Mybayes 构建系统发生树方法
贝叶斯网络构建算法

贝叶斯网络构建算法贝叶斯网络(Bayesian Network)是一种概率图模型,用于表示和推断变量之间的因果关系。
构建一个准确、有效的贝叶斯网络需要采用相应的构建算法。
本文将介绍几种常用的贝叶斯网络构建算法及其应用。
一、完全数据集算法完全数据集算法是贝叶斯网络构建中最简单、最常用的方法之一。
它假设已有一个完整的数据集,其中包含了所有要构建贝叶斯网络所需的信息。
该算法的主要步骤如下:1. 数据预处理:对数据进行清洗、归一化等预处理操作,确保数据的准确性和一致性。
2. 变量分析:根据数据集对变量之间的关系进行分析,确定要构建贝叶斯网络的变量。
3. 贝叶斯网络结构初始化:将变量之间的关系表示为图的结构,可以使用邻接矩阵或邻接链表等数据结构进行存储。
4. 结构学习:利用数据集中的频数统计等方法,通过学习训练数据集中的概率分布来确定贝叶斯网络结构中的参数。
5. 参数学习:在确定了贝叶斯网络结构后,进一步学习网络中各个变量之间的条件概率分布。
6. 结果评估:使用评估指标如准确率、精确率和召回率等来评估生成的贝叶斯网络模型的性能。
完全数据集算法的优点是能够利用完整数据构建准确的贝叶斯网络模型,但它的缺点是对于大规模的数据集,计算成本较高。
二、半监督学习算法半监督学习算法是一种使用有标记和无标记数据进行贝叶斯网络构建的方法。
这种方法可以在数据集不完整的情况下也能获得较好的贝叶斯网络模型。
以下是半监督学习算法的主要步骤:1. 数据预处理:对有标记和无标记数据进行预处理,清洗、归一化等操作。
2. 初始化:使用有标记数据初始化贝叶斯网络结构,可以采用完全数据集算法。
3. 标记传播:通过标记传播算法,将有标记数据的标签扩散到无标记数据中,这样可以在无需标记大量数据的情况下获得更多的有关因果关系的信息。
4. 参数学习:在获得了更多的有标记数据后,使用这些数据进行参数学习,并更新贝叶斯网络模型。
5. 结果评估:使用评估指标对生成的贝叶斯网络模型进行评估。
建系统发育树步骤
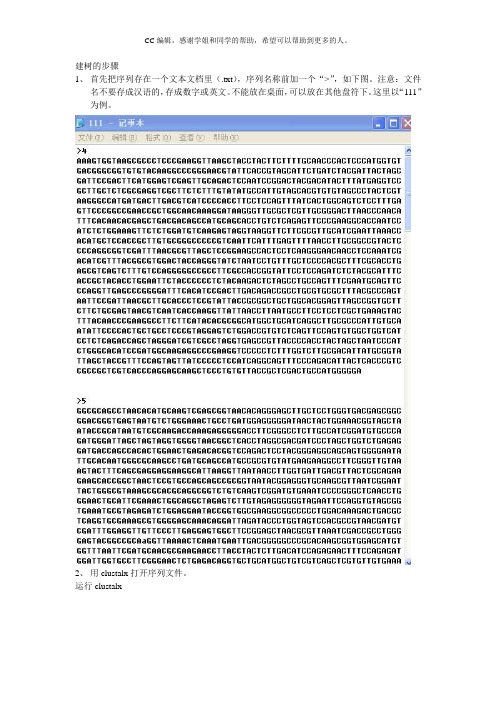
建树的步骤1、首先把序列存在一个文本文档里(.txt),序列名称前加一个“>”,如下图。
注意:文件名不要存成汉语的,存成数字或英文。
不能放在桌面,可以放在其他盘符下。
这里以“111”为例。
2、用clustalx打开序列文件。
运行clustalx点击“File”,选择Load Sequences,会弹出查找对话框,选择111所在的路径后打开。
如下图点击Aligment 选择Do Complete Aligment,选择输出文件路径,默认和111文本文档在同一个目录下。
点OK即可执行完Aligment后,序列被对齐(如果明显没有对齐,说明有反向或者互补序列,用DNAMAN或者DNASTAR进行序列的调整后再进行对齐),并产生两个文件“111.aln”“111.dnd”。
关闭clustalx。
运行MEGA点击File,选择convert to MEGA format,打开“111.aln”点OK 出现拉动滚动条到最后将#*都删去然后点工具栏的保存然后关闭两个窗口看到MEGA主界面选择找到MEGA文件格式文件 111点打开,出现下图我们选择第一个核酸序列点OK出现下列对话框是否是蛋白质编码的核酸序列我们点“NO”出现点击关闭,返回主窗口点击选择bootstrap test of tree 选择我们要建树的类型以NJ (neighbor-joining)树为例点可以修改参数,修改好后点compute计算机会自动测算,生成一个树如果对树不满意,可以用左侧的工具来修改CC编辑,感谢学姐和同学的帮助,希望可以帮助到更多的人。
点可以保存为MEGA 识别的文件,点击可以保存为其他格式。
贝叶斯法构建系统发育树

贝叶斯法构建系统发育树1.打开PAUP软件,打开目标文件和primates文件,将目标文件修改成primates文件格式。
2. 用modeltest3.7软件分析模型参数。
3. 打开mrbayes软件,文件输入。
命令:>execute 文件名.nex4. 设置参数,模型(上面modeltest3.7软件分析模型参数)。
命令:>lset nst=6/2 rates =gamma/invgamma/propinv,若要检查模型的参数,输入命令showmodel。
若设定lset nst=2,需输入命令report tratio=dirichlet。
3.1 >mcmc ngen=100000(1000000) (samplefreq=10(100)),注意:代数可以先设为10000,以便估计时间的长短。
>help mcmc来确认设置。
3.2 运行结束前,标准误差要小于0.01,否则增加代数,继续运行4.1 >sump burnin=250(2500);抽样的25%划为老化样本,舍去。
PSRF值需约等于1.0,否则要运行更长时间。
4.2 >sumt burnin=250(2500),输出所得的进化树,可用treeview打开.Modeltest 3.7基本操作步骤(中文)Moedltest是进行似然法计算必须的软件之一,它可以帮助大家为所获数据选择最佳的模型进行计算,得到最优的结果。
目前该软件的这里介绍一下Modeltest3.7的基本操作步骤:1. 下载Modeltest3.7软件和模型文件modelblockPAUPb10.txt;2. 将序列同源排序后保存为XXX.nex文件;全部拷贝到C盘。
3. 打开模型文件,将文件内容拷贝到XXX.nex文件的末尾,可以将该文件另存为XXX.test.model.nex,保留原来的*.nex文件;;4. 打开PAUP4.0应用程序,将XXX.test.model.nex文件拖入PAUP窗口,然后在命令行输入:execute XXX.test.model.nex,回车后PAUP就开始对数据进行模型估计,结果将保存为model.scores文件和modelfit两个文件,文件位于PAUP4.0软件的文件夹中;5. 将model.scores文件拷贝到Modeltest3.7.win.exe所在的文件夹中。
构建系统发生树的方法

构建系统发生树的方法构建系统发生树是一种对于系统进行分析和优化的有效方法,在实践中有许多种方法可以构建系统发生树。
以下是10条关于构建系统发生树的方法,并对每条方法进行详细描述。
1. 系统流程图系统流程图是一种常见的构建系统发生树的方法。
通过对系统的主要流程进行图形化的描述,可以更好地了解系统的组成部分以及它们之间的关系。
系统流程图往往是由开始和结束节点、处理节点和决策节点组成的。
前者用来表示系统的输入和输出,后者则用来表示系统的核心过程和逻辑判断。
2. 系统分层结构图系统分层结构图是将系统按照层次进行组织和描述的一种方法。
通过将系统分解为多个层次,并描述这些层次之间的关系,可以更好地了解系统的组成和结构。
这种方法通常用于处理大型和复杂的系统,能够帮助开发人员更好地管理和优化系统。
3. 系统模块图系统模块图是一种用于展示系统模块和它们之间关系的图形化表示方法。
系统模块图通常由多个模块和模块之间的输入和输出组成。
每个模块通常都对应一个特定的功能或业务逻辑。
通过了解系统中各个模块之间的关系和作用,可以更好地理解系统的架构和逻辑。
4. 系统数据流图系统数据流图是一种用来描述系统数据传输流程的图形化表示方法。
该方法通常由多个数据流和与这些数据流相关的处理过程组成。
每个数据流都对应一个特定的数据,而每个处理过程通常都包含两个或多个数据流。
通过了解系统中各个数据流之间的关系和流动过程,可以更好地理解系统的功能和性能。
5. 系统性能图系统性能图是一种用于展示系统性能指标和变化趋势的图形化表示方法。
该方法通常包括多个参数和变量,比如系统响应时间、吞吐量、并发数等。
通过了解系统性能参数的表现和变化趋势,可以更好地理解系统的性能瓶颈和瓶颈优化的方向。
6. 事件序列图事件序列图是一种用于展示系统中事件和处理过程之间关系的图形化表示方法。
该方法通常由一个或多个故障事件和与之相关的处理过程组成。
通过了解系统中各个事件和处理过程之间的关系,可以更好地了解系统的运行过程和故障排查过程。
重建系统发育树(PAUP的ML法和贝叶斯法)

重建系统发育树(PAUP的ML法和贝叶斯法)1 多重序列比对将待比对的序列以fasta格式保存,利用clustalx2.1或MEGA中的clustalW 软件进行多序列比对。2 保守区的选择将1得到的序列提交Gblock在线服务器(http://www.phylogeny.fr/one_task.cgi?task_type=gblocks),得到保守区的序列.fasta,并通过MEGA软件将其转换为.nex;3 核苷酸替换饱和度检测用DAMBE 软件验证替换饱和。只要比较ISS和ISS.c 值大小及显著与否,即可。当ISS小于ISS.c 且p=0.0000(极显著),就说明没序列替换未饱和,可以建树。4 核苷酸替换模型的选择在进行系统发育分析过程中,建树序列的进化模型选择是至关重要的一步,尤其对进化模型敏感的ML法和BI法。通过MrMTgui 软件选择核酸替代模型。4.1 安装PAUP、ModelTest (或MrModelTest) 软件,然后再安装MrMTgui 软件。配置MrMTgui,分别设置PAUP、ModelTest和MrModelTest路径。4.2 运行PAUP点击Run Paup,选择2中.nex文件。当模型参数值计算完毕,程序会提示是否立即启动分析,选择“否”,先保存scores文件。然后选择,运行MrModeltest,就得到模型数据了。一种是基于hLRT 标准选择的模型,另一种是基于AIC标准选择的模型,一般选择AIC标准。4.3添加模型参数,添加到建树的文件 .nex。5 使用PAUP软件重建ML树(运行时间较长)将用AIC标准选择的模型参数直接拷贝到Nexus文件的最后。参数设置:set criterion=likelihood 转化为似然法。outgroup 1 2 …….设定外类群bootstrap nreps=1000 keepall=yes brlens=yes 此命令设定循环次数为1000次(具体次数可根据实际情况自定),保存枝长。describetrees 1/plot=both brlens=yes 此命令设定了描述树的方式,即phylogram和cladogram均显示,显示枝长。最后用 savetrees from=1 to=1000 保存树。6 贝叶斯树6.1在Nexus文件的最后加入一个MrBayes block。(MEGA输出Nexus格式文件不能被Mrbayes识别,因此要进行修改)格式修改前:格式修改后:6.2运行mrbayes.exe,在命令行界面中输入转换或者修改的Nexus文件,点击回车,最后生成 *.tre,即最终的BI树。用Figtree 查看生成 .tre在运行1000代后都会显示 Average standard deviation of split frequencies。注:当这个值 < 0.01 时,说明两次运行的结果差异显著,Convergence 已经达到,这时可以输入 no 终止运行;这个值<0.05也可以,但不能>0.05。
贝叶斯法系统树

贝叶斯法系统树全文共四篇示例,供读者参考第一篇示例:贝叶斯法系统树(Bayesian System Tree,BST)是一种基于贝叶斯方法的机器学习模型,它结合了贝叶斯网络和决策树的优点,能够有效地处理复杂的分类和回归问题。
BST模型不仅具有较高的准确率和鲁棒性,还能够提供对模型推理过程的可解释性,因此在各个领域都取得了广泛的应用。
BST模型的核心思想是将贝叶斯网络和决策树进行结合,通过后验概率的计算来表示不同特征之间的关联性,并通过递归划分特征空间来构建树结构。
在进行预测时,BST模型会同时考虑先验知识和数据信息,从而得到更加精确和可靠的结果。
在构建贝叶斯法系统树模型时,首先需要确定节点的分裂准则。
一般而言,可以采用信息增益或基尼系数等指标来评估节点的分裂效果,从而选择最优的分裂点。
接着,需要确定每个叶子节点的概率分布,一般可以采用最大后验估计或极大似然估计等方法来估计参数值。
通过计算后验概率来选择最优的划分方式,从而得到最终的贝叶斯法系统树模型。
贝叶斯法系统树模型具有许多优点。
它具有较高的准确率和泛化能力,能够有效地处理复杂的分类和回归问题。
BST模型能够提供对模型推理过程的可解释性,使得用户可以清晰地了解模型的判断依据。
BST模型还可以处理缺失数据和噪声数据,具有较好的鲁棒性和稳定性。
在实际应用中,贝叶斯法系统树模型已经被广泛应用于各个领域。
在医疗领域,BST模型可以帮助医生进行疾病诊断和预测治疗效果;在金融领域,BST模型可以帮助分析师进行股票价格预测和风险评估;在智能驾驶领域,BST模型可以帮助自动驾驶汽车进行交通预测和路径规划。
第二篇示例:贝叶斯法系统树是一种基于贝叶斯理论的决策树算法,它将贝叶斯理论和系统树结合起来,能够有效地处理不确定性和推理问题。
这种算法在机器学习和数据挖掘领域有着广泛的应用,可以用于分类、回归和聚类等任务。
贝叶斯法系统树的基本原理是将特征空间划分成若干个区域,每个区域内的数据点都具有相同的特征。
贝叶斯法系统树

贝叶斯法系统树全文共四篇示例,供读者参考第一篇示例:贝叶斯法系统树的基本原理是基于贝叶斯定理,通过表示变量之间的依赖关系来建立模型。
在一个贝叶斯法系统树中,每个节点表示一个变量,每条边表示一个变量之间的依赖关系。
每个节点还包含一个概率分布,用于描述该节点给定其父节点的条件概率。
通过这种方式,可以构建一个树形结构,描述多个变量之间的复杂关系。
在进行推理或预测时,可以利用这个模型计算后验概率,从而得到最可能的结果。
贝叶斯法系统树在许多领域都有广泛的应用。
在医疗诊断领域,可以利用贝叶斯法系统树建立疾病诊断模型,从而帮助医生更准确地做出诊断。
在金融领域,可以利用贝叶斯法系统树进行信用评估,预测客户的信用评分和违约风险。
在自然语言处理领域,可以利用贝叶斯法系统树进行文本分类、情感分析等任务。
贝叶斯法系统树在处理不确定性信息和复杂关系时具有很大的优势,被广泛应用于各种领域。
贝叶斯法系统树的优点在于可以表示复杂的概率关系,处理不确定性信息,同时具有较强的推理和预测能力。
它可以利用概率分布描述变量的不确定性,通过条件概率计算后验概率,从而得到最可能的结果。
贝叶斯法系统树还具有参数少、计算简单、结构清晰等优点,适用于大规模数据和复杂模型。
贝叶斯法系统树也存在一些缺点,如模型训练过程需要大量数据、难以处理高维数据等。
在未来的研究中,需要进一步提高模型的效率和准确性,解决这些问题。
第二篇示例:贝叶斯法系统树(Bayesian network)是一种用概率和图形结构描述复杂系统的方法。
它是基于贝叶斯定理的一种概率图模型,能够对不同变量之间的关系进行建模,并利用条件概率来推断未知变量的概率分布。
贝叶斯法系统树最早由美国学者Pearl在1988年提出,是一种用来表示变量之间概率关系的有向无环图(DAG)。
在这种图中,节点表示随机变量,边表示变量之间的依赖关系。
每个节点都代表一个随机变量,它的父节点则表示影响该节点的因素。
贝叶斯法系统树通常由两部分组成:一个结构模型和一个概率模型。
系统发育树构建方法优劣

2.最大简约法最大简约法(maximum parsimony method,MP)最早是基于形态特征分类的需要发展起来的,具体的算法有许多不同版本,其中有些已被广泛地应用于分子进化研究中。
利用MP方法重建系统发生树,实际上是一个对给定OTUs其所有可能的树进行比较的过程。
对某一个可能的树,首先对每个位点祖先序列的核苷酸组成做出推断,然后统计每个位点用来阐明差异的核苷酸最小替换数目。
在整个树中,所有信息简约位点最小核苷酸替换数的总和称为树的长度(常青和周开亚,1998)。
MP法是一种优化标准,这种标准遵循“奥卡姆剃刀原则(Occam’S Razor principle)”:对数据最好的解释也是最简单的,而最简单的所需要的特别假定也最少。
MP法基于进化过程中所需核苷酸(或氨基酸)替代数目最少的假说,对所有可能正确的拓扑结构进行计算并挑选出所需替代数最小的拓扑结构作为最优系统树,也就是通过比较所有可能树,选择其中长度最小的树作为最终的系统发生树,即最大简约树(maximum parsimony tree)。
与其他建树方法相比,MP法无需引入处理核苷酸或者氨基酸替代时所必需的假设(替代模型)。
同时,MP法对于分析某些特殊的分子数据(如插入序列和插入/缺失)有用。
在分析的序列位点上没有回复突变或平行突变,且被检验的序列位点数很大的时候,MP法能够获得正确的(真实)系统树。
但MP法推导的树不是唯一的,在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会出现建树错误。
故MP法适用于序列残基差别小,具有近似变异率,包含信息位点比较多的长序列。
3.最大似然法最大似然法(maximum likelihood method,MI。
)是20世纪60年代末期由于对地生物信息学分析实践震波和水声信号等处理的需要而发展起来的一种非线性谱估计方法。
最早由凯佩用这种方法对空间阵列接收信号进行频率波数谱估值,后来推广到对时问信号序列的功率谱估值。
构建系统发育树的三大方法

构建系统发育树的三大方法
1、距离法:基于距离的方法,首先通过各个物种之间的比较,根
据一定的假设(进化距离模型)推导得出分类群之间的进化距离,构建一个进化距离矩阵。
进化树的构建则是基于这个矩阵中的进化距离关系。
2、特征法:基于特征的方法,不计算序列间的距离,而是将序列
中有差异的位点作为单独的特征,并根据这些特征来建树。
3、简约法:基于简约的方法,通过构建一棵由所有可能的子树组
成的树,然后从这个树中选择一个最优的子树作为进化树。
系统发育树的详细构建方法

构建系统发育树需要注意的几个问题1 相似与同源的区别:只有当序列是从一个祖先进化分歧而来时,它们才是同源的。
2 序列和片段可能会彼此相似,但是有些相似却不是因为进化关系或者生物学功能相近的缘故,序列组成特异或者含有片段重复也许是最明显的例子;再就是非特异性序列相似。
3 系统发育树法:物种间的相似性和差异性可以被用来推断进化关系。
4 自然界中的分类系统是武断的,也就是说,没有一个标准的差异衡量方法来定义种、属、科或者目。
5 枝长可以用来表示类间的真实进化距离。
6 重要的是理解系统发育分析中的计算能力的限制。
任何构树的实验目的基本上就是从许多不正确的树中挑选正确的树。
7 没有一种方法能够保证一颗系统发育树一定代表了真实进化途径。
然而,有些方法可以检测系统发育树检测的可靠性。
第一,如果用不同方法构建树能得到同样的结果,这可以很好的证明该树是可信的;第二,数据可以被重新取样(bootstrap),来检测他们统计上的重要性。
分子进化研究的基本方法对于进化研究,主要通过构建系统发育过程有助于通过物种间隐含的种系关系揭示进化动力的实质。
表型的(phenetic)和遗传的(cladistic)数据有着明显差异。
Sneath和Sokal(1973)将表型性关系定义为根据物体一组表型性状所获得的相似性,而遗传性关系含有祖先的信息,因而可用于研究进化的途径。
这两种关系可用于系统进化树(phylogenetictree)或树状图(dendrogram)来表示。
表型分枝图(phenogram)和进化分枝图(cladogram)两个术语已用于表示分别根据表型性的和遗传性的关系所建立的关系树。
进化分枝图可以显示事件或类群间的进化时间,而表型分枝图则不需要时间概念。
文献中,更多地是使用“系统进化树”一词来表示进化的途径,另外还有系统发育树、物种树(species tree)、基因树等等一些相同或含义略有差异的名称。
系统进化树分有根(rooted)和无根(unrooted)树。
系统发育树构建的三种方法

系统发育树构建的三种方法
系统发育树(Systems 发育 Tree,简称Stree)是一种用于描述生物系统进化的图形化工具,通常用于模拟生物系统行为的演化过程。
以下是三种构建系统发育树的方法:
1. 基于规则的方法:这种方法使用预定义的规则和偏好来构建
系统发育树。
例如,可以使用遗传算法或人工神经网络等机器学习方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法需要大量
的人工工作,但可以生成较为准确的演化树。
2. 基于统计方法的方法:这种方法使用统计学方法来推断物种
之间的演化关系。
例如,可以使用最大似然估计或贝叶斯推断等方法,来预测一个物种的遗传特征或行为演化轨迹。
这种方法不需要人工工作,但需要更多的计算资源和时间,才能得到比较准确的演化树。
3. 基于模型的方法:这种方法使用已经建立的模型和数据来构
建系统发育树。
例如,可以使用层次结构模型(如生物进化树、社会网络模型等)来预测一个物种的遗传特征或行为演化轨迹。
这种方法可
以快速构建系统发育树,但需要更多的人工工作来验证模型的准确性。
使用贝叶斯方法构建系统发育树mrbayes

使用贝叶斯方法构建系统发育树—MrBayesmrBayes需要的比对文件格式为:nex,可以在比对是选择输出此种文件格式mtBayes可以在命令提示符里面运行在CMD里面输入mrBayes,出现如下界面在界面内输入exe file(或者execute file,其中file为序列文件名),得到如下界面如果没有错误,则说明数据文件格式是正确的。
设置替换模型参数可以使用help lset查看lset设置的参数Nucmodel: 指的是核酸的类型。
4by4指的是不区分序列上的位点。
而codon指的是使用密码子模型。
这时序列上每个位点的替换速率会根据密码子模型来推断。
Doublet通常用于具有协同进化效应的序列。
一般情况下可以使用4by4,如果是编码序列的话,最好使用codon Nst:核酸替换模型。
1 是JC69模型,即单参数模型。
2为F81模型。
6为GTR模型。
在mrBayes 中,可以尝试分别使用三个模型运行,以选择最优的结果。
Code: 指的是密码子编码的规律。
Universal指的是通用密码子使用规律。
如果是推测线粒体内的基因,需要使用Metmt,叶绿体则需要使用Mycoplasma Ploidy: 物种是单倍体还是二倍体。
Rates:指定序列上每个位点的替换速率。
Equal表示替换速率都是一致的。
Gamma表示用gamma来确定序列上的替换速率。
Ngammacat:配合上面的参数,如果替换速率设置为Gamma、Invgamma、Adgamma,则需要设置此选项。
Nbetacat:同上。
使用lset Nst=6 Rate=gamma 类似命令设置参数。
设置模型的相关先验信息使用help prset查看相关参数及其说明一般情况下,需要关注的参数有:Tratiopr:指定转换和颠换的比例。
可以使用fixed指定,也可以使用beta分布来模拟产生。
Revmatpr:指定GTR模型里面替换速率的先验分布。
课件:用实例演示最大简约法构建系统发生树

d3 G
树1
A
G
d2 A
d4 G
d1 A
树2
G
d3 G
d2 A G
d4 G
d1 A
树3
G
d4 G
d2 A G
d3 G
第2列与第1列情况一样 第3列各碱基在3种进化树中的替换情况如下:P247
d1 C
树1
T
d2 T
d3 C T
d4 Td1 CFra bibliotek树2C
d3 C
d2 T T
d4 T
d1 C
树3
T
d4 T
d2 T T
用实例演示最大简约法构建系 统发生树
生技第五组 蔡苑颖 陈晓兰 陈观芝 张满桥 张
明红 陆丽平
系统进化树:用来表示被认为具有共同祖先
的各物种间演化关系的树。是一种亲缘分支 分类方法。在树中,每个节点代表其各分支 的最近共同祖先,而节点间的线段长度对应 演化距离(如估计的演化时间等)
最大简约法:根据信息位点提供的个序列间 的替换情况,在所有可能的树中找出替换数 最小的树的方法。
d3 C
④ 找出替换总数最小的数
树1
树2
树3
第1列替换数
1
2
2
第2列替换数
1
2
2
第3列替换数
2
1
2
替换总数
4
5
6
因为树1的替换总数最小,因此取树1作为这4个序列的进化树
信息位点:指能把所有可能的树区别出来的 位点,即核苷酸序列存在差异的点
步骤:序列比对 写出所有可能的树
分析信息位点 将每棵树信息位点 上的字符替换数相加,找出总替换数最小的 树
贝叶斯法建树原理

贝叶斯法建树原理
贝叶斯法建树是一种有效的决策树学习算法。
它的基本思想是利用贝叶斯规则,使用数据训练出树模型。
贝叶斯决策树主要由两个部分组成:训练过程和预测过程。
在贝叶斯决策树的训练过程中,每一个内部节点都有一个变量,它的值决定了该节点的分支点。
每个分支都具有一组可能的值,即每个节点的“描述器”。
在训练数据中,我们会遍历每一个样本,收集做出决策的各种值作为特征,这些值会用来建立树枝上的结点。
具体而言,特征值在贝叶斯规则中被额外赋予了一种权重。
这个子集中的特征值包含在贝叶斯公式中,用于决定分类的最优属性。
根据贝叶斯规则,从平均概率的角度来看,每一个分类都有一个关联的概率。
如果一个分类的概率大于其他分类,那么就说明当前样本多倾向于这个分类,最终就会落在这个分类下的叶子节点上。
而这个过程也就是贝叶斯法建树的训练过程。
贝叶斯法建树的预测过程和训练过程类似,只是没有训练数据。
我们只需要给定测试样本,收集其中的特征,然后根据贝叶斯规则在每个分支上计算出关联的概率,选出概率最大的一个值,即可决定该样本最终分类的结果。
贝叶斯法建树有很多优点,比如分类准确率高、处理特征值不稳定的数据变得简单等。
贝叶斯法建树能够准确有效地学习得到高质量的决策树模型,因此在数据挖掘、机器学习等领域有着广泛的应用。
三种方法构建系统发育树学习笔记

三种方法构建系统发育树学习笔记所用数据为一个属内不同种不同群体的叶绿体基因组序列,数量为80条。
发现用全长序列建树的时候,不适合选用太多外类群,否则ML法中会导致属内分枝的枝长特别短。
原因应该是基因间隔区和内含子区域序列位点的差异较大。
枝长含义NJ:表示遗传距离;MP:性状状态变换的替换数;ML/BI:该分枝上的相对进化数量(遗传变异量);每个位点上的替换数(一般以每位点多少次核苷酸替换或氨基酸取代来表示)。
遗传距离大多数情况以序列来说遗传距离就是两个OTU(个体、群体、物种或基因家族)之间序列的差异值。
序列比对多序列比对用mafft得到的结果较为准确,muscle比对的速度较快。
多序列比对的绝大多数算法都是基于渐进比对的概念。
简单来说就是先从两个序列的比对开始,逐渐添加新序列,直到所有的序列都加入为止。
但是不同的添加顺序会产生不同的比对结果。
所以由最相似的两个序列开始比对,由近到远逐步完成最为可靠。
mafft --thread 15 --auto 80-AcoeOut.fasta > 80-AcoeOut_aln.fasta##比对时如果不清楚什么参数合适,加个参数--auto,软件可以自动帮你处理挑选保守位点进行下一步建树序列比对完后,用于建树的序列位点必须保证具有良好的同源性。
所以需要删除序列分歧很大的区域和gap区域。
我用的软件为Gblocks,主要目的是把有gap的位点全部去除,参数为-b5=n,其余的选项有-b5=h,h表示half 指去除在大于50%的序列中出现gap的位点。
Gblocks 80-AcoeOut_aln.fasta -t=d -b5=n最大简约法(软件PAUP)最大简约法的树长指所有性状在一棵树上的进化改变总数。
计算得到的结果可能会有许多树长相等的简约树,此时需要计算它们的一致树。
分为strict consensus和semistrict consensus等,strict表示100%,在所有简约树中都出现的分枝,才会出现在一致树中,否则为梳子。
回顾系统进化树构建的常见方法

回顾系统进化树构建的常见方法构建进化树的方法主要分为1.距离矩阵法(含 UPGMA、ME、NJ等)2.最大简约法(MP)3.极大似然法(ML)4.贝叶斯法(Bayesian)基于距离的进化树构建方法常用的距离法构建系统树:1.不加权算术平均对方法(Unweighted Pair-Group Method using Arithmetic average, UPGMA)2.最小进化距离法(Mnimal Evolution Method)3.邻接法(Neighbor-Joining Method, NJ)这一系列方法主要考量参数是:1.如何计算距离,节点间的距离;cluster之间的距离;校正的距离2.如何聚类?UPGMA 法本质上是“自下而上”或者说“聚合”的层次聚类(hclust)法,且距离的计算采用“平均距离法”。
一般绘制热图,常见的表达模式聚类方法也是 hclust,往往默认“最长距离法”。
两者只是cluster之间距离的计算方式不同。
当所有分支的突变率相同,UPGMA效果较好。
最小进化法(ME)寻找某一进化树的拓扑结构,使得全树枝长总和最短。
逻辑上需要对每一个拓扑结构进行评估,当序列增加时,计算量暴增。
这个与后续提到的最大简约法(MP)的最大区别是:(1)ME 法直接基于一个距离矩阵,计算的是最终进化树枝长总和最小;(2)MP法直接基于序列,计算的最终是在当前拓扑结构下,所有序列需要发生突变的位点的总和最少。
邻接法(NJ)与UPGMA几乎相反,UPGMA约等于层次聚类的聚合法;而NJ 法从拓扑结构的变化上来看,与层次聚类的分离法比较像。
当然还是有比较大的区别。
层次聚类的分离实现考量的是分离后两个cluster的内部距离总和最小。
NJ法考量的是分离出来的两个leaf node的校正距离最小。
这一校正距离综合考量了每个leaf node以及cluster的距离。
在距离计算上的实现,逻辑上比层次聚类的分离法要简单一些。
4.4系统发生树的构建

《生物信息学》第四章:分子进化与系统发生系统发生树的构建构建系统发生树的方法很多。
最常用的有基于距离的构建方法,包括非加权分组平均法(Unweighted Pair Group Method with Arithmetic mean,UPGMA),最近邻居法(Neighbor- Joining method,NJ),最小二乘法(Generalized Least Squares,GLS)等。
还有最大简约法(Maximum Parsimony,MP),最大似然法(Maximum Likelihood,ML),贝叶斯推断法(Bayesian Inference,BI)等。
我们并不打算一一讲解这些方法的具体原理。
你只要掌握何时选用何种方法就可以了,算法的事自由软件替你搞定。
从计算速度来看,最快的是基于距离的方法,几十条序列几秒钟即可完成。
其次是最大简约法。
最大似然法就要慢得多。
最慢的是贝叶斯法。
但是从计算准确度来看,算得最慢的贝叶斯法确是最准确,而算得最快的基于距离法结果确是最粗糙。
从实用的角度,建议使用最大似然法。
因为这种方法无论从速度还是准确度都比较适中。
最近邻居法虽然算得快,但是当序列多,彼此差别小的时候,这种方法不适合。
最大简约法,似乎是个掉空里的方法,高不成低不就,所以很少有人使用。
贝叶斯法不是所有的建树软件都提供,算法开发上还有待提高,而且计算时间过长。
目前流行的建树软件(表1),比如PHILIP和MEGA,基本能够包括上述所有算法。
如果想要构建ML树,也可以尝试专门构建ML树的PHYML。
贝叶斯的算法以MrBayes为代表,只是计算速度比较慢。
如果构建的系统发生树要用于发表生物信息学领域的文章,需要两种以上的构建方法锁定同一个结果才能审稿通过。
如果是用于发表以生物实验为主的文章用一种构建方法就可以了。
虽然软件可以快速自动地完成系统发生树的构建,但是对于基本算法的了解还是必不可少的。
以非加权分组平均法(UPGMA法)为例,介绍如何通过计算所有序列两两间的距离,再根据距离远近构建系统发生树。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
简单步骤
1 序列的比对,然后将比对好的序列转化成.nex格式
2 运行MrBayes,简单步骤如下:(依次输入命令,完成简单也最常用的分析):Execute filename.nex,打开待分析文件,文件必须和mrbayes程序在同一目录下。
Lset nst=6 rates=invgamma,该命令设置进化模型为with gamma-distributed rate variation across sites和a proportion of invariable sites的GTR模型。
模型可根据需要更改,不过一般无须更改。
3 mcmc ngen=10000 samplefreq=10,保证在后面的可能性分布中probability distribution至少取到1000个样品。
默认取样频率:every 100th generation。
4 如果分裂频率分支频率split frequencies的标准偏差standard deviation在100,000代generations以后低于0.01,当程序询问:“Continue the analysis?(yes/no)”,回答no;如果高于0.01,yes继续直到该值低于0.01。
5 sump burnin=250(在此为1000个样品,即任何相当于你取样的25%的值),参数总结summarize the parameter,程序会输出一个关于样品(sample)的替代模型参数的总结表,包括mean,mode和95 % credibility interval of each parameter,要保证所有参数PSRF(the potential scale reduction factor)的值接近1.0,如果不接近,分析时间要延长。
6 sumt burnin=250,总结树summarize tree。
程序会输出一个具有每一个分支的posterior probabilities的树以及一个具有平均枝长mean branch lengths的树。
这些树会被保存在一个可以由treeview等读取的树文件中。