配位化合物和配位平衡
普通化学 第九章 配位化合物与配位平衡

配合物的稳定性及配位平衡
2 中心离子的水解效应 若溶液酸度太小,金属离子易发生水解,金属离子 浓度减小,使配离子发生解离。
Fe3+ + 3C2O42- = Fe(C2O4)33+ 3OH– 3Fe(OH)3
Fe(C2O4)33- + 3OH– = 3Fe(OH)3 ↓ + 3C2O42-
配合物的稳定性及配位平衡
习惯上沿用
K3[Fe(CN)6] K4[Fe(CN)6]
铁氰化钾
亚铁氰化钾
配位化合物的基本概念
无机化合物 分子式 H 2 SO 4 NaOH KBr 名称 硫酸 氢氧化钠 溴化钾 分子式 H 2 [PtCl 6 ] [Cu(NH 3 ) 4 ](OH) 2 [Ag(NH 3 ) 2 ]Br [Cr(NH 3 ) 4 (H 2 O) 2 ]Cl 3 K 2 [HgI 4 ] K[Co(NO 2 ) 4 (NH 3 ) 2 ] K 2 SO 4 硫酸钾 [Cu(NH 3 ) 4 ]SO 4 [Co(NH 3 ) 2 (en) 2 ](NO 3 ) 3 [Pt(py) 4 ][PtCl 4 ]
配位化合物 名称 六氯合铂 (IV) 酸 氢氧化四氨合铜 (II) 溴化二氨合银 (I) 氯化四氨· 二水合铬 (III) 四碘合汞 (II) 酸钾 四硝基· 二氨合钴 (III) 酸钾 硫酸四氨合铜 (II) 硝酸二氨·二(乙二胺)合钴(III) 四氯合铂 (II) 酸四吡啶合铂 (II)
第二节
1 K = K fθ Kθ sp
θ
影响水解效应大小的因素: (1) KfӨ越小,配合物越易解离, (2)介质酸度越小,pH越高,
配位化合物的基本概念
配合物形成的原因
(1)内外界之间为离子键,配合物可解离。
普通化学 第八章 配位化合物和配位平衡

c(NH ) / c
3
2
0.10 0.10 1.9 103 {( x 2 0.10mol / L) / c }2
x = 2.5 mol/L, 即氨水的初始浓度至少为 2.5 mol· -1。 L
3
8.1.1 配合物的组成
[Ag(NH3)2]Cl
配合物 内界 外界
[Ag(NH3)2]+
中 心 离 子 配 位 原 子 配 位 体 配 位 体 数 配 离 子 电
Cl外 界 离 子
4
荷
常见配位体名称、配位原子
配位体 FClBrISCNNCSH2 O NH3 NH2OHCNS2O32配位体名 称 氟 氯 溴 碘 硫氰酸根 异硫氰酸 根 水 氨 氨基 羟基 氰 硫代硫酸 根 配位 原子 F Cl Br I S N O N N O C O 配位体 CO NO ONONO2CH3COOC2O42(ox)* 配位体名称 羰基 亚硝酰 亚硝酸根 硝基 乙酸根 草酸根 吡啶 联吡啶 甲胺 乙二胺 乙二胺四乙酸 根 配 位 原 子 C N O N O O N N N N N,O
Hongmei Wang , Zhiliang Liu, Caiming Liu, Deqing Zhang, et al
Inorganic chemistry, 2004, 43,4091-4098.
12
13
不饱和烃配合物 π 电子参与形成配位键的配合物。
14
冠醚类配合物
15
C60-配合物
4.31
Cu(NH 3 ) 2 NH 3 Cu(NH ) NH 3
2 Cu(NH 3 )3 NH 3
第八章 配位化合物

A·m2
(3)测定 磁矩可通过磁天平测定。 • 顺磁性:被磁场吸引
• 反磁性:被磁场排斥
• 铁磁性:被磁场强烈吸引 (如 Fe , Co , Ni)
..
..
..
..
N
S
(a)无磁场
N
S
(b)磁场打开
顺磁性的说明
(4)影响因素 未成对电子数越多,磁矩越高,配合物
的磁性越大。
(5)意义
• 根据未成对电子数求磁矩; • 根据磁矩求未成对电子数; • 判断杂化方式、空间构型、配合物类型。
未成对电子数 0 1 2 3 4 5
µ计 / B.M
0 1.73 2.83 3.87 4.90 5.92
例: 测定FeF63-的µ为5.90 B.M,可判断: Fe3+有5个未成对电子;
Ag+
4d
[Ag(NH3)2]+
4d
5s
5p
NH3 NH3
5s
5p
sp杂化
2. 配位数为4的配合物的杂化方式及空间构型
(1)[NiCl4]2-:Ni 3d84s2
sp3杂化
Ni2+
Ni2+ 3d8 外轨型
四面体
3d
[NiCl4]2-
3d
4s
4p
Cl-
Cl- Cl- Cl-
4s
4p
sp3杂化
[NiCl4]2-
NH2-CH2-CH2-H2N
说明:
少数配体虽然有两个配位原子,由于两 个配位原子靠得太近,只能选择其中一 个与中心原子成键,故仍属单齿配体。
硝基NO(2 N是配位原子) 亚硝酸根ONO- (O是配位原子) 硫氰根SCN (S是配位原子) 异硫氰根NCS (N是配位原子)
第9章配位化合物与配位平衡

第9章配位化合物与配位平衡9.1配合物的基本概念一.配合物的形成例:NH3(aq)蓝色NH3(aq)深蓝色溶液(1)[Cu(H2O)4]2+(左),[Cu(NH3)4]2+(右)CuSO4(aq)(1)+NaOH(aq)无Qi<Kp[Cu(OH)2](1)+BaCl2(aq)白色(BaSO4)(1)浓缩、冷却深蓝色晶体[Cu(NH3)4]SO4(c)[Cu(H2O)4]2++4NH3=[Cu(NH3)4]2++4H2O配位化学创始人AlfredWerner(1866-1919),1913NobelPrizeinChemitry.二.配合物与“简单化合物”和复盐的差别溶于水中电离情况:盐(简单化合物):CuSO4=Cu2++SO42复盐(明矾):KAl(SO4)212H2O=K++Al3++2SO42-+12H2O配合物[Cu(NH3)4]SO4含有“复杂离子”[Cu(NH3)4]2+和SO42-.―复杂离子”(配离子):[Cu(NH3)4]2+=[Cu(NH3)3]2++NH3K1=5.010-3[Cu(NH3)3]2+=[Cu(NH3)2]2++NH3……总的离解方程式:[Cu(NH3)4]2+=Cu2++4NH3K=4.810-14―配合物”与复盐之间无绝对界限.配位键-由配体单方面提供电子对给中心原子(离子)而形成的共价键[Cu(NH3)4]2+Cu2+NH3配位键(dp2,空)K[Pt(C2H4)Cl3](W.C.Zeiealt)Pt2+C2H4(dp2,空)配位键Fe(C5H5)2或Fe(cp)2二茂铁(Ferrocene)Fe2+C5H5-配位键(环戊二烯基阴离子C5H5-或cp-)(教材p.232,图10-1)中心原子(离子)是酸,配体是碱,配合物是酸碱加合物。
Fe(C5H5)2或Fe(cp)2二茂铁(Ferrocene)Fe2+C5H5-配位键(环戊二烯基阴离子C5H5-或cp-)四.配合物的组成例1.[Cu(NH3)4]SO4[内界]例2.[CoCl(NH3)5]Cl2(右下)中心离子配位体外界例3.[Co(NH3)6]Cl3(左下)CoCl36NH3+3AgNO33AgCl()[Co(NH3)6]Cl3(黄色晶体)CoCl35NH3+2AgNO32AgCl()[CoCl(NH3)5]Cl2(紫红色晶体)四.配合物的组成(续)(一)中心离子(或中心原子)——又称“配合物形成体”。
南昌大学大学化学第七章习题答案
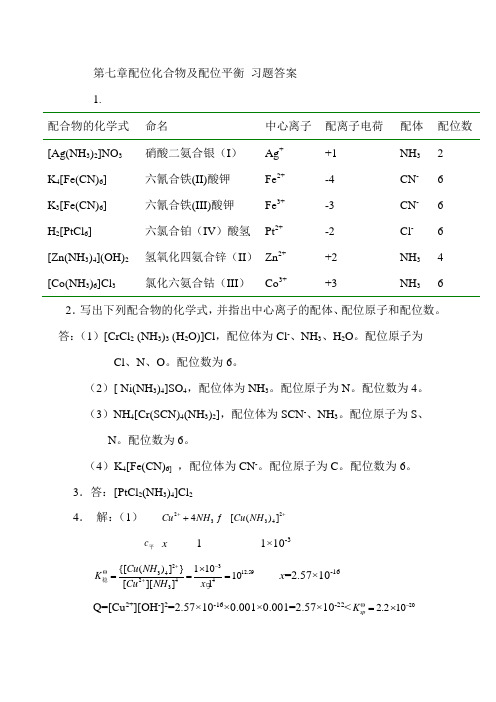
第七章配位化合物及配位平衡 习题答案 1.配合物的化学式 命名 中心离子 配离子电荷 配体 配位数 [Ag(NH 3)2]NO 3 硝酸二氨合银(I ) Ag + +1 NH 3 2 K 4[Fe(CN)6] 六氰合铁(II)酸钾 Fe 2+ -4 CN - 6 K 3[Fe(CN)6] 六氰合铁(III)酸钾Fe 3+-3 CN - 6 H 2[PtCl 6] 六氯合铂(IV )酸氢 Pt 2+ -2 Cl - 6 [Zn(NH 3)4](OH)2 氢氧化四氨合锌(II ) Zn 2+ +2 NH 3 4 [Co(NH 3)6]Cl 3氯化六氨合钴(III )Co 3++3NH 362.写出下列配合物的化学式,并指出中心离子的配体、配位原子和配位数。
答:(1)[CrCl 2 (NH 3)3 (H 2O)]Cl ,配位体为Cl -、NH 3、H 2O 。
配位原子为Cl 、N 、O 。
配位数为6。
(2)[ Ni(NH 3)4]SO 4,配位体为NH 3。
配位原子为N 。
配位数为4。
(3)NH 4[Cr(SCN)4(NH 3)2],配位体为SCN -、NH 3。
配位原子为S 、N 。
配位数为6。
(4)K 4[Fe(CN)6] ,配位体为CN -。
配位原子为C 。
配位数为6。
3.答:[PtCl 2(NH 3)4]Cl 24. 解:(1) 223344[()]Cu NH Cu NH +++ c 平 x 1 1×10-32312.59342443{[()]}11010[][]1Cu NH K Cu NH x +-Θ+⨯=== 稳x =2.57×10-16 Q=[Cu 2+][OH -]2=2.57×10-16×0.001×0.001=2.57×10-22<202.210sp K Θ-=⨯无沉淀。
(2) Q=[Cu 2+][S 2-]=2.57×10-16×0.001=2.57×10-19>366.310sp K Θ-=⨯,有沉淀。
高中化学竞赛课程 无机化学第十一章 配位化合物和配位平衡

Chapter 11 Coordination Compounds and Coordication Equilibrium
一、 配合物的基本概念
实验: 1. CuSO4(aq)
+ BaCl2 + NaOH
BaSO4 Cu(OH)2
有SO42有Cu2+
2. CuSO4(aq) + NH3.H2O 深蓝色aq + 乙醇 深兰色晶体
[Co(en)3][Cr(ox)3]和[Cr(en)3][Co(ox)3] [PtII(NH3)4][PtIVCl6]和[PtIV(NH3)4Cl2][PtIICl4]
配位体的种类、数目可以任意组合,中心离子、氧化态可以 相同,也可以不同。
d. 键合异构 组合相同,但配位原子不同的配体,如-NO2-和-ONO[CoNO2(NH3)5]Cl2 (黄褐色) [CoONO(NH3)5]Cl2 (红褐色)
[Co(en)3]2+ > [Co(NH3)6]2+
2. 化学式的书写原则
(1) 配合物中,阳离子在前,阴离子在后。 (2) 配离子中,按如下顺序:
形成体
阴离子配体
中性配体
例如: [Co(NO2)(NH3)5]SO4
3. 配位化合物的命名原则
遵循无机化合物的命名原则,不同点是配离子部分。
NaCl [Co(NH3)6]Cl3
d1~d3构型: 无高低自旋之分,无论强场还是弱场, 均形成内轨型配合物.
d8~d10构型: 无高低自旋之分,无论强场还是弱场, 均形成外轨型配合物.
稳定性:内轨型配合物 > 外轨型配合物
例: [Fe(CN)6]3-中CN-很难被置换,而[FeF6]3-中F-很容易被置换。
配合物的生成和性质 实验报告

1
实验7. 配合物的生成和性质
一、 实验目的
1. 加深理解配合物的组成和稳定性,了解配合物形成时的特性。
2. 初步学习利用配位溶解的方法分离常见混合阳离子。
3. 学习电动离心机的使用和固-液分离操作。
二、 实验原理
配位化合物与配位平衡
配位化合物的内、外层之间是靠离子键结合的,在水中是完全解离。
而配位个体在水中是部分的、分步的解离,因此就存在解离平衡。
配合物的标准平衡常数
f K ,也被称为稳定平衡常数。
f K 越大,表明配合物越稳定。
形成配合物时,常伴有溶液颜色、酸碱性、难溶电解质溶解度、中心离子氧化还原性的改变等特征。
利用配位溶解可以分离溶液中的某些离子。
三、实验内容
2
3
4
四、 注意事项
1.使用离心机时要注意安全。
2.及时记录实验过程中配合物的特征颜色。
3.节约药品,废液倒入废液缸。
五、思考题
1. 请应用“配合物的结构和性质”一章中的知识并结合实验现象,阐述实验CuSO4+NH3·H2O(过量);溶液分为2份,分别+2 mol·L-1 NaOH / 0.1mol·L-1 BaCl2的本质。
2. 如何正确使用电动离心机?
六、实验体会和建议
5。
中山大学无机化学第8章配位化合物与配位平衡习题及答案

第8章 配位化合物与配位平衡一、选择题8-1 下列配合物的命名不正确的是: ( ) (A) (B)(C)(D)答案: B8-2 下列离子都可以作为配合物的中心原子,但生成的配合物稳定性最差的是: ( ) (A) (B) (C) (D) 答案: D 8-3 的名称是: ( )(A) 三氯化一水二吡合铬(III ) (B) 一水合三氯化二吡合铬(III ) (C) 三氯一水二吡合铬(III ) (D) 一水二吡三氯合铬(III ) 答案: C8-4 下列哪种物质是顺磁性的: ( )(A)(B) (C)(D) 答案: B 8-5用溶液处理再结晶,可以取代化合物中的,但的含量不变,用过量处理该化合物,有氯含量的氯以沉淀析出,这种化合[]()3233K Co(NO )Cl III 三氯三硝基合钴酸钾[]()3233K Co(NO )Cl III 三硝基三氯合钴酸钾()()()2323Co OH NH Cl Cl III ⎡⎤⎣⎦氯化二氯一水三氨合铬[]()26H PtCl IV 六氯合铂酸3+Sc 3+Cr 3+Fe 3+La ()()232Cr py H O Cl ⎡⎤⎣⎦()234Zn NH +⎡⎤⎣⎦()336Co NH +⎡⎤⎣⎦[]4TiF ()336Cr NH +⎡⎤⎣⎦33CoCl 4NH ⋅24H SO 2-4SO -Cl 3NH 3AgNO 1/3AgCl物应该是: ( )(A)(B) (C)(D)答案: A 8-6 羰基合物的磁矩为零,它的空间构型为: ( )(A) 三角双锥形 (B) 四方形(C) 三角锥形 (D) 四方锥形 答案: A8-7 配离子的磁矩为: ( ) (A) 3.88(B) 2.83 (C) 5.0 (D) 0 答案: D8-8 配离子的稳定性与其配位键类型有关,根据价键理论,可以判断下列配合物稳定性的大小,指出正确的是: ( )(A)(B)(C) (D) 答案: B 8-9 化合物的磁矩为,而的磁矩为,对于这种差别可以用下列哪一项所叙述的理由来解释: ( )(A) 铁在这两种化合物中有不同的氧化数 (B) 氰离子比氟离子引起更多的轨道分裂 (C) 氟比碳、氮具有更大的电负性 (D) 氰离子是弱的电子授体 答案: B8-10 某金属中心离子形成配离子时,由于配体的不同,其电子分布可以有1个未成对电()324Co NH Cl Cl ⎡⎤⎣⎦()334Co NH Cl ⎡⎤⎣⎦()324Co NH Cl Cl ⎡⎤⎣⎦()334Co NH Cl ⎡⎤⎣⎦()5Fe CO ⎡⎤⎣⎦()32Cu NH +⎡⎤⎣⎦()B.M.()()33266Fe CN Fe H O -+⎡⎤⎡⎤<⎣⎦⎣⎦()()32266Fe CN Fe H O -+⎡⎤⎡⎤>⎣⎦⎣⎦()()322Ag CN Ag NH -+⎡⎤⎡⎤=⎣⎦⎣⎦()()322Ag CN Ag NH -+⎡⎤⎡⎤<⎣⎦⎣⎦[]36K FeF 5.9B.M.()36K Fe CN ⎡⎤⎣⎦2.4B.M.d d子,也可以有5个未成对电子,此中心离子是: ( ) (A) (B) (C) (D)答案: C8-11 根据晶体场理论,高自旋配合物的理论判据是: ( ) (A) 分裂能 > 成对能 (B) 电离能 > 成对能 (C) 分裂能 > 成键能 (D) 分裂能 < 成对能 答案: D8-12 某金属离子在八面体弱场中的磁矩为 4.9 B.M.,而它在八面体强场中的磁矩为零,该中心金属离子可能是: ( ) (A) (B) (C)(D) 答案: D二、计算题和问答题8-13 用晶体场理论判断配离子,,,(,Co(III) 的电子成对能)是高自旋还是低自旋,并计算配合物的磁矩以及晶体场稳定化能(CFSE )。
配位化合物的稳定性与配位平衡教案

配位化合物的稳定性与配位平衡教案引言:配位化合物是由中心金属离子与周围的配体通过配位键结合形成的化合物。
配位化合物的稳定性与配位平衡是理解和掌握配位化学的重要基础。
本文将从稳定性和配位平衡两个方面进行探讨,并提供一份配位化合物的稳定性与配位平衡的教案。
一、稳定性的影响因素1. 配体的性质配体的配位能力是影响配位化合物稳定性的关键因素之一。
通常,配体的配位能力与其配位原子的电性、大小和配位方式有关。
例如,迈克尔加合物(氮气配合物)由于配位原子的不同电性,形成的稳定性存在较大差异。
2. 配位键的强度配位键的强度直接影响配位化合物的稳定性。
通常,配位键的强度与配体的键长和键能有关,配位键愈强,配位化合物的稳定性就愈高。
例如,持键配体一般形成较稳定的配位化合物。
3. 中心金属离子的性质中心金属离子的性质对配位化合物的稳定性起着重要影响。
中心金属离子的电子结构、电荷以及配位数等因素都可以对配位化合物的稳定性产生影响。
二、配位平衡的影响因素1. 配位物浓度配位物浓度是影响配位平衡的一个重要因素。
配位物浓度的增加可以促进正向反应,使得配体与中心金属离子更容易结合形成配位化合物。
2. 配位物配位能力配位物的配位能力也是影响配位平衡的关键因素。
一般来说,配位物的配位能力越强,反应向右方向(生成配位化合物)进行的速度越快,平衡位置就会向配位化合物方向移动。
3. 配体交换速率配体交换速率是影响配位平衡的另一个重要因素。
当配体与配位化合物发生配位键交换时,交换速率的快慢将直接影响配位平衡的位置。
三、1. 教学目标通过本节课的学习,学生将能够了解配位化合物的稳定性与配位平衡的影响因素,掌握相关概念和基本理论知识。
2. 教学内容(1)稳定性的影响因素:配体的性质、配位键的强度和中心金属离子的性质。
(2)配位平衡的影响因素:配位物浓度、配位物配位能力和配体交换速率。
3. 教学方法(1)理论讲解:通过讲解配位化合物稳定性与配位平衡的影响因素,引导学生理解相关概念和理论。
基础化学配位化合物及配位平衡习题解答

13.解答:lgc
K
θ/ ZnY
=7.75>6,可以标定。
14.解答:lgc
K
θ/ ZnY
=9.21>6,可以滴定。
反式-二溴·四氨合钌(III)
顺式-四氨·二水合钴(III) 反式-四氨·二水合钴(III)
11 配位化合物及配位平衡
22.解答:顺式-二氯·二氨·二水合铬(III)结构式:
可能的异构体有:
其中(I)、(II)为反式二氨异构体。 23.解答:配体 en 比配体 F-具有更强的场强,F-引起的中心离子 Co3+d 轨道分裂能
小于 en 引起的中心离子 Co3+d 轨道分裂能。所以电子在[CoF6-]3-的 eg 与 t2g 之间跃迁需要的能量比[Co(en)3]3+的小,即[CoF6-]3-电子跃迁吸收的光波波 长比[Co(en)3]3+的长,[CoF6-]3-显示出的颜色对应的光波波长比[Co(en)3]3+ 的短。所以黄色溶液应该是[Co(en)3]3+的溶液,而[CoF6-]3-溶液成蓝色。 24.解答:(1) [CuBr4]2-、[Cu(H2O)6]2(2) 正 方 形 场 配 合 物 [CuBr4]2- 中 心 离 子 d 轨 道 分 裂 能 比 八 面 体 场 [Cu(H2O)6]2-配合物 d 轨道分裂能小,[Cu(H2O)6]2-的电子跃迁能大于 [CuBr4]2-,[Cu(H2O)6]2-显示的颜色波长大于[CuBr4]2-,[Cu(H2O)6]2-呈淡 蓝色,而[CuBr4]2-呈深紫色。
东北林业大学 无机化学 第六章配合物 周志强

配位原子
N Cl,N
配位数
4 6
[Cu(en)2]2+
en
双齿
N
4
单齿配体:配位数= 配体数 多齿配体:配位数≠配体数
16
4. 配离子的电荷
17
影响配位数的因素:
1. 中心离子的影响
中心离子电荷越多,则配位数越大。例如[PtCl4]2-中Pt2+
的配位数为4,而[PtCl6]2-中Pt4+的配位数为6。 中心离子半径越大,则配位数越大。例如[AlF6]3-中Al3+ 的半径为50pm,配位数为6,而[BF4]-中B3+的半径为20pm, 配位数为4。 中心离子电荷 +1 +2 +3 +4
配 合 物
配酸 H2[PtCl6]
配碱 [Cu(NH3)4](OH)2
配盐 [Cu(NH3)4]SO4 , K4[Fe(CN)6],[Cr(NH3)6][Co(CN)6] 8
CuSO4· 2O 5H
[Cu(H2O)]4][SO4(H2O)]
FeSO4· 2O、NiSO4· 2O、ZnSO4· 2O 7H 7H 7H CrCl3· 2O [Cr(H2O)4Cl2]Cl· 2O 6H 2H 暗绿色
5
配合物的形成
+
CuSO4+4NH3=[Cu(NH3)4]2++SO42将氨水逐滴加入浅蓝 色的CuSO4溶液中至 生成的沉淀刚好溶解 为止。将溶液分成三 份:
(1)加入BaCl2溶液,有白色BaSO4生成,说明
溶液中有自由的水合SO42-存在。
(2)加入适量NaOH溶液,既无Cu(OH)2蓝色沉淀
CuSO4 4NH3 [Cu(NH3 )4 ]SO4
第七章 配位化合物与配位平衡测验题与答案

第七章. 配位化合物与配位平衡测验题一、选择题1、欲用EDTA测定试液中的阴离子,宜采用: ()A.直接滴定法;B.返滴定法;C.置换滴定法;D.间接滴定法2、用EDTA测定Cu2+,Zn2+,Al3+中的Al3+,最合适的滴定方式是: ()A.直接滴定;B.间接滴定;C.返滴定;D.置换滴定(已知lg K CuY=18.8,lg K ZnY=16.5,lg K AlY=16.1)3、EDTA滴定Al3+的pH一般控制在4.0~7.0范围内。
下列说法正确的是: ()A.pH<4.0时,Al3+离子水解影响反应进行程度;B.pH>7.0时,EDTA的酸效应降低反应进行的程度;C.pH<4.0时,EDTA的酸效应降低反应进行的程度;D.pH>7.0时,Al3+的NH3配位效应降低了反应进行的程度4、在Fe3+,Al3+,Ca2+,Mg2+的混合液中,用EDTA法测定Fe3+,Al3+,要消除Ca2+,Mg2+的干扰,最简便的方法是采用: ()A.沉淀分离法;B.控制酸度法;C.溶液萃取法;D.离子交换法5、用指示剂(In),以EDTA(Y)滴定金属离子M时常加入掩蔽剂(X)消除某干扰离子(N)的影响。
不符合掩蔽剂加入条件的是: ()A.K NX<K NY;B.K NX>>K NY;C.K MX<<K MY;D.K MIn>K MX6、已知lg K BiY=27.9;lg K NiY=18.7。
今有浓度均为0.01mol⋅L−1的Bi3+,Ni2+混合试液。
欲测定其中Bi3+的含量,允许误差<0.1%,应选择pH值为: ()pH 0 1 2 3 4 5lgαY(H)24 18 14 11 8.6 6.6A.<1;B.1~2;C.2~3;D.>47、某配离子[M(CN)4]2-的中心离子M2+以(n-1)d、ns、np轨道杂化而形成配位键,则这种配离子的磁矩和配位键的极性将...........................................................................................()。
配位化合物与配位滴定法—配合物的解离平衡(基础化学课件)

K稳越大,Ksp越大沉淀越易溶解生成配离子。
3、氧化还原反应对配位平衡的影响
氧化还原反应可改变金属离子的浓度,使 配位平衡移动。
➢在氧化还原平衡体系中加入配位剂能与其中
的氧化剂或还原剂反应生成稳定的配合物,使 金属离子浓度发生改变(即电极电势E改变)而 改变氧化还原反应的方向。
例如:
在血红色的Fe(SCN)3溶液中加入SnCl2,血红色消失。
练习
例 : 若 只 考 虑 酸 效 应 , 计 算 pH=1.0 和 pH=6.0 时 PbY的lgK‘PbY值。
配位平衡移动
(配位平衡与其它平衡一样遵循吕·查德原理 )
1、酸度对配位平衡的影响 配位体的酸效应(配体与H+结合使配离子稳定性降低的作用)
[Cu(NH3)4]2+
Cu2+ + 4NH3
L MLn
M(L)
=
[M'] [M]
主反应 辅助配位效应引起的副反应
3、配合物的条件稳定常数(有效稳定常数)
配位反应 M + Y
MY
副反应系数
αY(H)
稳定常数
K MY
[MY ] [M ][Y ]
条件稳定常数 K 'MY [MY ] [M ][Y ']
lgK’MY = lgKMY - lg αY(H)
平衡移动方向
+ 4H+
4 NH4+
酸度↑(PH越低) →配位体浓度↓→配离子稳
定性降低(酸效应越强)。
水解效应(金属离子与OH-结合使配离子稳定性降低的作用)
[FeF6]3-
Fe3+ + 6F-
平衡移动方向
《配位化合物与配位平衡》部分习题解答

第八章《配位平衡与配位滴定》部分习题解答18-1、AgNO 3能从Pt(NH 3)6C14溶液中将所有的氯沉淀为AgCl ,但在Pt(NH 3)3Cl 4中仅能沉淀1/4的氯。
试根据这些事实写出这两种配合物的结构式,并命名。
解:注意配合物的内界与外界之间是离子键结合,易断裂。
中心离子与配位原子之间是配8-3354一种配合物的溶液中加入BaCl 2时产生BaSO 4沉淀,但加AgNO 3时不产生沉淀;而第二种配合物则与此相反。
写出这两种配合物的化学式,并指出钴的配位数和氧化数。
解:此题与8-1是同类型。
注意配合物的内界与外界之间是离子键结合,易断裂。
中心离子配离子的空间构型。
[Mn(H 2O)6]2+ ; [Ag(CN)2]- ; [Cd(NH 3)4]2+ ; [Ni(CN)4]2- ; [Co (NH 3)6]3+。
8-5、试确定下列配合物是内轨型还是外轨型,说明理由,并以它们的电子层结构表示之。
(1) K 4[Mn(CN)6]测得磁矩m /μB =2.00;(2) (NH 4)2[FeF 5(H 2O)]测得磁矩m /μB =5.78。
解:(1) K 4[Mn(CN)6],磁矩m /μB =2.00,只有一个未成对电子;25Mn 2+, 3d 54S 0, ↑↓ ↑↓ ↑ ,d 2sp 3杂化,内轨型;(2)(NH 4)2[FeF 5(H 2O)],磁矩m/μB =5.78,有五个未成对电子;26Fe 3+ , 3d 54S 0, ↑ ↑ ↑ ↑ ↑ ,sp 3d 2杂化,外轨型。
8-8、(1)、(0.0592(lg ()a b V c E E n c θ=+氧化态还原态)氧化态)还原态 B/C/D 中氧化态浓度是减少的,故A 最大。
选A(2)、例如:AgCl(s) + 2NH 3= [Ag (NH 3)2]+ + Cl -3232223332[Ag NH ](Cl )[Ag NH ](Cl )(Ag )c(NH )c(NH )(Ag ),[Ag NH ],Ag f sp c c c c c K c K K Clθθθ+-+-+++==⨯=⨯()()() 要有利于沉淀的溶解,即是K 要大,所以选B无机及分析化学学习指导2 8-9、H 2O ;过氧化氢(HO —OH);H 2N —CH 2CH 2一NH 2;联氨H 2N —NH 2;解:有效的螯合剂为H 2N —CH 2CH 2一NH 2有效的螯合剂是一个配体中含两个及以上的配位原子,而且配位原子间要相隔2~3个其它原子,故只有H 2N —CH 2CH 2一NH 2满足。
第12章 配位化合物与配位平衡

O
N
C
CH3NH2 甲胺
N
阴离子 F- Cl- Br- I- OH- CN- NO2配体 氟 氯 溴 碘 羟基 氰 硝基
配位原子 F Cl Br I O
C
N
阴离子 配体
ONO-
SCN-
亚硝酸根 硫氰酸根
NCS异硫氰酸根
配位原子
O
S
N
分子式
O
O
CC
-O O-
常见多齿配体 名称
草酸根
乙二胺
N N N N
外轨型配合物:由外轨配键形成的配合物
单电子数多
Co(CN)63-
3d
d2sp3
内轨配键:由次外层(n-1)d与最外层ns、
np轨道杂化所形成的配位键 内轨型配合物:由内轨配键形成的配合物
单电子数少
b. 影响内轨型和外轨型的因素
(i) 中心离子的电子构型
离子的电子 形成配合物类型 构型
d10
外轨型
[Cu(NH3)4]2+ 四氨合铜(Ⅱ)离子 [Co(NH3)6]3+ 六氨合钴(Ⅲ)离子 [CrCl2(H2O)4]+ 二氯·四水合铬(Ⅲ)离子
2. 配位体命名原则 不同配体之间用“”隔
(1) 先阴离子,后中性开分子
[PtCl5(NH3)] 五氯·(一)氨合铂(Ⅳ)离子
(2) 先无 机配体,后有 机配体
② 中心离子的价层轨道首先杂化, 杂化类型决定于 a.中心离子的价层电子构型 b.配位数 c.配位体的配位能力
③ 中心离子的价层空轨道与配体的含孤对电子的 轨道重叠,成键形成配合物。
即M L
④中心离子的杂化类型决定配合物的空间构型。
2. 中心离子的杂化轨道
大学化学-配合物与配位平衡总结

大学化学 | 配合物与配位平衡总结●7.1 配位化合物的基本概念●中心离子(或原子):具有接受孤对电子或不定域电子的空位的原子或离子●具有9~17电子构型的d区金属离子,如Fe3+、Co3+;具有18电子构型的ds区金属离子,如Cu+、Ag+ 等;●s区金属离子如叶绿素中Mg2+、p区高氧化态的非金属元素,如BF4-中B(Ⅲ)、SiF62-中的Si(Ⅳ)、PF6-中的P(Ⅴ);●0价金属或负价金属,金属羰基配合物Ni(CO)4、Fe(CO)5 和[Ti(CO)6]2−、[M(CO)4]2− (M = Fe, Ru, Os,氧化数-2)。
●配体:能给出孤对电子或多个不定域电子的离子或分子●单齿配体:一个分子只有一个配位原子的配体●氢氰酸:H一C≡N、异氢氰酸:H-N ≡ C●硫氰根以S为配位原子;异硫氰根以N作配位原子。
●多齿配体:含有两个或两个以上配位原子的配体,如乙二胺四乙酸根,其中含有2个N、4个O均可配位,是六齿配体,1个配体可形成多个配位键●桥联配体:如OH-、Cl-不止1对孤对电子,可提供两对电子作桥,桥联多个中心离子。
主要在多核配合物中。
●配位原子:配体中与中心离子直接键合的原子,即提供孤对电子的原子。
常见配位原子是电负性较大的非金属原子,如O、S、N、X等●配位数,C.N.:配位原子的个数●配位数一般为偶数(2、4、6、8),其中最常见的是4和6●配位数大小与中心离子的电荷、半径以及配体的电荷、半径有关●规律●中心离子电荷数越高、半径r越大:内层空轨道越多、周围能容纳的配体就越多,C.N.越大●配体半径 r 越小(在中心离子周围能容纳的配体就越多),电荷数越少(配体之间的斥力小), C.N.越大●配合物生成条件:配体浓度越高、温度越低, C.N.越大●●配位单元:由中心离子和配体通过配位共价键结合形成的单元,用[ ]标出●配位键:一个原子提供孤对电子,一个原子或离子的空轨道接受孤对电子形成的共价键●内界:即配位单元,由中心原子(离子)和配体构成的离子●外界:带有与内界异号电荷的离子●如:在配合物[Co(NH3)6]Cl3中,内界[Co(NH3)6]3+,外界Cl-; K4[Fe(CN)6]?中性配位单元作为配合物的[Ni(CO)4]则无外界●7.1.3配合物的命名:中文名的写法●内、外界之间先写阴离子,后阳离子。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
中性原子Ni(CO)4 Fe(CO)5 极少数负离子[HCo-1 (CO)4]
2、 配体与配位原子
(1)配位体--可以给出孤电子对或键电子的分子或阴离子。
中性分子 NH3 H2O CO en PPH3 阴离子 CN- SCN- NO2- S2O32- C2O42- X-
6、掌握的意义,重点掌握与或 ,与,与之间的关系,会进行有关配合物方面的计算。
前面有关内容的学习,我们时常提及配合物这个名词,对配合物涉及的内容有了一点较为浅显的认识:配合物就是一个中心离子与配位体以配位键形成的稳定的一类特殊应用价值的化合物。由于配合物的形成,很多物质的性质发生了改变
(1)、物质溶解度的改变 Ag(NH3)2+
H2N NH2 M
CH2 CH2
C2O42- Phen bipy
多齿配体:EDTA 乙二胺四乙酸
(1) 配体的分类
①无机配体和有机配体
②单齿配体和多齿配体 只含一个配位原子的配体(大部分无机配体)。
多齿配体--含有两个或两个以上配位原子,同时与一个中心原子结合形成两个或两个以上配键的配体,它与中心原子或离子形成具有环状结构的配合物--螯合物。如:
二齿配体:乙二胺(en) CH2 CH2 2+ O O
(2) 配体有孤对电子或键电子去填充中心离子的空价轨道。
(3) 中心离子与配体以一定的组成和空间构型结合为配位单体--存在配位键。
2.配合物的定义:
凡是可以给出孤对电子或键电子的一定数目的分子或离子(配体)和具有接受孤对电子或键电子的空价轨道的原子或离子(中心离子),以配位键方式按一定的组成和空间构型所形成的结构单元--配位单体,含有配位单体的化合物--配合物。
(4)、 改变了物质的氧化还原性
(5)、改变了物质的生物活性。
由于配合物这部分内容不仅是无机化学的中心课题(是高校学生必学的专业课),而且对分析化学、有机化学、生物化学、环境化学、材料化学以及对人体至关重要的超分子化学的研究都有重要的意义。因此学习配合物的有关内容至关重要。
Chapter9 配位化合物和配位平衡
学习要求:
1、掌握配合物的概念和配位键的本质,学会配合物的命名,会书写结构式。
2、熟记配位数、螯合物、螯合效应的概念,会确定中心离子的配位数CN,理解CN与杂化轨道类型和空间构型的关系。
3、了解配合物的异构化现象,重点会确定平面方形、八面体几何异构体的数目。
注:只有结构确定的配合物才能正确地确定其配位数CN。如:
Cs3CoCl5 CN=4 熔融AlCl3中AlCl4- CN=4
而熔融的 AlF3中AlF63- CN = 6 2Ca(CN)2CN = 6
CN=4
含有6个配位原子,六齿配体,往往与中心原子或离子1:1螯合形成CN = 6的五个五元环的螯合物,它是很好的多酸螯合试剂在分析上有重要作用和价值。
③按配位原子与中心原子结合的方式分类:
A、 配体--Werner配体(F- H2O OH- NH3)
B、酸配体(d-d反馈键)--配体除了提供孤对电子外,还存在空的MO或空的d轨道。如:CO CN- Cl- Br- PPh3 C2O42- 等。
如: [Pt(en)2]Cl2 CN = 22=4 K[Cr(C2O4)2(H2O)2] CN = 22+2=6
[Cr(en)3]3+ CN = 23=6 Cr(EDTA)- CN= 16=6
§9-2配合物的有关内容和涉及的基本概念
2-1配合物的概念
随着配位化学的飞速发展,配合物数目与日俱增,应用前景十分广阔,因特征给出一个比较清楚、比较确定的定义还是可能的。
1. 配合物的特征
(1)中心离子有空的价电子层轨道,可以接受配体的孤对电子和键电子。
注意:(1)配离子与配合物往往不必区分。
(2)配离子即存在于晶体中,又存在于溶液中。
2-2 配合物的组成
1、 中心离子(或原子)--配合物的形成体,它具有价电子层轨道可以接受孤电子对或键电子。一般是d区,ds区的离子:Ag+ Cu2+ Zn2+ Cd2+ Hg2+ Ni2+ Pt2+Mn2+ Fe3+Fe2+Pd2+ Co2+
键电子的CH2=CH2 芳香烃
(2)配位原子--配位体上直接与中心原子或离子相连接的原子或离子形成配键。配位原子一般在p区,共计14个。 IVA:C (CO CN-) IA: N(NH3 NO2-) P(PR3) As(Sb)
VIA:O(H2O OH- ,C2O42-) S(SCN- ) Se(Te) F- Cl- Br- I- H-(BH4-)
4、熟练掌握用HOT解释配合物的形成和空间构型,会判断配合物的类型:内轨、外轨 高自旋、低自旋配合物,会比较配合物的相对稳定性和磁性大小,会解释含有反馈键配合物稳定性的原因。
5、了解CFT的要点,掌握Oh、Td及平面方形场对d轨道分裂的数目,会用CFT解释八面体结构配合物的稳定性、磁性、配合物的颜色及吸收光谱,会熟练计算CFSE。
(1)、大多数元素具有两种形式的价键--主价和副价(现代术语中,主价--氧化数;副价--配位数)。
(2)、每个元素倾向于满足它的主价和副价。 (3)、副价在空间有确定的位置,即配合物有确定的空间结构,据Werner配位理论中的6个NH3在空间以八面体形式排布。 NH3
NH3
Werner提出的副价理论,弥补了当时不完善的原子价理论,这是它的重要贡献之一,而它创造性地把有机化学的空间构型推广至无机化学领域,奠定了配合物的立体化学的基础,这是它的又一重大贡献。由于它的突出贡献1913年获诺贝尔化学奖,成为获此殊荣的第一个无机化学家(它是配位化学的奠基人)。从此配位化学进入空前的发展时期,处于21世纪现代化学的中心地位.(有下列化学家在配位化学领域获得诺贝尔化学奖:1912年Grignard--格林雅试剂;1913年Werner--配位化学理论; 1963年Ziegler-Natta--金属烯烃催化剂;1976年lipscomb--硼烷和碳硼烷;1981年Hoffmann--配合物的等瓣理论;1983年Toube--金属配合物电子转移反应机理。)
Cu(en)22+ H2N NH2 C C
Cu O O
(2)配位数CN的确定 ①若中心原子或离子与单齿配体或混合单齿配体形成配合物.
CN = 配体的数目。如:Co(NH3)6Cl3 CN = 6 [Cr(NH3)3Cl3] CN =6
[Cr(SCN)3Br2 H2O]2-CN = 6
②多齿配体与中心离子或原子形成螯合物 CN = 配体的个数X齿数
(2)、改变了物质的颜色。
(3)、改变了物质的酸碱性。 H2C-O O-CH2
CHOH B CHOH
H2C-O O-CH2
H3N NH3
Cl3
H3N NH3
C、配体--配体提供键电子,分子中还存在空的MO与中心原子可形成另一种键--d-反馈键。如:CH2=CH2 、芳烃。
3. 配位数(用CN表示)
(1)定义:直接与中心原子或离子配位的配位原子的数目或中心原子接受孤电子对的数目或形成配键的数目。配位数一般为CN =2,3,4,5,6,7,8,12。常见且主要的是2,4,6。
3、常见的配位单体:
配位阴离子单体 Cu(CN)43- Ag(CN)2- Fe(CN)63-
配位阳离子单体 Cu(NH3)42+ Ag(NH3)2+ Co(NH3)63+
中性分子单体 Ni(CO)4 Fe(CO)5 [Pt(NH3)2Cl2]
沉淀Co2+进行重量分析测Co的含量,实验过程中NaOH溶液用完了,它就用,结果过量时只得到土黄色的溶液,未得到Co(OH)2沉淀。次日它回到实验室时,发现溶液只析出橙黄色晶体,分析其组成为: Co(NH3)6Cl3。起初它认为是加合物,进行实验时既无游离的Co3+,又无游离的NH3 (但却存在游离的Cl-),它这一成果公布后,立即引起众多无机化学的重视,纷纷进行科学研究,研究发现不仅Co3+与形成这类化合物 Ag+ 、Cu2+、、 Zn2+、 Cd2+、Ni2+ 、Pt2+ 等均于生成此类化合物,并且除外X- CN- SCN- S2O32-等亦能发生类似的反应。但Tassert得到的这类化合物难以用当时的理论加以解释。因为是很稳定的,它们又是如何结合为更稳定的化合物呢?当时人们提出很多理论试图来解释这类化合物,但均未获得公认。大约经过100多年,即1893年,仅有26岁的瑞士苏黎士大学的Werner总结了前人和自己大量的工作,发表了一篇"关于无机化合物结构问题"的论文--Werner的配位化学理论,其要点有三:
§9-1 配位化学发展的简单历史
配位化学这门学科的诞生和发展是人类长期生产实践活动、逐步了解一些自然现象加入总结和发展的结果,很难准确地说何时第一次合成配合物,但或许最早记载是Prussian blue
。
它是1704年柏林的颜料技师Dissbach用动物的血、皮和草木灰在铁锅中强烈煮沸得到的(因氨基酸水解产生CN- K4Fe(CN)6黄血盐。它于Fe3+立即生成 Prussian blue)。它的这个成果被老板占有20年后才公布于世。但最早对配合物进行深入研究的是1798年发现的第一个化合物Co(NH3)6Cl3。1798年,德国分析化学家Tassert在研究HCl条件下用NaOH