如何处理无序(晶体解析)
Olex2单晶的可视化结构解析与精修

单晶的可视化结构解析与精修之Olex2软件的使用目录1.1 Olex2简介1.2关于Olex2的安装、引用和更新1.3 Olex2的图形化界面1.4 文件的建立第2章晶体解析和精修2.1 前言2.2 打开结构2.3 解粗结构2.4 原子指认2.5 各项异性精修2.6 加氢2.7 精修权重2.8 历史记录2.9 结构整理原子重命名排序2.10其他问题第3章无序处理3.1 无序处理方法3.2 无序类型及实例第4章结构验证及画图4.1 完善CIF信息生成CIF文件4.2 结构验证4.3 生成图像第5章总论5.1 空间群转换5.2 如何用olex2画叠合图5.3 解析的合理性5.4 绝对构型的问题5.5 解析实例第1章Olex2介绍1.1 Olex2简介Olex2是由英国杜伦大学化学系Dolomanov教授开发的一款具有解析、精修、画图等多功能的可视化单晶解析软件。
软件基于Python语言,现在已更新至1.2.7版本。
Olex2具有美观的图形界面,可以用鼠标操作,使用方便快捷。
而Shelxtl 大多数时候只有一个黑洞洞的屏幕,且需要使用键盘输入命令,略繁琐。
Olex2具有方便的数据回滚功能,当进行多次尝试时可以直接回滚,无需手动保存。
Olex2扩展性强,可以方便调用多种解析和精修软件,而且可以直接调用platon。
Shelxtl只能使用自带的XS和XL软件。
Olex2自带多种实用工具,如solvent mask和twinning等。
Shelxtl所带的工具较少,仅有Xprep。
Olex2是一个免费且不断更新的程序。
本教程意在使大家熟悉Olex2视图画界面并使用该程序。
1.2关于Olex2的安装、引用和更新1.2.1安装首先需要到网站(这个网站现在需要注册)上下载Olex2的最新版本,推荐下载绿色版本,直接解压缩就可以使用,且32位系统下载32位版本,64位版本下载64位版本。
然后将压缩包解压,放置在一个没有中文路径名的文件夹里,如D:\c\olex2-win64中。
Olex2单晶的可视化结构解析与精修

单晶的可视化结构解析与精修之Olex2软件的使用目录第1章前言1.1 Olex2简介1.2关于Olex2的安装、引用和更新1.3 Olex2的图形化界面1.4 文件的建立第2章晶体解析和精修2.1 前言2.2 打开结构2.3 解粗结构2.4 原子指认2.5 各项异性精修2.6 加氢2.7 精修权重2.8 历史记录2.9 结构整理原子重命名排序2.10其他问题第3章无序处理3.1 无序处理方法3.2 无序类型及实例第4章结构验证及画图4.1 完善CIF信息生成CIF文件4.2 结构验证4.3 生成图像第5章总论5.1 空间群转换5.2 如何用olex2画叠合图5.3 解析的合理性5.4 绝对构型的问题5.5 解析实例第1章Olex2介绍1.1 Olex2简介Olex2是由英国杜伦大学化学系Dolomanov教授开发的一款具有解析、精修、画图等多功能的可视化单晶解析软件。
软件基于Python语言,现在已更新至1.2.7版本。
Olex2具有美观的图形界面,可以用鼠标操作,使用方便快捷。
而Shelxtl 大多数时候只有一个黑洞洞的屏幕,且需要使用键盘输入命令,略繁琐。
Olex2具有方便的数据回滚功能,当进行多次尝试时可以直接回滚,无需手动保存。
Olex2扩展性强,可以方便调用多种解析和精修软件,而且可以直接调用platon。
Shelxtl只能使用自带的XS和XL软件。
Olex2自带多种实用工具,如solvent mask和twinning等。
Shelxtl所带的工具较少,仅有Xprep。
Olex2是一个免费且不断更新的程序。
本教程意在使大家熟悉Olex2视图画界面并使用该程序。
1.2关于Olex2的安装、引用和更新1.2.1安装首先需要到网站(这个网站现在需要注册)上下载Olex2的最新版本,推荐下载绿色版本,直接解压缩就可以使用,且32位系统下载32位版本,64位版本下载64位版本。
然后将压缩包解压,放置在一个没有中文路径名的文件夹里,如D:\c\olex2-win64中。
单晶结构解析技巧

单晶结构解析技巧1. 通常,H原子的处理方法作者要给出:(1)一般通过理论加H,其温度因子为固定值,可通过INS等文件查看(2) 水分子上H原子可通过Fourier syntheses得到(3)检查理论加上的H原子是否正确,主要看H原子的方向。
若不正确则删去再通过Fourier syntheses合成得到(4) 检查H原子的键长、键角、温度因子等参数是否正常。
通过检查分子间或分子内的H键是否合理最易看出H键的合理性(5) 技巧:有时通过Fourier syntheses得到的H原子是正确的,可一计算其温度因子等参就变得不正常,则可以固定其参数后再精修(如在INS中的该H原子前用afix 1,其后加afix 0)(6) 各位来说说方法与心得?2.胡老师,下面的问题怎么解决啊?谢谢您。
220_ALERT_2_B Large Non-Solvent C Ueq(max)/Ueq(min) ... 3.70 Ratio222_ALERT_3_B Large Non-Solvent H Ueq(max)/Ueq(min) ... 4.97 Ratio342_ALERT_3_B Low Bond Precision on C-C bonds (x 1000) Ang (49)B 级提示当然得重视了。
建议你先把H撤消,精修到C的热椭球不太变形和键长趋正常。
如做不到就要看空间群?衍射点变量比太小?以至追查到原始数据的录取参数和处理等。
这些粗略意见仅供参考,如何?3.在XP中画图时,只有一部分,想长出另外的对称部分。
我是envi完了,然后sgen长出来的,可是和symm显示的对称信息不一样。
比如:我根据envi的结果用sgen O1 4555得到的是O1A而不是O1D,这跟文献中标注的不一样啊,怎么统一呢?很困扰,忘达人指教。
xp里是按顺序编号的,第一个sgen出的的统一为A,依次标号。
你如果想一开始就统一D的话,重新name一下4.高氯酸根怎么精修呀?我用的SHETXL6.1版的,最好告诉我怎么用其中的XSHELL来做,我觉得他好用!Method 1DFIXDfix 1.42 0.02 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Dfix 1.42 0.02 O1 O2 O1 O3 O1 O4 O2 O3O2 O4O3 O4Method 2SADISadi 0.01 Cl1 O1 Cl1 O2 Cl1 O3 Cl1 O4Sadi 0.01 O1 O2 O1 O3 O1 O4 O2 O3 O2 O4 O3 O45. 晶体的无序是怎么造成的呀,是晶体培养的问题吗?如果无序太多,在解单晶的时候怎么办?我指的是很多的点,没有结构,他们的峰值都大于了0.5大于0.5没什么的,解完后都在1以下就可以了。
晶体结构无序

晶体结构无序引言晶体是由原子、离子或分子按照一定的规则排列而形成的固体物质。
晶体结构是指晶体中原子、离子或分子的排列方式和规律。
在大多数情况下,晶体结构都具有高度的有序性,即原子、离子或分子按照固定的空间位置和方向排列。
然而,在某些特殊情况下,晶体结构可能呈现出一定程度的无序性。
无序晶体结构的定义无序晶体结构是指晶体中原子、离子或分子的排列方式缺乏明显规律,呈现出随机性和混乱性。
这种无序性可以在不同尺度上存在,包括局部无序和全局无序。
局部无序局部无序是指在晶体中存在着一些小区域,其中原子、离子或分子的排列方式没有明显规律。
这些小区域可以是缺陷、杂质或其他形成无序结构的因素导致的。
局部无序可以通过X射线衍射、电镜等实验技术来观察和表征。
全局无序全局无序是指整个晶体结构中原子、离子或分子的排列方式没有明显规律,呈现出随机性和混乱性。
全局无序的晶体结构通常由于温度升高、压力变化或化学反应等因素导致。
全局无序可以通过热力学模拟、分子动力学模拟等计算方法来研究和描述。
无序晶体结构的特点无序晶体结构具有以下几个特点:1.非周期性:与有序晶体结构不同,无序晶体结构缺乏明确的周期性重复单元。
2.随机性:无序晶体结构中原子、离子或分子的排列方式是随机的,没有明显的规律可循。
3.多样性:由于无序晶体结构缺乏规则和限制,因此可以存在多种不同类型的无序结构。
4.动态性:无序晶体结构通常具有较高的热运动,原子、离子或分子会不断发生位置变化。
无序晶体结构的形成机制无序晶体结构可以通过多种方式形成,以下是一些常见的形成机制:1.温度升高:当温度升高到一定程度时,晶体中的原子、离子或分子会发生较大的热运动,导致结构的无序化。
2.压力变化:压力的改变可以导致晶体结构发生相变,从有序结构转变为无序结构。
3.化学反应:某些化学反应可以破坏晶体中的有序排列,形成无序晶体结构。
4.外界扰动:如辐射、机械振动等外界扰动也可以导致晶体结构发生无序化。
无序结构及其精修-1

无序结构及其精修-1以下内容整理自《单晶结构分析原理与实践》第二版当一种物质大部分结构长程有序(long-range ordered),仅有小部分结构无序(disordered)时,其晶体结构可以用通常的方法进行解析。
但是,无序部分可能出现原子位移参数太大,其结构在化学上不合理的情况。
无序的种类有多种,这里只讨论常见的情况。
1. 位置占有率无序一种比较常见的情况是,晶体中某些位置同时被多种原子占据。
例如在一些矿物或一些金属掺杂无机材料中经常出现某个位置被两种(或两种以上)半径相似的原子以统计分布、无序的方式占据。
假定同时占据同一位置的原子有两种,可以在结构模型中同一位置同时设置这两种原子,并使两者的位移参数相同。
这时,其中一种原子的位置占有率(site occupancy)定k,另一种为(1–k)。
通过精修k值,得出两种原子各自在该位置的实际占有率,这样就解决了这种位置占有率无序(site occupancy disorder)的问题。
位置占有率无序还可以表现为原子(或基团)在某一位置上的位置占有率小于1,即该位置在整个空间中,只有部分被这个原子(或基团)占据,而另一部分是空的。
这种情况常见于晶体中的结晶水分子和溶剂分子等,处理起来也很简单,只要设置该占有率在结构精修过程中浮动,就可以达到最优化。
当然,最好有其它分析数据,如元素分析等,以便对照和确定这些无序结构的位置占有率,保证占有率数据的准确性。
2. 位置无序和取向无序如果晶体中分子的部分基团、甚至整个分子按统计方式分布在两个或者更多的位置上,就叫位置无序(positional disorder)。
取向无序(orientation disorder)则是指部分基团、甚至整个分子位于旋转轴、镜面或对称中心上,分布在两个相互联系的方向上。
取向无序的基团或分子的两种取向往往有一个共同的重心,在含有球形基团的结构中比较常见。
这些球形基团包括NH4+, ClO4–, BF4–, PF6–, CCl4, NEt4+, SO42–等等,也常见于近似球形的基团,如-CMe3, ClO3–, NO3–, PR3, CHCl3等等。
晶体结构无序

晶体结构无序晶体结构无序是什么意思?晶体结构无序是指晶体中原子或离子排列无规则或有缺陷。
具有无序晶体结构的物质通常具有一些特殊性质,例如强磁性和导电性。
无序晶体结构的形成可以有多种原因。
例如,在晶体生长过程中,由于外部条件的变化或晶体体系的非稳定性,晶体中的原子或离子排列可能会发生变化,从而导致晶体结构无序。
此外,在化学反应中,也有可能发生原子间跃迁,进而打破原本有序的晶体结构。
然而,许多无序晶体具有重要的应用价值。
例如,无序硬磁体可以被用作电动机等电力设备的永久磁性材料;无序导电材料可应用于薄膜晶体管等电子器件。
因此,无序晶体结构的研究与应用具有广泛的前景。
无序晶体结构的研究由于无序晶体结构具有高度复杂性和多样性,研究其结构与性质具有一定的难度。
历史上,人们曾经通过X射线衍射等手段对晶体结构进行分析,但有序晶体结构直接出现的特征谱线在无序晶体结构中通常不可观测到,因此无序晶体结构的解析常常需要借助于高分辨率的电镜等先进技术。
在研究无序晶体结构的过程中,人们还通常会运用计算晶体学的方法。
通过现代计算机技术,科学家们可以模拟各种晶体结构的稳定性与物理性质,并且还可以探究无序晶体的熵、热容等重要参数。
这些模拟计算为进一步研究无序晶体结构的物理和化学行为提供了有力的工具。
无序晶体结构的应用无序晶体结构意味着物质的性质不同寻常。
通过研究无序晶体结构,人们可以获取更深入的了解原子之间的相互作用、能量转移和传递等动力学过程,从而在材料科学、物理学和化学等领域中开发出更加先进的应用。
对于无序磁体,它们具有超强的磁性能力,常常被应用于电力设备的永久磁性材料。
而对于无序导电材料来说,其高导率和热稳定性等特性,可以用于制备微处理器正交编码器芯片、高能量密度电池等电子器件以及工业雷达和通讯设备等应用。
此外,在生物学和医药领域,无序晶体结构也具有重要意义。
例如,许多生物分子、当阻塞药物等都可能形成无序凝聚态,进而导致疾病。
如何处理无序晶体解析 (2)

11
Disorder of 15-crown-5: an odd number of atoms packing in a over a center-of-inversion
O OO
OO
1 Use a ‘PART –1’ command to suppress the calculation of bonds/angles. 2 Assign ½ occupancies to all atoms. 3 DFIX C–C = 1.50, C–O = 1.41, C…O = C…C = 2.37 Å.
Because total charge is 8, there are should be 1 Cu(II) and 2 Fe(III) for the 3 sites.
Refine each site as ⅓Cu⅔Fe.
F 3C
CF3
OO
O
O
THF
THF
Or, use SUMP to tie the 3 Cu atoms
如何处理无序晶体解析晶体管剧情解析晶体结构解析蛋白质晶体结构解析晶体解析软件数字图像处理疑难解析晶体契约如何设置晶体如何判断java事务处理全解析晶体管
Some Strategies for the Refinement of Disordered Crystal Structures
Ng Seik Weng
Rotation axis through the mid-points of the 1,2 and 4,5 carbon atoms.
Asymmetric unit has 2 aromatic and 1 carboxyl carbon atoms, and 1 oxygen atom only. High R, which is an artifact of the disorder and not because the data are bad.
数据检查cif文件时常见问题及解决办法一、需要修改参数和添加命令

数据检查cif文件时常见问题及解决办法一、需要修改参数和添加命令进行精修的问题1、Mu(calc) and Mu(CIF) Ratio Differs from 1.0 by . 77.73 %问题:理论计算的单胞线性吸收系数与cif文件中不一致2、Check R eported M olecular Weight ................ 78.48问题:分子式不正确。
3、Reported F000 D iffers f r om C alcd (o r M issing)... ? Check问题:单胞中的电子数不正确。
解决方法:根据空间群修改res文件中晶胞内原子个数及Z值。
4、Minimum Crystal Dimension M issing (o r E rror) ... ? D o !5、Medium Crystal D imension M issing (o r E rror) ... ? D o !6、Maximum Crystal D imension M issing (o r E rror) ... ? D o !问题:Cif文件中没有晶体尺寸。
解决办法:在res文件中加入SIZE命令和晶体尺寸进行精修。
7、CIF Contains no X-H Bonds ...................... ? C heck8、CIF Contains no X-Y-H or H-Y-H Angles .......... ? C heck问题:cif文件中没有与氢原子有关的键长和键角。
解决办法:在res文件中添加BOND $H 和CONF 命令进行精修。
9、Centre of G ravity not Within U nit Cell: R esd. # 1问题:分子不在晶胞内。
解决办法:将分子移到晶胞内进行精修。
10、The absolute value of parameter shift to su ratio > 0.2011、Absolute value of the parameter shift to su ratio given 5.41312、Additional refinement cycles may be required.Maximum Shift/Error ............................ 5.41问题:参与精修的参数没有收敛。
单晶结构解析技巧

(5) 技巧:有时通过Fourier syntheses得到的H原子是正确的,可一计算其温度因子等参就变得不正常,则可以固定其参数后再精修(如在INS中的该H原子前用afix 1,其后加afix 0)
反过来讲如果一个结构报告把 H参数都准确列出,我们可以认定这是一篇高水平的研究。
理论加 H是基于分子几何构型指定 H的辅助方法,水和甲基等等都不是它可应付得来的。
看来是介绍能量优化理论计算来指定 H的时候了,将请国武老师贴出两篇好文章供分享。
有关的计算程序已在论文中列出并可在网上下载,希望这种“理论加氢”方法得以推广。
如果无序太多,在解单晶的时候怎么办?我指的是很多的点,没有结构,他们的峰值都大于了0.5
大于0.5没什么的,解完后都在1以下就可以了。特殊的比较大的在重原子附近也没有关系
5.
比较确切的定义是单胞中你测定的或你设想的“化学式”的数目。
在分子晶体中,Z 是分子数,在其它各类晶体中则为化学式个数。
2)氢键,水分子的H应位于能形成合适的氢键位置上,而不是随意的位置。
基于以上两点考虑,在用SHELXTL程序精修时,在主体骨架都确定之后把残余峰的数量改为50,甚至更大(PLAN 50),然后在O周围的残余峰中仔细辨认,把位置合适的残余峰定为H。
从残余峰中得到H原子,键长一般不是理想的键长,而且位置在经修过程可能会发生改变,为了解决这些问题,我们可以这样来做。
(6)各位来说说方法与心得?
2.
胡老师,下面的问题怎么解决啊?谢谢您。
220_ALERT_2_B Large Non-Solvent C Ueq(max)/Ueq(min) ... 3.70 Ratio
萘环-穿过对称元素的无序处理实例1
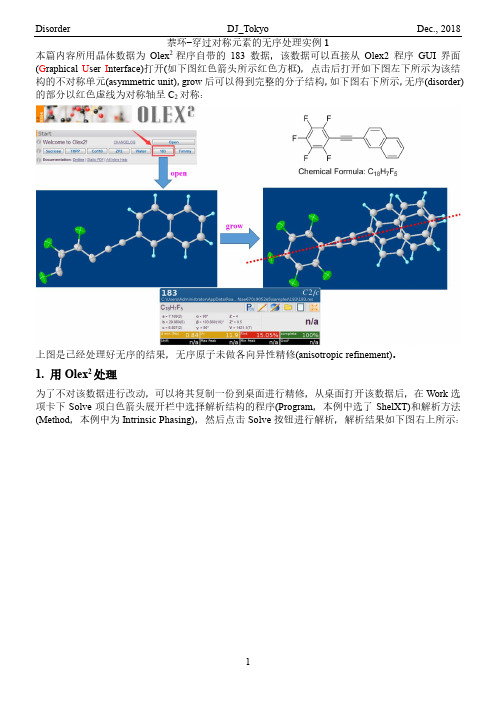
萘环-穿过对称元素的无序处理实例1本篇内容所用晶体数据为Olex2程序自带的183数据,该数据可以直接从Olex2程序GUI界面(G raphical U ser I nterface)打开(如下图红色箭头所示红色方框),点击后打开如下图左下所示为该结构的不对称单元(asymmetric unit),grow后可以得到完整的分子结构,如下图右下所示,无序(disorder)的部分以红色虚线为对称轴呈C2对称:上图是已经处理好无序的结果,无序原子未做各向异性精修(anisotropic refinement)。
1. 用Olex2处理为了不对该数据进行改动,可以将其复制一份到桌面进行精修,从桌面打开该数据后,在Work选项卡下Solve项白色箭头展开栏中选择解析结构的程序(Program,本例中选了ShelXT)和解析方法(Method,本例中为Intrinsic Phasing),然后点击Solve按钮进行解析,解析结果如下图右上所示:精修几轮后,隐藏Q峰,然后将结构grow,如下图左上所示,删除所选原子(选中后原子为绿色高亮标记),得到萘环骨架(下图右下红色方框所示),此时只需要是两个并六元环即可,形状不好看没关系:选中这个萘环中的所有原子(按住Shift键不放,再按住鼠标左键不放的情况下拖动鼠标,将这个萘环圈在鼠标所画出的方框中即可),然后在指令输入区输入指令“part -1 -c”(c要小写),敲回车后输入“fuse”再敲回车回到不对称单元状态,发现多了一个原子,将其删除:然后选中萘环所有原子,在指令输入区输入指令“part -1 10.5”,回车后Olex2程序会将该萘环分到part -1并赋予所有原子占有率为10.5,然后再选中萘环所有原子,在指令输入区输入指令“afix 116”对萘环进行刚性限制,回车后精修,萘环看起来就正常很多了(下图右上所示):各项异性精修,以下图左所示两种方式中的任意一种方式进行各项异性精修,结果如下图右所示:炔基的两个碳原子也需要进行无序处理,选中这两个碳原子,在指令输入区输入指令“split”后回车,程序将其分为part 1 21和part 2 -21两部分,将方向偏离萘环的两个原子删除,剩下的两个原子归入part -1 10.5,然后精修,并加上各项异性精修,精修数轮后结果如下图右上所示:最后加氢精修至数据收敛即可:2. 用shelxtl处理同样,将该数据复制到桌面,然后用shelxtl程序打开:打开数据后,点击XL下拉菜单中的XL进行精修,点击XSHELL打开视图查看结构,由于Q峰的存在,结构看着比较乱,可以点击Edit下拉菜单中的Kill all Q-Peaks将Q峰删除,即可看到清晰的结构:在Atoms下拉菜单中点击Grow,或者在空白处单击鼠标右键,在弹出的菜单栏中单击Grow即可查看完整结构,如下图右所示:这是处理好的数据,为重新展现处理过程,可以先将无序的原子删除,先以下图所示任意一种方式回到不对称单元状态:选择原子:有三种方式方式1:在Select下拉菜单中选择Select,在弹出的Select Atoms对话框中Atom List中选择要删除的原子方式2:将鼠标放到要选择的原子上,单击鼠标右键,在弹出的菜单中点击Select即可选中该原子;点击Delete可以直接删除该原子方式3:按住Shift键不放,按住鼠标左键并拖动鼠标画出一个矩形,矩形内的所有原子将会被选中以上述三种方式中的任意一种方式将选中要删除的原子,然后点击Select下拉菜单中的Kill Selected 将选中的原子删除:点击菜单栏中的Refine按钮,在弹出的Refinement Control对话框中设置精修方法、精修轮数以及Q峰数目等,然后点击OK执行,程序会弹出一个精修过程对话框,如下图右下所示,结束后点击OK即可:可以看到与C13相连的Q5和Q12应该是炔基的两个碳原子,选中这两个Q峰,以下图所示两种方式的任意一种打开Atom对话框,在Type中直接输入元素符号(或者点击其后的El按钮,在弹出的周期表中选择元素类型),在Name中输入编号,比如C12(不要和已有原子重复即可),Occupancy 中可以设置占有率,一般按默认的即可,在Hybridization中设置杂化方式,最后点击OK即可,更改后结果如下图右下所示:依照上述步骤找出大致结构,如下图所示:重新命名一下:从Grow的情况可以看出,需要把C7-C15的原子的对称操作原子给找出来,这时就需要用到XP 了,打开XP后,输入“fmol”然后回车,再输入“info”后回车,让程序读取各项信息:接着输入“envi C7 C8 C9 C10 C11 C12 C13 C14 C15”来查看这些原子的对称信息:从上图可以看出,C7-C15的等效原子是通过对称操作码5655得到的,因此输入“sgen C7 C8 C9 C10 C11 C12 C13 C14 C15 5655”将C7-C15的等效原子生长出来(下图所示带A的原子),然后将这些原子重命名:最后输出文件,输入File name,比如本例中输入“File a”,即输出的文件的名字为“a”,然后输入原来文件的名称,本例中为183,最后输入quit退出程序:此时在该晶体数据文件夹中即会产生一个a.ins文件,然后将原来的hkl(本例中为183.hkl)复制一份,并将其重命名为a:然后用XShell打开,由于XShell默认打开res文件,因此将文件名后面的下拉菜单中选择All Files 才能打开ins文件:打开后如下图左边所示,删除不要的原子,保留一个萘环,然后重命名,并对原子进行排序,如下图右所示:打开ins文件:将C5-C16归入part -1,并将占有率均改为10.5,将C7-C16加上AFIX 116进行限制:保存后精修至收敛即可。
晶体化学教学中单晶结构解析存在的问题
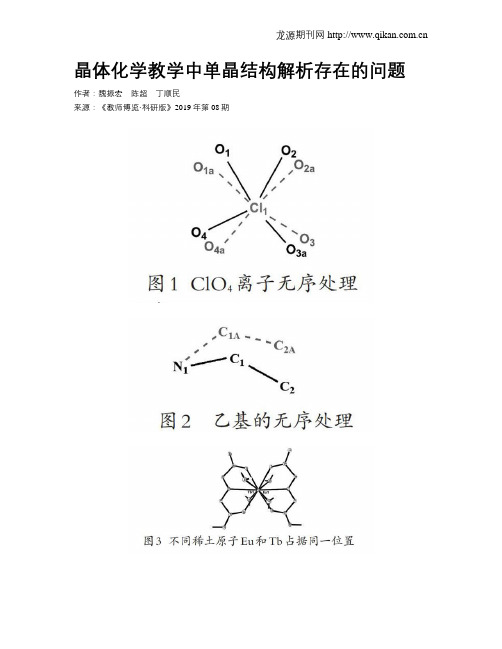
晶体化学教学中单晶结构解析存在的问题作者:魏振宏陈超丁顺民来源:《教师博览·科研版》2019年第08期[摘要] 晶体是由原子、分子或离子在三维空间按一定规则呈周期性排列而成,在外形上表现为具有一定形状的几何多面体。
晶体内部结构可以通过X-射线单晶衍射实验及相应的程序解析获得,但除了经验丰富的晶体学家,一般的科研工作者对单晶解析都感到束手无策。
晶体化学中,要顺利解析出单晶结构,关键在于初始套的确定、无序的精修、加氢三个方面。
[关键词] 晶体化学;晶体结构;单晶解析晶体化学是研究晶体在原子水平上的结构,揭示晶体的化学组成、结构和性能三者之间内在联系的学科。
自1966年以后,由于计算机控制的自动单晶衍射仪和与之匹配的晶体结构分析软件的迅速发展和普及,X-射线单晶衍射测试成为获取分子立体结构和键参数最权威的技术手段。
截至目前,剑桥晶体数据库(CCDC)已收录的无机化合物、有机化合物和金属配合物的晶体结构数量已超过100万种,并且仍以每年超过5万的数量在增加。
这些大量的晶体化学结构信息为广大科研工作者深入研究反应机理、指导合成和深入探讨物质分子构型与分子化学活性间的内在联系提供了可靠的依据。
目前,国内高校和科研机构已经基本配备了X-射线单晶衍射仪,势必要求每位与之相关的研究人员都掌握X-射线单晶结构的解析方法,因此晶体化学成为高校化学专业研究生的选修课程。
然而,一线科研工作者,特别是初学者,在解析晶体结构时经常感到困惑,不知从何下手。
其实,我们知道解析晶体结构是一门经验技术活,教师除了需要在课堂上详细介绍教材上的晶体解析的步骤和方法外,还需要把自己多年在解析单晶时积累的经验、技巧和心得告诉学生。
通过最近几年晶体化学的教学,我们发现研究生经常对初始套的解析、无序的精修和如何加氢感到困难重重,因此,我们有必要将这些问题进行归纳总结,希望利用积累的经验帮学生熟练掌握晶体解析这门技术。
一、初始套的确定在解析结构前,我们需要评判衍射数据的好坏。
晶体结构分析中的无序、绝对结构和

三、无心空间群与绝对结构 1、绝对结构
对于无心晶体,晶体中的分子或分子片断相对于晶轴可有不同的取向,这称为 晶体的绝对结构。属于晶体学的概念。
a
a
2、绝对构型 对于手性化合物(无任意次旋转反映轴),具有互为镜像的两种结构,称为化
合物的绝对构型。属于立体化学的概念。
Br
Me
Cl
OH
Br
Cl Me
HO
3、反常散射 Friedel定律:|Fhkl| = |F-h-k-l| (对于三斜晶系) 反常散射(anomalous scattering): f = fo + ∆f’+i∆f” 对于中心对称结构,反常散射效应相互抵消,能严格满足 Friedel 定律。 非中心对称结构的反常散射不能互相抵消,因而不严格服从 Friedel 定律。可
6、如何区分结构解析中的真无序和假无序? 若无序现象可通过降低空间群的对称性得到消除,即为假无序。由于真无序
结构中无序部分呈统计性排列,因此无法通过降低空间群的对称性来消除。
二、如何将结构由低对称性空间群转换为高对称性空间群(如 Cc 到 C2/c)? 由于无心群出现的概率非常小,因此当一个结构按无心群解出后,必须尝试
5、结构解析中的假无序:分子结构的大部分在所选空间群下有序,而小部分呈 现规律性无序。这是由于晶体结构的假对称性,导致错误地选择了对称性过 高的空间群导致的。
固体物理学基础晶体的有序态与无序态转变

固体物理学基础晶体的有序态与无序态转变晶体是由原子、分子或离子以高度有序的方式排列而成的固体材料。
在固体物理学中,研究晶体的有序态与无序态转变是一个重要的课题。
本文将探讨晶体的有序态和无序态,以及它们之间的相互转变过程。
一、有序态有序态是指晶体中原子、分子或离子按照严格的空间排列规则形成的有序结构。
这种有序结构通常具有周期性,可以通过几何和数学方法进行描述和分析。
有序态的排列规则主要有以下几种:1. 晶格结构:晶体中原子、分子或离子的排列方式形成了一种特定的晶格结构,比如立方晶系、正交晶系等。
晶格结构的特征决定了晶体的物理性质和热力学行为。
2. 排序规则:原子、分子或离子在晶格中的位置和排列有着一定的规则,可以通过晶体学中的透射电子显微镜等技术进行观察和研究。
3. 对称性:有序态的晶体通常具有高度的对称性,可以通过对称元素和晶体中的点群操作来描述。
对称性可以在晶体的结构中反映出来,如轴对称、面对称等。
有序态的晶体在不同的温度和压力条件下,可能发生相变,转变为无序态或其他有序态结构。
二、无序态无序态是指晶体中原子、分子或离子的排列没有一定的规律,呈现出无序或准无序的状态。
无序态通常具有较低的对称性,难以通过传统的晶体学方法进行描述和分析。
无序态常见的形式包括:1. 非晶体:也被称为无定形固体,其原子、分子或离子的排列没有周期性,呈现出均匀无序的状态。
非晶体通常具有高度的各向同性,物理性质在各个方向上基本相同。
2. 准晶体:准晶体是介于有序态和非晶体之间的一种结构,具有局部的有序性,但整体上没有完全的周期性。
准晶体是一种特殊的结构,由于其非周期性的特点,具备一些独特的物理性质。
无序态与有序态之间的转变可以通过一系列的热力学和动力学过程来实现。
其中一种常见的转变形式是熔化,即晶体在一定温度范围内转变为液体。
三、有序态与无序态转变有序态与无序态之间的转变是一个复杂的过程,涉及到原子、分子或离子在晶格中的位置变化以及晶体结构的重组。
傅里叶变换 晶体无序

傅里叶变换晶体无序傅里叶变换是一种广泛应用于信号处理与图像处理领域的数学工具,而晶体无序是指晶体结构的无规则排列。
尽管这两个概念看似毫无关联,但通过深入探索,我们可以发现它们之间的奇妙联系。
傅里叶变换是法国数学家傅里叶在19世纪提出的一种变换方法,它能够将一个函数在时域上的表示转化为频域上的表示。
通过傅里叶变换,我们可以将一个信号分解成不同频率的正弦波组成的谱,从而更好地理解信号的性质。
傅里叶变换的数学表达式和计算方法虽然复杂,但在实际应用中却发挥着重要作用,例如在图像处理中,傅里叶变换可以将图像转换为频谱图,进而实现图像的滤波、增强等操作。
晶体无序是指晶体的原子或分子在空间中的排列无规则,缺乏周期性。
晶体是由原子或分子按照一定规则排列而成的,具有规则的晶格结构。
然而,由于各种因素的干扰,晶体结构可能会受到扭曲、杂质、缺陷等因素的影响,从而导致晶体无序。
晶体无序在材料科学中是一个重要的研究领域,对于理解材料的性质和开发新材料具有重要意义。
那么,傅里叶变换与晶体无序之间的联系是什么呢?首先,傅里叶变换可以提供频域上的信息,通过分析信号的频谱特征,我们可以了解信号中各个频率成分的贡献及其重要性。
类似地,晶体无序也可以被看作是一种频域上的信息,晶体结构的无规则排列可以通过对晶体的衍射图谱进行分析而得到。
衍射图谱是晶体结构的“指纹”,通过分析衍射图谱的特征,我们可以了解晶体中原子或分子的排列方式以及晶体的无序程度。
其次,傅里叶变换可以将一个信号从时域转换到频域,而晶体无序可以看作是晶体结构在空间中的无规则排列。
这种转换的类比可以帮助我们更好地理解晶体无序的性质。
在傅里叶变换中,时域上的信号可以看作是频域上的正弦波的叠加,而在晶体中,有序的晶格结构可以看作是无序排列的原子或分子的叠加。
通过傅里叶变换的思想,我们可以将晶体结构视为由不同程度的无序排列构成的,从而更好地理解晶体无序的本质。
最后,傅里叶变换在信号处理和图像处理中有广泛的应用,可以提取出信号或图像中的有用信息。
晶界 无序结构

晶界⽆序结构晶界是固体材料中最重要的结构单元之⼀,它对材料的物理、化学和机械性能具有显著的影响。
然⽽,晶界往往呈现出⼀种⽆序的结构状态,这种⽆序状态在理解晶界的性质以及在材料科学和⼯程领域中的应⽤都具有重要意义。
⾸先,我们要明确什么是晶界。
简单来说,晶界是晶体材料中不同晶粒之间的边界。
在多晶体材料中,晶界⼴泛存在,它们分割着不同取向的晶粒。
晶界的结构复杂,⼀般情况下,我们可以通过X射线或电⼦衍射等⽅法研究其结构。
尽管晶界具有复杂的结构,但是⼤多数情况下,晶界都被认为是⼀种⽆序的结构状态。
那么,为什么晶界会出现⽆序的结构状态呢?这主要是因为晶界的形成过程与晶体内部的⽣⻓过程不同。
在晶体内部,原⼦按照⼀定的规律排列,形成有序的晶体结构。
然⽽,在晶界处,由于受到温度、压⼒、杂质等因素的影响,原⼦排列的规律被打乱,导致晶界呈现出⼀种⽆序的结构状态。
此外,由于晶界的能量较⾼,⼀些⾼能晶界还可能出现类似液态的⽆序结构。
那么,这种⽆序的结构状态对晶界的性质有哪些影响呢?⾸先,⽆序的结构状态使得晶界的原⼦排列与晶体内部不同,这导致了晶界处原⼦的平均⾃由程减⼩,使得晶界的导热性、导电性和扩散系数等物理性质与晶体内部存在明显的差异。
此外,⽆序的结构状态还使得晶界的机械性能与晶体内部不同,例如,晶界往往是材料脆性的来源之⼀。
了解晶界的⽆序结构对理解其性质和应⽤具有重要的意义。
在材料科学和⼯程领域中,我们需要了解材料的物理、化学和机械性能以便进⾏合理的设计和有效的应⽤。
因此,深⼊探讨晶界的⽆序结构可以为我们提供更多的信息和思路来理解材料的性质和⾏为。
对于材料设计和制备⽽⾔,理解晶界的⽆序结构有助于我们更好地控制材料的微观结构和性能。
例如,通过控制材料的制备条件,我们可以调整晶界的数量、⼤⼩和分布,从⽽优化材料的性能。
此外,⼀些特殊的应⽤可能需要利⽤晶界的⽆序结构来实现。
例如,⼀些⾼能晶界具有类似液态的⽆序结构,这使得它们在润滑、减磨和⾃修复材料等领域具有潜在的应⽤价值。
位置无序处理及其简单理解

位置无序处理及其简单理解位置无序是无序中的“正常现象”:某个原子占据的位置布置一个,这些位置可以在同一个晶胞内(动态无序——固体中的一种真正运动),也可以分布于不同的晶胞中(静态无序,固体中的类运动状态)。
如下图所示,与金属配位的乙醇溶剂分子末端甲基C22——由其各向异性原子位移参数(ADPs, Atomic Displacement Paramerters)得到的椭球形状(拉长成香蕉状)可能是不合理的,因为程序试图将两个或多个原子位置仅用一个椭球来描述。
另外该结构中最大残余电子峰Q1出现在该原子附近。
种种迹象表明C22原子可能存在无序。
这一点也可以从位于精修过程记录文件lst文件中的原子位移参数主均方值U (Principal mean square atomic displacements U)列表中找到相关证据。
在Olex2中可以在命令行输入命令edit lst来查看lst文件,或者在晶体数据所在文件夹找到name.lst文件并以记事本的方式将其打开,如下图所示:在lst文件中按下Ctrl+F快捷键打开查找对话框,以Principal为关键词进行搜索,如下图所示:就可以找到原子位移参数主均方值U (Principal mean square atomic displacements U)列表,如下图所示:在该表格中找到C22,如下图所示:可以看到在C22的后面有一句话:may be split into 0.6430 0.8982 0.2186 and 0.5684 0.9036 0.2326,这句话的意思是C22原子可能需要劈裂为两个原子,其坐标分别为0.6430 0.8982 0.2186和0.5684 0.9036 0.2326,也就表明C22原子可能存在无序。
那么这时就需要用两个位置来描述C22原子,这可以通过两种方法来进行,介绍如下:方法一:在Olex2中,选中Q1,并用name c22a指令将Q1命名为c22a,如下图所示:然后选中C22,并用指令part 1 21将C22分配到part 1中,并将其位置占有因子sof (site occupancy factor)设置为21(意思是指其sof指向第二个自由变量),如下图所示:然后选中c22a,并用指令part 2 -21将c22a分配到part 2中,并将其sof设为-21(意思是指:若part 1的sof为x,则part 2的sof为1-x。
无序材料微观结构分析的新方法及其应用

精选文档报告题目:无序资料微观构造剖析的新方法及其应用报告人:复旦大学光科学与工程系王松有教授报告纲要:与晶态资料对比,无序资料因为缺少长程有序性,所以实验上没法利用X射线衍射等手段确立其微观构造,而理论上对无序资料构造的研究手段也相当缺乏。
本报告将在综述无序资料微观构造剖析的理论和实验方法基础上,联合几个应用实例,给出一种新的剖析无序资料的新方法。
参照文件:1、S.Y.Wang,C.Z.Wang,etal,Phys.Rev.B,78,184204(2008)2、S.Wu,S.Y.Wang,etal,J.Appl.Phys.,2011,110,1035183、X.W.Fang,etal.Phys.Rev.B,82,184204(2010)4、S.Wu,S.Y.Wang,C.Z.Wangetal.Phys.Rev.B,2011,84,1342085、X.W.Fang,etal,Nature,SCIENTIFIC REPORTS|1:194|DOI:10.1038/srep00194报告时间:2013年1月5日下午15:00报告地址:29-414王松有教授简介:光科系党支部书记,博士生指导老师。
1987年获郑州大学物理系学士学位,2000年10月获复旦大学物理系凝集态物理专业博士学位。
2000年7月在复旦大学光科学与工程系任教。
2003年以来多次到美国能源部Ames实验室和衣阿华州立大学物理系从事接见科学家研究,美国能源部Ames实验室国外兼职研究员(2006~2015年)。
2003年获教育部提名国家技术发明一等奖(第三获奖人),2005年当选上海市“浦江人材计划”,主持两项国家自然科学基金项目,作为主要成员参加了多项国家要点基础研究发展计划、国家自然科学基金和上海市科委要点基金项目。
已在国内外学术期刊上发布论文100余篇。
研究方向:(1)、光功能资料构造光学及磁学性质的理论和实验研究;(2)、非晶态资料微观构造及动力学性质的理论研究;(3)新资料理论设计。
晶界 无序结构

晶界无序结构
晶界,是晶体结构中的一个概念,它指的是晶体内部不同晶粒之间的边界或界面。
晶界的存在使得晶体内部形成了一种无序的结构,与晶粒内部的有序排列形成鲜明的对比。
晶界在晶体材料中起着重要的作用。
首先,它可以影响晶体的力学性能。
晶界是晶体中的缺陷,会影响晶体的强度和韧性。
晶界的存在会导致晶体内部的应力集中,从而影响晶体的机械性能。
其次,晶界还可以影响晶体的导电性和热导性。
晶界是晶体中的能量势垒,会对电子、离子和热传导产生阻碍作用,从而影响晶体的导电性和热导性。
然而,晶界的无序结构也为晶体材料带来了一些问题。
首先,晶界的存在会导致晶体的强度下降。
晶界是晶体中的缺陷,会导致晶体的强度降低。
其次,晶界还会对晶体的电子和离子传输产生阻碍作用,影响晶体的导电性和离子传输性能。
此外,晶界还会对晶体的热传导性能产生影响,导致晶体的热导率降低。
针对晶界的问题,科学家们进行了大量的研究,希望能够找到解决方案。
一种常见的方法是通过调控晶体生长过程中的温度和压力条件,来控制晶界的形成和分布。
另一种方法是通过合金化或掺杂等手段,来改变晶界的结构和性质。
此外,还有一些研究致力于开发新的材料或设计新的结构,以减少晶界对材料性能的影响。
晶界是晶体内部的一种无序结构,它对晶体的力学性能、导电性和热导性等方面产生重要影响。
科学家们正在不断研究晶界的形成机制和调控方法,希望能够解决晶界带来的问题,进一步提高晶体材料的性能。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Disorder in the para substituents, and disorder in the halides
SUMP
Me
to tie three p-Cl to 2 site occupancy, along with distance restraints.
Ph Cl Sn O P Ph
1
Disorder of 15-crown-5: an odd number of atoms packing in a over a center-of-inversion
O O O O O
1 2 3
Use a ‘PART –1’ command to suppress the calculation of bonds/angles. Assign ½ occupancies to all atoms. DFIX C–C = 1.50, C–O = 1.41, C…O = C…C = 2.37 Å.
The vibration of some atoms 2 - 3 times those of other atoms
of the same molecular fragment.
Peaks in the difference map that are too close to existing
OH O O O O H O O O HO O
H constrained by symmetry (C2/c, Z = 4) O…H = 1.25 Å
H N N H
H restrained by DFIX (P21/c, Z = 4) O–H = 0.85 Å
OH O O
O
O
H
O O O HO
O
H N N H
Because total charge is 8, there are should be 1 Cu(II) and 2 Fe(III) for the 3 sites. Refine each site as ⅓Cu⅔Fe.
THF
F 3C O O O
CF3
O
THF M M O O
Or, use SUMP to tie the 3 Cu atoms to unit occupancy (0.73, 0.15, 0.12). The 0.12 occupancy M atom is coordinated by water.
F
F
F
F
O O Sn O
O
Sn
A phenylene ring disordered along the C1–C4 axis
O O O O O O
As the anion lies on a center-of-inversion, ‘AFIX 66’ cannot be used. DFIX pairs of distances, and FLAT some atoms.
and 4,5 carbon atoms.
Asymmetric unit has 2 aromatic and 1 carboxyl carbon atoms, and 1 oxygen atom only. High R, which is an artifact of the disorder and not because the data are bad.
Point group 2
Point group mmm
Twinning Case 2: Monoclinic with nearly equal a(12.479 Å) and c- (12.597 Å) axes
Look for water near the ion.
Disorder over a center-ofinversion
O O O O O O O O O O
Use a ‘PART –1’ command to suppress symmetry. 2 Refine the ring with ‘AFIX 66’. 3 Some atoms to have full and other half occupancy.
Which SHELX commands to use depend on the disorder. And it is very messy to treat disorder.
The energy barrier to rotation of CO2CH2CH3 radical is of the order of kT – there is nothing you can do about the data.
separated by more than 0.5 Å. Pyridyl rings can also be fitted as hexagons.
‘AFIX 66’ for phenyl rings
Can be used for noncentric space groups because there are fewer reflections, or if there is a spread of C—C distances.
Twinning Case 1: Monoclinic emulating orthorhombic Br = 90.05(1)° Im Im
Im Cu Im
TWIN 1 0 0 0 -1 0 0 0 -1 BASF 0.47 The diffraction pattern emulates orthorhombic but is not because the intensities cannot be averaged in orthorhombic (Rint is large).
Disorder in a cyclopentyl ring
DFIX the C—C to 1.50 Å and the C…C…C distance to 2.45 Å so that the angle is 109.5°.
M
Perchlorate ions are usually disordered: any molecular fragment that is spherical will be disordered.
HO H CH3
O
O
Disorder in the heavy atom only
Ag is 3-coordinate in major component and 4-coordinate in minor component in centric structure. Complicated as two N atoms are disordered with two C–H units. The correctly refined structure should not have any Ag…H interactions.
O O
respect to the C6H6 ring.
Sn
The entire anion is disordered but the [Ph2P(CH2)3PPh2]2Ag cation is ordered.
O
O
F
F F
Refinement of more than two atoms sharing the same site: The SUMP command
Some Strategies for the Refinement of Disordered Crystal Structures
Ng Seik Weng
Department of Chemistry University of Malaya Kuala Lumpur Malaysia
The assumption of perfect periodicity is a basic assumption of crytallography. However, real crystals are not perfect.
atoms to form plausible bonds.
Atoms that are too close to symmetry elements in the space
group to produce chemically-reasonable bonding.
Anyway, the data are mothballed because the structures cannot be published.
Br
A simple idea of twinning
A brick with top and bottom indentations, embossed with ‘London Brick’ on the top only.
Turning brick upside down uses an operation of mmm.
H N H O N H O
H N H O N H O
Disorder in four-atom, pyramidal fragments, e.g., CHCl3 and CH3CH(OH)CO2
H C Cl Cl Cl
The methine –CH fragments are above and below the carboxyl plane.
Disorder imposed by mm2 symmetry
DMF =
H
N O
N
H
O
Disorder imposed by mm2 symmetry
O O O O
Mirror plane passing through ring. Mirror plane perpendicular to ring. Rotation axis through the mid-points of the 1,2