pymol使用教程
pymol使用指南

pymol使用指南
PyMOL是一款用于分子可视化的强大工具,它可以帮助科学家
和研究人员可视化蛋白质、分子和其他化合物的三维结构。
下面我
将从安装、基本操作和高级功能三个方面来介绍PyMOL的使用指南。
首先是安装。
PyMOL可以在官方网站上下载,它支持Windows、Mac和Linux系统。
安装完成后,你可以启动PyMOL并开始使用。
接下来是基本操作。
在PyMOL中,你可以通过命令行或者图形
用户界面来操作。
你可以加载分子结构文件,比如PDB文件,然后
可以旋转、平移和放大分子结构。
你还可以选择不同的渲染方式来
显示分子,比如线框模式、球棍模式和表面模式。
此外,你可以使
用命令来选择特定的原子或残基,并对它们进行操作,比如着色、
标记或者测量距离。
最后是高级功能。
PyMOL还提供了许多高级功能,比如分子对接、电荷分布计算、分子动力学模拟等。
你可以使用PyMOL来进行
分子对接研究,寻找分子间的相互作用。
你还可以计算分子的电荷
分布,以及模拟分子的运动和构象变化。
总的来说,PyMOL是一个功能强大的分子可视化工具,它可以帮助科研人员直观地理解分子结构和相互作用。
通过学习和使用PyMOL,你可以更好地进行分子建模、药物设计和生物研究。
希望这个简要的指南可以帮助你开始使用PyMOL,并在科研工作中发挥作用。
pymol做图简单教程
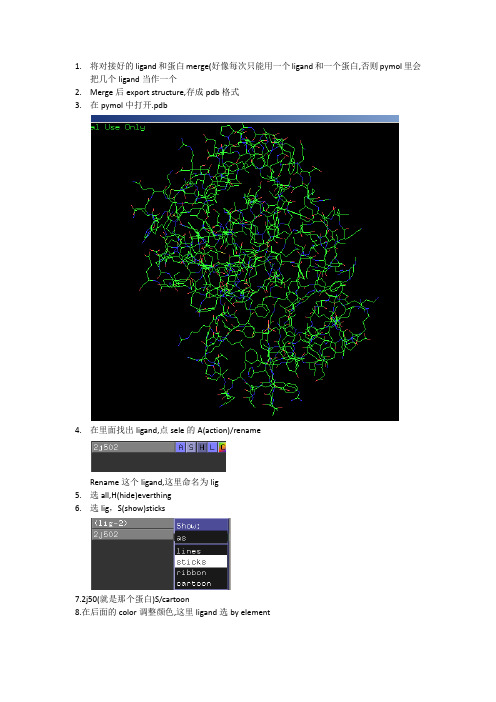
1.将对接好的ligand和蛋白merge(好像每次只能用一个ligand和一个蛋白,否则pymol里会把几个ligand当作一个2.Merge后export structure,存成pdb格式3.在pymol中打开.pdb4.在里面找出ligand,点sele的A(action)/renameRename这个ligand,这里命名为lig5.选all,H(hide)everthing6.选lig,S(show)sticks7.2j50(就是那个蛋白)S/cartoon8.在后面的color调整颜色,这里ligand选by element2j50选white9.下面显示氢键,点击2j50的Action,选择find/polar contacks/to other atoms in object,氢键就出来了10.然后再找氢键作用的残基,可以在上面直接点选,但最好找出氨基酸代码来选取(因为这里面点不准)Rename它们,以便以后好找,这里rename为res,show它们为line,color为by element11.再选display,将background改成white12.然后分别点选这几个res(因为颜色不同,所以很容易找出来),右键label/residuesbel出来后,还可以调整它们的位置,点一下左图viewing,就变成了右图的editing,这时就可以按住ctrl键拖动label的位置了12.大体已经完成了,再进行一些修饰Setting/transparency/cartoon/50% 蛋白的透明度Setting/label/size 18调节字体的大小Setting/edit allcartoon的颜色改回白色(在filter里输cartoon可以快点找出来,改完后按回车) //或者输入命令Pymol>set cartoon_color white在改氢键的线型,在dash gap length width进行调整把虚线改好看点Stick也可以改细点13.电子密度1)首先,登陆http://eds.bmc.uu.se/eds/搜索你的蛋白2)download maps3)选ccp4格式,generate map4)下载后解压,重命名为*p45)在pymol里打开,在打开的.map,action/mesh,color/blue6)选中lig,在PyMOL输入isomesh map,2j50.map,2.0,sele,carve=1.67)这样就从原来的map里iso出lig周围的蛋白的电子密度,把2j50.map_mesh关了就可以看见了然后再调整它的粗细电子密度周围的线好像有些奇怪,可能设置里哪个地方还没调好吧.另外,ray 1900,980就能生成1900*980分辨率的图片,然后file/save image as输出.png。
pymol使用教程中文

pymol使用教程中文PyMOL是一款用于分子建模的强大工具,许多化学生物学和生物化学研究人员使用它来可视化蛋白质、核酸和其他生物分子的三维结构。
本文将介绍PyMOL的基本用法,包括如何加载分子结构、调整视图和颜色、创建图片和动画等功能。
### 1. 安装与启动首先,您需要下载PyMOL软件并安装到您的计算机上。
安装完成后,双击图标启动PyMOL。
您可以在终端中输入`pymol`命令启动PyMOL。
### 2. 加载分子结构在PyMOL中加载分子结构非常简单。
点击菜单栏中的“File”,然后选择“Open”或者直接拖拽您的分子结构文件到PyMOL窗口中即可加载。
常见的分子结构文件格式包括PDB、XYZ、MOL等。
### 3. 调整视图与颜色PyMOL提供了丰富的功能来调整分子的视图和颜色。
您可以使用鼠标滚轮缩放、移动视图,也可以通过菜单栏中的“Edit”来选择不同的视图模式。
同时,您还可以通过选择不同的颜色方案为分子上色,以突出不同的氨基酸或核苷酸。
### 4. 选择和操作分子在PyMOL中,您可以使用鼠标来选择分子中的原子、氨基酸或核苷酸等部分。
通过单击可以选择一个原子,通过按住Shift键加单击可以选择多个原子。
您还可以使用命令行来选择特定的部分,例如`select resi 10`选择氨基酸编号为10的氨基酸。
### 5. 创建图片与动画PyMOL还支持创建高质量的图片和动画。
您可以调整分子的角度和视角,然后点击菜单栏中的“File”-“Save Image”来保存图片。
如果您想要创建动画,可以使用PyMOL的命令行来逐帧绘制动画,并将其导出为视频文件。
### 总结在本教程中,我们介绍了PyMOL的基本用法,包括加载分子结构、调整视图和颜色、选择和操作分子、创建图片和动画等功能。
希望这些内容能够帮助您更好地使用PyMOL进行分子建模和可视化工作。
如果您想进一步了解PyMOL的高级功能,可以查阅PyMOL的官方文档或参考其他教程。
pymol教程

pymol教程
Pymol是一种用于分子可视化的软件工具,它可以让科学家和
研究人员更好地理解和展示分子结构和相互作用。
本教程将引导您使用Pymol进行一些基本的分子可视化操作。
1. 安装Pymol
在开始之前,您需要在计算机上安装Pymol软件。
您可以从
官方网站上下载适用于您的操作系统的最新版本。
安装完成后,启动Pymol。
2. 加载分子数据
在Pymol中,您可以加载各种不同格式的分子数据文件,包
括PDB、MOL2、XYZ等。
您可以从菜单栏中选择“File”-
>“Open”来加载一个分子文件。
3. 调整分子的显示样式
一旦分子文件加载完成,您可以使用Pymol的各种工具来调
整分子的显示样式。
例如,您可以选择隐藏或显示原子、调整原子的颜色和大小等。
4. 旋转和平移分子
Pymol允许您通过鼠标来旋转和平移分子。
按住鼠标左键并
拖动可以旋转分子的视角,按住鼠标右键并拖动可以平移分子的位置。
5. 添加和调整分子的标签
Pymol还支持添加和调整分子上的标签。
您可以选择一个原
子或残基,并在菜单栏中选择“Label”->“Atom”或“Residue”,然后在弹出的对话框中输入您想要显示的标签。
6. 创建和编辑分子图形
Pymol还提供了一些工具来创建和编辑分子图形。
您可以使用线条、球棒、面片等工具来可视化分子的结构和相互作用。
以上是使用Pymol进行基本分子可视化的简要教程。
希望这对您有所帮助!。
pymol使用教程

pymol使用教程PyMOL是一种图形化的分子可视化工具,它可以用于展示和分析蛋白质、DNA、RNA等分子结构。
下面是一份PyMOL使用教程,帮助你了解如何使用PyMOL进行分子可视化。
1.安装PyMOL2.打开和导入结构在PyMOL中,你可以通过点击菜单中的“File”->“Open”来打开一个已有的结构文件。
PyMOL支持多种结构文件格式,包括PDB、MOL2、SDF 等。
你也可以通过在命令行中输入“load 文件路径”来导入结构文件。
3.调整视图PyMOL提供了多种视图设置选项,可以通过鼠标右键点击图像来进行操作。
你可以旋转、平移和缩放结构,以便更好地观察分子。
此外,你还可以通过鼠标滚轮或点击菜单中的“View”->“Zoom”来放大或缩小结构。
4.显示分子PyMOL允许你显示分子的不同组成部分,例如原子、氢键、键、半径等。
你可以通过点击菜单中的“Display”进行选择。
你还可以通过命令行输入“show 选项”来显示特定的分子部分,例如“show sticks”用于显示分子键。
5.颜色设置PyMOL允许你为分子的不同组成部分设置不同的颜色。
你可以通过点击菜单中的“Color”进行设置。
例如,你可以将蛋白质的α-螺旋设置为红色。
“color 红色, 选择项”命令可以实现相同的效果。
6.制作表面PyMOL还支持制作分子表面,以更好地展示分子的形状和结构。
你可以点击菜单中的“Action”->“Surface”来创建分子表面。
你还可以通过命令行输入“show surface”来实现相同的效果。
7.保存和导出在PyMOL中,你可以点击菜单中的“File”->“Save Image”将当前视图保存为图片文件。
此外,你还可以使用命令行输入“pn g 文件路径”将图片保存为指定的路径。
PyMOL还支持导出结构文件,你可以点击菜单中的“File”->“Export”来导出结构文件。
非常好的pymol教程

非常好的pymol教程Pymol是一款强大的分子可视化工具,它能够帮助生物学家、化学家和结构生物学家分析和可视化蛋白质、小分子和复杂化合物的结构。
本教程将向你展示Pymol的基本功能和操作步骤,以便你能够更好地理解和使用这个工具。
1. 安装Pymol:2.打开和导入分子结构:打开Pymol软件后,你将看到一个主界面。
你可以通过点击菜单栏上的“File”选项或使用快捷键Ctrl+O来打开分子结构文件。
Pymol支持多种主流的分子结构文件格式,如PDB、CIF和XYZ等。
选择你想要导入的分子结构文件,然后点击“Open”按钮。
3.调整视图和颜色:一旦成功导入分子结构,你可以通过使用鼠标右键和滚动调整视图。
拖动鼠标右键可以旋转分子,滚动滚轮可以缩放分子。
此外,你还可以在菜单栏的“Display”选项下调整分子的颜色、图形和材质。
4.选择和操作分子的部分结构:要选择和操作分子的部分结构,你可以使用菜单栏上的“Select”选项或使用快捷键Ctrl+Shift+A。
在打开的“Selection”对话框中,你可以输入选择命令来选择具体的分子结构,比如选择指定的氨基酸残基或原子。
选择完成后,你可以对所选结构进行操作,如旋转、平移、放大等。
5.显示分子间的非共价相互作用:Pymol能够计算并显示分子间的非共价相互作用,比如氢键、离子键和范德华力等。
你可以在菜单栏的“Display”选项下选择“Nonbonded”来显示非共价相互作用。
这将帮助你更好地理解和分析分子的结构和性质。
7.绘制分子的电子密度图:Pymol允许你绘制分子的电子密度图,以更好地理解其结构和性质。
你可以在菜单栏的“Display”选项下选择“Map”来打开“Maps”功能。
然后选择你想要的地图类型和参数,点击“Apply”按钮来生成电子密度图。
你可以使用鼠标右键和滚动调整绘制的电子密度图。
通过以上步骤,你已经了解了Pymol的基本功能和操作。
pymol基础教程

pymol基础教程Pymol是一种分子建模和可视化软件,主要用于生物和化学领域的分子结构的可视化。
它提供了许多工具和功能,方便用户进行分子结构的操作和分析。
本教程将介绍Pymol的基本功能和用法。
Pymol的界面分为几个主要区域,包括主菜单、工具栏、命令行窗口和3D视图窗口。
主菜单包括常用操作和功能的快捷方式,工具栏提供了一些常用工具的图标,命令行窗口用于输入Pymol的命令,3D视图窗口用于显示分子结构。
第二步是加载分子结构。
Pymol可以加载多种格式的分子结构文件,包括PDB、MOL、SDF等。
在主菜单中选择"File",然后选择"Open",浏览并选择需要加载的分子结构文件,点击"Open"按钮即可加载分子结构。
加载分子结构后,可以使用鼠标左键拖动分子结构进行旋转,使用鼠标右键拖动进行平移,使用鼠标滚轮进行缩放。
第三步是选择和操作分子结构。
在Pymol中,可以使用命令行窗口或主菜单中的"Select"工具选中分子结构的特定部分。
例如,输入命令"select a, resi 10-20"将选择分子结构中氨基酸残基编号为10到20的部分。
选中分子结构后,可以使用命令行窗口或主菜单中的"Display"工具对其进行操作。
例如,输入命令"hide lines, a"将隐藏所选部分的结构连线。
除了对分子结构进行选择和操作外,Pymol还提供了一些可视化功能。
例如,可以使用命令行窗口或主菜单中的"Color"工具对分子结构进行着色。
例如,输入命令"color red, a"将所选部分的颜色设置为红色。
另外,Pymol还提供了一些高级功能,如分子对接、蛋白质构象分析和分子动力学模拟等。
这些功能超出了本教程的范围,可以在Pymol的官方网站上查找更多关于这些功能的资料。
PyMOL使用入门

PyMOL使用入门安装完成后,启动PyMOL。
PyMOL可以通过命令行启动,也可以通过图形用户界面(GUI)启动。
启动后,会出现一个主窗口,其中包含菜单栏、工具栏和视图窗口。
在视图窗口中,可以使用鼠标右键进行旋转、平移和缩放的操作。
熟练掌握这些操作可以帮助我们更好地查看和分析分子结构。
要加载一个分子结构,可以使用菜单栏中的“File”->“Open”命令,或者直接拖拽文件到视图窗口中。
PyMOL支持多种常见的分子文件格式,如PDB、MOL2和XYZ。
加载分子后,可以使用命令行或菜单栏中的工具进行各种操作。
例如,可以使用“Actions”菜单中的“Align”命令将两个分子进行结构对齐,或者使用“Colors”菜单中的“Spectrum”命令将蛋白质按照氨基酸序列进行上色。
PyMOL还提供了许多高级功能,如图像渲染、分析和可视化脚本编写。
通过菜单栏的“Wizard”->“Visualization”可以打开一个可视化向导来创建各种高级效果,如球面细胞、水分子和草图效果。
例如,可以使用Python脚本来计算两个分子之间的距离、绘制氢键网络、生成蛋白质表面等。
可以在PyMOL的官方网站或在线社区中找到许多示例脚本和教程,以帮助学习和使用这些功能。
总之,PyMOL是一个功能强大且灵活的分子可视化工具,可以帮助科研人员和学生进行生物分子的可视化和分析工作。
通过掌握基本操作和使用高级功能,可以更好地利用PyMOL来研究和理解分子结构。
希望通过本文的介绍,读者能够对PyMOL有一个初步的了解,并能够开始使用和探索这个强大的工具。
pymol常用命令

pymol常用命令Pymol是一款强大的分子可视化软件,常用于生物医药领域的蛋白结构和分子模拟等研究。
下面是一些Pymol常用命令的相关参考内容。
1. 启动Pymol要启动Pymol,只需要在终端或命令行中输入pymol的命令即可。
启动后,Pymol的交互界面会出现,用户可以通过命令行输入命令来操作。
2. 导入和加载分子结构要导入或加载一个分子结构,在Pymol的命令行中输入load命令,后面跟上分子结构文件的路径。
例如,load protein.pdb就会加载一个名为protein.pdb的PDB文件。
3. 显示和调整分子结构的外观在Pymol中,可以使用多种命令来显示和调整分子结构的外观。
例如,使用show命令可以显示分子结构的所有原子和键,使用hide命令可以隐藏特定类型的原子或键。
还可以使用color命令来改变分子结构的颜色,使用spectrum命令可以根据原子属性自动上色。
4. 选择和操作特定的分子片段在Pymol中,可以使用select命令来选择特定的分子片段。
例如,select protein, chain A就会选择链A中的所有原子。
选择完成后,可以使用一系列的命令来操作选定的分子片段,如移动、旋转、缩放等。
5. 创建和编辑分子结构在Pymol中,可以使用多种命令创建和编辑分子结构。
例如,使用create命令可以创建一个新的分子对象,使用alter命令可以编辑分子结构中的原子属性,使用bond命令可以在分子结构中创建新的键等。
6. 绘制图形和标签Pymol提供了丰富的绘图和标签命令,以帮助用户更好地可视化分子结构。
例如,使用label命令可以在分子结构中添加注释标签,使用cartoon命令可以绘制蛋白质的二级结构等。
7. 输出和保存可视化结果在Pymol中,可以使用多种命令将可视化结果输出和保存为不同的文件格式。
例如,使用png命令可以将当前屏幕截图保存为PNG图像文件,使用ray命令可以渲染高质量的分子图像,使用save命令可以将分子结构保存为PDB或其他格式的文件。
pymol教程

pymol教程PyMOL⽤户指南⽬录⼀、⿏标操作⼊门4(这个数字是超链接,ctrl+左键)1.启动41)通过⿏标42)通过命令⾏42.PyMOL窗⼝41)Virewer窗⼝42)外部GUI窗⼝53.下载PDB⽂件54.操控视图61)基本⿏标控制62)虚拟滚动球旋转73)移动截⾯74)改变旋转中⼼点85)简单回顾9⼆、命令⾏操作⼊门91.记录结果92.载⼊数据93.操控对象(Object)101)原⼦选择112)对象和选择的着⾊123)对象和选择的on/off134.改变视点135.保存⼯作141)脚本和⽇志⽂件142)图像⽂件153)会话⽂件166.命令⾏快捷键161)⽤TAB键完成命令162)⽤TAB键完成⽂件名173)⾃动推理177.其他命令和帮助17注:页⾯背景和页脚的图像分别是1GCL、111D的cartoon显⽰三、命令句法和原⼦选择181.语法181)选择表达182)原⼦选择命名193)单字选择符4)属性选择符205)选择代数226)宏指令232.从PyMOL中读取Python 24四、卡通表⽰251.背景251)可达性252)美化和精确262.定制化281)卡通类型282)精美螺旋313.⼆级结构归属32五、光线追踪331.重要设置332.保存图⽚34六、⽴体效果341.⽀持的⽴体模式342.制作⽴体图⽚343.相关命令34七、动画351.概念352.重要命令351)Load2)Mset3)Mdo4)Mmatrix3.简单举例364.复杂举例365.预览ray-traced动画图⽚37 1)Cache_frames 2)mclear6.保存动画37⼋、⾼级⿏标控制371.选择原⼦和键 372.“pk”原⼦选择的应⽤举例383.“lb”和“rb”选择 384.构象编辑 38九、晶体应⽤381.晶体对称性381)Load2)Symexp2.电⼦密度图391)Load2)Isomesh和isodot⼗、汇编图形对象(CGO)和Molscript ribbons 401.简介 402.Molscript ribbons 401)Load2)Using Molscript3.创建CGOs 414.CGO参考 41NOTES:本教程以PyMOL user’s guide为蓝本翻译⽽来,并引⽤了其他资料。
pymol使用教程

基本的鼠标操作里主要介绍一下Pymol的基本操作,包括窗口菜单、加载文件、图像的基本鼠标操作等等。
当你打开Pymol后,你将会看到如下图所示的界面:该界面分为2窗口,上面的外部GUI窗口(External GUI)和下面的Viewer Window。
Viewer Window又分为左右两块,左边用来显示结构图像的(Viewer),右边则是一个内部GUI窗口(Internal GUI)。
Viewer自身包含一个命令行(如图中左下方的PyMOL>提示符),可以用来输入Pymol命令;在Inernal GUI中则可以选定一些特定的对象并完成一些操作。
External GUI则包含一个标准菜单、一个输出区、一个命令行输入区以及右边的一些常用命令按钮。
请注意,标准的“复制、剪切和粘贴”操作只能在External GUI中完成,并且必须使用“Ctrl+C、Ctrl+X以及Ctrl+V”来完成,这也是这个所谓的外部GUI的最重要的优点。
加载文件,有二种方法:1.在External GUI中选择File - Open2.使用命令行:load<filename>例如我们现在从上下载了一个离子通道蛋白的pdb文件(PENTAMERIC LIGAND GATED ION CHANNEL FROM ERWINIA CHRYSANTHEMI),名字为2vl0.pdb,然后用pymol打开它:load 2vl0该蛋白质的结构就被显示出来啦,如下图:基本的图像操作:是不是觉得上面的那个三维结构图看起来乱七八糟的阿,那是因为蛋白质分子都是由成千上万个原子组成的,而Pymol打开pdb文件时是默认把所有的原子都显示在那个小小的Viewer窗口里面的,当然看起来就很乱了。
这时候就需要我们对这个图像进行一些操作,来得到漂亮的清晰的蛋白质三维结构图。
先说说鼠标吧。
∙任意旋转图像:对准图像的任意处点住鼠标左键然后移动鼠标。
pymol使用教程中文

pymol使用教程中文PyMOL是一款用于生物分子可视化的强大软件工具。
它提供了许多功能和特性,可以帮助科学家和研究人员在研究蛋白质、核酸以及其他生物分子时进行结构分析和可视化展示。
本教程旨在向初学者介绍PyMOL的基本用法和操作技巧,帮助读者快速上手并熟练使用这一工具。
一、安装和启动PyMOL首先,您需要下载适用于您的操作系统的PyMOL安装程序。
在官方网站上获取安装程序,并按照提示进行安装。
安装完成后,您可以通过点击桌面上的PyMOL图标或者输入命令来启动PyMOL。
二、PyMOL界面和基本操作PyMOL的界面由几个部分组成,包括菜单栏、工具栏、命令行和视图窗口等。
在视图窗口中,您可以加载并可视化生物分子结构。
在PyMOL中,您可以使用鼠标和键盘进行交互操作,如旋转、平移、缩放等。
1. 加载分子结构文件在PyMOL中,您可以通过点击菜单栏中的“文件”选项,然后选择“打开”来加载分子结构文件。
常见的分子结构文件格式包括PDB、XYZ和MOL2等。
选择合适的文件并加载它们到PyMOL中。
2. 调整视图和可视化选项PyMOL提供了许多视图和可视化选项,可以帮助您呈现和展示生物分子结构。
您可以通过菜单栏或者命令行来调整视图和可视化选项,如改变颜色、显示盒子、添加标签等。
3. 交互操作和结构分析通过鼠标和键盘的交互操作,您可以旋转、平移、缩放和选择生物分子结构。
鼠标左键用于旋转,中键用于平移,右键用于缩放。
您还可以使用命令行来执行更高级的结构分析操作,如计算距离、测量角度等。
三、PyMOL高级功能除了基本操作外,PyMOL还提供了许多高级功能和特性,可以扩展其应用范围和功能。
以下是一些PyMOL的高级功能的简要介绍:1. 脚本和自动化通过编写脚本,您可以实现自动化操作和批量处理多个结构文件。
PyMOL的脚本语言非常灵活和强大,您可以使用它来实现各种自定义功能和工作流程。
2. 插件和扩展PyMOL支持插件和扩展,可以帮助您增强其功能和工作效率。
pymol 蛋白互作结构

pymol 蛋白互作结构使用`Pymol`软件可以观察和分析蛋白质之间的相互作用结构。
以下是一个示例,展示了如何使用`Pymol`来观察蛋白质互作结构:1. 导入蛋白质结构文件:使用`Pymol`的`load`命令或点击菜单中的"File"->"Load",将蛋白质结构文件(例如 PDB 文件)导入到`Pymol`中。
2. 显示蛋白质结构:在`Pymol`的图形界面中,可以看到导入的蛋白质结构。
你可以使用鼠标拖动、旋转和缩放来查看不同角度和放大倍数下的结构。
3. 选择互作的蛋白质分子:使用`Pymol`的选择工具(例如,点击"Select"工具或使用快捷键),选择你感兴趣的蛋白质分子。
4. 观察蛋白质互作:在`Pymol`中,你可以使用各种显示样式和颜色来突出显示蛋白质之间的相互作用。
例如,你可以使用"Surface"显示样式来查看蛋白质的表面,或者使用"Cartoon"显示样式来查看蛋白质的卡通模型。
5. 分析蛋白质互作:`Pymol`提供了一些分析工具,帮助你研究蛋白质之间的相互作用。
例如,你可以使用"Distance Measurement"工具来测量蛋白质分子之间的距离,或者使用"Contacts"工具来查看蛋白质之间的接触点。
这只是一个简单的示例,`Pymol`提供了许多其他功能和工具,可根据具体需求进行更深入的分析和可视化。
请注意,在使用`Pymol`进行蛋白质互作结构分析时,需要一定的蛋白质结构知识和生物信息学背景。
如果你对蛋白质结构分析不熟悉,建议先学习相关的基础知识和教程。
pythonmolecularviewer使用方法

pythonmolecularviewer使用方法Python Molecular Viewer (PyMol)是一个用于分子可视化和模拟的Python程序。
它提供了一套强大的工具和函数,可以在二维和三维空间中显示和操作分子结构。
PyMol安装方法:2.安装二进制文件,并确保将其添加到系统的路径中。
3. 在终端或命令提示符下,输入“pymol”命令,以启动PyMol。
现在,让我们来探索PyMol的使用方法:1.打开分子结构:- 使用命令`load`加载已经存在于本地的分子文件。
例如,`load molecule.pdb`。
2.导航和旋转:-使用鼠标左键点击并拖动以旋转分子。
-使用鼠标中键或滚轮放大或缩小分子。
-使用鼠标右键点击并拖动以平移分子。
3.选择和高亮分子:- 使用命令`select`选择特定的原子。
例如,`select protein, chain A`将选择名为“chain A”的蛋白质链。
- 使用命令`zoom`或`center`聚焦和居中选择的分子部分。
例如,`zoom protein`将放大和居中显示选择的蛋白质链。
4.修改分子显示样式:- 使用命令`show`显示特定的分子组件。
例如,`show cartoon`将显示蛋白质的卡通样式。
- 使用命令`hide`隐藏特定的分子组件。
例如,`hide lines`将隐藏分子的线框模式。
- 使用命令`color`为分子组件着色。
例如,`color red, protein`将蛋白质链的颜色设置为红色。
5.添加分子表面:- 使用命令`show surface`显示分子的表面。
- 使用命令`set transparency`设置表面的透明度。
例如,`set transparency, 0.5`将表面的透明度设置为50%。
- 使用命令`color`为表面着色。
例如,`color blue, surface`将表面的颜色设置为蓝色。
6.创建视图和图像:- 使用命令`orient`将分子结构重新定向到初始位置。
pymol基础教程

p y m o l基础教程(总25页)--本页仅作为文档封面,使用时请直接删除即可----内页可以根据需求调整合适字体及大小--简介&安装Pymol是一个开放源码,由使用者赞助的分子三维结构显示软件,由Warren Lyford DeLano编写,并且由DeLano Scientific LLC负责商业发行。
Pymol被用来创作高品质的分子(特别是生物大分子如蛋白质)三维结构。
据软件作者宣称,在所有正式发表的科学论文中的蛋白质结构图像中,有四分之一是使用Pymol来制作的。
Pymol名字的来源:“Py”表示该软件基于python这个计算机语言,“Mol”则是英文分子(molucule)的缩写,表示该软件用来显示分子结构。
由于实验需要,本人正在学习该软件,在这里把学习过程记录下来,希望对有需要的朋友有所帮助。
今天先来说说安装吧。
自2006年8月1日起,DeLano Scientific 对事先编译好的PyMOL执行程序(包括beta版)采取限定下载的措施。
目前,只有付费用户可以取得。
不过源代码目前还是可以免费下载,供使用者编译。
如果你和我一样,不想为此花钱的话:1.如果你是Windows用户,首先下载Pymol的源代码。
然后安装CygWin,并且确保正确安装以下模块:C++ (gcc or g++ package name)PythonOpenGLPNG然后在源代码目录里面依次运行:2.如果你是Linux用户,首先确保以下东东已安装:PythonPmwOpenGL driver(我用的是NVdia)libpngSubversion client(下载源代码需要)然后下载Pymol的源代码$ mkdir pymol-src$ svn co pymol-src然后进入源代码目录# cd pymol-src开始依次编译# python install# python install拷贝执行脚本到某个$PATH,安装就搞定了# cp ./pymol /usr/bin如果运行时得到错误信息"ImportError: No module named Pmw",那么你应该运行# python install pmw如果你在使用Gentoo,请确保编译python时添加了tcl/tk支持,否则运行是会提示错误"ImportError: No module named _tkinter"# USE="tcl tk" emerge python好了,下面我们就可以进入Pymol的世界了。
pymol用法(教程二)

[转载]PyMOL用法(教程二)基础Pymol命令这里主要介绍一下Pymol的一些基本命令操作。
就像Linux 一样,要想更好的操作Pymol,掌握一些常用的命令是必不可少的。
Pymol是区分大小写的,不过目前为止Pymol还是只用小写,所以记住,所有的命令都是使用小写字母的。
当你开始用Pymol来完成一个项目时,你也许想会让Pymol自动保存你所有输入过的命令,以方便日后你再次读取并修改。
这个可以通过创建一个log文件来达到,该文件的后缀名应为.pml,记住,Pymol像Linux一样支持Tab键命令补全:Pymol> log_open log-file-name.pml如果你想终止记录,只需要键入:Pymol> log_close好了,现在载入pdb文件(继续前用的pdb文件):Pymol> load 2vlo.pdb现在Pymol就创建了一个叫2vlo的对象,你可以在内部GUI窗口里面看见这个项目的名字。
但是你也可以自己定义该项目的名字(如test):Pymol> load 2vlo.pdb, test下面说说如何来操作你新建的对象。
首先:Pymol> show representationPymol> hide representation其中representation可以为:cartoon, ribbon,dots, spheres, surface和mesh。
使用这2个命令可以让Pymol以不同的方式显示蛋白质结构。
例如当我们键入:Pymol> hide linesPymol> show ribbon我们将得到如下结果:也许你已经注意到结构中有2个一模一样的蛋白质分子,只是方向不同而已,那么如何让Pymol只显示当中的一个分子呢?首先输入如下命令:Pymol> label all, chains这个命令的作用是让Pymol给蛋白质结构中的“链”编号,你会发现,第一个分子由“链”A-E组成,第二个则由F-J组成。
pymol使用教程

pymol使用教程Pymol是一款用于分子建模和可视化的强大工具,可以帮助研究人员更好地理解和探索分子结构。
下面是一个简单的Pymol使用教程,帮助您入门:1. 安装Pymol:首先,您需要从Pymol官方网站下载并安装Pymol软件。
根据您的操作系统版本选择合适的安装程序,并按照提示进行安装。
2. 启动Pymol:安装完成后,您可以双击Pymol图标启动软件。
一旦启动,您将看到一个Pymol的主窗口。
3. 导入分子结构:要导入您的分子结构,可以从Pymol菜单栏中选择“File”(文件)>“Open”(打开),然后浏览并选择您的分子文件。
4. 显示分子结构:一旦成功导入分子结构,Pymol会在主窗口中显示分子。
您可以使用鼠标左键旋转和移动分子结构,使用鼠标右键放大和缩小分子结构。
5. 改变分子显示方式:Pymol提供许多不同的分子显示方式,如线框模型、球棍模型、表面模型等。
您可以通过点击Pymol菜单栏中的“Display”(显示)来选择不同的显示方式,并在下拉菜单中选择适当的选项。
6. 调整颜色和透明度:您可以使用Pymol菜单栏中的“Color”(颜色)和“Transparency”(透明度)选项来调整分子的颜色和透明度。
您可以选择预设的颜色和透明度设置,或者自定义您喜欢的设置。
7. 添加标签和注释:如果您想在分子结构中添加标签和注释,则可以使用Pymol菜单栏中的“Label”(标签)和“Annotation”(注释)选项。
您可以选择分子中的特定原子或残基,并添加您想要显示的标签和注释。
8. 导出图像或动画:一旦您完成了分子结构的可视化,您可以使用Pymol菜单栏中的“File”(文件)>“Export”(导出)选项将图像或动画保存为常见的图像或视频格式。
这是一个简单的Pymol使用教程,它会帮助您开始使用Pymol 进行分子建模和可视化。
随着您的实践与探索,您将能够更深入地了解Pymol的各种高级功能和功能。
pymol使用指南

pymol使用指南Pymol是一款用于可视化蛋白质和分子的开源软件。
它提供了许多功能和工具,使用户能够准确而美观地展示和分析分子结构。
下面是Pymol的使用指南。
1. 下载和安装:首先,从Pymol的官方网站(2. 打开Pymol:安装完成后,打开Pymol软件。
界面会显示一个空白画布,以及菜单栏和工具栏。
用户可以使用菜单栏中的各个选项,通过命令行或者Python 脚本来控制Pymol的操作。
3. 导入分子结构:使用Pymol之前,需要导入要分析的分子结构。
用户可以将分子结构从PDB(蛋白质数据库)文件中导入,或者使用命令行或Python脚本导入其他文件格式,如XYZ或MOL。
4. 显示分子结构:在Pymol中,用户可以调整和控制分子结构的显示方式。
例如,用户可以选择显示线框模型、球棍模型、表面模型或半透明模型。
通过菜单栏中的"Display"选项,用户可以调整颜色、透明度、线宽和渲染方式等参数。
5. 选择和标记:Pymol提供了各种选择和标记分子结构的方法。
用户可以通过命令行、Python脚本或图形界面来选择分子中的特定原子、残基或链。
用户还可以给分子中的特定部分添加标签、箭头或球。
6. 组合和编辑:Pymol允许用户组合多个分子结构,并进行编辑。
用户可以使用命令行或Python脚本来操作和修改分子结构的位置、旋转、缩放和翻转等。
此外,Pymol还提供了多种对称操作和变换功能,如对称翻转和镜像。
7. 分析和计算:Pymol还提供了一些强大的分析和计算工具,用于分析分子结构的性质和相互作用。
例如,用户可以计算蛋白质的二级结构、表面积、分子对接能力和相互作用能量等。
通过菜单栏中的"Analyze"选项,用户可以选择所需的分析功能。
8. 创建动画和图片:使用Pymol,用户可以创建分子结构的动画和图片。
用户可以通过命令行或Python脚本来设置动画的帧数、速度和循环次数等参数。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
简介&安装Pymol是一个开放源码,由使用者赞助的分子三维结构显示软件,由Warren Lyford DeLano编写,并且由DeLano Scientific LLC负责商业发行。
Pymol被用来创作高品质的分子(特别是生物大分子如蛋白质)三维结构。
据软件作者宣称,在所有正式发表的科学论文中的蛋白质结构图像中,有四分之一是使用Pymol来制作的。
Pymol名字的来源:“Py”表示该软件基于python这个计算机语言,“Mol”则是英文分子(molucule)的缩写,表示该软件用来显示分子结构。
由于实验需要,本人正在学习该软件,在这里把学习过程记录下来,希望对有需要的朋友有所帮助。
今天先来说说安装吧。
自2006年8月1日起,DeLano Scientific 对事先编译好的PyMOL执行程序(包括beta版)采取限定下载的措施。
目前,只有付费用户可以取得。
不过源代码目前还是可以免费下载,供使用者编译。
如果你和我一样,不想为此花钱的话:1.如果你是Windows用户,首先下载Pymol的源代码。
然后安装CygWin,并且确保正确安装以下模块:▪C++ (gcc or g++ package name)▪Python▪OpenGL▪PNG然后在源代码目录里面依次运行:2.如果你是Linux用户,首先确保以下东东已安装:▪Python▪Pmw▪OpenGL driver(我用的是NVdia)▪libpng▪Subversion client(下载源代码需要)然后下载Pymol的源代码$ mkdir pymol-src$ svn co https:///svnroot/pymol/trunk/pymolpymol-src然后进入源代码目录# cd pymol-src开始依次编译# python setup.py install# python setup2.py install拷贝执行脚本到某个$PATH,安装就搞定了# cp ./pymol /usr/bin如果运行时得到错误信息"ImportError: No module named Pmw",那么你应该运行# python setup2.py install pmw如果你在使用Gentoo,请确保编译python时添加了tcl/tk支持,否则运行是会提示错误"ImportError: No module named _tkinter"# USE="tcl tk" emerge python好了,下面我们就可以进入Pymol的世界了。
基本的鼠标操作里主要介绍一下Pymol的基本操作,包括窗口菜单、加载文件、图像的基本鼠标操作等等。
当你打开Pymol后,你将会看到如下图所示的界面:该界面分为2窗口,上面的外部GUI窗口(External GUI)和下面的Viewer Window。
Viewer Window又分为左右两块,左边用来显示结构图像的(Viewer),右边则是一个内部GUI窗口(Internal GUI)。
Viewer自身包含一个命令行(如图中左下方的PyMOL>提示符),可以用来输入Pymol命令;在Inernal GUI中则可以选定一些特定的对象并完成一些操作。
External GUI则包含一个标准菜单、一个输出区、一个命令行输入区以及右边的一些常用命令按钮。
请注意,标准的“复制、剪切和粘贴”操作只能在External GUI中完成,并且必须使用“Ctrl+C、Ctrl+X以及Ctrl+V”来完成,这也是这个所谓的外部GUI的最重要的优点。
加载文件,有二种方法:1.在External GUI中选择File - Open2.使用命令行:load <filename>例如我们现在从上下载了一个离子通道蛋白的pdb文件(PENTAMERIC LIGAND GATED ION CHANNEL FROM ERWINIA CHRYSANTHEMI),名字为2vl0.pdb,然后用pymol打开它:load 2vl0该蛋白质的结构就被显示出来啦,如下图:基本的图像操作:是不是觉得上面的那个三维结构图看起来乱七八糟的阿,那是因为蛋白质分子都是由成千上万个原子组成的,而Pymol打开pdb文件时是默认把所有的原子都显示在那个小小的Viewer窗口里面的,当然看起来就很乱了。
这时候就需要我们对这个图像进行一些操作,来得到漂亮的清晰的蛋白质三维结构图。
先说说鼠标吧。
∙任意旋转图像:对准图像的任意处点住鼠标左键然后移动鼠标。
∙放大/缩小图像:对准图像的任意处点住鼠标右键然后移动鼠标:向上是缩小,向下则是放大。
∙移动图像:对准图像的任意处点住鼠标中键或者滚轮,然后移动鼠标。
∙设定图像旋转中心: Ctrl+Shift+鼠标中键或滚轮。
∙移动剪切平面: Shift+鼠标右键。
鼠标上下移动:调整前剪切平面(离你近的);鼠标左右移动:调整后剪切平面(离你远的)。
最后一项“移动剪切平面”有点不容易理解,需要多试几次。
配合下面的示意图你会发现Pymol的这项设定其实很方便。
今天没时间了,明天还要出远门,就学到这里吧,用下面这个图作为结束,其实就是用cartoon的形式显示了上面的那个蛋白质,不过还比较难看。
By wei luPyMOL用法(教程二)基础Pymol命令这里主要介绍一下Pymol的一些基本命令操作。
就像Linux一样,要想更好的操作Pymol,掌握一些常用的命令是必不可少的。
Pymol是区分大小写的,不过目前为止Pymol还是只用小写,所以记住,所有的命令都是使用小写字母的。
当你开始用Pymol来完成一个项目时,你也许想会让Pymol自动保存你所有输入过的命令,以方便日后你再次读取并修改。
这个可以通过创建一个log文件来达到,该文件的后缀名应为.pml,记住,Pymol像Linux一样支持Tab键命令补全:Pymol> log_open log-file-name.pml如果你想终止记录,只需要键入:Pymol> log_close好了,现在载入pdb文件(继续前用的pdb文件):Pymol> load 2vlo.pdb现在Pymol就创建了一个叫2vlo的对象,你可以在内部GUI窗口里面看见这个项目的名字。
但是你也可以自己定义该项目的名字(如test):Pymol> load 2vlo.pdb, test下面说说如何来操作你新建的对象。
首先:Pymol> show representationPymol> hide representation其中representation可以为:cartoon, ribbon, dots, spheres, surface和mesh。
使用这2个命令可以让Pymol以不同的方式显示蛋白质结构。
例如当我们键入:Pymol> hide linesPymol> show ribbon我们将得到如下结果:也许你已经注意到结构中有2个一模一样的蛋白质分子,只是方向不同而已,那么如何让Pymol只显示当中的一个分子呢?首先输入如下命令:Pymol> label all, chains这个命令的作用是让Pymol给蛋白质结构中的“链”编号,你会发现,第一个分子由“链”A-E组成,第二个则由F-J组成。
好了,如果我们想把一个蛋白质分子去掉,那么只要把“链”A-E或者F-J去掉即可:Pymol> hide ribbon, chain f+g+h+i+j上面的东东还可以这样完成:Pymol> select test, chain f+g+h+i+jPymol> hide ribbon, test上面的第一句命令的作用是选择“链”F-J,并命名为test,然后在第二句命令中隐藏它。
这样做的好处是,一旦你选择并命名了某个目标,你可以在后面随时对它进行各种操作。
并且你在右边的控制面板里面也可以看到你选定的目标,并可以对其进行操作。
比如你可以:Pymol> hide everything, testPymol> show cartoon, test这样你会得到:说到这里就提到了Pymol的一个比较重要的东东,就是选择并命名目标,它的基本语法就是:Pymol> select selection-name, selection-expression其中名字可以由字母[A/a-Z/z],数字[0-9]已经下划线[_]组成,但是要避免使用:! @ # $ % ^ & * ( ) ' " [ ] { } \ | ~ ` <> ? /如果你要删除你选定的目标或者整个对象,你可以:Pymol> delete selection-namePymol> delete object-name下面讲讲如何给对象以及目标改变颜色。
预定义的颜色名字可以在外部GUI窗口的Settings - Colors中找到:Pymol> color color-namePymol> color color-name, selection-expression比如我们可以:Pymol> color red, ss hPymol> color yellow, ss sPymol> color green, ss l+""其中“ss”代表secondary structure,“h”代表Helix,“s”代表Beta sheet,l+""代表Loop和所以其他结构。
这3句的作用分别是把所有的Helix变成红色;把所有的Beta sheet变成黄色;把所有的Loop以及其他部分变成绿色,于是我们得到:Pymol可以同时打开多个pdb文件:Pymol> load object-name-1.pdbPymol> load object-name-2.pdb如果你想暂时关闭/打开某个对象,可以这样:Pymol> disable object-name-1Pymol> enable object-name-1你也可以用disable命令去除最后一个选择的目标上出现的粉红色的小点,但是该命令并不会使你选定的目标不可见。
Pymol> disable selection-name使用鼠标通常是改变图像视角的最方便的办法,不过命令如zoom,orient等等有时候使用起来也是很有用的,它们提供了另一种改变图像视角的办法。
放大选定目标:Pymol> zoom selection-name定向选定目标,可以使选定目标最大的尺寸水平显示,次大的尺寸竖直显示::Pymol> orient selection-name你也可以用view命令保存你目前的视角,注意,该命令只保存视角,并不保存你的对象显示方式:Pymol> view key, action其中“key”是你随便给当前视角定的名字,“action”可以为:store或者recall。