一次性医疗器械化学检验
常见医疗器械化学性能及评价

蒸发残渣
Eva 测定浸提液中的非挥发物。
Purpose: To determine the non-volatile matter in the extract.
The measurement objects of atomic absorption spectrophotometry are metal elements and some non-metal elements in atomic state. The characteristic spectrum line emitted by the lamp of the element to be tested passes through the atomic vapor generated by the atomization of the test sample, and is tested in the vapor. The ground state atom of the element is absorbed, and the content of the element to be tested in the test product is obtained by measuring the degree of weakening of the radiant light intensity.
方法二(硫化钠法) Method two (sodium sulfide method) 原理 principle 在碱性溶液中,铅、铬、铜、锌等重金属能与硫化钠作用生成不溶性有色硫化物。以铅为代表制备 标准溶液进行比色,测定重金属的总含量。 In alkaline solution, heavy metals such as lead, chromium, copper and zinc can react with sodium sulfide to form insoluble colored sulfides. A standard solution was prepared with lead as a representative for colorimetry to determine the total content of heavy metals.
一次性无菌医疗器械化学检验讲稿

氯化物
检验液如带颜色,采用标准加入法消除干扰 检验液两份,分置50mL纳氏比色管中 一份加硝酸银试液1.0mL,摇匀,放置10分钟, 如显浑浊,可反复过滤至滤液完全澄清,再加规 定量的标准氯溶液与水适量使成50mL,摇匀,在 暗处放置5分钟,作为对照液; 另一份中加硝酸银试液1.0mL与水适量使成50mL, 摇匀在暗处放置5分钟,作为供试液。 为防止过滤过程中引入滤纸中的氯产生污染,先 用热去离子水洗涤滤纸几次,再过滤检验液。
还原物质(易氧化物) 还原物质(易氧化物)
2.1.6 结果计算
V= (VS − V0 )CS C0
式中:V—消耗高锰酸钾标准滴定液的体积,mL; Vs—检验液消耗滴定液高锰酸钾标准溶液的体积,mL; Vo—空白液消耗滴定液高锰酸钾标准溶液的体积,mL; Cs—滴定液高锰酸钾标准溶液的实际浓度,mol/L; Co—标准中规定的高锰酸钾标准溶液的浓度,mol/L。
还原物质(易氧化物) 还原物质(易氧化物)
试验步骤 精确量取检验液20mL置于锥形瓶中,精确加 入产品标准中规定浓度的高锰酸钾标准滴定液 3mL,硫酸溶液5mL,加热至沸并保持微沸 3mL 5mL 10min,稍冷后精确加入对应浓度的草酸钠溶 液5mL,置于水浴上加热至75℃~80℃。用产 品标准中规定浓度的高锰酸钾标准滴定液滴定 至显微红色,并保持30s不褪色为终点,同时 与同批空白对照液相比较。
医疗器械质量管理体系-纯化水化学性能检测记录
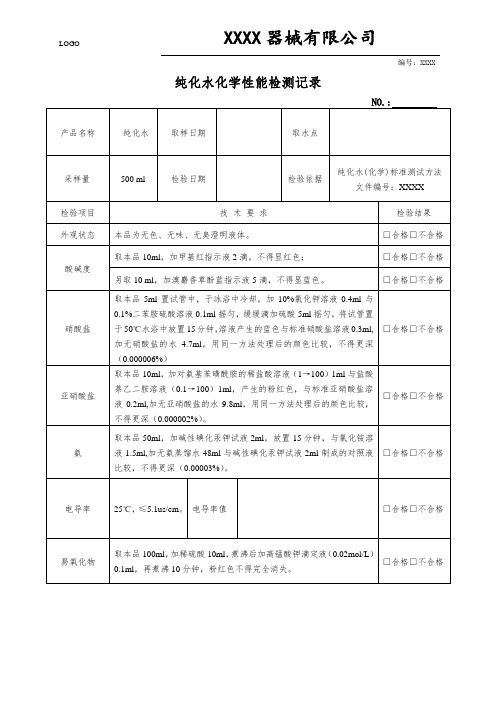
XXXX器械有限公司
编号:XXXX
纯化水化学性能检测记录
NO.:
产品名称
纯化水
取样日期
取水点
采样量
500ml
检验日期
检验依据
纯化水(化学)标准测试方法
文件编号:XXXX
检验项目
技术要求
检验结果
外观状态
本品为无色、无味、无臭澄明液体。
□合格□不合格
酸碱度
取本品10ml,加甲基红指示液2滴,不得显红色;
□合格□不合格
亚硝酸盐
取本品10ml,加对氨基苯磺酰胺的稀盐酸溶液(1→100)1ml与盐酸萘乙二胺溶液(0.1→100)1ml,产生的粉红色,与标准亚硝酸盐溶液0.2ml,加无亚硝酸盐的水9.8ml,用同一方法处理后的颜色比较,不得更深(0.000002%)。
□合格□不合格
氨
取本品50ml,加碱性碘化汞钾试液2ml,放置15分钟,与氯化铵溶液1.5ml,加无氨蒸馏水48ml与碱性碘化汞钾试液2ml制成的对照液比较,不得更深(0.00003%)。
□合格□不合格
不挥发物
空蒸发皿重
残渣不得过1mg
□合格□不合格
空蒸发皿+残渣
结论:□合格□不合格
备注:“在结果判断”一栏,若合格,打“√”,若不合格“X”。
检验人/日期:
复核人/日期:
□合格□不合格
另取10 ml,加溴麝香草酚蓝指示液5滴,不得显蓝色。
□合格□不合格
硝酸盐
取本品5ml置试管中,于冰浴中冷却,加10%氯化钾溶液0.4ml与0.1%二苯胺硫酸溶液0.1ml摇匀,缓缓滴加硫酸5ml摇匀,将试管置于50℃水浴中放置15分钟,溶液产生的蓝色与标准硝酸盐溶液0.3mБайду номын сангаас,加无硝酸盐的水4.7ml,用同一方法处理后的颜色比较,不得更深(0.000006%)
医疗器械型式检验、检测生物、化学送样数量说明
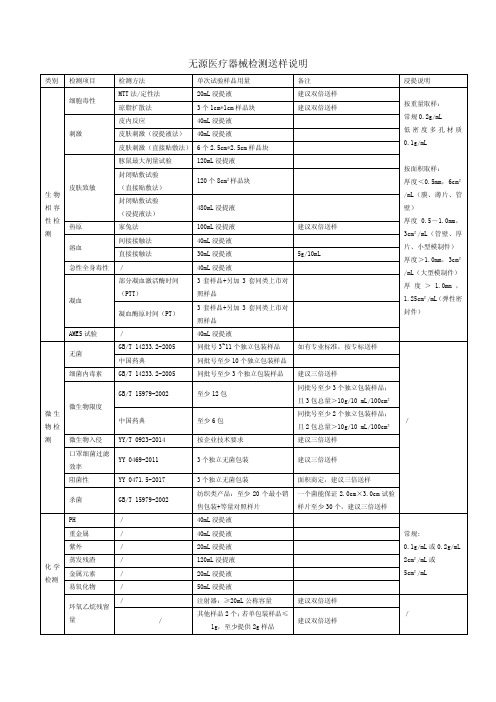
类别 检测项目
检测方法
单次试验样品用量
备注
浸提说明
细胞毒性 刺激
MTT 法/定性法
20mL 浸提液
琼脂扩散法
3 个 1cm*1cm 样品块
皮内反应
40mL 浸提液
皮肤刺激(浸提液法) 40mL 浸提液
皮肤刺激(直接贴敷法) 6 个 2.5cm*2.5cm 样品块
建议双倍送样 建议双倍送样
1g,至少提供 2g 样品
480mL 浸提液
100mL 浸提液
建议双倍送样
40mL 浸提液
30mL 浸提液
5g/10mL
40mL 浸提液
3 套样品+另加 3 套同类上市对
照样品
3 套样品+另加 3 套同类上市对
照样品
按面积取样: 厚度<0.5mm,6cm² /mL(膜、薄片、管 壁) 厚度 0.5~1.0mm, 3cm²/mL(管壁、厚 片、小型模制件) 厚度>1.0mm,3cm² /mL(大型模制件) 厚 度 > 1.0mm , 1.25cm²/mL(弹性密 封件)
AMES 试验
/
40mL 浸提液
无菌
GB/T 14233.2-2005 中国药典
同批号 3~11 个独立包装样品 同批号至少 10 个独立包装样品
如有专业标准,按专标送样
细菌内毒素 GB/T 14233.2-2005
同批号至少 3 个独立包装样品 建议三倍送样
微生 物检
微生物限度
GB/T 15979-2002 中国药典
效率
ቤተ መጻሕፍቲ ባይዱ
3 个独立无菌包装
建议三倍送样
阻菌性
YY 0471.5-2017
医疗器械产品检验检测工作内容

医疗器械产品检验检测工作内容一、医疗器械产品检验检测工作概述医疗器械产品检验检测工作是指对生产出来的医疗器械产品进行各种技术性能和安全性能的测试,以确保其符合国家和行业标准的要求,同时保障使用者的健康和安全。
医疗器械产品检验检测工作是一个复杂而又严谨的过程,需要专业人员进行操作,严格遵循相关规定和标准。
二、医疗器械产品检验检测工作流程1.样品接收与登记样品接收与登记是医疗器械产品检验检测工作的第一步。
在接收到样品后,需要对样品进行登记,并记录相关信息,如样品名称、型号、规格、数量等。
2.外观质量检查外观质量检查是对医疗器械产品外观是否完好无损以及是否符合相关标准要求进行评估。
主要包括外观缺陷、尺寸偏差、表面处理等方面。
3.物理性能测试物理性能测试是对医疗器械产品在使用过程中可能发生的力学变化进行测试。
主要包括耐压、耐拉、耐弯曲等方面。
4.化学性能测试化学性能测试是对医疗器械产品的成分和化学特性进行分析和测试。
主要包括材料成分、溶解度、酸碱度等方面。
5.生物相容性测试生物相容性测试是对医疗器械产品与人体组织接触时是否会引起过敏反应进行评估。
主要包括皮肤刺激试验、黏膜刺激试验、注射试验等方面。
6.微生物检测微生物检测是对医疗器械产品是否存在致病菌进行检测。
主要包括细菌培养试验、真菌培养试验等方面。
7.电气安全测试电气安全测试是对带有电气部件的医疗器械产品是否符合相关电气安全标准进行评估。
主要包括绝缘电阻测试、漏电流测试等方面。
8.辐射安全检测辐射安全检测是对带有放射性元素的医疗器械产品是否符合相关辐射安全标准进行评估。
主要包括辐射剂量测试、辐射污染检测等方面。
9.环境适应性测试环境适应性测试是对医疗器械产品在不同环境条件下的使用情况进行测试。
主要包括温度、湿度、气压等方面。
10.功能性能测试功能性能测试是对医疗器械产品的功能是否符合相关标准要求进行评估。
主要包括使用寿命、稳定性、精度等方面。
11.报告编制与审核报告编制与审核是医疗器械产品检验检测工作的最后一步。
医疗器械化学检验操作规程

取2支纳氏比色管,分别加入25ml检验液、25ml铅标准液,每支试管再加入醋酸盐缓冲液(PH3.5)2ml,硫代乙酰胺试液2ml,摇匀,置白色背景下观察,比较颜色深浅。检验合格要求:颜色不得更深。
试剂配置:
1)醋酸盐缓冲液(pH 3.5):取醋酸铵25g,加水25ml溶解后,加7mol/L盐酸溶液38ml,用2mol/L盐酸溶液或5mol/L氨溶液准确调节pH值至3.5(电位法指示),用水稀释至100ml。
4)7mol/L盐酸:浓盐酸 294mL,定容至500ml。
5)2mol/L盐酸:7mol/L盐酸稀释3.5倍。
6)1mol/L氢氧化钠:40g氢氧化钠定容至1L。
7)硝酸为10%硝酸:71ml98%硝酸定容至1L,115ml65%硝酸定容至1L。
8)5mol/L氨溶液:350毫升浓氨水定容至1L。
医疗器械化学检验操作规程
1、目的Purpose:
为检验人员提供正确的医疗器械化学分析操作依据,保证检验程序的规范化、标准化。
2、范围Scope:
适用于本公司产品一次性使用医疗器械产品的化学分析。
3、职责Responsibility:
1
2
3
质量管理部相关工作人员按本规程操作。
4、定义Definition
2)硫代乙酰胺试液:取硫代乙酰胺4g,加水使溶解成100ml,置冰箱中保存。临用前取混合液(由1mol/L氢氧化钠溶液15ml、水5.0ml及甘油20ml组成)5.0ml,加上述硫代乙酰胺溶液1.0ml,置水浴上加热20秒,冷却,立即使用。
3)标准铅溶液:称取硝酸铅0.160g,置1000ml量瓶中,加硝酸10ml(1+9)溶解后,用水稀释至刻度,摇匀,作为贮备液。精密量取贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10µg的Pb)。本液仅供当日使用。
认可的医疗器械受检目录生化分析仪等

YY0033-2000无菌医疗器具生产管理规范
《药品生产质量管理规范》(1998年修订),GB/T 16292(16293、16294)-1996医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法
YBB00412004药品包装材料生产厂房洁净-室(区)的测试方法
13.
传统型血袋
全性能
GB 14232.1-2004/ISO 3826-1:2003
人体血液及血液成分袋式塑料容器,第一部分:传统型血袋
14.
与血液相互作用试验
部分性能
GB/T 16886.4-2003
医疗器械生物学评价第4部分:与血液相互作用试验选择
限做溶血
15.
体外细胞毒性试验
全性能
GB/T 16886.5-2003
医疗器械生物学评价第5部分:体外细胞毒性试验
16.
植入后局部反应试验
4.
一次性使用输血器
全性能
GB 836全性能
GB 18671-2002一次性使用静脉输液针
6.
天然胶乳橡胶避孕套
全性能
GB 7544-2004天然胶乳橡胶避孕套技术要求和试验方法
7.
医用脱脂棉
全性能
YY0330-2002医用脱脂棉
8.
医用脱脂纱布
全性能
GB/T18990.3-2003黄体生成素(LH)检测试纸第3部分:“安全期”避孕试纸及现行有效标准
21.
尿液检测(分析)试纸
全性能
YY/T0478-2004干化学尿液分析试纸条通用技术条件及现行有效标准
22.
临床化学体外诊断试剂
全性能
WS/T124-1999临床化学体外诊断试剂盒质量检验总则及现行有效标准
无菌医疗器械生化检验

常用浸提液制备方法
见附页
化学分析
1.浊度和色泽的测定
1.1 浊度的测定 1.1.1 仪器 a)分析天平:精确至0.1mg; b)分光光度计。 1.1.2溶液的配制 a)浊度标准贮备液的制备:称取于105℃干燥恒重的硫酸肼1.00g,
置于100mL容量瓶中,加水适量使溶解,必要时可先置于洁净烧杯中 在40℃的水浴中温热溶解,再用水转移至100mL容量瓶中,并稀释至 刻度,摇匀,放置4h~6h,取此溶液与等容量的10% 六亚甲基四胺 (乌洛托品)溶液混合,摇匀,于25℃避光静置24小时。标准贮备液 应置冷处避光保存,在两个月内使用,用前摇匀。 b) 浊度标准原液的制备:取浊度标准贮备液15.0mL,置1000mL容 量瓶中,加水稀释至刻度,摇匀,取适量,置1cm吸收池中,在 550nm处测定其吸光度值,结果应在0.12~0.15范围内。标准原液应 在48小时内使用,用前摇匀。
解,冷却后稀释定容。 cm)ocl/(LN草a2酸C2钠O4标)=0准.0溶05液m适o量l/L,草加酸水钠准标确准稀溶释液1:0倍用。前取0.05 d酸) 钾C(标1准/5溶KM液n:O4取)3=.30g.1高m锰ol/酸L 钾[c(,KM加n水O41)0=500.0m2Lm,o煮l/L沸]高1锰5
学研究设计 GB/T 16886.17-2005 医疗器械生物学评价 第17部分:可滤沥物允许限量的建立 GB/T 16886.20-200 医疗器械生物学评价 第20部分:医疗器械免疫毒理学试验原理和
方法 GB/T 16175-2008 医用有机硅材料生物学评价试验方法
GB14233.2-2005 医用输液、输血、注射器具检验方法 第2部分:生物学试验方法
(推荐医学)医疗器械的常见理化性能要求及检测

第三节 医疗器械的灭菌及环氧乙烷残留量检测P220
目前常用的灭菌方法有:湿热灭菌、环氧乙烷 灭菌、辐射灭菌,国家也颁布了3个标准: GB 18278(ISO11134)医疗保健产品灭菌确认和
常规控制要求工业湿热灭菌 GB18279-2000(ISO11135)医疗器械环氧乙烷灭菌
确认和常规控制 GB18280-2007(ISO11137)医疗保健产品灭菌 确认
2
第五章 医疗器械的常见理化性能要求及检测
医疗器械包括无源医疗器械和有源医疗器械, 无源医疗器械是指不依靠电能或其他能源驱动直 接由人体或重力产生的能源来发挥其功能的医疗 器械,主要包括植入性医疗器械和无菌医疗器械 等。
植入性医疗器械是指任何通过外科手段达到下 列目的的医疗器械:全部或部分插入人体或自然 腔道中或为替代上表皮或表面用的,并使其在体 内至少保留30d,且只能通过内科或外科手段取出。
10
第一节 医疗器械的常见物理性能要求
物理性能方面个人经验(小结) 1、多数产品需要规定外观、尺寸,基本参数一 般不要放在要求中;如3.1 外观;3.2尺寸 2、对于有粘合剂粘接的产品需要规定连接牢固 度; 3、根据产品特点规定流速、流量、密封性、硬 度、畅通性、压力、体积、行程; 4、无菌产品多数需要规定微粒污染指标; 5、透明度、含量(非化学成分)等。
制备检验液所用的方法应尽量使样品所有被测表 面都被萃取到。
推荐在表1中选择检验液制备方法
14
第二节 医疗器械的常见化学性能要求
一、溶出物及可渗出物含量的化学分析(P207) 1、检验液制备 推荐在表1中选择检验液制备方法 化学性能检验液制备方法GB14233.1.doc GB T14233.1-200X(报批稿).doc
国家食品药品监督管理局关于认可河南省医疗器械检验所对一次性使
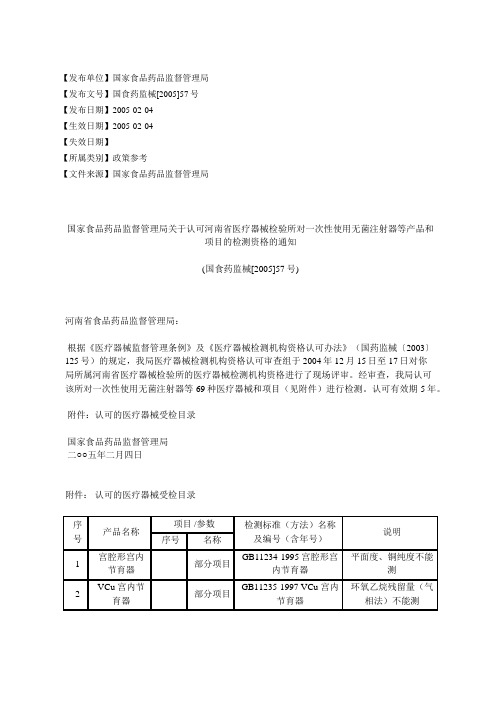
【发布单位】国家食品药品监督管理局【发布文号】国食药监械[2005]57号【发布日期】2005-02-04【生效日期】2005-02-04【失效日期】【所属类别】政策参考【文件来源】国家食品药品监督管理局国家食品药品监督管理局关于认可河南省医疗器械检验所对一次性使用无菌注射器等产品和项目的检测资格的通知(国食药监械[2005]57号)河南省食品药品监督管理局:根据《医疗器械监督管理条例》及《医疗器械检测机构资格认可办法》(国药监械〔2003〕125号)的规定,我局医疗器械检测机构资格认可审查组于2004年12月15日至17日对你局所属河南省医疗器械检验所的医疗器械检测机构资格进行了现场评审。
经审查,我局认可该所对一次性使用无菌注射器等69种医疗器械和项目(见附件)进行检测。
认可有效期5年。
附件:认可的医疗器械受检目录国家食品药品监督管理局二○○五年二月四日附件:认可的医疗器械受检目录本内容来源于政府官方网站,如需引用,请以正式文件为准。
出师表两汉:诸葛亮先帝创业未半而中道崩殂,今天下三分,益州疲弊,此诚危急存亡之秋也。
然侍卫之臣不懈于内,忠志之士忘身于外者,盖追先帝之殊遇,欲报之于陛下也。
诚宜开张圣听,以光先帝遗德,恢弘志士之气,不宜妄自菲薄,引喻失义,以塞忠谏之路也。
宫中府中,俱为一体;陟罚臧否,不宜异同。
若有作奸犯科及为忠善者,宜付有司论其刑赏,以昭陛下平明之理;不宜偏私,使内外异法也。
侍中、侍郎郭攸之、费祎、董允等,此皆良实,志虑忠纯,是以先帝简拔以遗陛下:愚以为宫中之事,事无大小,悉以咨之,然后施行,必能裨补阙漏,有所广益。
将军向宠,性行淑均,晓畅军事,试用于昔日,先帝称之曰“能”,是以众议举宠为督:愚以为营中之事,悉以咨之,必能使行阵和睦,优劣得所。
亲贤臣,远小人,此先汉所以兴隆也;亲小人,远贤臣,此后汉所以倾颓也。
先帝在时,每与臣论此事,未尝不叹息痛恨于桓、灵也。
侍中、尚书、长史、参军,此悉贞良死节之臣,愿陛下亲之、信之,则汉室之隆,可计日而待也。
1类医疗器械检验标准

1类医疗器械检验标准摘要:1.1类医疗器械的定义和范围2.1类医疗器械的检验标准概述3.1类医疗器械的具体检验项目和方法4.1类医疗器械检验标准的实施和监管5.1类医疗器械检验标准的意义和作用正文:一、1类医疗器械的定义和范围1类医疗器械是指具有较低风险的医疗器械,主要包括以下类别的产品:- 常规手术器械(如手术刀、镊子、剪刀等);- 一次性使用医疗器械(如注射器、输液器、采血器等);- 医疗设备(如体温计、血压计、心电图机等);- 康复器材(如轮椅、拐杖、助行器等);- 医疗耗材(如棉签、棉球、敷料等);- 体外诊断试剂和设备(如生化分析仪、免疫分析仪等)。
二、1类医疗器械的检验标准概述1类医疗器械的检验标准主要包括以下几个方面:- 产品技术要求:包括功能性、安全性指标以及与质量控制相关的其他指标;- 原材料检验:对原材料的化学成分、物理性能、生物学性能等进行检验;- 生产工艺检验:对生产工艺的稳定性、可重复性等进行检验;- 成品检验:对成品的性能、质量、安全性等进行检验。
三、1类医疗器械的具体检验项目和方法1类医疗器械的具体检验项目和方法依据产品的类别和特性而定,以下是一些常见的检验项目和方法:- 物理性能检验:如硬度、韧性、耐磨性等;- 化学性能检验:如腐蚀性、毒性、过敏性等;- 生物学性能检验:如细胞毒性、致敏性、刺激性等;- 功能性检验:如设备的功能、精度和稳定性等;- 安全性检验:如电气安全、机械安全、生物安全等。
四、1类医疗器械检验标准的实施和监管1类医疗器械检验标准的实施和监管主要依靠国家和地方食品药品监督管理部门。
企业应按照标准要求进行产品检验,确保产品质量符合标准要求。
监管部门会对企业进行定期检查和监督,确保企业遵守检验标准。
五、1类医疗器械检验标准的意义和作用1类医疗器械检验标准对于确保医疗器械的安全性、有效性和质量具有重要意义。
它有助于:- 规范医疗器械的生产和质量控制;- 保障患者和用户的使用安全;- 促进医疗器械行业的健康发展;- 维护国家的公共卫生安全。
医疗器械的质量标准及检验方法

医疗器械的质量标准及检验方法近年来,随着医疗器械的不断更新和广泛应用,人们对医疗器械的质量和安全性的要求日益提高。
医疗器械的质量标准及检验方法成为了一个备受关注的话题。
本文将从医疗器械的质量标准和检验方法两个方面进行探讨。
首先,医疗器械的质量标准是保障其安全性和有效性的基础。
医疗器械的质量标准由国家相关机构制定,并依法实施。
这些标准旨在确保医疗器械在设计、生产和使用过程中能够满足一定的技术要求,以确保患者的安全和治疗效果。
医疗器械的质量标准涵盖了各个方面,包括设计要求、生产工艺、材料选择、产品性能等。
例如,对于一种外科手术器械,其质量标准要求机械结构稳定、使用寿命长、材料无毒无害、易于清洁和消毒等。
而对于一种医用导管,其质量标准则要求产品内外表面光滑、耐化学性能好、可靠性高等。
其次,医疗器械的质量标准需要进行严格的检验。
只有通过检验,才能确定医疗器械是否符合质量标准。
医疗器械的检验方法有多种,包括物理检验、化学检验、生物学检验等。
物理检验主要是对医疗器械的尺寸、外观等进行检查。
例如,对于一种医用注射器,物理检验会检查其长度、直径、注射针的锋利度等。
物理检验的目的是确保医疗器械的尺寸符合要求,外观无明显瑕疵。
化学检验则是对医疗器械中的化学成分进行分析。
这种检验方法主要用于检测或排除医疗器械可能存在的有害物质,如重金属、有毒溶剂等。
化学检验需要使用专业的设备和试剂,确保分析结果的准确性和可靠性。
生物学检验主要是对医疗器械的生物相容性进行评估。
通过进行细胞培养、动物实验等方法,确定医疗器械对组织和生物体的刺激性、毒性等。
生物学检验的目的是排除医疗器械可能对人体产生的不良影响,确保其安全性。
除了物理、化学和生物学检验外,还有一些特殊的检验方法,如放射性检验、病原体检验等,针对不同类型的医疗器械有不同的检验要求。
总之,医疗器械的质量标准及检验方法是确保其安全和有效的关键。
只有通过严格的检验,才能确定医疗器械是否符合质量标准,从而保障患者的生命健康。
一次性使用无菌医疗器械混装EO验证方案

一次性使用无菌医疗产品混合装载经环氧乙烷灭菌工艺验证方案1 验证名称:一次性使用输液器和带针可吸收性外科缝合线、一次性使用医用透明贴膜、一次性使用自粘式伤口敷料混合装载经环氧乙烷灭菌工艺验证.2 验证目的:确定混合灭菌过程及灭菌工艺参数设定的适用性、有效性。
3验证项目:负载装载模式确认、灭菌过程确定、灭菌效果测试、灭菌参数设定。
4验证类别:此次实施的验证是对一次性使用无菌医疗产品混合装载经环氧乙烷灭菌进行过程确认,即工艺验证.5验证依据:欧盟EN550《医疗器械环氧乙烷灭菌确认和常规控制》(和国家GB18279—2000《医疗器械环氧乙烷灭菌确认和常规控制》)标准。
6验证方案适用灭菌产品:一次性使用无菌医疗产品。
7参加验证部门:技术、质量、设备、生产、灭菌车间、检测中心。
8验证实施部门: 技术、灭菌车间、检测中心。
9 验证文件资料保管部门:技术、检测中心。
10 验证场所:灭菌车间、检测中心11 验证小组的构成根据GB18279—2000和EN550的标准规定,确定了从事环氧乙烷灭菌验证过程中的方案制定及实施操作、微生物检验、设备管理及计量管理工作的人员,再根据设备维护操作、计量器具检定、物理性能及微生物性能测定作了分工,建立了《实施环氧乙烷灭菌验证人员资格确认表》。
见下表:12 收集文献资料需收集:⑴灭菌器及相关设备的主要技术资料。
⑵计量器具的相关的合格证明材料。
⑶环氧乙烷灭菌剂供方的相关资料和用于此次灭菌验证的环氧乙烷灭菌剂的质检单、合格证等相关资料。
⑷灭菌产品的技术文档及图纸。
a.所有有关验证的资料、所取得的数据、表单和验证报告由使用(用户)单位妥善保管、存档.b。
建立《实施环氧乙烷混合灭菌验证附件资料明细表》。
13 计量器具的校验灭菌设备上的主要计量器具,如:温控仪、压力表、湿度表、计时器及相应的传感器,在灭菌验证过程中要保证其相应的准确性,其精度符合等级要求。
计量器具必须在规定的检定周期内使用。
医疗器械环氧乙烷残留量的测试方法(气相色谱法)

医疗器械环氧乙烷残留量的测试方法(气相色谱法)环氧乙烷残留量检测意义:环氧乙烷是一种有机化合物,为一种最简单的环醚,化学式是C2H4O,是一种有毒的致癌物质。
环氧乙烷具有顽强的扩散和穿透能力,对细菌芽孢、真菌和病毒等各种微生物均有灭杀作用,属于广谱灭菌剂,现在被广泛的应用于洗涤,制药,印染等行业消毒使用。
环氧乙烷灭菌装置是一次性使用无菌医疗器械生产企业的关键设备,安装操作、使用管理有其特殊要求,使用环氧乙烷做灭菌剂,可在常温下杀灭各种微生物,包括芽孢、结核杆菌、细菌、病毒、真菌等。
但因环氧乙烷本身是有毒的致癌物质,对人体毒性伤害非常大,所以医疗器械行业相关标准对一次性使用无菌医疗器械产品的环氧乙烷残留量指标有着严格要求。
检测适用产品范围:适用于所有采用环氧乙烷灭菌装置消毒灭菌的一次性医疗用品进行环氧乙烷残留量检测。
部分产品列举如下:环氧乙烷残留量检测执行标准:GB/T 14233.1-2008 医用输液、输血、注射器具检验方法第1部分:化学分析方法GB/T 16886.7-2001 医疗器械生物学评价第7部分:环氧乙烷灭菌残留量环氧乙烷残留量检测—气相色谱法:1. 试验设备:气相色谱仪(品牌:济南兰光)2. 测试原理:在一定温度下,用萃取剂-水萃取样品中所含环氧乙烷(EO),用顶空气相色谱法测定环氧乙烷含量。
3. 标准贮备液制备方法(称重法):取外部干燥的50ml容量瓶,加水约30ml,加瓶塞,称重,精确到0.1mg。
用注射器注入约0.6ml环氧乙烷,不加瓶塞,轻轻摇匀,盖好瓶塞,称重,前后两次称重之差,即为溶液中所含环氧乙烷的重量。
加水至刻度再将此溶液稀释成1× 10-2g/L作为标准贮备液。
4. 测试方法:4.1 取环氧乙烷标准品适量,制成六个浓度的标准溶液,各取10ml,制备六个浓度的标准品试样。
4.2 当标准品试样达到气液平衡时,不同浓度的液体对应于不同浓度的气体,取平衡后的气体,注入进样器,记录环氧乙烷的峰高(或面积)。
GB15980-2009一次性使用医疗用品卫生标准-医疗器械注册

ICSCGB中华人民共和国国家标准GB15980-2009一次性使用医疗用品卫生标准Hygienic standard of disinfection for single use medical products(征求意见稿)2009-××-××发布 2009-××-××实施中华人民共和国卫生部发布中国国家标准化管理委员会前言本标准除5.1.3、8.2、10.1.1.4、10.4为推荐性条款外,其余条款均为强制性。
本标准代替GB15980-1995。
本标准与GB 15980-1995相比主要修改内容:——按GB/T 1.1、GB/T 1.2的要求和医疗用品的特性修改了标准结构和章的名称;——适用范围增加了一次性使用医疗用品经营、使用单位及生产企业消毒灭菌车间;——更新了规范性引用文件;——增加了术语和定义中有关内容、分类;——增加了接触性创面敷料的阻菌性能及其检验方法;——增加了原辅材料卫生要求及包装材料性能检验方法、贮存和运输卫生要求;——增加了压力蒸汽消毒或灭菌效果评价标准及其检验方法;——增加了消毒或灭菌效果确认及其方法;——增加了医疗用品包装的成形和密封性能要求;——修改了产品卫生质量要求及其检验方法;——修改了生产卫生要求、包装标识要求;——修改了环氧乙烷和电离辐射消毒或灭菌效果评价标准的检验方法;——删除了附录C初始污染菌数检测、附录D无菌检查法。
本标准附录A、附录B、附录C、附录D、附录E是规范性附录,附录F、附录G是资料性附录。
本标准由中华人民共和国卫生部提出并归口。
本标准由中华人民共和国卫生部负责解释。
本标准主要起草单位:江苏省卫生监督所、上海市疾病预防控制中心、山东省卫生厅卫生监督所、无锡宇寿医疗器械有限公司。
本标准主要起草人:顾健、沈伟、叶蓉春、袁青春、时玉昌、冯忠、徐萍。
一次性使用医疗用品卫生标准1 范围本标准规定了一次性使用医疗用品的术语和定义、分类、产品卫生质量要求,还规定了一次性使用医疗用品的消毒或灭菌质量控制要求、消毒或灭菌效果评价标准、原辅材料、生产、包装标识、贮存和运输的卫生要求以及检验方法。
医疗器械化学检验操作规程 14233.1-2020

医疗器械化学检验操作规程14233.1-2020
《医疗器械化学检验操作规程》14233.1-2020是一份针对医疗器械的化学检验操作进行规范的文件。
该规程的目的是确保医疗器械的化学检验过程符合相关法规和标准,以保证产品的质量和安全性。
具体而言,这份规程可能包括以下内容:
1.检验前准备:包括实验室环境要求、检验设备的校准和维护、试剂和溶液的制
备等。
2.检验方法:详细描述了各种化学检验的具体步骤和方法,如pH值测定、重金
属检测、残留溶剂检测等。
3.样品处理:规定了样品的采集、保存、处理和标识等要求,以确保样品的真实
性和代表性。
4.质量控制:包括检验过程中的质量控制措施,如重复性检验、空白试验、标准
物质对比等。
5.数据处理和报告:规定了检验数据的记录、处理、分析和报告的要求,以确保
数据的准确性和可靠性。
6.安全和防护:强调了实验室安全和防护的重要性,提出了相应的安全措施和应
急处理方案。
请注意,以上仅为可能的内容概述,具体的规程内容可能因实际情况而有所不同。
在实际操作中,应严格按照规程要求进行,以确保检验结果的准确性和可靠性。
此外,关于这份规程的更多具体信息,建议参考相关的官方文件或咨询专业人士。
医疗器械化学表征测试

医疗器械化学表征测试ISO 10993-18:2005《医疗器械生物学评价第18部分:材料化学表征》已经等同转化为GB/T 16886.18-2011.GB/T 16886.1对生物安全性进行评估的结构流程框架,给出了应用于材料和器械的生物学评价总则。
总则指出,在选择用于器械生产的材料时,首先考虑的是材料与器械用途相一致。
这就需要考虑材料的特征与性能,其中包括化学性能、毒理学性能、物理性能、电性能、形态学以及机械性能。
这些信息在任何生物学评价之前都是很必要的。
按该标准中规定的要求应该能获得如下信息,这些信息将有助于预测材料的生物学反应:(1)生产过程中所用物料的化学成分,包括加工助剂和残余物,如微量化学物质、清洗剂、消毒剂和检验试剂、酸性和富含物质。
(2)医疗器械生产中所用材料的特性,以及器械的最终形式。
(3)医疗器械材料成分的鉴定。
(4)医疗器械材料在生产过程中释放物质或分解产物的可能性。
生产工艺的变化或生产过程控制不足导致材料结构的变化。
生产材料的组成特性主要由材料的供应商控制,而其他特性主要受最终医疗器械的要求和生产工艺的要求影响。
一范围本标准为医疗器械的区分和定义提供了框架,从而为生物安全性评价提供了信息。
这一过程包括区分材料中的化学成分,并确定其潜在的生物暴露。
它包括确定相关的物理化学、机械和形态特征。
该标准不涉及与标准实施相关的性能条款,仅限于生物安全评价。
它也不涉及降解产物的任何定性和定量分析。
该标准可用于材料供应商、医疗器械制造商和其他需要评估生物安全性的人员。
如果材料或仪器与人体直接或间接接触,则应适用本部分的标准。
所获得的化学表征信息可以用于一些重要的应用中,例如判断你所使用的材料与临床上已建立的材料之间的等效性。
确定最终仪器和原型仪器之间的等效性,并检查用于支持最终仪器评估的原型仪器数据的相关性。
筛选适合医疗器械预期临床应用的新材料。
GB/T 16886的本部分不涉及降解产物的定性和定量,关于这方面的内容见ISO 10993-9、ISO 10993-13、ISO 10993-14和ISO 10993-15。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
酸碱度
目的
保证产品在使用时有合适的pH环境
测定方法
pH计 滴定法
评价
与空白液做对比
中性范围
易氧化物(还原物质)
目的
可以作为医疗器械浸提液的污染指数,与浸提液中有机
物单体残留物、微生物的数量成比例关系
检验方法 间接滴定法 直接滴定法
间接碘量法的原理:水浸提液中的还原 物质在酸性条件下加热时,被 KMnO4 氧 化,过量的 KMnO4 将 KI 氧化成 I2 ,而 I2 被 Na2S2O3还原。 MnO4-+8H++5e=Mn2++4H2O MnO4-+5I-+8H+= Mn2++4H2O +5/2I2 I2+2S2O32-=2 I-+ S4O62-
检测条件
炉温:100℃,保持5min;
进样口温度:200℃;检测器温度 210℃色谱
柱:AT-624 30×0.25×1.4; 载气:氦气,99.999%; 检测器:ECD 柱流量:1.0mL/min, 电流:1nA 样品加热温度:70℃, 加热时间:20min
单体残留
国家食品药品监督管理局提出:
质谱 离子源:EI,测定方式:SCAN,扫描范围:30-500u, 溶剂切除时间:1.6min
方法1
方法2
GC/MS的其它应用:鉴定未知峰
uV(x10 ,0 00) 2.5 Chromatog ram 2.0 1.5 1.0 0.5 0.0 0.5 1.0 1.5 2.0 2.5 min
EO检测所需仪器
顶空进样器(HS)
气相色谱仪(GC)
氢火焰离子化检测器(FID)
EO标准液配制
储备液
在100ml容量瓶中预先加入适量水(60ml左右),
称量,冷冻室放置2~3分钟,低温条件下向瓶内 滴加EO 0.1克左右,再次称重,然后加水定容, 作为储备液。
标准液
用纯化水依次稀释储备液,浓度范围最好在
重金属总量
目的
医疗器械中的有毒金属,包括铅、铬、铜、锌等,通常
都属于致癌物,化合态的毒性一般要大于单质的毒性。
检验方法 通常采用比色法检查重金属
方法1:弱酸性溶液 方法2:碱性溶液
评价 样品管与空白管做颜色对比
微量金属离子
原子吸收分光光度法
外标法
钡、铬、铜、铅、锡 镉离子
1~20μg/ml
样品浸提
模拟浸提:如注射器(37℃放置1h) 极限浸提:水浸提顶端空间气体分析
检测条件
炉温:100℃,保持3 min; 进样口温度:120℃;检测器温度 260℃ 色谱柱:AT-624,30m×0.25mm×1.4μm;
载气:氦气,99.999%;
检测器:FID 气流量:氦气:1.0mL/min, 氢气:40ml/min 空气:
检验液制备 酸碱度 易氧化物 重金属总量 微量金属离子 蒸发(水、有机溶剂)残渣 紫外吸光度 氯化物 硫酸盐 溶剂残留
检验液制备
浸提原则 尽量模拟产品实际使用时的条件 浸提介质 水 浸提条件 温度 时间 样品与浸提介质的比例 注意事项 浸提完成后浸提液须与样品及时分离
评价 还原物质的结果用消耗0.002mol.l-1 KMnO4 标准溶液的 体积来表示,每个样品平行滴定两次,以两个样品的算 术平均值报告结果
易氧化物测定注意事项
加入KI后,应立即快速滴定,尽可能减少I2
的损失。 滴定液变为淡黄色出现在 Na2S2O3 溶液还差 2~3ml到终点时,此时加入5滴淀粉指示液继 续滴定至蓝色消失。 还原物质测定实验须在浸提液制备后尽快完 成,浸提液如需存放,须密封后冷藏。
蒸发残渣
目的
测定浸提液中的非挥发物。
检验方法
减重法
评价
样品液的蒸发残渣与空白液的蒸发残渣相减
注意事项
恒重
紫外吸光度
目的 测定浸提液中含不饱和键的小分子化合物。 检验方法 紫外分光光度计 评价 用空白液做基线校正,对样品液的吸光度值进行限定
注意事项 过滤 当天测试
对一次性血路导管、一次性心脏停跳灌注管等含
光固化胶的一次性医疗器械需进行是否有单体残 留的验证实验。
单体残留检测所需仪器
顶空进样器(HS)
气相色谱仪(GC)
质谱检测器(MS)
样品浸提 (举例:紫外固化胶)
. 方法一:取试片一片(0.5克左右),使用100mL
生理盐水浸泡胶片,37℃±1℃水浴加热6小时,冷 却至室温后取出胶片,所获浸提液作为供试液,取 供试液1.0ml于顶空瓶中,密封,然后进行GC-MS 测试。
一次性医疗器械的化学检验
内容主要依据:付步芳研究员 奚廷斐 主任 中国药品生物制品检定所第3期医疗器械培训班讲义
概述
一次性医疗器械系指只能使用一次的医疗器
械( one use only)
一次性医疗器械约占整个医疗器械市场份额
的15%~18%, 是使用范围广泛、与人体疾病 康复息息相关的一类医疗器械产品。
紫外吸光度
步骤
以空白液做基线校正,在250 nm~320 nm波长范
围内扫描,测其吸光度。
结果
溶剂残留
环氧乙烷
2-氯乙醇 单体残留
环氧乙烷
EO性质
灭菌能力强,是广谱灭菌剂
目前一次性医疗器械广泛采用环氧乙烷灭菌
沸点10.8℃,常温下是易燃易爆的危险气体 已知的有毒物质,致癌,致突变
一次性医疗器械的化学性能检验是其安全性
评价的重要内容之一。
检验依据
产品标准
----GB 8368 一次性使用输液器 ----GB 15810 一次性使用注射器
----GB 14232 一次性使用血袋
分析方法标准
GB/T 14233.1医用输液、输血、注射器具检验方
法
企业标准审查
重金属总量
步骤 取上述检验液25mL置于比色管中,另取Pb2+ (100μg/mL)标准液 mL加入 对照比色管内,加 入蒸馏水稀释至25 mL,于上述两比色管中分别 加入1.0mL的醋酸盐缓冲液(pH 3.5)和1.0mL 硫代乙酰胺试液,摇匀放置2分钟,置白色背景 上,自上而下观察颜色深浅。 结果 检验液所呈现的颜色不深于标准管的颜色
400ml/min 样品加热温度:60℃, 加热时间:20 min
2-氯乙醇
性质
无色透明液体。熔点-62.6℃,沸点128.7℃,
51-52℃(2.93kPa),29℃(1.33kPa), 相对密度1.2045(20/4℃),沸点95-98℃ (97.8kPa)。与水、乙醇能按任意比例混溶。
EO测定时未检出EO,检出其它峰
GC/MS鉴定未知峰
毒性及来源
毒性
能刺激身体表面,具有急性毒性,可被皮肤快速
吸收,有微弱的致突变性,有潜在的致胎儿毒性 和致畸性 能对肺、肾、中枢神经系统和心血管系统造成损 伤。
产生
EO与空气中的氯离子或器械本身释放的氯离子
反应生成
ECH检测所需仪器
顶空进样器(HS)
气相色谱仪(GC)
电子捕获检测器(ECD)
酸碱度
仪器校准 两点法 测量
校准完毕后,取上述检验液适量,插入测量电极和温度
校正电极进行测量。空白液和样品液分别平行测二次, 结果取平均值
结果 空白:6.483,6.483 均值6.483 样品:6.364,6.362 均值6.363 差值:0.120
易氧化物
步骤 取检验液10mL置于三角瓶中,加入稀硫酸1.0 mL和 0.0020mol/L高锰酸钾10ml,用0.0103mol/L硫代硫酸钠 溶液滴定至浅黄色,然后加入0.25 mL 1 %淀粉溶液, 滴定至蓝色消失。同样条件下滴定空白对照液。空白液 和样品液分别平行测二次,结果取平均值 结果 空白:11.20ml,11.18ml 均值11.19ml 样品:11.10ml,11.10ml 均值11.10ml 计算:(11.19-11.10)*0.0103/0.01=0.09(ml)
一次性医疗器械 基础化学项目检测举例
检品名称
内窥镜胆道支架及输送系统
检验液制备方法
取3套样品的输送系统加入纯化水250mL,于
37 ℃浸提24 h。同时同条件作空白对照液。
该检品的检验项目
酸碱度 水浸提液与同批空白对照液PH值之差不超过1.5
易氧化物 检验液与空白液耗用0.002mol/L高锰酸钾溶液的体积差 不应超过2.0 mL 重金属总量 水浸提液中重金属总量不应超过1μg/mL 蒸发残渣 在50 mL检验液中非挥发物总量不得超过2.0 mg 紫外吸光度 检验液在250nm~320nm波长范围内吸光度应不大于0.1
蒸发残渣
步骤 取以上检验液50mL至已恒重过的烧杯中,于水 浴中蒸干,并于105℃下烘干至恒重,同法测定 空白对照液。
结果/单位:克
前 均值 后 空白:54.7499 54.7498 54.7501 54.7497 54.7503 样品:49.9984 49.9983 49.9988 49.9982 49.9988 结果:0.0005-0.0004=0.0001即0.1mg 均值 54.7502 49.9988 差值 0.0004 0.0005
•企标的指标要求应等 于或高于国标或行标 •检验方法一致 •无漏项
若该产品有相应的国标或行标,则企标的规
定应与国标或行标一致。
若该产品尚无相应的国标或行标,则借鉴类