC-FID的测定结果进行比较,顶空GC-ECD方法前处理简单、检测灵敏度高,大幅提升了明胶空心胶囊中2-CE含量的检测效率.1 试验部分1.1 仪器与试剂GC-2010Plus岛津气相色谱仪,配备HS-20顶空进样器(日本岛津公司);ECD检测器及FID检测器均为日本岛津公司;色谱柱DB-624(30 m×0.320 mm×1.8 µm)、色谱柱HP-5(30 m×0.320 mm×0.25µm)、色谱柱HP-INNOWAX(30 m×0.320 mm×0.25µm)均产自美国Agilent公司;ML304T电子天平(梅特勒托利多公司);Milli-Q超纯水器(美国Millipore公司).2-CE对照品溶液(广州佳途科技股份有限公司,批号:202209010035,浓度:5 000 mg/L水溶液);NaCl(国药集团化学试剂有限公司,批号:20220107,纯度:99.5%);Na2SO4(国药集团化学试剂有限公司,批号:20221209,纯度:99.0%);NH4Cl(国药集团化学试剂有限公司,批号:20220213,纯度:99.5%);超纯水;10批来自不同厂家生产的明胶空心胶囊样品.1.2 试验方法1.2.1 顶空和色谱条件顶空条件:顶空平衡温度为90 ℃,定量环温度为95 ℃,传输线温度为100 ℃,顶空平衡时间30 min.色谱条件:气相毛细管色谱柱DB-624(30 m×0.320 mm×1.8 µm);色谱柱温度105 ℃,维持20 min;检测器温度300 ℃;载气采用氮气;色谱柱流量0.3 mL/min;吹扫流量3.0 mL/min;分流比10∶1.1.2.2 溶液配制空白溶液的配制:取NaCl固体150 g,精密称定,置于500 mL烧杯中,加水至500 mL,搅拌至完全溶解,取5 mL混合溶液于20 mL顶空瓶中作为空白溶液.对照品溶液的配制:精密量取2-CE对照品溶液40 µL,置于50 mL量瓶中,采用含30% NaCl的水溶液稀释至刻度,振摇,取5 mL混合溶液于20 mL顶空瓶中,即得对照品溶液.供试品溶液的配制:取明胶空心胶囊样品1.0 g,压扁,精密称定,置于20 mL顶空瓶中,加入5 mL 含30% NaCl的水溶液,作为供试品溶液.300分析测试技术与仪器第 29 卷纯水对照溶液的配制:精密量取2-CE对照品溶液40 µL,置于50 mL量瓶中,用水稀释至刻度,摇匀,作为纯水对照溶液.基质溶液的配制:取3份明胶空心胶囊样品各1.0 g,压扁,精密称定,置于20 mL顶空瓶中,加入5 mL纯水对照溶液,作为基质溶液.2 结果与讨论2.1 基质效应分别取3份基质溶液,与纯水对照溶液同时进样,结果如表1所列.表 1 纯水溶液中的基质效应Table 1 Matrix effect of pure aqueous solution溶液峰面积与纯水对照溶液的峰面积比值纯水对照溶液 5 353 1.00基质溶液-1 3 8290.71基质溶液-2 3 8660.72基质溶液-3 3 7710.70由表1可见,明胶空心胶囊溶于水后基质效应明显,2-CE在胶囊溶液中被明胶所包埋,导致检测时响应信号降低,无法将2-CE完全释放. 作为溶剂,纯水也存在沸点低、伤害检测器的问题. 因此,本试验采用饱和盐溶液对明胶空心胶囊溶液中的明胶蛋白盐析,使其无法维持三螺旋结构,从而使胶囊溶液失去胶体性质,阻止其包埋2-CE,同时可提高溶剂沸点,减小基质效应的影响.2.2 盐的选择按1.2.2项下盐对照溶液处理方法,由于30%盐在水溶液中基本饱和,因此分别选择30% NaCl、30% Na2SO4、30% NH4Cl作为盐析剂,研究对水基类基质样品的盐析效果. 结果如图1所示.由图1可见,三种盐溶液中NaCl对明胶胶囊盐析作用效果明显,可有效提高回收率,又考虑到NaCl溶液获取简单、容易配制、价格低,因此本试验选择NaCl溶液为盐析试剂.2.3 顶空条件优化2.3.1 顶空平衡温度优化根据《中华人民共和国药典》2020版0861残留溶剂测定法[10]项下说明,顶空平衡温度一般应比溶解供试品所用溶剂的沸点低10 ℃及以下,能满足检测灵敏度即可. 鉴于NaCl溶液沸点近似为110 ℃,因此采用90 ℃作为顶空平衡温度.2.3.2 顶空平衡时间优化取1.2.2项下对照溶液各12份,分别在90 ℃下保温10、20、30、60 min,每组3份,按1.2.1项下方法进样,记录2-CE的峰面积. 以峰面积为纵坐标,时间为横坐标作柱状图,结果如图2所示.6 0007 0005 0004 00010403020t/minPeakarea/(μV·s)图2 2-CE顶空时间考察Fig. 2 Study on headspace times of 2-CE 由图2可见,2-CE的峰面积随顶空时间增长而增大,且30 min前测定结果的相对标准偏差(RSD)偏大,可能是顶空瓶中未达到平衡导致结果差距较大,说明2-CE对照品在90 ℃下,可以在30~60 min 达到平衡. 由于30 min已达到检测要求,因此综合考虑选择30 min为平衡保温时间.2.4 色谱条件优化2.4.1 色谱柱选择本方法选用3 种不同类型的色谱柱对2-CE的分离度、响应和峰形等方面进行评估比较,探究适5 0006 0004 0003 000NaCl NH4ClPeakarea/(μV·s)Na2SO4ControlSample spiking图1 30% NaCl、30% Na2SO4、30% NH4Cl溶液对照与加标峰面积比较Fig. 1 Comparison of spiked peak areas of 30% NaCl,30% Na2SO4, 30% NH4Cl salt solutions第 3 期程磊,等:顶空气相色谱-电子捕获检测器法测定明胶空心胶囊中2-氯乙醇残留量301合该检测方法的色谱柱. 分别采用弱极性HP-5毛细管柱、中等极性DB-624毛细管柱、强极性HP-INNOWAX 毛细管柱在相同分析条件下,对同一浓度的对照品溶液进行测定. 结果显示,HP-INNOWAX 柱对2-CE 保留效果较强、出峰时间慢,不符合高效检测的改进理念. HP-5柱中,对2-CE 几乎不保留,出峰最快,但与溶剂峰相近. 而DB-624柱能在15 min 内分离2-CE ,且峰形尖锐、响应值高,并且可以准确定量. 因此,本试验选用DB-624毛细管柱对2-CE 进行分析测试,其色谱图如图3所示.2 0001 5001 000500010.012.515.02-CEt /min强度17.520.0图3 20 µg/g 2-CE 对照溶液谱图色谱条件: DB-624(30 m × 0.320 mm ×1.8 µm )色谱柱,105 ℃柱温,ECD 检测器温度为300 ℃,色谱柱流量为0.3 mL/minFig. 3 Chromatogram of 20µg/g 2-CEchromatographic conditions: chromatographic column used as DB-624 (30 m×0.320 mm×1.8 µm), column temperature of 105 °C, temperature of ECD detector of 300 °C, flow rateof 0.3 mL/min2.4.2 检测器选择本方法比较了2 种不同类型的检测器对2-CE的信号灵敏度,分别使用FID 与ECD 在相同参数条件下,对同一样品溶液进行测定,结果如图4、5所示.由图4、5结果可见,样品溶液在FID 下存在杂峰,且杂峰与FID 中2-CE 对照品出峰时间接近,影响结果的判断. 相反,样品溶液在ECD 下无明显杂峰,且同浓度的2-CE 对照品在ECD 检测器中峰形尖锐且明显. 因此采用ECD 检测明胶空心胶囊中2-CE 方法较可靠.2.5 方法学2.5.1 线性关系考察取对照品溶液,按10~35 µg/g 配制5份不同浓度的对照品标准曲线溶液. 以浓度为横坐标(X ),峰面积为纵坐标(Y ),绘制标准曲线,得回归方程:Y =254.3X +921,相关系数r 为 0.999 4. 表明2-CE 在10~35 µg/g 范围内呈良好线性关系.2.5.2 检测限将对照品溶液逐步用水稀释,依法进样测定.分别以信噪比(S/N )为10和3确定定量限和检出限,其分别为10和5 µg/g.2.5.3 仪器精密度取对照品溶液连续进样6次,记录2-CE 峰面积,结果显示:2-CE 峰面积的RSD 为1.90%,表明仪器精密度较好.2.5.4 重复性取6份明胶空心胶囊样品各1.0 g ,精密称定,添加100%浓度对照品溶液,进样分析,记录2-CE 峰面积. 结果显示:2-CE 峰面积的RSD 为1.94%,1 000500010.012.515.0t /min强度17.520.0图4 样品溶液在ECD 中色谱图色谱条件: 色谱柱DB-624(30 m × 0.320 mm ×1.8 µm ),柱温105 ℃,ECD 检测器温度为300 ℃,色谱柱流量为0.3 mL/minFig. 4 Chromatogram of sample solution in ECD chromatographic conditions: chromatographic column used as DB-624 (30 m×0.320 mm×1.8 µm), column temperature of 105 °C, temperature of ECD detector of 300 °C, flow rateof 0.3 mL/min1 5001 000500010.012.5123ba 15.02-CEt /min强度17.520.0图5 2-CE 及样品溶液在FID 中色谱图(a )2-CE ,(b )样品,(1)~(3)均为杂峰色谱条件: DB-624(30 m × 0.320 mm ×1.8 µm )色谱柱,105 ℃柱温, FID 检测器温度为300 ℃,色谱柱流量为0.3 mL/minFig. 5 Chromatograms of 2-CE and samplesolution in FID(a) 2-CE, (b) sample, (1)~(3) all impure peakschromatographic conditions: chromatographic column used as DB-624 (30 m×0.320 mm×1.8 µm), column temperature of 105 °C, temperature of FID detector of 300 °C, flow rateof 0.3 mL/min302分析测试技术与仪器第 29 卷说明重复性较好.2.5.5 回收率精密称取明胶空心胶囊样品各1.0 g ,分别添加低、中、高浓度的对照品溶液,每个水平做3次平行试验,按1.2.2项下供试品溶液配制条件,按照1.2.1项下条件进样测定,计算平均回收率,结果如表2所列.表 2 2-CE 回收率测定结果Table 2 Determination results of recovery rates of 2-CE 编号样品质量/µg 加入质量/ µg 测得质量/ µg 回收率/%平均回收率/%RSD/%10.016.016.4102.298.04.420.016.016.099.730.016.014.792.140.020.019.597.597.81.350.020.019.999.660.020.019.396.570.024.022.493.598.53.780.024.024.0100.090.024.024.5102.02.6 样品测定取明胶空心胶囊样品1.0 g ,压扁,精密称定,置于20 mL 顶空瓶中,加入5 mL 含30% NaCl 的水溶液,然后立即压盖密封,置于顶空炉内,按照1.2.1项下条件进样测定,计算样品含量. 结果表明所有样品均未检出2-CE ,与现有药典方法检测结果一致.3 结论本文建立了GC-ECD 法对明胶空心胶囊中2-CE 残留量进行测定,对现有2-CE 测定法进行优化提高,建立科学合理的2-CE 测定方法. 优化提高后的2-CE 测定方法能快速、灵敏测定明胶空心胶囊中2-CE 的含量,该研究为2-CE 的测定标准提高以及明胶空心胶囊的质量控制提供参考.参考文献:Gullapalli R P, Mazzitelli C L. Gelatin and non-gelat-in capsule dosage forms [J ]. Journal of Pharmaceutic-[ 1 ]al Sciences ,2017,106 (6):1453-1465.庞小刚, 郭捷, 王昊. 双柱气相色谱法测定明胶空心胶囊中环氧乙烷的残留量[J ]. 化学分析计量,2016,25(1):38-40. [PANG Xiaogang, GUO Jie, WANG Hao. Determination of ethylene oxide in gelatin hol-low capsules by dual capillary column gas chromato-graphy [J ]. Chemical Analysis and Meterage ,2016,25(1):38-40.][ 2 ]Wang D D, Zhang A J, Guo W X, et al. Identificationof residues in ethylene oxide sterilized hard gelatin capsule shells by gas chromatography-mass spectro-metry and development of a simple gas chromato-graphy-flame ionization detector method for the de-termination of residues [J ]. Journal of Chromato-graphy Open ,2022,2 :100061.[ 3 ]Borroto J, Castoldi A F, Chiusolo A, et al. Statementon the BfR opinion regarding the toxicity of 2-chloro-ethanol [J ]. EFSA Journal ,2022,20 (2):e07147.[ 4 ]Allemang A, Lester C, Roth T, et al. Assessing thegenotoxicity and carcinogenicity of 2-chloroethanol through structure activity relationships and in vitro testing approaches [J ]. Food and Chemical Toxicology ,2022,168 :113290.[ 5 ]国家药典委员会. 中华人民共和国药典(四部 2020年版)[M ]. 北京: 中国医药科技出版社, 2020: 687.[ 6 ]Jensen K G. Determination of ethylene oxide residuesin processed food products by gas-liquid chromato-graphy after derivatization [J ]. Zeitschrift Für Lebens-mittel-Untersuchung Und Forschung ,1988,187 (6):535-540.[ 7 ]Gilsbach W, Weeren R D. Ringuntersuchungen zurvalidierung einer gaschromatographischen methode zur bestimmung von rückstnden an ethylenoxid und 2-chlorethanol in gewürzen aus paprika und chili [J ].Deutsche Lebensmittel-Rundschau, 1999.[ 8 ]Bessaire T, Stroheker T, Eriksen B, et al. Analysis ofethylene oxide in ice creams manufactured with con-taminated carob bean gum (E410)[J ]. Food Addi-tives & Contaminants Part A, Chemistry, Analysis,Control, Exposure & Risk Assessment ,2021,38 (12):2116-2127.[ 9 ]国家药典委员会. 中华人民共和国药典(二部 2020年版)[M ] . 北京: 中国医药科技出版社, 2020: 796.[ 10 ]第 3 期程磊,等:顶空气相色谱-电子捕获检测器法测定明胶空心胶囊中2-氯乙醇残留量303。