Remodelin hydrobromide_1622921-15-6_DataSheet_MedChemExpress
MK2-IN-1_hydrochloride_SDS_MedChemExpress

Inhibitors, Agonists, Screening LibrariesSafety Data Sheet Revision Date:Jul.-24-2017Print Date:Jul.-24-20171. PRODUCT AND COMPANY IDENTIFICATION1.1 Product identifierProduct name :MK2-IN-1 (hydrochloride)Catalog No. :HY-12834ACAS No. :1314118-94-91.2 Relevant identified uses of the substance or mixture and uses advised againstIdentified uses :Laboratory chemicals, manufacture of substances.1.3 Details of the supplier of the safety data sheetCompany:MedChemExpress USATel:609-228-6898Fax:609-228-5909E-mail:sales@1.4 Emergency telephone numberEmergency Phone #:609-228-68982. HAZARDS IDENTIFICATION2.1 Classification of the substance or mixtureNot a hazardous substance or mixture.2.2 GHS Label elements, including precautionary statementsNot a hazardous substance or mixture.2.3 Other hazardsNone.3. COMPOSITION/INFORMATION ON INGREDIENTS3.1 SubstancesSynonyms:MK2 InhibitorFormula:C27H26Cl2N4O2Molecular Weight:509.43CAS No. :1314118-94-94. FIRST AID MEASURES4.1 Description of first aid measuresEye contactRemove any contact lenses, locate eye-wash station, and flush eyes immediately with large amounts of water. Separate eyelids with fingers to ensure adequate flushing. Promptly call a physician.Skin contactRinse skin thoroughly with large amounts of water. Remove contaminated clothing and shoes and call a physician.InhalationImmediately relocate self or casualty to fresh air. If breathing is difficult, give cardiopulmonary resuscitation (CPR). Avoid mouth-to-mouth resuscitation.IngestionWash out mouth with water; Do NOT induce vomiting; call a physician.4.2 Most important symptoms and effects, both acute and delayedThe most important known symptoms and effects are described in the labelling (see section 2.2).4.3 Indication of any immediate medical attention and special treatment neededTreat symptomatically.5. FIRE FIGHTING MEASURES5.1 Extinguishing mediaSuitable extinguishing mediaUse water spray, dry chemical, foam, and carbon dioxide fire extinguisher.5.2 Special hazards arising from the substance or mixtureDuring combustion, may emit irritant fumes.5.3 Advice for firefightersWear self-contained breathing apparatus and protective clothing.6. ACCIDENTAL RELEASE MEASURES6.1 Personal precautions, protective equipment and emergency proceduresUse full personal protective equipment. Avoid breathing vapors, mist, dust or gas. Ensure adequate ventilation. Evacuate personnel to safe areas.Refer to protective measures listed in sections 8.6.2 Environmental precautionsTry to prevent further leakage or spillage. Keep the product away from drains or water courses.6.3 Methods and materials for containment and cleaning upAbsorb solutions with finely-powdered liquid-binding material (diatomite, universal binders); Decontaminate surfaces and equipment by scrubbing with alcohol; Dispose of contaminated material according to Section 13.7. HANDLING AND STORAGE7.1 Precautions for safe handlingAvoid inhalation, contact with eyes and skin. Avoid dust and aerosol formation. Use only in areas with appropriate exhaust ventilation.7.2 Conditions for safe storage, including any incompatibilitiesKeep container tightly sealed in cool, well-ventilated area. Keep away from direct sunlight and sources of ignition.Recommended storage temperature:Powder-20°C 3 years4°C 2 yearsIn solvent-80°C 6 months-20°C 1 monthShipping at room temperature if less than 2 weeks.7.3 Specific end use(s)No data available.8. EXPOSURE CONTROLS/PERSONAL PROTECTION8.1 Control parametersComponents with workplace control parametersThis product contains no substances with occupational exposure limit values.8.2 Exposure controlsEngineering controlsEnsure adequate ventilation. Provide accessible safety shower and eye wash station.Personal protective equipmentEye protection Safety goggles with side-shields.Hand protection Protective gloves.Skin and body protection Impervious clothing.Respiratory protection Suitable respirator.Environmental exposure controls Keep the product away from drains, water courses or the soil. Cleanspillages in a safe way as soon as possible.9. PHYSICAL AND CHEMICAL PROPERTIES9.1 Information on basic physical and chemical propertiesAppearance Pink to red (Solid)Odor No data availableOdor threshold No data availablepH No data availableMelting/freezing point No data availableBoiling point/range No data availableFlash point No data availableEvaporation rate No data availableFlammability (solid, gas)No data availableUpper/lower flammability or explosive limits No data availableVapor pressure No data availableVapor density No data availableRelative density No data availableWater Solubility No data availablePartition coefficient No data availableAuto-ignition temperature No data availableDecomposition temperature No data availableViscosity No data availableExplosive properties No data availableOxidizing properties No data available9.2 Other safety informationNo data available.10. STABILITY AND REACTIVITY10.1 ReactivityNo data available.10.2 Chemical stabilityStable under recommended storage conditions.10.3 Possibility of hazardous reactionsNo data available.10.4 Conditions to avoidNo data available.10.5 Incompatible materialsStrong acids/alkalis, strong oxidising/reducing agents.10.6 Hazardous decomposition productsUnder fire conditions, may decompose and emit toxic fumes.Other decomposition products - no data available.11.TOXICOLOGICAL INFORMATION11.1 Information on toxicological effectsAcute toxicityClassified based on available data. For more details, see section 2Skin corrosion/irritationClassified based on available data. For more details, see section 2Serious eye damage/irritationClassified based on available data. For more details, see section 2Respiratory or skin sensitizationClassified based on available data. For more details, see section 2Germ cell mutagenicityClassified based on available data. For more details, see section 2CarcinogenicityIARC: No component of this product present at a level equal to or greater than 0.1% is identified as probable, possible or confirmed human carcinogen by IARC.ACGIH: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by ACGIH.NTP: No component of this product present at a level equal to or greater than 0.1% is identified as a anticipated or confirmed carcinogen by NTP.OSHA: No component of this product present at a level equal to or greater than 0.1% is identified as a potential or confirmed carcinogen by OSHA.Reproductive toxicityClassified based on available data. For more details, see section 2Specific target organ toxicity - single exposureClassified based on available data. For more details, see section 2Specific target organ toxicity - repeated exposureClassified based on available data. For more details, see section 2Aspiration hazardClassified based on available data. For more details, see section 212. ECOLOGICAL INFORMATION12.1 ToxicityNo data available.12.2 Persistence and degradabilityNo data available.12.3 Bioaccumlative potentialNo data available.12.4 Mobility in soilNo data available.12.5 Results of PBT and vPvB assessmentPBT/vPvB assessment unavailable as chemical safety assessment not required or not conducted.12.6 Other adverse effectsNo data available.13. DISPOSAL CONSIDERATIONS13.1 Waste treatment methodsProductDispose substance in accordance with prevailing country, federal, state and local regulations.Contaminated packagingConduct recycling or disposal in accordance with prevailing country, federal, state and local regulations.14. TRANSPORT INFORMATIONDOT (US)This substance is considered to be non-hazardous for transport.IMDGThis substance is considered to be non-hazardous for transport.IATAThis substance is considered to be non-hazardous for transport.15. REGULATORY INFORMATIONSARA 302 Components:No chemicals in this material are subject to the reporting requirements of SARA Title III, Section 302.SARA 313 Components:This material does not contain any chemical components with known CAS numbers that exceed the threshold (De Minimis) reporting levels established by SARA Title III, Section 313.SARA 311/312 Hazards:No SARA Hazards.Massachusetts Right To Know Components:No components are subject to the Massachusetts Right to Know Act.Pennsylvania Right To Know Components:No components are subject to the Pennsylvania Right to Know Act.New Jersey Right To Know Components:No components are subject to the New Jersey Right to Know Act.California Prop. 65 Components:This product does not contain any chemicals known to State of California to cause cancer, birth defects, or anyother reproductive harm.16. OTHER INFORMATIONCopyright 2017 MedChemExpress. The above information is correct to the best of our present knowledge but does not purport to be all inclusive and should be used only as a guide. The product is for research use only and for experienced personnel. It must only be handled by suitably qualified experienced scientists in appropriately equipped and authorized facilities. The burden of safe use of this material rests entirely with the user. MedChemExpress disclaims all liability for any damage resulting from handling or from contact with this product.Caution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
芦荟素对酪氨酸酶活性的抑制作用

目前国内关于芦荟素对酪氨酸酶的作用方面研究较少,该实验以L-多巴为底物,以氢醌为对照,研究芦荟素对酪氨酸酶的活性影响,并为临床上芦荟素或使用含有大量芦荟素的芦荟产品的综合利用提供实验依据。
酶在医药、食品、化工和环保等领域应用广泛。
酪氨酸酶是一种以双铜离子为活性单元的氧化还原性酶,在动物、植物体内广泛存在,对黑色素细胞的合成具有调控作用,是黑色素形成的限速酶[1]。
黑色素细胞是生物体内黑色素的合成主要场所,在黑色素的合成过程中,酪氨酸酶对整个黑色素的合成具有重要催化作用,酪氨酸酶主要存在于黑色素细胞中。
酪氨酸酶的活性升高,则黑色素的形成增加;酪氨酸酶的活性降低,则黑色素的生成减少,因此,酪氨酸酶活性的强弱与黑色素形成的多少存在正相关关系。
临床上酶氨酶活性过强、或者过弱均会导致临床疾病的发生,如黄褐斑、白化病、雀斑、白癜风等异性性色素性皮肤病[2]。
国内外学者常利用抑制酪氨酸酶活性试验,研究筛选治疗皮肤色素沉着症的药物[3]。
该实验采用蘑菇酪氨酸多巴速率氧化法,研究活性单体化合物芦荟素对酪氨酸酶的影响[4]。
1材料与方法1.1试剂与仪器L-多巴胺(L-DOPA),蘑菇酪氨酸酶(美国Sigma公司);芦荟素(中国药品生物制品检定所);氢醌(天津市巴斯夫化工有限公司);其他试剂均为市售分析纯。
UV-1800紫外可见分光光度计(日本岛津);AUY220电子分析天平(日本岛津)。
1.2方法采用pH6.8磷酸缓冲溶液,将L-DOPA、芦荟素、氢醌分别配制成0.1%的溶液,将蘑菇酪氨酸酶配制成100U/mL的溶液。
取上述磷酸缓冲240μL,加入0.1%L-DOPA反应液80μL,蘑菇酪氨酸酶80μL混合均匀,常温静置5min,于分光光度计上在400~600nm扫描。
可见在475nm处有最大吸收峰。
同法分别测定L-DOPA、蘑菇酪氨酸酶均未见有吸收。
采用酪氨酸酶多巴速率氧化法,在pH6.8磷酸缓冲液中,用酪氨酸酶催化多巴变成多巴醌,有肾上腺素红色,利用分光光度计在475nm处的特定吸收测定生成多巴醌时的吸收度:A=A(药+L-DOPA+酶)—A(药+酶)。
氧化芳基偶联和Scholl反应的比较
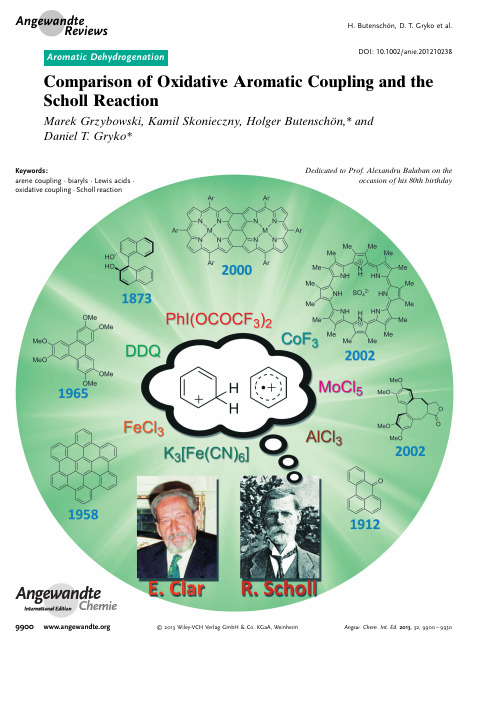
DOI:10.1002/anie.201210238Comparison of Oxidative Aromatic Coupling and the Scholl ReactionMarek Grzybowski,Kamil Skonieczny,Holger Butenschçn,*and Daniel T.Gryko*AngewandteChemieKeywords:arene coupling ·biaryls ·Lewis acids ·oxidative coupling ·Scholl reactionDedicated to Prof.Alexandru Balaban on theoccasion of his 80th birthday.Angewandte ReviewsH.Butenschçn,D.T.Gryko et al.99002013Wiley-VCH Verlag GmbH &Co.KGaA,WeinheimAngew.Chem.Int.Ed.2013,52,9900–99301.IntroductionThe first example of an oxidative dimerization of aromatic compounds was published in 1871,[1]and 39years later Roland Scholl reported that a similar effect can be achieved by heating certain aromatic compounds with AlCl 3.[2]For many years,these two reactions were distinguishable,and when Balaban and Nenitzescu published their fundamental review on the Scholl reaction,there was still a clear demar-cation between them.[3]Nowadays,however,there is a mix-up in the literature,and the oxidative coupling of electron-rich aromatic substances is often called the Scholl reaction.The aim of this Review is to summarize the development of both reactions,to discuss their mechanisms,and to show their current scope.We will present the historical origin of both of these processes in Section 2,followed by a discussion of their mechanisms in Section 3.In that section we will discuss the similarities and differences between reactions mediated by AlCl 3and reactions mediated by typical oxidants in terms of scope and electronic requirements.[4]In the following two sections we will present representative examples of the Scholl reaction (Section 4),intramolecular oxidative coupling,and intermolecular oxidative coupling reactions (Section 5).We will focus on the most important examples,with special emphasis given to the recent literature.Palladium-catalyzed oxidative aromatic cross-coupling and dehydrogenative cou-pling by C ÀH activation by organometallic catalysts are not included in this Review.[5]2.Historical Development2.1.Oxidative Aromatic CouplingThe first known example of the oxidative coupling of aromatic compounds,the formation of ellagic acid (2)from gallic acid (1),was published in 1868(Scheme 1).[1]The reaction was mediated by H 3AsO 4or Ag 2O;however,the yield was not reported.Other examples quickly followed,and in the 1870s it was shown that a variety of phenols and phenyl ethers can be coupled oxidatively using one-electron oxidants such as FeCl 3or K 3[Fe(CN)6].Seminal examples include the synthesis of 1,1’-bi-2-naphthol (binol,4)by the oxidation of 2-naphthol (3)with FeCl 3reported in 1873by Dianin (Scheme 2).[6b]Progress continued into the 20th century and accelerated after the discovery of the role of oxidative aromatic coupling in biogenesis.[7]The early literature has been summarized by[*]M.Grzybowski,K.Skonieczny,Prof.Dr.D.T.GrykoInstitute of Organic Chemistry,Polish Academy of Sciences Kasprzaka 44/52,Warsaw (Poland)E-mail:dtgryko@.pl Prof.Dr.H.ButenschçnInstitut für Organische Chemie Leibniz Universität,HannoverSchneiderberg 1B,30167Hannover (Germany)Prof.Dr.D.T.GrykoFaculty of Chemistry,Warsaw University of Technology 00-664Warsaw (Poland)D oes the dehydrogenative coupling of aromatic compounds mediatedby AlCl 3at high temperatures and also by FeCl 3,MoCl 5,PIFA,or K 3[Fe(CN)6]at room temperature proceed by the same mechanism in all cases?With the growing importance of the synthesis of aromatic compounds by double C ÀH activation to give various biaryl structures,this question becomes pressing.Since some of these reactions proceed only in the presence of non-oxidizing Lewis acids and some only in the presence of certain oxidants,the authors venture the hypothesis that,depending on the electronic structure of the substrates and the nature of the “catalyst”,two different mechanisms can operate.One involves the intermediacy of a radical cation and the other the formation of a sigma complex between the acid and the substrate.The goal of this Review is to encourage further mechanistic studies hopefully leading to an in-depth understanding of this phenomenon.From the Contents1.Introduction99012.Historical Development 99013.Mechanistic Considerations 99034.Scholl Reaction—Scope,Limitations,and Utilization 99065.Oxidative Aromatic Coupling 99076.Summary and Outlook9926Scheme 2.Scheme 1.Biaryl Synthesis9901Angew.Chem.Int.Ed.2013,52,9900–9930 2013Wiley-VCH Verlag GmbH &Co.KGaA,WeinheimHeuben,[8]and later examples have also been extensively reviewed.[9,10]2.2.Scholl ReactionThe Scholl reaction was first mentioned as early as 1910,when Scholl and Mansfeld reported the transformation of quinone 5to the p -extended quinone 6by treatment with an excess of neat anhydrous aluminum chloride for 45min at 140–1458C as a clean reaction,although no yield was given (Scheme 3).The authors mentioned that such a reaction had been observed earlier,for example in the formation of 1,1’-binaphthalene by heating naphthalene with aluminum chlo-ride,but they emphasized that the observation of the formation of quinone 6under comparatively mild reaction conditions was new.[2]In a subsequent publication,this reaction was applied to the synthesis of perylene (8)from 1,1’-binaphthalene (7;Scheme 4).Interestingly,Homer de-scribed the same reaction at the same time,but without the true formula of the product 8.[11]Perylene (8)was also obtained from naphthalene (9)without isolation of the intermediate 7;however,because of some decomposition,the yield of 8was poor.[12]Later,the reaction of 4,4’-dicyano-1,1’-binaphthalene was reported to give the corresponding coupling product in 72%yield.[13]The method was further elaborated and led to the synthesis of benzanthrone (11)from ketone 10and of compound 13from 12(no yield was given in the last case,Scheme 5).[14,15]Numerous other examples were published by Scholl and co-workers in the following years.[16,17]Possible mechanisms were discussed in some of the early reports on the dehydrogenative coupling of aromatic com-pounds by treatment with anhydrous aluminum chloride;these mechanisms were mainly based on similarities to Friedel Crafts reactions.[15]The original procedure by Scholl required baking the organic substrate with AlCl 3.This procedure was soon replaced by Kränzlein and Vollmann,who used an equimolar mixture of AlCl 3and NaCl,which is liquid above 1008C at a low vapor pressure,[18]and this procedure predominates in the later literature.Since the early 1920s,this reaction has been utilized in the industrial synthesis of many antraqui-none-derived dyes.It is difficult to overestimate the technical importance of the Scholl reaction.Indanthrene khaki 2G (15)Scheme 3.Scheme 4..Angewandte ReviewsH.Butenschçn,D.T.Gryko et al.2013Wiley-VCH Verlag GmbH &Co.KGaA,Weinheim Angew.Chem.Int.Ed.2013,52,9900–9930has been produced in millions of kilograms from 14(Scheme 6).[3,19]After World War II,the focus shifted and the Scholl reaction started to be used in the synthesis of extended aromatic hydrocarbons.This new direction sparked methodo-logical developments.In 1971,Wick revealed that the cyclization of di-(1-anthraquinonyl)amine into 1,2;7,8-di-phthaloylcarbazole (C.I.Vat Yellow 28)proceeds more efficiently in the presence of an AlCl 3/pyridine complex than in an AlCl 3/NaCl melt.[20]Other variants include the use of AlCl 3in high-boiling solvents such as dichlorobenzene and trichlorobenzene,[21]low-melting complex AlCl 3/SO 2,[22]or of ZrCl 4.[23]Needless to say,many protocols involve the addition of a certain amount of oxidant such as air,oxygen,nitro-benzene,or potassium m -nitrobenzene sulfonate.[24]In 1961,Kovacic and Kyriakis introduced new conditions for the oxidative polymerization of benzene (AlCl 3/CuCl 2/neat),[25a]which were later modified by Müllen and co-workers (AlCl 3/CuCl 2/CS 2[25b]and AlCl 3/Cu(OTf)2/CS 2),[25c]thus allowing the reaction temperature to be lowered to 258C.3.Mechanistic ConsiderationsNumerous examples of dehydrogenative coupling reac-tions of aromatic compounds in the presence of various Lewis acids have recently been published.[26]In most cases,they are assigned as Scholl reactions.[27]Here,we come to the critical question of whether there is a difference between oxidative aromatic coupling and the Scholl reaction.The typical oxidative aromatic coupling relates to the reaction of electron-rich aromatic compounds such as phe-nols,alkyl aryl ethers and the like.The broadly accepted mechanism is shown in Scheme 7.It involves the formation ofa radical cation from one molecule of the substrate followed by substitution at the neutral second molecule and finally convergence to the biaryl product.This mechanism implies that:1)the substrate is reasonably electron rich and 2)the attack of the electrophilic radical cation occurs at the most electron-rich position of the second substrate molecule.Other mechanistic pathways have also been discussed for the coupling of phenols,such as oxidation to the radical followed by dimerization and radical substitution.[10]According to the definition proposed in very early reviews,the Scholl reaction is a dehydrogenation of aromatic nuclei under the influence of aluminum chloride that results in the formation of a condensed ring system.[28]Balaban and Nenitzescu reformulated it as “the elimination of two aryl-bound hydrogens accompanied by the formation of an aryl–aryl bond under the influence of Friedel–Crafts catalysts”.[3]Baddeley was the first to propose that the mechanism of the Scholl reaction involves the formation of a s complex between the Lewis acid with the aromatic compound followed by the formation of an arenium cation,an electrophilic attack,and finally a dehydrogenation.[29]This hypothesis was further reformulated by Nenitzescu and Balaban.[30]Kenner,on the other hand was the first to propose a radical cation mechanism for the Scholl reaction.[31]This concept was soon supported by Rooney and Pink [32]and later by Clover.[33]The arenium cation mechanism implies the protonation of the aryl species,for example,7,to form an electrophilic s complex 7’(shown with H +for simplicity,but this couldalsoScheme5.Scheme6.Scheme 7.Biaryl Synthesis9903Angew.Chem.Int.Ed.2013,52,9900–9930 2013Wiley-VCH Verlag GmbH &Co.KGaA,Weinheimbe a s complex with a Lewis acid;Scheme 8).The attack of the latter species at the other aromatic ring to form a new carbon–carbon bond (7’’)followed by hydrogen elimination regenerates the aromatic system,finally giving 8.Numerous experimental results supported this mecha-nism.Several research groups observed that dehydrogenation of certain aromatic compounds can occur not only in the presence of AlCl 3and similar Lewis acids,but also in media such as anhydrous HF [34,35]or PhSO 3H,[30]while radical cations cannot be formed under such conditions.Baddeley and Kenner observed that the presence of hydrogen chloride is essential for the synthesis of benzanthrone (11)to occur.[29]Electronic and steric effects in various positions play a con-siderable role in intramolecular cases.Remarkably,benzo-phenone does not yield fluorenone upon heating with AlCl 3at 180–2208C,[15]most likely because this would involve an electrophilic aromatic substitution at the ortho position relative to the carbonyl group.On the other hand,milder reaction conditions (1008C)allowed the synthesis of phenan-threnequinone from benzil.[14]The third step,that is,dehy-drogenation/aromatization,is the most controversial one,since studies have shown that only a substoichiometric amount of H 2was evolved when ketones were subjected to Scholl reaction conditions.To the best of our knowledge,no such study has so far been performed on hydrocarbons.There is an unquestionable positive influence on the yield of products from the Scholl reaction when hydrogen acceptors are added to AlCl 3.Various examples include O 2(conversion of 3,8-dibenzoylpyrene into pyranthrone,25%versus 80%;[36]conversion of 1,5-dibenzoylnaphthalene into 2,3;7,8-dibenzo-pyrene-1,6-quinone),[10]and nitrobenzene (intermolecular reaction of ethyl 1-naphthyl ether,0%versus 70%).[37]Importantly,the carbonyl group present in many Scholl reaction precursors can serve as a temporary oxidizing agent.In such cases,the corresponding secondary alcohol can be an intermediate,which is then reoxidized by O 2to a ketone.Balaban and Nenitzescu argue that this may be the reason for the high yields of the intramolecular Scholl reactions with these ketones,despite the electron-withdrawing (hence deactivating)character of the carbonyl group.[3]While discussing possible pathways of rearomatization,one has to remember that AlCl 3itself can catalyze the dehydrogenation of compounds such as 9,10-dihydroanthracene.[37]The second mechanism advocated by Kenner,Rooney,and Clover implies the formation of radical cations (Scheme 9).Although the mechanism has been studied only occasionally,the schism continued,and in recent years an intense discussion has evolved,prominently between the research groups of King and Rathore.The differentiationbetween the two mechanisms is clearly not a trivial issue.One of the key difficulties lies in the fact that most of the Lewis acids used in the Scholl reaction are also milder or stronger oxidants.Moreover,aromatic hydrocarbons can also form paramagnetic species in the presence of non-oxidizing AlCl 3.[32]The problem with the most commonly used oxidant,namely FeCl 3,is that this compound is both a Lewis acid and an oxidant capable of catalyzing or mediating a variety of reactions.For this reason,experiments with FeCl 3or MoCl 5cannot give the definitive answer to the key mechanistic question.One of the iron complexes broadly used in oxidative aromatic coupling,[9]which cannot be considered a Lewis acid,is K 3[Fe(CN)6],but its relatively low oxidation potential does not allow for direct comparative studies in many cases.King and co-workers published a series of reports that presented both computational and experimental evidence supporting the arenium ion mechanism.[38]Computational studies led to the conclusion that the mechanistic pathway involving the arenium cation is thermodynamically favored under both vacuum and solvated conditions,because of the transition states have lower energy than those found in the radical cation pathway.Additionally,the authors presented computational evidence showing that in the case of the oxidation of hexaphenylbenzene to hexa-peri -hexabenzocor-onene,the formation of the first C ÀC bond is the slowest.This explains the lack of observed intermediates in this process.One has to emphasize,however,that the interaction of the reagents with the organic substrate was not taken into account in these calculations.Given that this would probably bring about considerable energy differences,more advanced com-putational methods have to be applied to gain a more in-depth understanding of this process.Rathore and co-workers studied the reaction of electron-rich aromatic compounds in the presence of various oxidants in detail,focusing on DDQ-MeSO 3H.[39]The same system had been used previously by these authors to efficiently synthesize a number of triphenylenes and hexa-peri -hexabenzocoro-nenes under mild conditions.[40]They presented some impor-tant evidence suggesting that,for many o -terphenyls such as 19,the reaction indeed proceeds via radical cation inter-mediates such as 19 and 19 (Scheme 9).First theyshowedScheme8.Scheme 9..Angewandte ReviewsH.Butenschçn,D.T.Gryko et al.2013Wiley-VCH Verlag GmbH &Co.KGaA,WeinheimAngew.Chem.Int.Ed.2013,52,9900–9930that various Scholl precursors with oxidation potentials <1.7V versus the SCE readily undergo oxidative C ÀC bond formation with DDQ/H +as the oxidant,whereas those with oxidation potentials >1.7V versus the SCE do not react.Additionally,they noted that the reaction does not occur in mixtures of CH 2Cl 2and various acids.These authors also claimed that the necessity of using strong oxidants for this reaction to occur is inconsistent with the arenium ion mechanism,since oxidation of dihydro intermediates such as 19a (formed through the arenium ion mechanism,Scheme 9)can easily be accomplished even with molecular oxygen.They did not,however,extend their study further to include compounds with lower oxidation potentials and/or larger aromatic systems.In our opinion,one of the most important compounds with a behavior that may help to understand the difference between typical oxidative aromatic coupling and the Scholl reaction is 2,2’-dihydroxy-1,1’-binaphthyl (4).This compound is formed by the oxidative aromatic coupling of 2-naphthol (3,Scheme 2).Subjecting 4to further portions of FeCl 3is ineffective,regardless of the conditions.In contrast,as early as in 1922,Zinke and Dengg performed the reaction of both 2,2’-dihydroxy-1,1’-binaphthyl (4)and 2,2’-dimethoxy-1,1’-binaphthyl (21)with AlCl 3,which resulted in the formation of perylene-1,12-diol (22),thus involving a cleavage of the ether functions in the case of 21(Scheme 10).[41]The authors preferred to start from the dimethoxy derivative 21as thereaction was initially much more sluggish when starting from the respective diol 4,presumably because of side reactions of the hydroxy functions with AlCl 3.The reaction conditions are remarkable,because neat 4/21and a fourfold excess of AlCl 3are mixed and melted at 140–1508C for 1h,with no solvent being used.Phenol 22undergoes oxidation by basic solutions in air with the formation of the respective quinone 23.[42]Later,the authors reported the synthesis of crystalline 23by oxidation of the crude product of quinone 22with lead(IV)oxide,which was purified by crystallization.Subsequent reduction of 23with zinc dust or sodium dithionite affords 22in pure form as bright yellow leaves,which become green on standing in air over longer periods of time.[43]It is clear that the highest electron density in compound 4is present at positions 3and 3’,and indeed electrophilic aromatic substi-tutions were performed at these positions.Oxidative aromatic coupling proceeding through the radical cation mechanism is very sensitive to the distribution of the electron density within the molecule.Numerous examples show that it proceeds:1)only if the overall electron density of the aromatic molecule is relatively high and 2)only at the position wherethe electron density is the highest.Consequently,the reaction of diol 4with FeCl 3,which tends to form radical cations,cannot proceed with carbon–carbon bond formation at positions 8and 8’(which possess rather moderate electron density).On the other hand,the reaction in the presence of AlCl 3is apparently less sensitive.It can proceed even when the overall electron density is significantly lower (the trans-formation of ketone 10into benzanthrone 11is a good example),and it can also proceed at positions which are not the most electron rich ones in the molecule (especially in an intramolecular fashion).The preparation of 1,12-dihydroxy-perylene (22)from (1,1’-binaphthalene)-2,2’-diol (4)through the action of AlCl 3was later patented by various authors.[44,45]The reactivity of naphthylisoquinolines is another exam-ple that emphasises this fundamental mechanistic difference stated above.We recently discovered the synthesis of 1-azaperylene (25)by anion radical coupling of two regioiso-meric naphthylisoquinolines,24and 26.[46]These substrates seemed to us to be perfect models to study the interrelation between AlCl 3-mediated reactions and FeCl 3-mediated reac-tions,since the oxidation potentials of naphthalene and isoquinoline are rather high.Indeed,both substrates are inert in the presence of stoichiometric or excess amounts of FeCl 3at 258C and 808C.[47]On the other hand,compound 24reacts in a AlCl 3/NaCl (5:1)melt at 1608C to afford 1-azaperylene (25)in 68%yield (Scheme 11).[47]In the case of 8-(naph-thalen-1-yl)isoquinoline (26),such a reaction would require an electrophilic attack at position 1of the isoquinoline moiety,which is very electron poor.Hence,submitting compound 26to Scholl reaction conditions does not result in the formation of 1-azaperylene (25).[47]An analogous example from our research group is the reaction of compound 27in the presence of AlCl 3/NaCl to afford p -extended coumarin 28in 30%yield (Scheme 12).[48]Again,the reaction of coumarin 27with FeCl 3does not proceeded,most likely because of the electronic effectsScheme 11.Scheme 12.Scheme 10.Biaryl Synthesis9905Angew.Chem.Int.Ed.2013,52,9900–99302013Wiley-VCH Verlag GmbH &Co.KGaA,Weinheimimparted by the OH group,which does not activate a suitable position for reaction to occur.In the context of the recent discussion concerning the mechanism of the dehydrogenation of aromatic compounds under various conditions,it seems that processes that occur at room temperature with a well-known one-electron oxidant (sometimes mild Lewis acids such as FeCl 3or MoCl 5)and processes which proceed at 120–1608C in the presence of AlCl 3(a strong Lewis acid)most probably undergo a different mechanism.We venture to say that the radical cation mechanism is operating in the first case and the arenium cation mechanism in the second case.The experiments by Nenitzescu and Balaban that show,in some cases,that dehydrogenation proceeds in the presence of a Brønsted acid (and nitrobenzene as the co-oxidant)supports this line of thought.[30]The reaction mediated by AlCl 3can generally proceed with substrates less electron rich than those for oxidative aromatic coupling.4.Scholl Reaction—Scope,Limitations,and UtilizationThe earliest examples of the Scholl reaction focused on the synthesis of large polyaromatic ketones and quinones.1-Benzoylpyrene is cyclized in an AlCl 3/NaCl melt to furnish dibenzo[def ,qr ]chrysene-8-one in approximately 40%yield.[36]The first example of multiple dehydrogenative coupling was reported by Scholl and Seer in 1913.The authors treated 4,4’-dibenzoyl-1,1’-binaphthyl (29)with AlCl 3for 8.5h at 95-1008C and obtained the nonacyclic dione “violanthrone”(30);however,again no yield was reported (Scheme 13).Oneof the largest systems ever synthesized by the Scholl reaction is quinone 32prepared from ketone 31(no yield given,Scheme 14).[49]The dehydrogenation of aromatic hydrocarbons can also take place upon melting in a vacuum (no yield given,Scheme 15).[50]Müllen and co-workers published a very interesting study showing that 3-(1-naphthyl)perylene (36)undergoes oxida-tive coupling to either 35or 37depending on the reaction conditions (Scheme 16).[51]Compound 36in the presence ofFeCl 3in dichloromethane forms 37in 46%yield,while an analogous reaction with AlCl 3in chlorobenzene gives rise to terrylene (35)in 43%yield.This result further emphasizes that the cyclization of aromatic hydrocarbons in the presence of oxidizing and non-oxidizing Lewis acids usually proceeds by different mechanisms.Cyclization of 12-(1-naphthyl)benz[a ]anthracene (39)in an AlCl 3/NaCl melt furnishes benzo[def ]naphtha[1,2-p ]chrys-ene (38;10%).However,if AlCl 3/SnCl 4is used,the main product is naphtho[1,2-a ]perylene (40;60%,Scheme 17).[52]These results further indicate that the nature of the reactive intermediates can vary quite significantly on changing the reaction conditions slightly.Scheme 13.Scheme 14.Scheme 15.Scheme 16.Scheme 17..Angewandte ReviewsH.Butenschçn,D.T.Gryko et al.2013Wiley-VCH Verlag GmbH &Co.KGaA,WeinheimAngew.Chem.Int.Ed.2013,52,9900–99302,3,4,5-Tetraphenylthiophene (41),when subjected to classical Scholl conditions (AlCl 3/NaCl,4:1),gives flavophen (42)in 26%yield (Scheme 18).[53]The reaction can be applied to heterocyclic systems,as has impressively been shown in an American patent disclosing the reaction of N -acylurea 43to afford pentacycle 44(no yield given,Scheme 19).[54]Weitzenbçck and Seer reported that dinaphtho[2,1-b :1’,2’-d ]furan undergoes cyclization to peryleno[1,12-bcd ]furan in the presence of AlCl 3,with the concomittent formation of perylen-1-ol.[13]Polyphosphoric acid has been used to induce the cyclization of precursor 45(Scheme 20).[55]Amino-substituted benzo[g ,h ,i ]perylene 46forms in excellent yield in a domino reaction involving an electrophilic sub-stitution followed by a Scholl reaction.5.Oxidative Aromatic Coupling5.1.Intermolecular Oxidative Aromatic CouplingAs stated above (see Section 3and Scheme 7),the reactions of reasonably electron-rich arenes with oxidants that lead to the formation of biaryls usually proceed through the radical cation mechanism.The term “oxidative coupling”will be used in this and the following sections for such reactions.The simplest variant of the oxidative coupling reaction is intermolecular oxidative homocoupling,in whichtwo molecules of one aromatic compound react to form a biaryl.The oxidative cross-coupling of aromatic compounds (the reaction between two different arenes)is much more difficult to achieve.This is due to the poor selectivity of the reaction,which strongly depends on steric factors as well as on the electron density of both aromatic molecules.The con-ditions must carefully be selected to avoid homocoupling.[56]Nonetheless,significant progress has been made in the field of intermolecular oxidative cross-coupling over recent years,and many interesting examples have been published.Some representative examples of intermolecular oxidative homo-and cross-coupling reactions of arenes from the recent literature are presented in the following sections.5.1.1.Homocoupling of Naphthalene DerivativesThe oxidative coupling of aromatic compounds often leads to the formation of axially chiral products.Structures of many widely used optically active catalysts are based on biaryl units.[57]Among them,binaphthyl and its derivatives are particularly important.1,1’-Bi-2-naphthol (4),whose first synthesis was described in Section 2.1,is a precursor of many important ligands used in asymmetric catalysis,with binap being the most prominent example.[58]The syntheses of binol (4)and other binaphthyls by the oxidative coupling of the corresponding naphthalenes were achieved under various conditions,and racemic products as well as pure enantiomers could be obtained.Many different catalytic systems,oxidants,and metal complexes were examined for this purpose.Besides the classical systems with FeCl 3,reasonable yields of binaphthyls were also achieved using reactants such as thallium(III)and mercury(II)trifluoroacetates,Pb(OAc)2,and CoF 3,[59]titanium(IV)chloride,[60]or CuCl 2in the presence of amines.[61]Particularly interesting are the cata-lyzed homocoupling reactions of naphthalenes to binaphthyls,for example,peroxidase-catalyzed oxidation with H 2O 2,[62]copper(I)-or copper(II)-catalyzed oxidation under oxygen or air,[63]as well as the oxidation catalyzed by methyltrioxo-rhenium.[64]The syntheses of various binaphthyls have been summarized in recent reviews.[65]Only a few recent examples are presented below.Shaw and co-workers employed a vanadium catalyst in the air oxidation of the chiral naphthol 47to binaphthol 48,a key intermediate in the synthesis of (À)-viriditoxin,which is a promising inhibitor of bacterial cell division.[66]The reaction in the presence of [VO(acac)2]as the catalyst gave the product 48in 67%yield with rather low diastereoselectivity (76:24).When [VO(acac)2]was replaced by the enantiopure chiral catalysts 49,which is a binol derivative,both the reaction yield and the diastereoselectivity substantially increased (Scheme 21).Many interesting copper complexes for the oxidative coupling of naphthols were developed by Kozlowski and co-workers,and and have successfully been applied in the total syntheses of numerous chiral,natural binaphthyl derivatives with high enantioselectivity.[65e,67]Wang and co-workers recently reported two new systems which are efficient in the oxidation of various 2-naphthols into the corresponding racemic binaphthols (Table 1).In thefirstScheme18.Scheme19.Scheme 20.Biaryl Synthesis9907Angew.Chem.Int.Ed.2013,52,9900–99302013Wiley-VCH Verlag GmbH &Co.KGaA,Weinheim。
盐酸普萘洛尔 分子量

盐酸普萘洛尔分子量盐酸普萘洛尔(C16H21NO2·HCl)是一种用于治疗高血压和心脏疾病的药物。
它能够通过阻断心脏β受体来减慢心率,降低血压,并改善心脏功能。
本文将详细介绍盐酸普萘洛尔的分子结构、药理作用、用途和副作用等方面的知识。
一、分子结构盐酸普萘洛尔的化学式为C16H21NO2·HCl,它是一种白色结晶性粉末。
其分子量为331.80 g/mol,密度为1.06 g/cm³。
盐酸普萘洛尔属于β受体阻滞剂药物。
二、药理作用盐酸普萘洛尔通过选择性阻断β1受体,降低心脏收缩力和心率,从而减轻心脏的负担,降低血压。
此外,它还能抑制交感神经系统的活性,减少肾素-血管紧张素-醛固酮系统的活性,从而进一步降低血压。
三、用途盐酸普萘洛尔主要用于治疗高血压和心脏疾病,如心绞痛、心律失常和心肌梗死后的心力衰竭等。
它可以减轻心脏负荷,改善心脏功能,并降低心脏病发作的风险。
此外,盐酸普萘洛尔还可用于预防偏头痛和手颤等疾病。
四、副作用使用盐酸普萘洛尔可能会引起一些副作用。
常见的副作用包括疲劳、头晕、低血压、心跳过缓、胃肠道不适、睡眠障碍等。
少数患者可能会出现心律失常、呼吸困难、皮疹、肌肉无力等严重副作用。
因此,在使用盐酸普萘洛尔时,应根据医生的建议进行使用,并定期进行相关检查。
五、禁忌和注意事项盐酸普萘洛尔在以下情况下禁用:1. 对该药物过敏的患者;2. 心脏传导阻滞或心动过缓的患者;3. 有严重心功能不全的患者;4. 孕妇、哺乳期妇女和儿童。
在使用盐酸普萘洛尔时,还需要注意以下事项:1. 长期使用时,应定期监测心电图、血压和心率等指标;2. 不可突然停药,应逐渐减量停药;3. 患者在服药期间应避免饮酒,以免增加药物的不良反应。
六、药物相互作用盐酸普萘洛尔与其他药物可能发生相互作用,例如:1. 与胰岛素或口服降糖药合用,可能会增加低血糖的风险;2. 与利尿剂合用,可能会增加低血压的风险;3. 与钙通道阻滞剂合用,可能会增加心脏传导阻滞的风险;4. 与抗凝剂合用,可能会增加出血的风险。
氢溴酸达非那新

适应证
氢溴酸达非那新用于膀胱过度刺激引起的尿频、尿急、尿失禁。
禁忌证
1.对氢溴酸达非那新及其中成分过敏者禁用。 2.尿潴留、胃潴留及未控制的闭角型青光眼患者禁用。 3.重度肝功能损害患者不推荐使用。
注意事项
1.由于尿潴留的可能,有明显膀胱尿道阻塞症状的患者使用时应谨慎。 2.氢溴酸达非那新具有抗胆碱作用,能降低胃肠道动力,胃肠道阻塞性疾病患者有胃潴留的可能,使用时应 谨慎。严重便秘、溃疡性结肠炎和重症肌无力患者慎用。 3.已控制的闭角型青光眼患者慎用。 4.氢溴酸达非那新生殖毒性分级为C,只有当对母体的益处高于对胎儿的危险时方可用于孕妇。 5.氢溴酸达非那新可经大鼠乳汁分泌,尚不知氢溴酸达非那新是否经人乳汁分泌,哺乳期妇女应慎用。
用法用量
口服,推荐剂量为7.5mg,1次/d,整片服下,不得嚼碎、掰开或压碎,可单服或与食物同服。根据个人临床 反应,剂量可增至15mg。中度肝功能损伤患者及与CYP3A4抑制剂(如酮康唑、伊曲康唑、利托那韦、奈非那韦、 克拉霉素、奈法唑酮)同服时,剂量不得超过7.5mg。
药物相互作用
1.氢溴酸达非那新主要经CYP2D6和CYP3A4代谢,CYP3A4抑制剂(酮康唑、伊曲康唑、利托那韦、奈非那韦、 克拉霉素、奈法唑酮)可使氢溴酸达非那新代谢减少,日剂量不应超过7.5mg。
尿失禁治疗药物是一个潜力巨大但尚未完全开发的市场,临床特征均是在24h内需要小便数不少于十次。据 世界卫生组织(WHO)有关人员估计,全球约有10%~15%中年人(50岁以下)和40%~70%老年人不同程度地受到此病 困扰。膀胱过动症一般没有神经源性损伤或疾病,可由膀胱的快速充盈、体位改变、甚至行走、咳嗽诱发。估计 全世界约有4~5亿名尿失禁患者,女性的发生率为男性的2倍。男性的发生率随着年龄的增长而升高,是一种常 见和令人痛苦的疾病。(另有一组数据估计世界7个主要国家受影响的人群达1.54亿,其中0.73亿人被分类为明 显尿失禁症。)。
胶原中氢键变化的红外光谱、拉曼光谱分析

第49卷第1期 2020年1月Vol. 49 No. 1Jan. 2020中©皮革CHINA LEATHER胶原中氢键变化的红外光谱、拉曼光谱分析张奇\唐春雪、丁克毅、何达海2a t(1.西南民族大学化学与环保工程学院,四川成都610041;2.西南民族大学药学院,四川成都610041)摘 要:通过红外光谱和拉曼光谱分析了皮股原经过铝、铬、四羟甲基氯化磷鞣制前后的氢键变化,以及这3种鞣法得到的皮股原湿热收缩前后的氢键变化。
红外光谱和拉曼光谱的分析结果均显示,鞣制使得胶原分 子中的氢键数量增加;鞣制过的皮肢原在湿热收缩过程中伴随着三股螺旋中酰胺羧基氢键的断裂;拉曼光谱 还可以观察到脯氨酸、羟脯氨酸氢键的断裂。
这一结果支持了氢键在维持股原(包括鞣制改性的肢原)三股螺 旋结构稳定的重要性,同时也为进一步研究鞣制改性皮股原的湿热收缩机理提供了试验依据。
关键词:红外光谱;拉曼光谱;鞣制股原;湿热收缩;氢键中图分类号 TS57 文献标识码 A D01:10.13536/ k i.issn l001-6813. 2020-001-003 Analysis for hydrogen bond changing of collagen byF T-IR and Raman spectrumZHANG Q i\T A N G Chunxue1,DING KeyV,HE Dahai(1. College of Chemistry & Environmental Engineering, Southwest Minzu University, Chengdu 610041, C hina;2. College of Pharm acy, Southwest Minzu University, Chengdu 610041, China) Abstract: Hydrogen bond changing of hide powder before and after tanned by chrome salt,aluminum salt and tetra- hydromethyl phosphorus chloride ( THPC) , tanned collagen before and after hydrothermal shrinkage were analyzed by FTIR and Raman spectroscopy. Both the results of FTIR and Raman spectroscopy indicate that the quantity of hydrogen bonds in collagen molecular increases after tanning process. There are hydrogen bonds breaking of amide carbonyl groups from triple helix of collagen during hydrothermal shrinkage. In addition, the breaking of hydrogen bonds of proline and hydroxyl-proline are also observed in Raman spectroscopy. These experimental results support the important role of hydrogen bonds in maintaining the stability of triple helix structure of collagen including tanned collagen, and provide experimental basis for further study of the hydrothermal shrinkage mechanism of tanned collagen.Key words:F T-IR;Raman spectrum;tanned collagen;hydrothermal shrinkage;hydrogen bond点,在有机化合物官能团结构辨认中起着重要作用。
四氢呋喃脱水

关于钠+二苯甲酮的除水问题【最近一直在做除水,有些资料和经历分享一下】★★★小木虫(金币+1):奖励一下,鼓励发有价值的话题秋天白云(金币+2):谢谢分享!2021-10-11 22:39:27一:二苯甲酮的物理性质英文名:Benzophenone, diphenylketone.化学名称:苯甲酮别名:二苯酮,苯甲酰基苯分子量:182.21 外观:白色片状结晶,微有玫瑰香味熔点:47-49℃/2k/Pa. 沸点:170℃. 相对密度?:1.095-1.099 溶解性:不溶于水,能溶于乙醇,醚和氯仿。
二:钠+二苯甲酮的除水原理说法一:二苯甲酮做为指示剂在合成实验中用途广泛,可以用来做为处理甲苯、苯、THF、乙腈等的指示剂;参加后假设出现了漂亮的蓝色,就可以蒸馏使用了,最好保存在钠中,但为何生成蓝色,有很多说法,莫宗一世,不尽一样。
现从一本国外的讲反响的机理书上摘录其原因:译成中文的大概意思是:‘由酮生成的自由基阴离子叫作羰基自由基,二苯甲酮做指示剂是二苯甲酮中的氧原子夺取了钠中的电子,生成了暗蓝色羰基自由基;该自由基在立体上、电性方面是稳定的,主要用来指示‘无氧条件’!用途广。
’。
参加二苯甲酮后,溶液越蓝,说明溶液中的氧越少,间接说明水分很少。
但是否变蓝,与参加的二苯甲酮和处理的溶剂的量有关,THF〔300ml)含水多,要回流约6 小时以上,当然与处理的溶剂量有关,越多那么时间越长,甲苯、苯等含水少的回流时间少,这样时间会缩短。
说法二:二苯甲酮和金属钠反响生成一个显蓝色的中间体1,如果溶剂中有水,继续反响生成无色的化合物2.如果没水了就停留在中间体1 的蓝色状态.黄色不大好解释,可能是有机物碱性条件下少量被破坏产生的杂质显色,另外,如果二苯甲酮长时间〔数天〕在金属钠环境中回流也会被破坏而导致不能显蓝色。
二苯甲酮的作用相当于酸碱滴定分析过程的指示剂,少量存在与蒸馏残液中,与残液一起做一般废液或废渣处理,不需要特殊的处理方式。
多奈哌齐杂质标准品

多奈哌齐杂质——孟成科技(上海)有限公司名称信息结构式多奈哌齐杂质Donepezil Impurity分子式/Molecular Formula :C24H29NO3分子量/Molecular Weight :379.50多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C24H24BrNO3分子量/Molecular Weight :454.36CAS#:231283-82-2多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C31H35NO3分子量/Molecular Weight :469.63多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C18H23NO4分子量/Molecular Weight :317.38CAS#:1808997-65-0多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C26H33NO4分子量/Molecular Weight :423.56多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C24H27NO3分子量/Molecular Weight :377.49多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C10H10O2S分子量/Molecular Weight :194.25多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C24H27NO3分子量/Molecular Weight :377.48多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C24H27NO3分子量/Molecular Weight :377.48多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C17H15NO4分子量/Molecular Weight :297.31多奈哌齐杂质Donepezil Impurity 分子式/Molecular Formula :C24H29NO3.HCl分子量/Molecular Weight :415.96。
百灵威核磁耗材产品

NMR Consumables and Accessories
客服热线:400-666-7788
全球 NMR 耗材 引领 60 年
百灵威
百灵威科技有限公司成立于 1992 年,始终以“为科研和生产提供世界一流的产品和服务”为宗旨,致力于超精细化学品的研发与 制造。经过近二十年的发展,百灵威已具备为化学、分析、生物、材料、物理及药物研发等领域提供近五十万种产品和专业服务 的能力。 百灵威拥有一支强大的具有丰富经验和创新能力的研发团队,在江苏、河北设立的两个研发中心可迅速研发出毫克至数百公斤级 的医药、生化、材料等中间体及特殊高端化学品,并可为客户定制合成各类产品,尤其擅长小分子药物中间体以及催化剂配体的 合成。 百灵威人坚信“发展民族科技”的理念,坚持依靠中国人自己的智慧和力量, 不断建设和发展位于潮白河畔的现代化工业生产基地,发挥百灵威在尖端技术研究、敏捷制造和系统性物流管理等方面的突出优 势,积极地将中国的各种高端化合物推荐给国际同行,为促进中国化学事业发展,推动世界文明与和谐进步而奋斗不息。 百灵威的使命 促进科技和工业发展,造福人类……
管壁厚度(mm) 平均凸度(µm)
包装
0.27
<60>
50只/塑料筒装
0.27
<60>
50只/塑料筒装
0.43
<60>
50只/塑料筒装
0.43
<60>
100只/纸盒装
0.43
<60>
50只/塑料筒装
0.43
<60>
100只/纸盒装
0.60
<60>
50只/塑料筒装
SampleJet®核磁管
喹啉合成
1/12
2014-10-11 04:21:14
Cl N Br O Br OH N N O
O N O
1
Rx-ID: 23335695 View in Reaxys Yield Conditions & References
93 %
Example Name 1.II.1 To a solution of 7 (15 g, 0.056 mol) in acetonitrile (80 mL) and DIPEA (15.9 g, 0.123 mol), was added POCI3 (17.1 g, 0.112 mol) dropwisely at O0C. The reaction temperature was slowly raised to 1000C for 2 hours. The mixture was cooled and poured onto ice-water. After Neutralized with aq NaHCC>3, extracted with ethyl acetate, and dried over Na2SO4, the crude product was obtained by evaporating of solution to dryness (15 g, 93percent) as a brown solid. MS (m/z) (M++H): 287, 289. With N-ethyl-N,N-diisopropylamine, trichlorophosphate in acetonitrile, Time= 2h, T= 0 - 100 °C Patent; PROGENICS PHARMACEUTICALS, INC.; WO2009/155527; (2009); (A2) English; WO 2009/155527 A2 View in Reaxys
羟基自由基氧化降解水中二-甲基异莰醇

中国环境科学 2017,37(11):4166~4172 China Environmental Science 羟基自由基氧化降解水中二-甲基异莰醇成建国1,白敏冬2*,余忆玄1,田一平1,张芝涛1*(1.大连海事大学轮机学院环境工程研究所,辽宁大连116026;2.厦门大学环境与生态学院海洋生物资源开发利用协同创新中心,福建厦门 361102)摘要:二-甲基异莰醇(2-MIB)是一种由蓝绿藻以及放线菌等微生物产生,具有桥环结构的饱和叔醇,在水中具有令人厌恶的土霉味,常规水处理工艺难以对其氧化降解.利用大气压强电离放电生成羟基自由基(·OH),对2-MIB进行氧化降解,确定了其氧化剂剂量效应、时间效应关系,并利用GC-MS对2-MIB氧化降解过程中间产物的分析,推断其氧化降解机制.结果表明:对初始浓度为150,300ng/L的2-MIB,分别投加总氧化剂TRO 1.8,2.3mg/L,接触反应6s去除率分别为96%和97.6%,处理后残余浓度低于10ng/L (低于人类嗅阈值).在2-MIB水样中加入•OH淬灭剂叔丁醇(TBA)后,2-MIB的去除效果明显降低,证明氧化降解2-MIB的主要为•OH.另外通过对氧化降解过程中间产物分析表明,•OH 能破坏2-MIB的桥环结构,并最终矿化生成CO2和H2O.关键词:二-甲基异莰醇;羟基自由基;氧化降解;矿化中图分类号:X131.2 文献标识码:A 文章编号:1000-6923(2017)11-4166-07Degradation of 2-methylisoborneol in water by hydroxyl radical. CHENG Jian-guo1, BAI Min-dong2*, YU Yi-xuan1, TIAN Yi-ping1, ZHANG Zhi-tao1* (1.Environmental Engineering Institute, Marine engineering college, Dalian Maritime University, Dalian 116026, China;2.Collaborative Innovation Center for Exploitation and Utilization of Marine Biological Resources, College of Environment and Ecology, Xiamen University, Xiamen 361102, China). China Environmental Science, 2017,37(11):4166~4172Abstract:2-methylisoborneol (2-MIB) produced by cyanobacteria and actinomycetes is a saturated bicyclic-tertiary alcohol, which can cause earthy/musty taste and odor in surface water. Moreover, 2-MIB is usually difficult to be decomposed and removed by conventional water treatment process. In this paper, hydroxyl radical (•OH) generated by a strong ionization discharge process at atmosphere pressure was used to degrade 2-MIB in water, of which the removal efficiency including dose effects and contact reaction time were investigated. The intermediate products formed in •OH treatment process were analyzed by GC-MS, and the oxidative degradation mechanism of 2-MIB by •OH was discussed. Results show that the removal rate for 2-MIB with initial concentration of 150and 300ng/L could reach 96% and 97.6% within 6.0s, while the total reactive oxidant (TRO) dose were 1.8and 2.3mg/L, respectively. After •O H treatment, the concentration of 2-MIB in water was lower than 10ng/L (lower than the human olfactory threshold). The degradation effects of 2-MIB were obviously reduced by the •OH scavengers tertiary butyl alcohol (TBA), indicating that •OH should be the main oxidant for 2-MIB oxidative degradation. By analyzing the intermediates produced in the oxidative degradation process, it was found that the bridge ring structures of 2-MIB could be destroyed by •O H and finally mineralized to CO2 and H2O.Key words:2-methylisoborneol;hydroxyl radical;oxidative degradation;mineralization我国饮用水来源以大的河流湖泊为主,全国70%以上的河流湖泊遭受了不同程度污染.在水华爆发时藻类大量繁殖、死亡过程中会产生致嗅物质导致饮用水变臭,水质质量下降.其中由蓝绿藻以及放线菌产生的次级代谢产物2-MIB是导致饮用水水体产生令人厌恶的土霉味的原因之一[1-3].在夏季和初秋季节饮用水中2-MIB浓度可达到100ng/L或更高,武汉东湖2-MIB最高时收稿日期:2017-04-07基金项目:国家科技支撑计划项目(2013BAC06B01,2013BAC06B02);国家重大科研仪器研制项目(NSFC:61427804);科技部创新人才推进计划重点领域创新团队(2015RA4008);辽宁省重点实验室基础研究项目(LZ2015008)* 责任作者, 白敏冬, 教授, mindong-bai@; 张芝涛, 教授, newzhangzhitao@11期成建国等:羟基自由基氧化降解水中二-甲基异莰醇 4167达317ng/L[4].日本霞浦水库污染严重时2-MIB 含量超过500ng/L[5].我国新《生活饮用水卫生标准(GB5749 2006)》[6]中规定2-MIB检出限值为10ng/L.2-MIB是一种桥环结构的饱和叔醇,化学结构稳定极难氧化降解,常规的饮用水处理工艺:混凝沉淀、沙滤、氯消毒等都无法有效去除2-MIB,另外常规氧化剂如:O3、HClO、ClO2、KMnO3、H2O2也难以有效的氧化降解2- MIB[7-8].研究表明,Cl2、ClO2对2-MIB去除能力很弱,反应72h去除率低于50%[9];另有研究表明, KMnO4、H2O2以及NaClO对2-MIB的氧化降解效果均较差[10].近年来,高级氧化技术在水中嗅味物质的去除研究较多.研究表明,以羟基自由基为主的高级氧化技术能有效的氧化降解2-MIB.例如李学艳等人报道得O3/H2O2高级氧化技术[11],Tran等人报道得TiO2/UV光催化技术[12],都能对水中2-MIB进行有效的氧化降解.以羟基自由基(•OH)为主的高级氧化剂技术,其优势为•OH的氧化还原电位高(2.80V),是仅次于氟的强氧化剂,而且•OH与2-MIB反应速率常数高达109M-1s-1,它可以断开2-MIB的桥环结构对其快速氧化降解.本文采用大气压强电离放电电解氧高效生成•OH的方法,对水中嗅味物质2-MIB进行快速氧化降解.研究了不同初始浓度2-MIB的去除氧化剂剂量效应和时间效应,并通过利用GC-MS对2-MIB氧化降解过程中间产物的分析进一步深入研究了•OH氧化降解2-MIB机制.1 实验原理与方法1.1 实验原理羟基自由基快速氧化降解2-MIB实验原理如图1所示,首先O2经过减压阀通入到等离子体源中,在高频高压电源(7kV,10kHz)激励条件下, 在等离子体源窄间隙(0.2mm)中形成微流柱和微辉光强电离放电[13].通入的O2在窄间隙放电通道内被电离和电解生成O2+、O(1D)、O、O2-、O2(a1∆g)、O3等氧活性粒子[14-16],然后氧活性粒子经过检测仪和流量计,利用射流器注入水中.注入过程中在快速射流和空穴效应下氧活性粒子与水分子迅速反应生成氧化能力更强的•OH及其它氧自由基包括H2O2、HO2¯、O2·¯、O3·¯、HO3·等,氧活性粒子在水中等离子体反应过程如式(1)~式(9)所示[14,17-19].O2+ + H2O + M → O2+·H2O + M (1)O2+·H2O + H2O → HO3+ + ·OH + O2 (2)O3 + H2O → O2 + H2O2(3)H2O2 → H+ + HO2−(4)O3 + HO2-→ O2-· + ·OH + O2(5)O2- ·+ O3 → O3-· (6) O3-· + H2O → ·OH +OH- + O2 (7)O3 + H2O + M → H2O2 + O2 + M (8)O3 + H2O2 → HO2-· + ·OH+ O2(9)含2-MIB水样由水泵泵入管路中,在射流器中与生成的氧自由基接触反应,生成的•OH对2-MIB进行快速氧化降解(见式(10)),管路中设置6个取样点,每个取样点接触反应时间间隔1s.另外为了研究•OH对2-MIB氧化降解的贡献,选择叔丁醇(TBA)作为•OH淬灭剂[20],在含2-MIB水样中加入不同浓度TBA,然后水样按照图1实验流程进行处理.·OH+2-MIB →产物 (10)集水桶图1 •O H氧化降解2-MIB装置示意Fig.1 Apparatus schematic of oxidative degradation2-MIB by •OH1.2 分析方法溶于水中的氧自由基统称总氧化剂用TRO4168 中国环境科学 37卷(mg/L)表示,测试方法采用KI–4-氨基-N,N-二乙基苯胺 (DPD)分光光度法,该方法依据美国EPA 330.5标准,由紫外可见分光光度仪测定(美国EPA Method 330.5, US)[15].水中2-MIB(Wako, Japan)采用HP-SPME吸附浓缩后用GC-MS分析.取处理后水样20mL 移入40mL带聚四氟乙烯涂层的顶空瓶中,加入6g NaCl(使用前450℃烘烤4h),插入SPME萃取头(聚二甲基硅氧烷/二乙烯基苯涂层纤维65µm PDMS/DVB) (Supelco, USA),于65℃、搅拌速度1200r/min条件下进行样品富集30min,然后拔出萃取头迅速插入GC进样口对样品解析进行分析[21].GC-MS(Agilent 7890A-5975C, USA)条件: 采用不分流进样模式,进样口温度250℃,解析时间3min(1min进样时间, 2min吹扫清理萃取头).毛细管柱(HP-5MS;30m×0.250mm× 0.25μm): 载气高纯He,流量1.0mL/min,升温条件: 40℃ (保持2min)然后升高到140℃(4℃/min)继续升高到280℃(10℃/min)保持5min.MS四级杆150℃、离子源230℃.定性分析:质谱全扫描m/z 30-500, 2-MIB特征离子95、108、168定量分析:选择性离子扫描2-MIB选择性特征离子m/z:95[22].2 实验结果与讨论2.1氧化剂剂量对2-MIB去除的影响按照图1所示,对不同浓度2-MIB水样进行处理.2-MIB初始浓度分别为150和300ng/L,处理后水样在第6个取样点取样检测,接触氧化降解时间为6s.2-MIB去除效果与氧化剂剂量效应关系如图2所示,随着注入的总氧化剂TRO浓度的增加,2-MIB氧化降解率也逐渐增加.当TRO为0.8mg/L时,初始浓度为150,300ng/L的2-MIB处理后分别剩余38,148ng/L;当TRO提高到1.8mg/L 时,150ng/L的2-MIB降解到6ng/L,降解率为96%;继续提高TRO到2.3mg/L, 300ng/L的2-MIB处理后残余浓度为7ng/L,低于人类嗅阈值.可见2-MIB初始浓度越高需要投加的TRO剂量也越高,最终处理后水中2-MIB残余低于《生活饮用水卫生标准(GB5749 2006)》[6]要求限值.0.00.5 1.0 1.5 2.0 2.5 3.0TRO浓度(mg/L)2-MIB浓度(ng/L)图2 不同浓度2-MIB的氧化降解结果Fig.2 Oxidative degradation result of differentconcentrations of 2-MIB2.2 •OH对2-MIB氧化降解的贡献在以往的研究中TBA作为•OH淬灭剂,用来研究高级氧化过程中•OH对有机污染物氧化降解作用,TBA与•OH反应速率常数k TBA, •OH 为6×108M-1s-1,在•OH 氧化降解2-MIB(k2-MIB, •OH =4.3×109M-1s-1)过程是有效的•OH淬灭剂[20].本实验选择在水样中加入0.1和4.0mmol/L的TBA 来研究•OH对2-MIB的氧化降解作用.将300ng/L的2-MIB水样通入到•OH氧化降解装置(图1)中,注入2.3mg/L的TRO, 在管路中1、2、3、4、5、6取样点取样检测(40mL取样品预先加入0.1mL 0.1mol/L的Na2S2O3用于瞬间终止反应),对应的•OH氧化降解时间为1~6s.另外用大烧杯取第6点样品避光保存,检测离开管路2-MIB的降解效果.•OH降解2-MIB的时间效应关系如图3所示,不加TBA时,对2-MIB有很好的氧化降解效果,接触反应时间6s对300ng/L的2-MIB去除率可达到98.7%,60s后可以完全氧化降解;加入TBA后对2-MIB的去除效果明显降低,当TBA浓度为0.1mmol/L时,反应6s与未加TBA相比去除率由98.7%降到59.7%,接触反应时间延长到180s对2-MIB去除效率增加到63.6%;当TBA浓度为4.0mmol/L时,反应6s对2-MIB去除率仅为8.3%,延长反应时间到180s 并无明显去除效果.可以得出在基于强电离放电11期 成建国等:羟基自由基氧化降解水中二-甲基异莰醇 4169生成氧自由基氧化降解水中2-MIB 过程中,对2-MIB 起氧化降解作用的为•OH,且•OH 对2-MIB 的氧化降解作用主要发生在管路气液混匀过程中.根据Chick -Watson 方程[23],用t 时刻2-MIB 含量(C t )与初始2-MIB(C 0)比值取对数值,对时间作图,即ln (C t / C 0) = -kc n t式中: k 为•OH 氧化降解2-MIB 反应速率常数,M -1s -1; t 为反应时间,s; c 为•OH 浓度,mg/L; n 为非一级拟合参量.从图3内小图ln(C t /C 0)对反应时间t 作图, R 2>0.98,6s 内线性拟合良好,所以•OH 对2-MIB 的氧化降解反应为一级反应动力学过程.与以往Y uan 等[24]研究的O 3法,Mizuno 等[25]研究的O 3/H 2O 2高级氧化技术,Bang 等[26]研究的H 2O 2/UV 方法以及Park 等[27]研究的H 2O 2/ FeO 42-等高级氧化相比,强电离放电生成•OH 技术氧化降解相同浓度水平的2-MIB 需要投加的氧化剂更少, 同时氧化降解速率更快.分析原因为,强电离放电过程生成的高浓度氧活性粒子溶于水时,与水分子的等离子体反应过程中能生成更多的•OH,同时射流器气液混匀提供的水力空化作用也进一步促进•OH 的生成,另外高速射流也大大提高了气液传质效率,所以经过图1实验装置处理可以快速的氧化降解水中嗅味物质2-MIB.2-M I B 浓度(n g /L )反应时间(s)图3 •O H 氧化降解2-MIB 的时间效应关系 Fig.3 Effects of reaction time on 2-MIB oxidativedegradation by •OH2.3 •OH 氧化降解2-MIB 机制选取浓度为1000ng/L 的2-MIB 进行•OH 氧化降解机制研究,在图1管路分别投加总氧化剂TRO 剂量为1.0,3.5mg/L,在第6取样点取样,用0.1mol/L 的 Na 2S 2O 3立刻终止反应.采用GC -MS 全扫面模式分析•OH 氧化降解2-MIB 过程生成的中间产物.实验结果如图4所示,TRO 为1.0mg/L 时,检测到9种中间产物;TRO 为3.5mg/L 降解2-MIB 时,处理后未检测到任何中间产物,TRO 为3.5mg/L 是降解1000ng/L 的2-MIB 阈值浓度,可以把2-MIB 完全氧化降解为小分子物质并最终完全氧化矿化生成CO 2和H 2O.中间产物与NIST11质谱数据库匹配结果如表1所示,检测到的中间产物匹配度均大于80%.024602460246响应值(×104)保留时间(min)图4 •O H 氧化降解2-MIB 过程气相色谱图 Fig.4 Gas chromatography for •OH oxidative degradationof 2-MIB process•OH 氧化降解2-MIB 过程主要步骤如下:第一步•OH 攻击2-MIB 侧链,经过脱水和脱甲基破坏其稳定结构,形成含酮基和环内双键的一级桥环产物,如I : 2-甲基-2-菠烯,II:樟脑,III:1,7,7-三甲基-2-亚甲基降冰片,IV:2-甲基环己稀-1-醛;第二步•OH 攻击双桥环产物,在C=C 位置上加成并转移自由电子到桥环上,导致桥环结构断裂形成二级单环醛、酮类产物,如VI:2-甲基环己稀-1-醛, VI :2,2-二甲基-1,3-环戊二酮等;第三步•OH 通过环内加成转移自由电子,使环状结构断4170 中 国 环 境 科 学 37卷键生产三级小分子醛类、酮类以及酸类产物,如X:4-甲基-2-己酮,XI:3-甲基丁醛, XI I :叔丁基乙酸以及XI V: 6-甲基-2,6-二烯醇,这与李学燕等人研究的H 2O 2/O 3氧化降解2-MIB 得出的结论较为相近[10].最后•OH 进一步氧化降解链状产物,最终矿化成CO 2和H 2O.表1 •O H 氧化降解2-MIB 过程中间产物匹配结果Table 1 Matching result for intermediates produced from •OH oxidative degradation 2-MIB process序号产物结构式保留时间(min)主要碎片离子(荷质比m /z )匹配度CAS 号一级桥环产物I 2-甲基-2-菠烯9.68 107(135) 94 72540-93-3II樟脑O10.98 95(152) 96 76-22-2III1,7,7-三甲基-2-亚甲基降冰片14.69 107(150) 96 27538-47-2IV葑醇OH17.91 81(135) 93 1632-73-1二级单环醛、酮类产物VI 2-甲基环己稀-1-醛 O 7.66 57(124) 90 102386-90-3VII2,2-二甲基-1,3-环戊二酮OO8.92 70(126) 91 3883-58-7三级小分子醛、酮、酸类产物 X 4-甲基-2-己酮 O5.45 43(114) 87 105-42-0XI 3-甲基丁醛O5.77 44(70) 82 590-86-3 XII 叔丁基乙酸 O4.86 57(101) 86 1070-83-3•OH 对2-MIB 氧化降解机制分析如图5所示:2-MIB 是一种桥环结构的饱和叔醇,在2号位有未配对电子,且电子云密度较高,而-OH 是唯一的亲水基团.•OH 具有极强的电负性和电子亲和性,首先攻击2号位-OH 夺取-H 生成H 2O,•OH 的自由电子转移到2号位侧链,破坏2-MIB 稳定结构.而且2-MIB 侧链2号位C -C 键解离能约为(81kcal/mol)低于桥环内C -C 解离能(88~90kcal/mol)更容易断键[28-29],在•OH 攻击下, 2-MIB 的2号位侧链会发生脱水和脱甲基,生成一级中间产物1,7,7-三甲基-2-亚甲基降冰片(III)和2-莰醇(V*),产物(III)异构化生成更稳定的2-甲基-2-菠烯(I),在形成2-莰醇(V*)过程中部分化学键重组生成葑醇(IV),其中部分V*侧链的-OH 与•OH 继续反应脱氢而生成樟脑(II).这与其它相关研究类似[8,11,30].然后•OH 进一步攻击桥环结构,进行亲和加成形成不稳定的过渡态中间体,中间体在更多•OH 存在条件下,断键破坏桥环结构,生成2-甲基环己稀-1-醛(VI)、2,2-二甲基-1,3-环戊二酮11期 成建国等:羟基自由基氧化降解水中二-甲基异莰醇 4171(VII))、2,2,3-三甲基-3-环戊烯-醛(VIII)以及2,2-二甲基-3环戊烯-醛(IX)等单环中间产物,这些单环中间产物大多数含有不饱和C=C,不饱和双键更容易被•OH 攻击.例如•OH 对一级中间产物2-甲基-2-菠烯(I)的氧化降解(如图6所示):首先在•OH 作用下,在3号位加成形成羟基,•OH 的自由电子转移到I 的2号位,由于含有自由电子,I 的过渡中间产物结构容易断键,桥环结构被破坏,同时侧链被多余的•OH 进攻,脱去H 2O 以及CO 2,而形成单环的二级中间产物如VII 、VIII*、IX*.图5 •O H氧化降解2-MIB 反应过程 Fig.5 Degradation process of 2-MIB by •OH*未检测到物质图6 •O H 对桥环结构中间产物I 的开环过程 Fig.6 The open bridge ring structure process ofintermediate I by •OH•OH 对单环的二级中间产物醛酮类物质的氧化降解过程.以VII 为例,VII 在•OH 的攻击下进行环内加成,在5号位生成羟基,同时•OH 自由电子转移到4号位,在自由电子作用下C -C化学键断裂,生成单链的小分子醛、酮、酸类物质,如3-甲基丁醛(XI)、叔丁基乙酸(XII),最后这些小分子化合物被•OH 矿化形成CO 2和H 2O. 图7 •O H 对单环中间产物VII 的开环及矿化过程 Fig.7 Ring opening and mineralization process ofmonocyclic intermediate VII by •OH 3 结论3.1 利用大气压强电离放电生成·OH 能快速有效的氧化降解水中嗅味物质2-MIB.对初始浓度为150,300ng/L 的2-MIB,投加1.8,2.3mg/L 的总氧化剂,接触反应时间6s,均可降到10ng/L 以下,处理后残余2-MIB 低于人类嗅阈值.3.2 分析不同浓度氧化剂氧化降解2-MIB 过程得到的中间产物表明,•OH 可以破坏2-MIB 稳定的桥环结构,对其进行断键开环并逐步氧化降解生成小分子有机物,最终矿化成为CO 2和H 2O 无任何有毒副产物残留.参考文献:[1] Su M, Yu J, Zhang J, et al. MIB -producing cyanobacteria(Planktothrix sp.) in a drinking water reservoir: distribution and odor producing potential [J]. Water Research, 2015,68:444-453. [2] Sun D, Yu J, An W, et al. Identification of causative compoundsand microorganisms for musty odor occurrence in the huang pu river, China [J]. Journal of Environmental Science -China, 2013, 25(3):460-465.[3] 邵 晨,黎 雷,于水利,等.产嗅藻类对东太湖某地原水中嗅味物质2-MIB 的贡献 [J]. 中国环境科学, 2014,34(9):2328-2333. [4] 徐 盈,黎 雯,吴文忠,等.东湖富营养水体中藻菌异味性次生代谢产物的研究 [J]. 生态学报, 1999,19(2):212-216.[5] Norio S, Kazunori N. Causative microorganisms for musty odoroccurrence in the eutrophic lake kasumigaura [J]. Hydrobiologia, 2000,434(1):145-150.[6] GB5749-2006 生活饮用水卫生标准 [S]. 中国人民共和国卫生部, 2006.[7] Zoschke K , Dietrich N, Bornick H, et al. UV -based advancedoxidation processes for the treatment of odour compounds: efficiency and by -product formation [J]. Water Research, 2012,4172 中国环境科学 37卷46(16):5365-5373.[8] Fotiou T, Triantis T M, K aloudis T, et al. Photocatalyticdegradation of water taste and odour compounds in the presence of polyoxometalates and TiO2: Intermediates and degradation pathways [J]. Journal of Photochemistry and Photobiology A: Chemistry, 2014,286(15):1-9.[9] Jung S W, Baek K H, Yu M J. Treatment of taste and odormaterial by oxidation and adsorption [J]. Water Science and Technology, 2004,49(9):289-295.[10] 李学艳,马军,陈忠林,等.若干氧化剂对水中嗅味物质2-MIB的氧化去除 [J]. 黑龙江大学自然科学学报, 2007,24(1):76-80.[11] 李学艳,高乃云,沈吉敏,等.O3/H2O2降解水中致嗅物质2-MIB的效能与机理 [J]. 环境科学学报, 2009,29(2):344-352.[12] Tran H, Evans G M, Yan Y, et al. Photocatalytic removal of tasteand odour compounds for drinking water treatment [J]. Water Science and Technology, 2009,9(5):477-483.[13] Zhang Y B, Bai M D, Chen C, et al. ·OH Treatment for killing ofharmful organisms in ship’s ballast water with medium salinity based on strong ionization discharge [J]. Plasma Chemistry and Plasma Processing, 2013,33(4):751-763.[14] Bai M, Zhang Z, Bai M. Simultaneous desulfurization anddenitrification of flue gas by ·OH radicals produced from O2+ and water vapor in a duct [J]. Environmental Science & Technology, 2012,46(18):10161-10168.[15] Bai M, Zheng Q, Tian Y, et al. Inactivation of invasive marinespecies in the proce ss of conveying ballast water using ·OH based on a strong ionization discharge [J]. Water Research, 2016, 96:217-224.[16] Lowke J J, Morrow R. Theoretical analysis of removal of oxidesof sulphur and nitrogen in pulsed operation of electrostatic precipitators [J]. IEEE Transactions on Plasma Science, 1995, 23(4):661-671.[17] Staehelin J, Hoigne J. Decomposition of ozone in water: rate ofinitiation by hydroxide ions and hydrogen peroxide [J].Environmental Science & Technology, 1982,16(10):676-681. [18] Yang Y, Jiang J, Lu X, et al. Production of sulfate radical andhydroxyl radical by reaction of ozone with peroxymonosulfate: a novel advanced oxidation process [J]. Environmental Science & Technology, 2015,49(12):7330-7339.[19] Bai M, Bai X, Zhang Z, et al. Treatment of red tide in ocean usingnon-thermal plasma based advanced oxidation technology [J].Plasma Chemistry and Plasma Processing, 2005,25(5):539-550. [20] Xie P, Ma J, Liu W, et al. Removal of 2-MIB and geosmin usingUV/ persulfate: contributions of hydroxyl and sulfate radicals [J].Water research, 2015,69:223-233.[21] 成建国,刘开颖,白敏冬,等.顶空固相微萃取-气相色谱-质谱联用测定饮用水中的2-甲基异莰醇和土臭素 [J]. 色谱, 2015, 33(12):1287-1293.[22] Watson S B, Brownlee B, Satchwill T, et al. Quantitative analysisof trace levels of geosmin and MIB in source and drinking waterusing headspace SPME [J]. Water Research, 2000,34(10):2818- 2828.[23] Gunten V U. Ozonation of drinking water: part II. Disinfectionand by-product formation in presence of bromide, iodide or chlorine [J]. Water Research, 2003,37(7):1469-1487.[24] Yuan B, Xu D, Li F, et al. Removal efficiency and possiblepathway of odor compounds (2-methylisoborneol and geosmin) by ozonation [J]. Separation and Purification Technology, 2013,117(30):53-58.[25] Mizuno T, Shiya O, Fumitake N. O3/H2O2 Process for bothremoval of odorous algal-derived compounds and control of bromate ion formation [J]. Ozone: Science & Engineering. 2011, 33(2):121-135.[26] Bang H, Slokar Y M, Ferrero G, et al. Removal of taste and odorcausing compounds by UV/H2O2 treatment: effect of the organic and inorganic water matrix [J]. Desalination and Water Treatment,2016,57(57):1-10.[27] Park G, Yu M, Go J, et al. Comparison between ozone and ferratein oxidising geosmin and 2-MIB in water [J]. Water Science & Technology, 2007,55(5):117-125.[28] Luo Y. Handbook of bond dissociation energies in organiccompounds [M]. United States of America: CRC Press LLC, 2003:170-210.[29] Smith M B, March J. March's advanced organic chemistry:reactions, mechanisms, and structure [M]. 6th edition. New Jersey: John Wiley & Sons Inc, 2007:28-35,1736-1750.[30] Qi F, Xu B, Chen Z, et al. Efficiency and products investigationson the ozonation of 2-methylisoborneol in drinking water [J].Water Environment Research, 2009,81(12):2411-2419.作者简介:成建国(1985-),男,山西晋中人,大连海事大学博士研究生,主要从事高级氧化技术应用研究.发表论文3篇.。
大孔树脂1课件

大孔吸附树脂的分离工艺:
原料 溶液
水溶 液
溶出剂
树 脂 柱
泵
检测器
再生剂
分段收集器
大致操作步骤如下: 树脂→预处理→上样→吸附→洗脱→收集洗脱液→回 收、浓缩→干燥→成品
1.树脂的预处理 树脂使用前,需根据使用要求进行程度不同的预处理, 目的是将树脂内孔残存的未聚合单体与致孔剂、分散剂、 防腐剂等有机残留物除去,提高树脂洁净度,提高树脂使 用的安全性。 ①用水除去水溶性杂质 ②用有机溶剂除去脂溶性杂志 ③再用吸附介质除去残留的其它溶剂,以免影响树脂的 吸附量
树脂再生的评价指标应从树脂的吸附性能、稳定性等方面综合判 断。具体可用比吸附量、比洗脱量、有效成份保留率及纯度等指标 评价。
再生条件
比表面积(m2/g-resin)
HP20未使用产品
约700
HP20已使用产品
96
99%甲醇(3BV)
246
95%异丙醇(3BV)
406
95%丙酮(2BV)
563
75%异丙醇+4%NaOH(4BV)
2.装柱与药液的上柱吸附
2.1 药液的上柱吸附 2.1.1 药液上柱前的预处理 为避免大孔树脂被污染堵塞,药液上柱前一般需经过滤
处理,除去较多的悬浮颗粒杂质,保证树脂的使用完全顺 利。
2.1.2 泄漏曲线与吸附容量的考察 树脂吸附容量=泄漏点前上柱样品体积(ml) × 样品浓 度(mg.L-1 ) 有人用大孔树脂D1300精制右归煎液时,对其泄漏曲线 做了如下考察研究
大孔树脂的一些性质:
1.大孔吸附树脂理化性质稳定,但在水和有机溶剂中可吸收溶剂而 膨胀,在室温下对稀酸、稀碱稳定
2.大孔吸附树脂通过物理吸附和分子筛作用而实现分离
四川氘代试剂类cas号

四川氘代试剂类cas号氘代试剂是一类重要的有机化学试剂,可以用于有机合成、材料科学、医药化学等领域。
氘代试剂与普通有机试剂的区别在于,其中的氢原子被氘原子取代,因此具有不同的化学性质和反应特征。
在四川省,也有许多企业和机构生产和销售氘代试剂,以下是一些常见的四川氘代试剂类CAS号。
1. 氘代二甲醚 CAS号:115-10-6氘代二甲醚又称氘代甲氧基甲烷,是一种无色透明的液体,化学式为CD3OD。
它是一种重要的溶剂,在有机合成中广泛应用。
氘代二甲醚可以用于核磁共振(NMR)等技术,可以用于确定化合物的结构和反应机理。
在医药化学中,氘代二甲醚可以用于药物代谢研究和药物开发。
2. 氘代三甲胺 CAS号:99-92-3氘代三甲胺又称氘代N,N-二甲基甲酰胺,是一种无色液体,化学式为CD3N。
它是一种常用的氢氧化剂和还原剂,在有机合成中广泛应用。
氘代三甲胺可以用于制备各种有机化合物,如醛、酮、酯等。
在有机合成中,氘代三甲胺可以用于制备氘代化合物,如氘代醇、氘代醛等。
3. 氘代甲醇 CAS号:811-98-3氘代甲醇是一种无色液体,化学式为CD3OH。
它是一种重要的溶剂,在有机合成中广泛应用。
氘代甲醇可以用于制备氘代化合物,如氘代醇、氘代醛等。
在医药化学中,氘代甲醇可以用于药物代谢研究和药物开发。
4. 氘代丙酮 CAS号:673-29-6氘代丙酮是一种无色液体,化学式为CD3COCH3。
它是一种常用的溶剂,在有机合成中广泛应用。
氘代丙酮可以用于制备各种有机化合物,如醛、酮、酯等。
在医药化学中,氘代丙酮可以用于药物代谢研究和药物开发。
5. 氘代二氯甲烷 CAS号:594-20-7氘代二氯甲烷又称氘代甲基氯,是一种无色液体,化学式为CD2Cl2。
它是一种常用的溶剂,在有机合成中广泛应用。
氘代二氯甲烷可以用于核磁共振(NMR)等技术,可以用于确定化合物的结构和反应机理。
在医药化学中,氘代二氯甲烷可以用于药物代谢研究和药物开发。
化合物名称

Oxygen ['ɔksidʒәn] 【氧】Nitrogen ['naitrәdʒәn] 【氮】Hydrogen ['haidrәudʒәn] 【氢】Urea ['juәriә]【尿素】Thiourea [ˏθaiәujuә'riә]【硫脲】Guanidine ['gwænәdi:n]【胍】Amidine ['æmiˏdi:n]【脒】Enamine [i'næmin]【烯胺】Furan ['fjuәræn]【呋喃】Imidazole [ˏimi'dæzәul]【咪唑】Imine['imi:n]【亚胺】Imide['imaid]【酰亚胺】Lactam['læktæm]【内酰胺】Lactone['læktәun]【内酯】Ether ['i:θә]【醚】Ester['estә]【脂】Carobohydrate['kɑ:bәu'haidreit]【碳水化合物】Acetal ['æsitæl]【缩醛】dichloromethane [daiˏklɔ:rә'meθein]【二氯甲烷】sulfone ['sʌlfәun] 【砜】sulfoxide [sʌl'fɔksaid]【亚砜】chloroform ['klɔ(:)rәfɔ:m]【氯仿】acidify [ә'sidifai]【酸化】saturated ['sætʃәreitid]【饱和的】sodium bicarbonate [bai'kɑ:bәnit]【碳酸氢钠】sodium carbonate【碳酸钠】Ketone['ki:tәun]【酮】Aldehyde['ældihaid]【醛】enol['i:nɔl]【烯醇】Enone ['i:nɔn]【烯酮】Dioxane [dai'ɔksein]【二噁烷】Acetone ['æsitәun]【丙酮】Ethyl acetate ['æsiˏteit]【乙酸乙酯】Petroleum [pi'trәuliәm] ether【石油醚】n-hexane [hek'sein]【正己烷】thiophene ['θaiәˏfi:n] 【噻吩】Glycine ['glaisi:n]【氨基乙酸】Glycoside ['glaikәˏsaid]【糖苷】thionyl['θaiәnil] chloride【氯化亚砜】Sulfur['sʌlfә]【硫】Proton ['prәutɔn]【质子】sodium borohydride [ˏbɔ:rә'haidraid]【硼氢化钠】lithium ['liθiәm]【锂】aluminum [ˏælju:'minjәm]【铝】sodium hydride [haidraid] 【氢化钠】glycol['glaikɔl] 【乙二醇】glycerol['glisәˏrɔl]; glycerin ['glisәrin]【甘油,丙三醇】Formic['fɔmik] acid 【甲酸】Chloroformate[ˏklɔ:rә'fɔ:meit]【氯甲酸脂】acetic [ә'sitik] acid 【乙酸】acetic anhydride[æn`haidraid] 【乙酸酐】trifluoroacetate [traifluәrә'æsiteit]【三氟乙酸酯(或盐)】anhydrous[æn`haidrɔs]【无水的】magnesium[mæg'ni:zjәm] sulfate['sʌlfeit]【硫酸镁】potassium[pә'tæsiәm] hydroxide [hai`drɔksaid]【氢氧化钾】sodium azide [`eizaid]【叠氮化钠】hydroxyl[hai`drɔksil] groups 【羟基】alkane['ælkein]【烷烃】alkene['ælki:n]【烯烃】alkyne['ælkain]【炔烃】halogen['hælәdʒәn]【卤素】halide['hælaid]【卤化物】fluorine['fluәri:n]【氟】fluorination[ˏflu:әri'neiʃәn]【氟代】chlorine['klɔ:ri:n]【氯】chlorinati on[ˏklɔ:ri'neiʃәn]【氯代】bromine['brәumi:n]【溴】bromination ['brәumineiʃәn]【溴代】iodine['aiәdain]【碘】iodination['aiәdineiʃәn]【碘代】linear['liniә]【直链的】sulfonation[ˏsʌlfә'neiʃәn]【磺化】chloromethylation[klɔ:rәˏmeθi'leiʃәn【氯甲基化】nitration[nai'treiʃәn]【硝化】reduction[ridʌkʃәn] 【还原】oxidation[ɔksi'deiʃәn] 【氧化】chlorosulfonation [ˏklәurәˏsʌlfә'neiʃәn]【氯磺化】alkylation [ˏælki'leiʃ(ә)n]【烷基化】acylation[ˏæsi'leiʃ(ә)n]【酰化作用】acrylic [ә'krilik] acid 【丙烯酸】phosphorus ['fɔsfәrәs] 【磷】phosphorous ['fɔsfәrәs] tribromide[tr ai'brәu maid]【三溴化磷】phosphorus oxychloride [ˏɔksi'klɔ:raid]【三氯氧磷】condensed sulfuric [sʌl'fjuәrik] acid【浓硫酸】fuming nitric ['naitrik] acid【发烟硝酸】poly phosphoric [fɔs'fɔrik] acid 【多聚磷酸】acid chloride【酰氯】amide ['æmaid]【酰胺】amine[ә`min] 【胺】ammonia ['æmәunjә]【氨(水)】oxime ['ɔksi:m]【肟】Methanol ['meθәnɔl]【甲醇】Methyl ['meθil]【甲基】Methoxyl [me'θɔksil]【甲氧基】Ethanol ['eθәnɔl]【乙醇】Ethyl ['eθil]【乙基】Triethylamine [traiˏeθәlә'mi:n]【三乙胺】aniline [`ænili:n]【苯胺】hydrazine [`haidrәˏzi:n]【肼】hydrazide [`haidrәˏzi:n]【酰肼】hydrazone [`haidrәˏzәun]【腙】nitrile [`naitrail]【腈】borane ['bәurein] 【硼烷】Sodium Hydrosulfite [ˏhaidrәu'sʌlfait]【连二亚硫酸钠、保险粉】sodium thiosulfate [ˏθaiәu'sʌlfeit]【硫代硫酸钠】Epoxide [e'pɔksaid]【环氧化物】Olefin ['әulәfin]【烯烃】Acetylene [ә'setili:n]【乙炔】Oxalic acid [ɔk'sælik]【草酸】Alkaline ['ælkәlain]【碱】Isocynate [aisәu'saiәneit]【异氰酸酯(或盐)】Isothiocynate [aisәuˏθaiәu'saiәneit]【异硫氰酸酯】Carbamate ['kɑ:bәmeit]【氨基甲酸酯(或盐)】Anisole ['æniˏsәul]【苯甲醚】Aziridine【氮杂环丙烷】epoxyethane [e'pɔksiθein]【环氧乙烷】Hypochlorite [ˏhaipәu'klɔ:rait] 【次氯酸盐】Enzyme ['enzaim]【酶】Guanine ['gwɑ:ni:n]【鸟嘌呤】Lead[li:d] acetate['æsiˏteit]【醋酸铅】Molecular [mәu'lekjulә]sieve[siv]【分子筛】Morphine['mɔ:fi:n]【吗啡】C ocaine[kә'kein]【可卡因】Morpholine['mɔ:fɔˏli:n]【吗啡啉】Nitromethane[ˏnaitrә'meθein]【硝基甲烷】Oligosaccharide[ˏɔligәu'sækәraid]【寡糖】Ortho-['ɔθә]【邻-,正-,原-】Meta-['metә]【间-, 偏-,次-】Para-['pærә]【对-,仲-,副-】Polymerization[ˏpɔlimәrai'zeiʃәn]【聚合】Phthalimide['θælimaid]【邻苯二甲酰亚胺】Selenium[si'li:niәm]【硒】Ultrasonication[ˏʌltrәˏsɔni'keiʃәn]【超声作用】Sulfonamide【sʌl'fɔnәmaid】【磺酰胺】Sulfonyl['sʌlfәnil]chloride【磺酰氯】Peptide ['peptaid]【肽】Barbituric [ˏbɑrbi`tjurik] acid【巴比妥酸】Thiocyanate[θaiәu'saiәneit]【硫氰酸酯】Thiolate ['θaiәuleit]【硫醇脂(或盐)】Transesterification [trænsәsˏterәfi'keiʃәn]【酯交换】Intramolecular [ˏintrәmә'lekjulә]【分子内的】Intermolecular [ˏintә:mә'lekjulә]【分子间的】Orthoformate[ˏɔθә'fɔmeit] 【原甲酸脂】Esterification[esˏterifi'keiʃәn]【酯化】Zinc[ziŋk]【锌】triflate[trifleit]【三氟甲磺酸酯(或盐)】Bulky['bʌlki]【体积较大的】Quaternisation[kwɔtәnai'seiʃәn]【季铵盐化】Benzylic[ˏben'zilik]【苄基】benzyloxy [ˏbenzi'lɔksi]【苄氧基的】Carbanion ['kɑ:bәnaiәn]【碳负离子】Carbocation [ˏkɑ:bә'keiʃәn]【碳正离子】Dipole['daipәul]【偶极】Electron rich【富电子】Electron deficency【缺电子】Electron donating group【供电基】Electron withdrawing group【吸电基】Steroid['stirɔid]【甾族化合物】i-Propanol['prәupænәl]【异丙醇】n-Butanol['bjutәnәul]【正丁醇】benzoyl ['benzәuil] peroxide [pә'rɔksaid]【过氧化苯甲酰】sodium nitrite ['naitrait]【亚硝酸钠】diazonium [ˏdaiә'zәuniәm]salt【重氮盐】azomethane [ˏæzәu'meθein]【偶氮甲烷】phenyl ['fenәl]【苯基】salicylate [sæ'lisileit]【水杨酸酯(或盐)】nitromethane [ˏnaitrә'meθein]【硝基甲烷】benzoic [ben'zәuik] acid 【苯甲酸】anthranilic [ænθ'rәnilik] acid【邻氨基苯甲酸】benzyl ['benzil] 【苄基】toluene ['tɔljui:n] 【甲苯】xylene ['zaili:n]【二甲苯】cuprous ['kju:prәs] cyanide['saiәnaid]【氰化亚铜】Sodium Citrate ['sitrit]【柠檬酸钠】Dimethyl sulfide['sʌlfaid]【二甲硫醚】Brine [brain]【盐水】Triphenylphosphine [traiˏfenәl'fɔsfi:n] oxide ['ɔksaid] 【三苯氧磷】Phenol ['fi:nәl]【苯酚】Irradiation [iˏreidi'eiʃәn]【光照】Radical ['rædikәl] initiator [i'niʃieitә]【自由基引发剂】Decarboxylation [ˏdikɑ:ˏbɔksә'leiʃәn]【脱羧】Phosgene['fɔzdʒi:n]【光气】Triphosgene【三光气】Carbonic [kɑ:'bɔnik] acid 【碳酸】cadmium ['kædmiәm] oxide氧化镉calcium ['kælsiәm] chloride【氯化钙】Tin(II) chloride【二氯化锡】Nickel ['nikl] 镍Palladium [pә'leidiәm]【钯】Carbonyl ['kɑ:bәnil]【羰基】Carboxylic [ˏkɑ:bɔk'silik]【羧基的】Organometallic[ˏɔ:gәnәumi'tælik]【有机金属的】quinoxaline [kwi'nɔksәli:n]【喹喔啉】quinoline ['kwinәli:n]【喹啉】isoquinoline【异喹啉】indole [' indәul]【吲哚】indazole ['indәzәul]【吲唑】carbazole ['kɑ:bәzәul]【咔唑】pyridine ['piridi:n]【吡啶】pyrimidine[piri'midi:n]【嘧啶】pyrrole ['piәrәul]【吡咯】pyrolidine[pi'rәuliˏdi:n] 【四氢吡咯】piperidine [pi'peridi:n]【哌啶】piperazine [pi'perәzi:n]【哌嗪】。
蛋白质PDB文件说明

字符集合只是一些非控制型字符,象空格和结束符,出现在PDB文件记录中。
也就是:abcdefghijklmnopqrstuvwxyzABCDEFGHIJKLMNOPQRSTUVWXYZ1234567890` - = [ ] \ ; ' , . / ~ ! @ # $ % ^ & * ( ) _ + { } | : " < > ?空格和结束符。
结束符根据系统而定,Unix用一行字符,而其他的系统可能就用一个回车来表示。
特殊字符希腊字母就详细的拼写出来。
比如:α, β, γ原子用DOT表示。
右箭头用-->表示。
左箭头用<--表示。
上标用两个等号表示开始和结束。
比如:S==2+==下标用一个等号来表示开始和结束。
比如:F=c=如果等号两边至少有一边有一个空格,那么这个字符就是表示等号。
比如:2 + 4 = 6逗号,冒号和括号用来表示文档中的分界苻,也就是下面几种中的一种:ListSListSpecification ListSpecification如果逗号,冒号或者括号在任何一片文档中使用不是作为分界苻的话,那么肯定有字符被漏掉了。
比如下边例子中第四行的"\":COMPND MOL_ID: 1;COMPND 2 MOLECULE: GLUTA THIONE SYNTHETASE;COMPND 3 CHAIN: NULL;COMPND 4 SYNONYM: GAMMA-L-GLUTAMYL-L-CYSTEINE\:GL YCINE LIGASECOMPND 5 (ADP-FORMING);COMPND 6 EC: 6.3.2.3;COMPND 7 ENGINEERED: YESCOMPND MOL_ID: 1;COMPND 2 MOLECULE: S-ADENOSYLMETHIONINE SYNTHETASE;COMPND 3 CHAIN: A, B;COMPND 4 SYNONYM: MA T, A TP\:L-METHIONINE S-ADENOSYLTRANSFERASE;COMPND 5 EC: 2.5.1.6;COMPND 6 ENGINEERED: YES;COMPND 7 BIOLOGICAL_UNIT: TETRAMER;COMPND 8 OTHER_DETAILS: TETRAGONAL MODIFICATION数据类型-------------------------------------该部分该部分主要用来描述试验和记录中该大分子的一些基本信息,有以下几种记录:HEADER,OBSLTE,TITTITLE,CA VEA T,COMPND,SOURCE,KEYWDS,EXPDTA,AUTHOR,REVDA T,SPRSDE,JRNL和REMARK部分。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
DMSO 44 mg/ml
Mechanisms: Pathways:Cell Cycle/DNA Damage; Target:HDAC Biological Activity: Remodelin HBr salt is a novel potent and selective inhibitor of the acetyl-transferase protein NAT10. IC50 value: Target: NAT10 inhibitor Remodelin can improve nuclear architecture, chromatin organization, and fitness of both human lamin A/Cdepleted cells and HGPS-derived patient cells, and decrease markers of DNA damage in these cells. Using a combination of chemical, cellular, and genetic approaches, acetyl-transferase protein NAT10 was identified as the target of Remodelin that mediated nuclear shape rescue in laminopathic cells via microtubule reorganization. Down-regulation and mutations of the nuclear-architecture proteins lamin A and C cause misshapen nuclei and altered chromatin organization associated with cancer and laminopathies, including the premature-aging disease Hutchinson-Gilford progeria syndrome (HGPS). Remodelin is a useful chemical tool to study ... References: [1]. Larrieu D, et al. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014 May 2;344(6183):527-32.
Caution: Not fully tested. For research purposes only Medchemexpress LLC
m o c . s s e r p x e m e h c d e Am S. Uw ,w 2 5w 8: 8b 0e JW Nm ,o n c o. i s t s c e nr up Jx he t m u e oh mc nd oe Mm D@ 2 0o f n 1i el : t i i a u Sm ,E e v i r D k r a P r e e 1 1
Product Data Sheet
Product Name: CAS No.: Cat. No.: MWt: Formula: Purity :
Remodelin hydrobromide 1622921-15-6 HY-16706A 363.28 C15H15BrN4S >98%
Solubility: