氟达17
关于CLL药物治疗的新观点

O'Brien和他的同事们报道了53%的总体应答 率,在10mg剂量组23个病人中9个应答(39 %),在30mg剂量组26个病人中17个应答 (65%)。大多数病人骨髓中的残留病灶被 清除,29个病人中11个病人(38%)有证据 显示获得了分子水平缓解。
可能对CLL最有效的药物治疗是最有效的单 个化学药物加上最有效的单克隆抗体:氟达 拉滨+阿仑单抗。
在一个针对CLL复发病人的二期实验中,氟 达拉滨和阿仑单抗的联合应用被证明是安全、 可行和非常有效的:
36个病人中OR率为83%(30/36),包括11 个CR(30%)和19个PR(53%)。此外1个 病人的病情达到稳定。经过3个月的随访,在 31个被研究的病人中16个获得了外周血MRD 阴性,并且在所有发病部位都可以看到病灶 的消退(尤其在血液、骨髓和脾)。
续贯的或同时的用药随机研究中结合应用了利妥昔 单抗和氟达拉滨,先前未治疗的CLL患者接受6个疗 程的氟达拉滨(用或不用利妥昔单抗)接着用了4
周,每周一次的利妥昔单抗,总体缓解率和完全缓 解率都比同时用药组高(90%、47% VS 77%、28
%)。接受氟达拉滨和利妥昔单抗治疗的病人,比
接受氟达拉滨单药治疗的病人有更好的无进展生存 率(PFS)和总体生存率(OS)。2年的无进展生 存率为67%VS45%,2年的总体生存率为93% VS81%。
然而,很久以来人们就已经认识到上述的
临床分期系统在预测个体的预后,尤其是在 疾病早期阶段(Binet A期和Rai 0-Ⅱ期)以 及年轻病人时,并不十分有效。因此,为了 更准确地预测CLL病人的预后,就要引入一 些其他参数。无论Rai分期和Binet分期如何, 这些参数能有效地预测预后,但只有分子生 物学在估计CLL病人对化疗反应上被证明是 有用的。
【国家自然科学基金】_氟达拉滨_基金支持热词逐年推荐_【万方软件创新助手】_20140802
2012年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13
科研热词 推荐指数 造血干细胞移植 2 高危 1 预处理方案 1 造血重建 1 血液病 1 慢性移植物抗宿主病 1 慢性淋巴细胞白血病 1 基因突变 1 前体细胞淋巴母细胞白血病淋巴瘤1 p53基因 1 p53 gene 1 mutation 1 chronic lymphocytic leukemia 1
2009年 序号 1 2 3 4 5 6 7 8
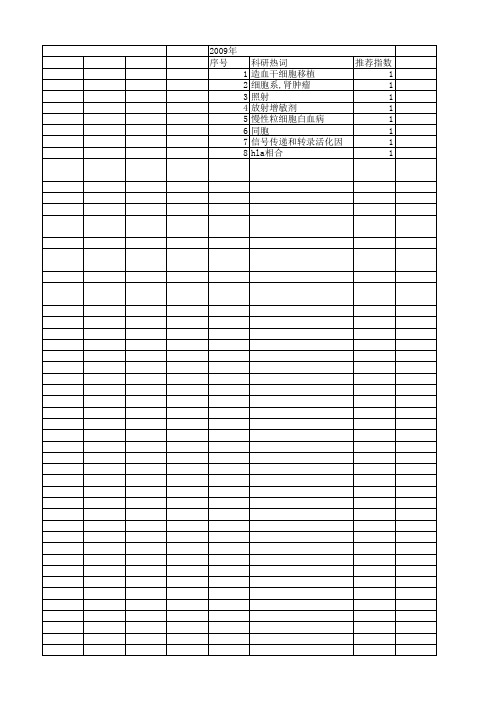
科研热词 推荐指数 造血干细胞移植 1 细胞系,肾肿瘤 1 照射 1 放射增敏剂 1 慢性粒细胞白血病 1 同胞 1 信号传递和转录活化因子 1 hla相合 1
2010年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
科研热词 高迁移率族蛋白b1 造血干细胞移植 自身免疫疾病 缺氧 移植预处理 白血病,淋巴细胞,慢性 白血病 环磷酰胺 治疗结果 氟达拉滨 急性髓系白血病 巩固强化 巨噬细胞 外周血干细胞移植 利妥昔单抗 信号转导和转录激活子 fa方案 cd34+细胞
推荐指数 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
2013年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
2013年 科研热词 推荐指数 移植物抗宿主病 2 异基因造血干细胞移植 2 骨髓移植 1 预处理 1 长期生存 1 造血干细胞移植 1 肠 1 移植预处理 1 移植后淋巴细胞增值性疾病 1 灌流 1 淋巴瘤,滤泡型 1 治疗结果 1 抗肿瘤联合化疗方案 1 慢性活动性eb病毒感染 1 心脏移植 1 多发性骨髓瘤 1 单倍体相合造血干细胞移植 1 利妥昔单抗 1 减低强度 1 ptld中枢侵犯 1 eb病毒 1
氟达拉滨联合化疗治疗初治滤泡型淋巴瘤分析

氟达拉滨联合化疗治疗初治滤泡型淋巴瘤分析(作者:___________单位: ___________邮编: ___________)摘要:目的:氟达拉滨联合化疗治疗初治滤泡型淋巴瘤分析。
方法:研究对象为本院确诊的40例初治滤泡型淋巴瘤患者,其中17例接受FC方案治疗,13例接受R-FC方案治疗。
所有患者都接受3-7个周期的化疗,平均化疗周期数为4个周期。
结果:40例患者中达达到完全缓解者26例,部分缓解者1例,总有效率为90.0%。
结论::氟达拉滨治疗初治滤泡型淋巴瘤的临床缓解率较高,无病生存时间较长,且不良反能够以耐受。
关键词:氟达拉滨;滤泡型淋巴瘤;初治滤泡型淋巴瘤在惰性非霍奇金淋巴瘤中最为常见的一种类型,直到现在为止最难以治愈的肿瘤。
FL大多数患者就诊时已经是Ⅲ或是Ⅳ,从前的化疗或是放疗方法均没有对患者的长期生存发挥有效改善。
虽然FL一般对最初阶段的化疗敏感,可是传统的化疗无法有效的缓解,具体表现为复发与缓解进展间相互交替过程[1]。
氟达拉滨属于嘌呤类似物,对于惰性的NHL有着较好的治疗效果,缓解能力较高。
目前氟达拉滨单药或是其他联合药物化疗是美国癌症综合网强烈推荐的治疗FL的首先方案。
国内以往对氟达拉滨的临床分析多数是针对惰性NHL进行的治疗,对于治疗FC的方案国内还内有形成系统性的报道。
本文主要探讨氟达拉滨联合化疗治疗初治滤泡型淋巴瘤的疗效。
现报告如下:1材料与方法1.1一般材料本院在2006-2010年收治的30例患者,其中男性患者14例,女性患者16例;病发年龄是23-78岁,平均年龄是54岁。
全部患者均进行了身体全面检查,主要包括血常规、红细胞降沉率、超声波检查、骨髓穿刺及胸、腹部及盆腔CT等。
1.2治疗方法患者中有17例接受了FC治疗方案实行化疗。
患者13例接受了R-FC治疗方案。
1.3近期效果与不良反应依据淋巴瘤恶性治疗效果的评价标准,评价本组患者的近期治疗效果,包括了完全缓解、部分缓解、疾病稳定和疾病发展,CR+PR所占的比例为整体有效率。
FLAG方案治疗难治-复发性急性髓细胞白血病临床观察论文

FLAG方案治疗难治/复发性急性髓细胞白血病的临床观察【摘要】目的:观察氟达拉滨(flud)联合阿糖胞苷(ara-c)、重组人粒细胞集落刺激因子(g-csf)组成的flag方案治疗难治/复发性急性髓细胞白血病的临床疗效及主要毒副反应。
方法:采用氟达拉滨(flud)50mg/d,静脉滴入,持续30min,d1-d5;阿糖胞苷(ara-c)2g/d,用flud后4h滴入,持续3h d1-d5;重组人粒细胞集落刺激因子(g-csf)300ug/d,皮下注射d0-d5,采用flag 方案治疗难治/复发性急性髓细胞白血病治疗17例,观察其疗效及毒副反应。
结论:17例患者完全缓解11例,完全缓解率为64.7%,部分缓解3例,未缓解1例,死亡2例,有效率82.4%。
主要副反应为骨髓抑制、感染、消化道症状、轻度肝损害。
结论:flag方案治疗治/复发性急性髓细胞白血病安全有效。
【关键词】flag方案;急性髓细胞白血病;难治/复发性【中图分类号】r733 【文献标识码】a 【文章编号】1004-7484(2013)06-0033-01急性髓细胞白血病(aml)是一种较常见的危及人类生命的血液系统恶性肿瘤。
虽然联合化疗的应用使aml的疗效明显提高,但仍有15%-30%初治患者不能达到完全缓解(cr),而且40%-50%以上已经获得cr的患者最终复发。
对于难治复发的急性白血病标准诱导方案缓解率低,预后极差。
早期复发以及原发耐药成为aml治疗失败的主要原因。
近几年来,国内部分学者采用福达拉宾+阿糖胞苷+重组人粒细胞集落刺激因子组成flag方案治疗难治/复发性急性髓细胞白血病,取得了较满意效果[1]。
我院采用该方案治疗17例难治/复发急性急性髓细胞白血病,也取得了较好效果,现报告如下。
1 资料与方法1.1 一般资料收集我院2009年3月-2012年4月收治的急性髓细胞白血病患者17例,男10例,女7例,年龄32-54岁,中位年龄42岁。
氟达拉滨联合环磷酰胺治疗慢性淋巴细胞白血病的临床疗效和不良反应

氟达拉滨联合环磷酰胺治疗慢性淋巴细胞白血病的临床疗效和不良反应发表时间:2016-06-02T15:16:59.113Z 来源:《中国医学人文》2016年第3期作者:樊婷婷[导读] 氟达拉滨联合环磷酰胺的治疗方案(FC)完全缓解率高,达到完全缓解时间短,不良反应无明显增加,是目前首选的 CLL 一线治疗。
樊婷婷(贵州省兴义市人民医院血液内科 562400)【摘要】目的:观察氟达拉滨联合环磷酰胺(FC)方案治疗慢性淋巴细胞白血病(CLL)的近期临床疗效和不良反应方法:回顾性分析 2010 年 2 月至 2016 年2 月就诊于我院经FC 方案治疗的 26 例 CLL 患者资料结果:26 例 CLL 患者的完全缓解(CR)率、部分缓解(PR)率及总缓解(OR)率分别为38.46%, 26.92% 及65.38%,治疗过程中出现的不良反应主要为不同程度的骨髓抑制、胃肠道反应和感染。
结论氟达拉滨联合环磷酰胺对 CLL 近期疗效较好,耐受性好,极大地改善拟CLL 的预后,提高DFS。
【关键词】氟达拉滨;环磷酰胺;慢性淋巴细胞白血病【中图分类号】 R2 【文献标号】 A 【文章编号】 2095-9753(2016)3-0369-02慢性淋巴细胞白血病(CLL/SLL)为淋巴细胞克隆性增殖的肿瘤性疾病,为血液系统恶性肿瘤之一。
发病时,成熟淋巴细胞在血液、骨髓、淋巴结、脾、肝及其他器官中进行性聚集为CLL,而病开始仅在淋巴结、脾、肝等淋巴器官中聚集为SLL。
CLL 的免疫表型可分为B 细胞CLL 和T 细胞CLL,大多数是B-CLL[1]。
CLL 多发于中老年人,临床进展缓慢,惰性的套细胞淋巴瘤 (M C L) 除外,可向侵袭巴瘤转化,治疗后可缓解,但难以治愈。
同时本病的异质性较大,临床表现、自然病程、实验室检查等差异性较大。
本病为西方国家发病率最高的成人白血病,在北美国家最高,占所有成人白血病22.6%,平均年发病率为2.7/100000. 但亚太地区发病率极低,如我国仅占所有白血病的1.7%,年发病率为0.05/100000。
十七氟癸基修饰的有机-无机杂化二氧化硅膜材料的制备及气体分离性能

十七氟癸基修饰的有机-无机杂化二氧化硅膜材料的制备及气体分离性能宋霖;韦奇;郝润秋;聂祚仁;李群艳【摘要】以十七氟癸基三乙氧基硅烷( PFDTES)和1,2-双(三乙氧基硅基)乙烷(BTESE)为前驱体,通过溶胶-凝胶法制备了十七氟癸基修饰的SiO2溶胶,采用浸渍提拉法在γ-Al2 O3/α-Al2O3多孔陶瓷支撑体上涂膜,然后在N2气氛保护下烧结成完整无缺陷的有机-无机杂化SiO2膜.利用扫描电子显微镜对膜材料的形貌进行观察,通过动态光散射技术对溶胶粒径及分布进行测试,利用视频光学接触角测量仪、红外光谱仪和热分析仪表征了十七氟癸基修饰对有机-无机杂化SiO2膜疏水性的影响.结果表明,十七氟癸基已经成功修饰到SiO2膜材料中,且随着PFDTES加入量的增大,溶胶粒径和膜材料对水的接触角不断增大.当n(PFDTES)∶n(BTESE)=0.25∶1时,溶胶粒径分布较窄,平均粒径为3.69 nm,膜材料对水的接触角为(112.0±0.4)°.在修饰后的有机-无机杂化SiO2膜中H2的输运遵循微孔扩散机理,在300℃时,H2的渗透率达到5.99×10-7mol·m-2·Pa-1·s-1,H2/CO和H2/CO2的理想分离系数分别达到9.54和5.20,均高于Knudsen扩散的理想分离因子,表明膜材料具有良好的分子筛分效应.%Hydrophobic organic-inorganic hybrid silica sols were prepared using 1,2-bis(triethoxysilyl) ethane (BTESE) and 1H,1H,2H,2H-perfluorodecyltriethoxysilane(PFDTES) as precursors under acidic condition, then the sols were deposited on γ-Al2O3/α-Al2O3 substrates by dip-coating under clean room condition. The cross-sectional morphology, particle size of the sols and hydrophobic property of the membranes were characterized by means of SEM, DLS, FTIR, water contact angle measurement and thermogravimetric analysis.The results show that the particle size of sols and the water contact angle of supported membranes increase with increasing the amount of PFDTES in the mixture. A narrow sol particle size distribution centered at 3. 69 nm and a water contact angle of (112.0±0.4) ° were obtained at aBTESE/PFDTES molar ratio of 0.25: 1. At 300 ℃, the membr ane shows a high H2 permeance of 5. 99×10-7mol·m-2·Pa-1·s-1 and a H2/CO andH2/CO2 permselectivity of 9. 54 and 5. 20, respectively, which were higher than the corresponding Knudsen value(H2/ CO=3.74, H2/CO2=4. 69). The H2 transport in the modified silica membranes is subjected to a micropore diffusion mechanism.【期刊名称】《高等学校化学学报》【年(卷),期】2012(033)008【总页数】6页(P1670-1675)【关键词】有机-无机杂化SiO2膜;溶胶-凝胶法;疏水性;气体分离【作者】宋霖;韦奇;郝润秋;聂祚仁;李群艳【作者单位】北京工业大学材料科学与工程学院,北京100124;北京工业大学材料科学与工程学院,北京100124;北京工业大学材料科学与工程学院,北京100124;北京工业大学材料科学与工程学院,北京100124;北京工业大学材料科学与工程学院,北京100124【正文语种】中文【中图分类】O613.7氢气分离技术是化石燃料制氢工艺中最重要的步骤之一.膜分离法具有投资少、能耗低、装置及操作简单、可在温和条件下实现连续分离及对环境友好等优点[1~3],极具发展前景.用于氢气分离的膜材料主要有分子筛膜、金属Pd膜和微孔陶瓷膜等,其中无定形的微孔SiO2膜被认为是最能接近工业应用的氢气分离材料之一[4~7].但SiO2膜的水热稳定性差,长期暴露于水热环境下会发生分解反应,从而导致膜破裂形成缺陷,影响气体分离效果[8].为了提高SiO2膜的水热稳定性,众多研究者开展了大量工作:(1)将过渡金属离子掺杂到SiO2骨架中以提高膜材料的稳定性,因为氧原子与过渡金属形成的共价键比硅氧键更稳定[9,10];(2)通过化学修饰在SiO2膜表面引入疏水基团,提高膜材料的疏水性,减少水分子在膜材料表面的吸附,降低水分子对Si—O—Si结构的破坏.1999年,de Vos等[11]首次将甲基修饰到SiO2膜表面,使其疏水性能得到显著提高.而后Wei等[12]制备了三氟丙基修饰的SiO2膜,在努力提高SiO2膜渗透率和分离系数的同时也提高了其抵抗水汽的性能.上述SiO2膜材料都是以正硅酸乙酯(TEOS)为前驱体制备的,膜材料的骨架依然是—Si—O—Si—网络结构,由于这种结构在水热环境下易水解,导致所制备的SiO2膜的长期水热稳定性不够理想.用1,2-双(三乙氧基硅基)乙烷(BTESE)作为前驱体制备有机-无机杂化SiO2膜,基于BTESE的膜材料骨架主要由—Si—C—C—Si—结构组成,与—Si—O—Si—结构相比,在水热环境下不会发生水解反应而导致膜材料产生缺陷.Kanezashi等[13]以BTESE为前驱体,在以α-Al2O3为基体、SiO2-ZrO2为过渡层的管式载体表面涂膜,于300℃焙烧30 min后得到具有较高He和H2渗透通量的有机-无机杂化SiO2膜.Castricum等[14,15]分别以甲基三乙氧基硅烷(MTES)和TEOS以及MTES和BTESE为前驱体制备了SiO2膜,结果显示,基于BTESE的有机-无机杂化SiO2膜与传统无机SiO2膜及甲基化SiO2膜相比,在正丁醇脱水的渗透汽化环境下,水热稳定性得到显著提高.尽管基于BTESE的有机-无机杂化SiO2膜比基于TEOS的无机SiO2膜具有更高的水热稳定性,但BTESE的水解缩聚反应过程也会在膜材料骨架中引入少量的—Si—O—Si—结构,加上有机-无机杂化SiO2膜还具有一定的亲水性[16],因而在水热环境下膜中的—Si—O—Si—结构还有可能受到水分子的攻击.为了减小水分子对有机-无机杂化SiO2膜的破坏,本文通过溶胶修饰的方法,用十七氟癸基三乙氧基硅烷(PFDTES)代替部分BTESE作为共前驱体,经共水解缩合反应制备有机-无机杂化SiO2膜,以提高膜的疏水性能,并研究了十七氟癸基修饰对溶胶结构的影响以及修饰后有机-无机杂化SiO2膜的氢气渗透和分离性能.1.1 实验原料1,2-双(三乙氧基硅基)乙烷(BTESE),分析纯,Sigma-Aldrich化学有限公司;十七氟癸基三乙氧基硅烷(PFDTES),分析纯,Alfa Aesar化学有限公司;硝酸,优级纯,广东汕头市西陇化工厂;无水乙醇(EtOH),分析纯,北京化工厂;去离子水,由优普ULUP-Ⅰ-10T型超纯水机制备.1.2 有机-无机杂化SiO2溶胶及膜材料的制备首先将BTESE和PFDTES在冰水浴中搅拌,使其混合均匀,然后加入无水乙醇继续搅拌,最后逐滴加入事先按比例混合好的浓硝酸和蒸馏水,再继续搅拌一段时间后,将此混合物于60℃水浴中剧烈搅拌3 h,最终混合物的摩尔比为n(BTESE)∶n(PFDTES)∶n(EtOH)∶n(H2O)∶n(HNO3)=1∶x∶10.5∶5.25∶0.1(x =0,0.11,0.25和0.43),反应结束后得到无色透明的十七氟癸基修饰的有机-无机杂化SiO2溶胶.将新制备的溶胶用无水乙醇以1∶19(体积比)稀释,然后通过浸渍提拉法(Dip-coating)将其涂覆在γ-Al2O3/α-Al2O3多孔陶瓷支撑体上.为了避免空气微颗粒在膜材料表面形成缺陷,整个涂膜过程在100级洁净室中进行.涂膜后于380℃煅烧4 h,为了防止氧化,在煅烧过程中以N2作为保护气.为了得到无缺陷的SiO2膜,上述涂膜和煅烧过程需重复4次.将剩余的SiO2溶胶干燥成凝胶,然后在380℃及N2气氛下煅烧4 h,得到无支撑的十七氟癸基修饰的有机-无机杂化SiO2膜.修饰的SiO2膜样品根据PFDTES的化学计量标记为(xPFDTES)BTESE(x=0,0.11,0.25和0.43).1.3 有机-无机杂化SiO2膜的表征有支撑有机-无机杂化SiO2膜的断面和表面形貌用JEOL JSM-7001F型扫描电镜观察.采用OCA20视频光学接触角测量仪(德国Dataphysics公司)测量膜材料的接触角(CA),室温下,将体积为2 μL的水滴以1 μL/s的速度滴在有支撑SiO2膜的表面,然后测量水滴在膜材料表面的接触角.热重分析在STA449C/1/G型热分析仪(德国耐驰公司)上进行,将(0PFDTES)BTESE样品和(0.25PFDTES)BTESE样品放入相对湿度为70%~80%、温度为40℃的水热环境中分别陈化20和40 d后进行热重分析,在30 mL/min的N2气吹扫下,以10℃/min的升温速度从室温加热到650℃.利用Nicolet 5700型傅里叶变换红外光谱仪(美国热电公司)测定(0PFDTES)BTESE和(0.25PFDTES)BTESE样品的红外光谱.通过动态光散射技术(DLS),在Zetasizer Nano ZS 90型激光粒度仪(英国马尔文公司)上进行溶胶粒径及分布测试.H2的渗透和分离性能测试在自制的气体渗透和分离装置上完成,有支撑有机-无机杂化SiO2膜采用柔性石墨垫片密封,膜材料两端的压力差用CWY100精密数字压力表(西安创威科技公司)测量,调节稳压阀使得膜材料两侧压差为0.1 MPa,随后加热渗透装置,待温度恒定后用电子气体流量计(Alicat Scientific Inc.公司)测量气体的质量流量.2.1 有支撑有机-无机杂化SiO2膜的形貌图1为有支撑有机-无机杂化SiO2膜的断面和表面的SEM照片.从图1(A)可见,膜体系主要由三层组成:顶层为有机-无机杂化SiO2膜,厚度约170 nm;中间为γ-Al2O3过渡层;底层为α-Al2O3多孔陶瓷.这说明有机-无机杂化SiO2膜已浸涂到γ-Al2O3/α-Al2O3衬体上.由图1(B)可见,该膜材料的表面平整光洁,无明显的裂纹和大孔等严重缺陷.2.2 有支撑有机-无机杂化SiO2膜的疏水性能图2是水滴在有支撑有机-无机杂化SiO2膜表面的照片.由图2可见,水滴在未修饰膜表面的接触角只有(49.0±0.5)°[图2(A)],随着PFDTES加入量的增大,接触角不断增大[图2(B)~(D)],且膜材料由亲水变成疏水,这是由于膜表面的部分羟基被疏水的十七氟癸基取代所致.从图2(C)可见,当n(PFDTES)∶n(BTESE)=0.25∶1时,接触角达到(112.0±0.4)°,与文献[17]中制备的三氟丙基修饰SiO2膜、文献[18]中制备的苯基修饰SiO2膜和文献[19]中制备的乙烯基修饰SiO2膜进行对比发现,当达到相同的接触角时,本文所需要修饰的十七氟癸基的量比三氟丙基、苯基和乙烯基少很多,说明修饰十七氟癸基比修饰三氟丙基、苯基和乙烯基具有更强的疏水效果.从图2(C)和(D)中还可以看出,水滴在(0.25PFDTES)BTESE和(0.43PFDTES)BTESE表面上的接触角分别为(112.0±0.4)°和(112.7±0.9)°,两者相差无几.这是因为修饰量达到一定程度后,修饰到膜材料表面的十七氟癸基密度增大,由于十七氟癸基是长链基团,占据较大的空间位阻,所以即使修饰量增大,十七氟癸基也较难取代更多的表面羟基,因而膜材料的疏水性能未能进一步提高.图3是无支撑有机-无机杂化SiO2样品在水热环境(温度40℃、相对湿度70% ~80%)处理前后的热重分析结果.可见,(0PFDTES)BTESE膜材料陈化20和40 d后有明显的失重,失重量分别为8%和10%,且随着陈化时间的延长,失重量逐渐增加.失重开始温度为50℃左右,这是物理吸附水的脱附引起的,到150℃左右这一阶段失重基本完成.而未陈化的(0PFDTES)BTESE膜材料在50~150℃基本未失重,说明在湿热环境的陈化过程中,膜材料表面未反应的硅醇悬键将水蒸气吸附在膜的表面,这会严重影响膜的结构和性能,导致孔径发生变化,从而影响气体分离效果[20,21]. 而(0.25PFDTES)BTESE膜材料无论是未陈化还是陈化20 d或40 d,在50~150℃基本没有失重,表明用疏水基团(十七氟癸基)修饰的膜材料在湿热环境下基本没有吸附水蒸气.与(0PFDTES)BTESE膜材料的失重行为相比,(0.25PFDTES)BTESE膜材料展现了优异的疏水性能.图4为修饰前后的有机-无机杂化SiO2膜的FTIR谱图.其中1048 cm-1处的吸收峰为Si—O—Si伸缩振动的特征峰,3455 cm-1处为物理吸附水的吸收峰,1628 cm-1处的吸收峰对应表面羟基Si—OH.对于十七氟癸基修饰的样品,1248和1150 cm-1处的吸收峰对应于C—F的振动峰,而未修饰的SiO2样品在上述波数处均未出现吸收峰,说明十七氟癸基已经成功修饰到SiO2膜材料中,且经过380℃的煅烧,C—F键并没有被破坏.从图4还可看出,未修饰的SiO2膜在3455 cm-1处归属于物理吸附水的吸收峰非常强烈,说明未修饰的膜材料疏水性较差;而十七氟癸基修饰后的膜材料吸附水的吸收峰则较弱,说明经修饰后膜材料的疏水性得到增强.同时还可以看出,随着PFDTES的加入,膜材料中Si—OH在1628 cm-1处的吸收峰明显减弱,表明Si—OH浓度逐渐降低,这是由于十七氟癸基取代膜材料中的部分羟基,增强了膜材料的疏水性能,这与图2接触角测量结果一致.2.3 溶胶粒径及分布基于BTESE的有机-无机杂化SiO2膜材料的结构主要由溶胶-凝胶反应所得的低聚物经过进一步组合而成,所谓的溶胶胶粒指的即是这些由Si—C—C—Si结构单元组成的低聚物.PFDTES的分子结构上存在一个没有反应活性的基团,即十七氟癸基,当BTESE和PFDTES进行共水解缩聚反应形成溶胶时,它并不参与反应,其结果是这些基团与硅原子形成悬键伸出低聚物的外表面,形成所谓的“分支”,低聚物通过这些分支的相互穿插,即可得到膜材料的微孔结构,因而这些低聚物的尺寸即溶胶胶粒的粒径对BTESE膜材料的孔结构起到重要的调控作用.本文利用十七氟癸基的修饰来提高膜材料的疏水性,必然对溶胶胶粒产生影响,因此必须综合考虑十七氟癸基对膜材料的疏水性及溶胶胶粒的影响.图5是不同修饰量的有机-无机杂化SiO2溶胶的粒径分布图.由图5可见,(0.11PFDTES)BTESE的粒径较小(平均1.74 nm),且粒径分布较窄(1.50~2.01 nm);随着PFDTES加入量的增大,SiO2溶胶的平均粒径逐渐增大,粒径分布也逐渐变宽;(0.43PFDTES)BTESE的平均粒径达到了12.73 nm,粒径分布也较宽(8.72~18.17 nm).这是因为十七氟癸基的分子链较长,当修饰量增大时,修饰到—Si—C—C—Si—网络结构上致使整个溶胶粒径变大.选择溶胶进行涂膜时,溶胶粒径大小最好与γ-Al2O3/α-Al2O3多孔陶瓷支撑体的孔径(约4 nm)相近,因为选择粒径大的溶胶(>10 nm)进行涂膜会得到较厚的SiO2膜层,导致膜层易形成裂纹或其它严重缺陷,从而影响膜层的分离效果[22].另外,过大的溶胶胶粒必然使得膜材料的孔径过大,不利于气体分子的分离;而粒径过小时,溶胶颗粒可能直接进入γ-Al2O3/α-Al2O3支撑体的孔隙中,也会使膜层形成缺陷.由图5可见,(0.25PFDTES)BTESE溶胶的平均粒径为3.69 nm,且粒径分布较窄,疏水性能也较好[对水的接触角为(112.0±0.4)°],因此本文选择(0.25PFDTES)BTESE溶胶涂膜并进行气体分离实验.2.4 单组分气体渗透和分离性能图6为(0.25PFDTES)BTESE膜体系在不同温度下H2,CO2,CO和SF6的渗透率,表1为不同温度下H2/CO,H2/CO2,H2/SF6,CO/SF6和CO2/SF6的理想分离系数及其 Knudsen扩散分离因子.从图6和表1可见,H2的渗透率随着温度的升高逐渐增大,300℃时达到5.99×10-7mol·m-2·Pa-1·s-1;H2/CO2,H2/CO和H2/SF6的理想分离系数分别达到了5.20,9.54和39.08,均高于各自的Knudsen扩散分离因子(H2/CO2=4.69,H2/CO=3.74,H2/SF6=8.54),说明H2在膜材料中的输运行为主要遵循微孔扩散机理,且膜材料在此温度下对H2/CO2,H2/CO和H2/SF6的分离起到了分子筛分的作用.H2,CO2,CO和SF6的分子动力学直径分别为0.289,0.33,0.376和0.55 nm,SF6只能在尺寸大于0.55 nm的微孔或介孔甚至微缺陷中输运.从图6可以看出,SF6的渗透率随着温度的升高而降低,说明SF6的输运受到Knudsen扩散机制的控制.而H2,CO2和CO气体分子既能进入到小于0.55 nm的微孔中输运,也能在大于0.55 nm的微孔或介孔甚至微缺陷中输运,因而理论上CO和CO2气体的输运既可能遵循微孔扩散机理,也可能受Knudsen扩散机制的控制.从图6可见,CO2和CO的渗透率随着温度的升高而降低,其气体分子的输运呈现出Knudsen扩散现象,然而从表1可见,在不同温度时CO/SF6和CO2/SF6的分离系数均大于各自的Knudsen扩散分离因子,这说明CO和CO2气体的输运确实也受到微孔扩散机理的控制.在温度为50℃时,膜材料对于H2/CO的理想分离系数低于其Knudsen扩散分离因子;在温度为50~250℃时,膜材料对于H2/CO2的理想分离系数低于其Knudsen扩散分离因子.这可能是由于在此温度下CO和CO2的输运主要受到Knudsen扩散机制的控制,导致其在温度降低时渗透率增加,从而使得H2/CO和H2/CO2的理想分离系数降低.综上所述,通过十七氟癸基三乙氧基硅烷(PFDTES)和1,2-双(三乙氧基硅基)乙烷(BTESE)的共水解缩聚反应可得到稳定的有机-无机杂化SiO2溶胶,溶胶的粒径随着PFDTES修饰量的增加而增大.当n(PFDTES)∶n(BTESE)=0.25∶1时,溶胶的平均粒径为3.69 nm,分布范围较窄,用此溶胶在γ-Al2O3/α-Al2O3多孔陶瓷支撑体上涂覆的有机-无机杂化SiO2膜[(0.25PFDTES)BTESE]对水的接触角可达(112.0±0.4)°.300 ℃时该膜材料的 H2 渗透率达到5.99×10-7mol·m-2·Pa-1·s-1,H2/CO2,H2/CO和H2/SF6的分离系数分别为5.20,9.54和39.08,均高于Knudsen扩散的理想分离因子,表现出良好的分子筛分效应.[1] Kawi S.,Lai mun.[J],1998,13:1047—1053 [2] Anita R.,Sunil A.,Yang S.S..Renew.Energ.[J],2010,35:2649—2655[3] Zahmakiran M.,Ayvali T.,Akbayrak S.,Cali?kan S.,elik D.,Ozkar S..Catalysis Today[J],2011,170(1):76—84[4] Araki S.,Imasaka S.,Tanaka S.,Miyake Y..J.Membr.Sci.[J],2011,380:41—47[5] Catalano J.,Baschetti M.G.,Sarti G.C..Int.J.Hydrogen.Energ.[J],2011,36(14):8658—8673[6] Pomerantz N.,Ma Y.H..J.Membr.Sci.[J],2011,370:97—108 [7] Varela-Gandia F.J.,Berenguer-Murcia A.,Lozano-Castello D.,Cazorla-Amoros D..J.Membr.Sci.[J],2011,378:407—414[8] Imai H.,Morimoto H.,Tominaga A.,Hirashima H..J.Sol-GelSci.Technol.[J],1997,10:45—54[9] Uhlmann D.,Liu S.,Ladewing B.P.,Diniz da Costa J.C..J.Membr.Sci.[J],2009,326(2):316—321[10] WEI Na-Na(魏娜娜),WEI Qi(韦奇),LI Zhen-Jie(李振杰),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).J.Inorg.Mater.(无机材料学报)[J],2010,25(10),1047—1052[11] de Vos R.M,Wilhelm F.M.,Henk V..J.Membr.Sci.[J],1999,158:277—278[12] Wei Q.,Wang F.,Nie Z.R.,Song C.L.,Wang Y.L.,LiQ.Y..J.Phys.Chem.B[J],2008,112:9354—9359[13] Kanezashi M.,Yada K.,Yoshioka T.,Tsuru T..J.Membr.Sci.[J].2010,348:310—318[14] Castricum H.L.,Sah A.,Kreiter R.,Blank D.H.A.,Vente J.F.,ten Elshof J.E..J.Mater.Chem.[J],2008,18:2150—2158[15] Castricum H.L.,Sah A.,Geenevasen J.A.J.,Kreiter R.,BlankD.H.A.,Vente J.F.,ten Elshof J.E..J.Sol-Gel.Sci.Technol.[J],2008,48:11—17[16] DUAN Xiao-Yong(段小勇),WEI Qi(韦奇),HE Jun(何俊),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).Chem.J.Chinese Universities(高等学校化学学报)[J],2011,32(10):2256—2261[17] WANG Fei(王飞),WEI Qi(韦奇),WANG Yan-Li(王艳丽),YU Chun-Xiao(于春晓),ZHONG Zhen-Xing(钟振兴),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).Acta Chim.Sin.(化学学报)[J],2008,66(1):44—48[18] LI Zhen-Jie(李振杰),WEI Qi(韦奇),WEI Na-Na(魏娜娜),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).Chem.J.Chinese Universities(高等学校化学学报)[J],2010,31(12):2482—2487[19] Wei Q.,Wang Y.L.,Nie Z.R.,Yu C.X.,Li Q.Y.,Zou J.X.,LiC.J..Microp.Mesop.Mater.[J],2008,11:97—103[20] Turov V.V.,Mironyuk I.F..Colloid Surface A[J],1998,134:257—263[21] WEI Qi(韦奇),LI Jian-Lin(李建林),SONG Chun-Lin(宋春林),LIU Wei(刘卫),CHEN Chu-Sheng(陈初升).J.Inorg.Mater.(无机材料学报)[J],2004,19(2):417—423[22] Castricum H.L.,Sah A.,Kreiter R.,Blank D.H.A.,Vente J.F.,ten Elshof mun.[J],2008,9:1103—1105【相关文献】[1] Kawi S.,Lai mun.[J],1998,13:1047—1053[2] Anita R.,Sunil A.,Yang S.S..Renew.Energ.[J],2010,35:2649—2655[3] Zahmakiran M.,Ayvali T.,Akbayrak S.,Cali?kan S.,elik D.,Ozkar S..Catalysis Today[J],2011,170(1):76—84[4] Araki S.,Imasaka S.,Tanaka S.,Miyake Y..J.Membr.Sci.[J],2011,380:41—47 [5] Catalano J.,Baschetti M.G.,Sarti G.C..Int.J.Hydrogen.Energ.[J],2011,36(14):8658—8673[6] Pomerantz N.,Ma Y.H..J.Membr.Sci.[J],2011,370:97—108[7] Varela-Gandia F.J.,Berenguer-Murcia A.,Lozano-Castello D.,Cazorla-Amoros D..J.Membr.Sci.[J],2011,378:407—414[8] Imai H.,Morimoto H.,Tominaga A.,Hirashima H..J.Sol-Gel Sci.Technol.[J],1997,10:45—54[9] Uhlmann D.,Liu S.,Ladewing B.P.,Diniz da Costa J.C..J.Membr.Sci.[J],2009,326(2):316—321[10] WEI Na-Na(魏娜娜),WEI Qi(韦奇),LI Zhen-Jie(李振杰),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).J.Inorg.Mater.(无机材料学报)[J],2010,25(10),1047—1052[11] de Vos R.M,Wilhelm F.M.,Henk V..J.Membr.Sci.[J],1999,158:277—278 [12] Wei Q.,Wang F.,Nie Z.R.,Song C.L.,Wang Y.L.,Li Q.Y..J.Phys.Chem.B[J],2008,112:9354—9359[13] Kanezashi M.,Yada K.,Yoshioka T.,Tsuru T..J.Membr.Sci.[J].2010,348:310—318[14] Castricum H.L.,Sah A.,Kreiter R.,Blank D.H.A.,Vente J.F.,ten ElshofJ.E..J.Mater.Chem.[J],2008,18:2150—2158[15] Castricum H.L.,Sah A.,Geenevasen J.A.J.,Kreiter R.,Blank D.H.A.,Vente J.F.,ten Elshof J.E..J.Sol-Gel.Sci.Technol.[J],2008,48:11—17[16] DUAN Xiao-Yong(段小勇),WEI Qi(韦奇),HE Jun(何俊),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).Chem.J.Chinese Universities(高等学校化学学报)[J],2011,32(10):2256—2261[17] WANG Fei(王飞),WEI Qi(韦奇),WANG Yan-Li(王艳丽),YU Chun-Xiao(于春晓),ZHONG Zhen-Xing(钟振兴),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).Acta Chim.Sin.(化学学报)[J],2008,66(1):44—48[18] LI Zhen-Jie(李振杰),WEI Qi(韦奇),WEI Na-Na(魏娜娜),LI Qun-Yan(李群艳),NIE Zuo-Ren(聂祚仁).Chem.J.Chinese Universities(高等学校化学学报)[J],2010,31(12):2482—2487[19] Wei Q.,Wang Y.L.,Nie Z.R.,Yu C.X.,Li Q.Y.,Zou J.X.,LiC.J..Microp.Mesop.Mater.[J],2008,11:97—103[20] Turov V.V.,Mironyuk I.F..Colloid Surface A[J],1998,134:257—263[21] WEI Qi(韦奇),LI Jian-Lin(李建林),SONG Chun-Lin(宋春林),LIU Wei(刘卫),CHEN Chu-Sheng(陈初升).J.Inorg.Mater.(无机材料学报)[J],2004,19(2):417—423[22] Castricum H.L.,Sah A.,Kreiter R.,Blank D.H.A.,Vente J.F.,ten Elshof mun.[J],2008,9:1103—1105(Ed.:N,K,M)。
化疗药物缩写

烷化剂:之阳早格格创做HN2:氮芥CTX:环磷酰胺IFO:同环磷酰胺MEL:苯丙氨酸氮芥NF:氮甲M-25:苦磷酰芥AT-1258:消瘤芥CB-1348:苯丁酸氮芥TSPA:塞替派BCNU:卡莫司汀CCNU:洛莫司汀Me-CCNU:司莫司汀ACNU:僧莫司汀FTM:祸莫司汀BUS:黑消安/马利兰DBD:两溴卫盾醇DDAG:两去火卫盾醇PM:泼僧氮芥ETM:雌两醇氮芥抗代开药物:MTX:氨甲蝶呤AG337:洛推直克6-GT:硫鸟嘌呤5-FU:氟尿嘧啶FT-207:替加氧UFT:劣祸定HCFU:卡莫氟5'-DFUR:脱氧氟尿苷希罗达:卡培他滨Ara-C:阿糖胞苷CC:环胞苷GEM:凶西他滨Raltitrexed/ZD1694:雷替直塞HMM:六甲蜜胺抗死素类ACTD/KSM:搁线菌素D MMC:丝裂霉素BLM:专去霉素PYM:仄阳霉素PEP/PLM:培洛霉素DNR:柔黑霉素ADM:阿霉素/多柔比星EPI:表阿霉素/表柔比星THP:吡喃阿霉素ACR:阿柔比星IDA:伊达比星MTH:灿烂霉素STZ:链脲霉素BST/ADD:比死群CMM:洋黑霉素MIT/MXT:米托葱醌动物药VLB:少秋碱VCR:少秋新碱VSD:少秋天辛NVB/VNR:少秋瑞滨VP-16:依托泊甙VM-26:替僧泊甙COLM:秋火仙碱酰胺HRT:三尖杉酯碱HCPT:羟基喜树碱CPT-11:伊坐替康/启普拓TPT:拓扑替康LT50100:甲基羟基玫瑰树碱PTX:紫杉醇TXT:泰索帝/多西紫杉醇Indirubin:靛玉黑激素类:PDN:泼僧紧PDNL:泼僧紧龙MePDNL:甲级泼僧紧龙DXM:天塞米紧HL-286:溴醋己烷雌酚Flutamide:氟他胺MPA:甲羟孕酮MA:甲天孕酮TAM:他莫昔芬TOR:托瑞米芬DRL:伸洛昔芬AG:氨鲁米特FMT:祸好坦Anastrozole:阿那直唑/瑞宁德Letrozole:去直唑LH-RH拮抗剂:诺雷德/抑那通纯类ASP:门冬酰胺酶PCZ/PCB:甲基苄肼DTIC:达卡巴嗪DDP:逆铂CBP:卡铂SHP:环硫铂L-OHP:奥沙利铂NDP:奈达铂Me-GAG:丙脒腙HU:羟基脲AT-1727:乙单吗啉O,P'DDD-:米托坦AMSA:胺苯丫啶死物治疗药物:INF:搞扰素IL-2:细胞黑介素2TNF:肿瘤坏死果子Lentinan:香菇多糖LMS:左旋咪唑Mabthera:好罗华Herceptin:赫赛汀Imatinib:伊马替僧/格列卫依照字母程序时常使用化疗药物中英文对于照AAclacinomycin ACLA 阿克推霉素 Alemtuznmab 阿去组单抗, 抗CD52嵌合抗体 All-trans retinoic acid, ATRA, RA 齐反式维甲酸, 维A酸 Aminoglutethimide AG 氨基导眠能, 氨鲁米特, 氨苯哌酮 Anastrozole, Arimidex 瑞宁得, 阿纳托唑, 阿那直唑 Arsenictrioxide AS2O3 三氧化两砷 Asparaginase L-ASP 门冬酰胺酶BBevacizumab, Avastin BEV 抗血管内皮死少果子单克隆嵌合抗体Bicalutamide, Casodex 比卡鲁胺Bleomycin BLM 专去霉素Busulfan, Myleran BUS, BSF 黑消安, 马利兰CCalichcamicin 人源化抗CD33单克隆抗体Camptothecin CPT 喜树碱Carboplatin, Paraplatin CBP 卡铂Carmofur HCFU 卡莫氟, 嘧祸禄Carmustine BCNU 卡莫司汀,卡氮芥Chlorambucil, Leukeran CLB 苯丁酸氮芥, 痛可宁Cisplatin PDD, CDDP 逆铂Cladribine, 2-chlorooxyadenosine CDA, 2-Cd A 2-氯脱氧腺苷, 克推伸滨Colchicine COL 秋火仙碱Colchicine amide COLM 秋火仙酰胺Compound Diphenoxylate Tablets 苯乙哌啶Cyclocytidine CCY 环胞苷, 安西他滨Cyclophosphamide CTX 环磷酰胺Cytarabine, Cytosine arabinoside Ara-C 阿糖胞苷 Dacarbazine DTIC 达卡巴嗪, 氮烯咪胺DDactinomycin, Actinomycin D ACD 搁线菌素D, 更死霉素Daunorubicin DNR 柔黑霉素, 正定霉素Dexamethasone DXM 天塞米紧Dexrazoxane 左雷佐死Diphenhy dramine 苯海推明Doxorubicin, Adriamycin ADM 阿霉素, 多柔比星 EElemene Emulsion 榄香烯乳Epirubicin, Epidoxorubicin EPI 表阿霉素, 表柔比星Estramustine EM 癌腺治, 雌两醇氮芥, 雌莫司汀 Etoposide VP-16 脚叶乙苷, 鬼臼乙叉甙, 依托泊苷, 脚叶已甙Exemestane EXE 依西好坦, 艾罗好新FFemara, Letrozole LTZ 去直唑Floxuridine FUDR 5-氟脱氧尿苷, 氟尿苷Fludarabine FA 氟达推宾Fluorouracil, Fluoracil 5-Fu 氟尿嘧啶Flutamide, Eulexin 氟硝丁酰胺, 慢退瘤Folic acid 叶酸Ftorafur, Tegafur FT207 替加氟, 喃氟啶,呋氟尿嘧啶FudR 氟苷GGemcitabine, Gemzar GEM 健择, 单氟胞苷, 凶西他滨Gemtuzumab Ozogamicin, Mylotarg TM CMA-676 卡偶霉素, 凶妥组单抗,人源化抗CD33嵌合抗体卡偶霉素免疫复合物Glivec, Imatinib STI 571 格列卫, 甲磺酸伊马替僧Homoharringtonine 下三尖杉酯碱Human granulocyte colony stimulating factor G-CSF沉组粒细胞集降刺激果子Human granulocyte-macrophage colony stimulating factor GM-CSF沉组巨噬细胞粒细胞集降刺激果子Hydrocortisone 氢化可的紧Hydroxycamptothecin HCPT 羟基喜树碱, 羟喜树碱Hydroxyurea, Hydroxycarbamide HU 羟基脲IIdarubicin IDA 去甲氧柔黑霉素Ifosfamide Ifosphamide IFO 同环磷酰胺Interferon IFN 搞扰素Interferon alfa IFNαα-搞扰素Interferon-βIFNββ-搞扰素Interlukin 2 rIL-2 黑细胞介素2Iressa, Gefitinib ZD1839 易瑞沙Irinotecan CPT-11 依利替康Leucovorin, Calcium folinate CF, LV 亚叶酸钙 Leuprolide 瘤破利得, 醋酸明丙瑞林Leuprolide depot 瘤破利得库Liposomal Doxorubicin 阿霉素脂量体Lomustine CCNU 洛莫司汀, 环已亚硝脲Loperamide, Imodium 洛哌丁胺, 易受停, 氯苯呱酰胺MMechlorethamine, Mustine, Chlormethine HN2 盐酸氮芥, 氮芥Megace MA 甲天孕酮Melphalan 好法仑Melphalan, Alkeran MEL 苯丙氨酸氮芥Mesna 好司钠Methotrexate MTX 甲氨蝶呤Methylprednisone MPED 甲基强的紧Mitomycin MMC 丝裂霉素Mitoxantrone, Novantrone MIT, NVT 米托蒽醌 Mitramycin MTH 灿烂霉素NNilutamide 僧鲁特米Nimustine ACNU 僧莫司汀, 嘧啶亚硝脲Nocardia rubra cell wall skeleton N-CWS 胞必好, 黑色诺卡氏菌细胞壁骨架OOxaliplatin L-OHP 草酸铂, 奥沙利铂PPemetrexed, Alimta 阿灵达Pingyangmycin PYM 仄阳霉素Prednisone PED, PDN 强的紧, 泼僧紧Procarbazine PCB, PCZ 甲基苄肼Provera MPA 甲孕酮, 乙酸甲羟孕酮RRaltitrexed, Tomudex 雷替直塞Retinoic acid RA 维甲酸Rituximab, Rituxan, Mabthera 好罗华, 利妥昔单抗TTamoxifen TAM 三苯氧胺Taxol, Paclitaxel TAX, PTX 紫杉醇, 泰素Taxotere, Docetaxel DOC 多西紫杉醇, 泰索帝Temozolomide, Temodal TEM, TMZ 替莫待我Teniposide VM-26 替僧泊苷,威猛Thalidomide 沙利度胺, 反应停Topotecan TPT 拓扑替康Trastuzumab, Herceptin 赫赛汀Tumor Necrosis Factor TNF 肿瘤坏死果子UUracil, UFT 劣祸定VVinblastine VLB 少秋花碱, 少秋碱Vincristine, Oncovin VCR 少秋新碱Vindesin VDS 少秋天辛Vinorelbine NVB 少秋瑞滨XXeloda, Capecitabine Cap, BOF-A2 希罗达, 卡培他滨,氟嘧啶氨甲酸酯ZZoladex, Goserelin depot 诺雷德库, 戈舍瑞林。
氟达拉滨联合米托蒽醌、地塞米松治疗惰性淋巴瘤的临床观察

氟达拉滨联合米托蒽醌、地塞米松治疗惰性淋巴瘤的临床观察王琛;夏堪冀;王玲;舒琦;何代英;徐诣芝【摘要】目的比较FND(氟达拉滨+米托蒽醌+地塞米松)与CHOP(环磷酰胺+阿霉素+长春新碱+强的松)方案治疗惰性淋巴瘤的疗效与安全性.方法临床观察的内容包括缓解率[总有效率和完全缓解(CR)率],无失败生存(FFS)及毒性反应.56例患者随机分组,FND和CHOP组各28例.FND方案:氟达拉滨30 mg·m-2·d-1d1~3静脉注射,米托蒽醌10 mg d1静脉注射,地塞米松20 mg d1~5静脉注射.平均接受6.7个化疗方案.CHOP方案:环磷酰胺600 mg·m-2·d-1d1静脉注射,阿霉素25 mg·m-2·d-1d1静脉注射,长春新碱1.4 mg·m-2·d-1d1静脉注射和强的松50 mg·m-2·d-1d1~5口服.平均接受7.1个化疗方案.以上两方案均为28 d一次.结果FND方案的完全缓解率和总有效率显著优于CHOP方案(总有效率82.1% vs57.1%,CR 53.6% vs 32.1%;P<0.01).2年无疾病进展生存(PFS)率FND组为86.7%而CHOP组为66.7%.两种治疗方案的耐受性均较好.结论 FND方案的CR率、总有效率均显著优于CHOP方案,并可有效改善预后.【期刊名称】《重庆医学》【年(卷),期】2010(039)012【总页数】2页(P1520-1521)【关键词】氟达拉滨;惰性淋巴瘤;米托蒽醌【作者】王琛;夏堪冀;王玲;舒琦;何代英;徐诣芝【作者单位】重庆市第三人民医院血液科,400014;重庆市第三人民医院血液科,400014;四川省疾病控制中心门诊部,成都,610041;重庆市第三人民医院血液科,400014;重庆市第三人民医院血液科,400014;重庆市第三人民医院血液科,400014【正文语种】中文【中图分类】R733.1;R730.53惰性淋巴瘤为一类恶性程度较低、进展缓慢的非何杰金淋巴瘤,在世界范围内占非何杰金淋巴瘤的22%。
国产氟多功能模块合成雌激素受体显像剂16α-[~(18)F]氟-17β-雌二醇
![国产氟多功能模块合成雌激素受体显像剂16α-[~(18)F]氟-17β-雌二醇](https://img.taocdn.com/s3/m/b77aac47852458fb770b56b2.png)
雌 激 素 受 体 显 像 剂 1 aL F 氟 1 1雌 二 醇 ( FF S 。结 果 显 示 : 成 的 F F S 不 校 正 合 成 效 率 为 6_ ] 7- 3 -E ) 合 —E ,
8 2 , 正 合 成 效 率 为 1 . ; 成 时 间 约 为 7 n 标 记 物 F F S放 化 纯 度 大 于 9 , 外 稳 定 性 良 . 校 28 合 O mi , —E 8 体
1F— a 0 C f r 1 n i e ld s se o o si y t e ie ,t e . 1 mo / 8 t1 5 o 5 mi n s ae y t m n d me tcs n h sz r h n 0 8 ml lI
HCld s o v d i . a e o t ie wa d d i hr e p r s t her a to e s lf r h i s l e n 7 2 mI c t ne r l s a de n t e a t o t e c in v s e o y— d o1 s s a d hy o yss r a to spe f me t 1 r y i n dr l i e c i n wa r or d a 05 ℃ f r 6 mi o n.Thefn lr a ton s l i a e c i o u—
好 。 以 上 结 果 表 明 , 产 氟 多 功 能 模 块 可 制 备 F F S溶 液 , 备 的 F E 国 -E 制 F S溶 液 符 合 放 射 性 药 物 的 质 量
要求 。
关 键 词 :: F F S 雌 激 素受 体 显 像 剂 ; 二 醇 ;国产 氟 多 功 能 合 成 模 块 E ; 雌
to a rfe y H PIC o g v F— i n w s pu i d b i t ie FES. Pr pa a i n o F— e r to f FES o o esi y he i ndm tc s nt s s
氟达拉滨联合方案治疗进展性非霍奇金淋巴瘤疗效观察

评价氟达拉 滨联合方案 ( 氟达拉 滨 +米托 蒽醌 +地 塞米松 ,MD) 疗进展性 非霍 奇金 淋 巴 F 治 所有病 例均 可作疗 效评价 , 有效 率( R+ C
瘤( HL 的疗效和安全性。方法 经组织 学确诊的既往接受过 C P等方案治疗 的进展性 N L共 1 N ) HO H 7例 , 均采用
主要包括乳源肽 、 卵蛋 白肽 、 蛋 白肽 、 畜 鱼蛋 白肽 和丝
蛋 白肽等 , 上述功能性短肽具有四大生物活性 : 于心 利 血管系统 , 主要包 括一些抗血栓肽 和抗 高血压肽 ; 于 利 神经系统 , 主要是 阿片肽 ; 于免疫 系统 , 利 主要 是一 些 免疫调节肽和抗菌肽 ; 于营养 系统 , 利 主要是利 于钙 吸
织修复 , 刺激和促进骨组织细胞合成 , 补充骨骼修复过 程 中需要的钙 、 、 、 镁 铁 锌等微量元素 , 具有改善骨折端
的血液循环 、 止痛 等作 用。本 院采 用古欣 肽骨 肽注射
液治疗风湿性关节炎疗效确切。
参 考 文 献
[ ] 何庆勇 , 1 张吉. 针药并用治 疗风湿性 关节炎 的疗 效观察 [ ] J.
例 为肺部 感染, 2例 未发现 感染病灶 , 另 经处理好转 。1例 出现轻度腹泻。结论
展 性 N L疗 效好 , 良反 应 轻 , 得 进 一 步 研 究 。 H 不 值
氟达拉滨杂质全套最新列表

中文名
磷酸氟达拉滨杂质1(磷酸氟 达拉滨EP杂质A)
CAS号 62314-92-5
用途
新药研发申报注册/ 鉴别、检查、含量测定
等 181+7164+1670
结构式
磷酸氟达拉滨杂质2(磷酸氟 达拉滨EP杂质B)
3373-53-3
新药研发申报注册/ 鉴别、检查、含量测定
等
磷酸氟达拉滨杂质3(磷酸氟 达拉滨EP杂质C)
7561-54-8
新药研发申报注册/ 鉴别、检查、含量测定
等
磷酸氟达拉滨杂质10(磷酸 氟达拉滨EP杂质J)
N/A
新药研发申报注册/ 鉴别、检查、含量测定
等
磷酸氟达拉滨杂质11 磷酸氟达拉滨杂质12 磷酸氟达拉滨杂质13 磷酸氟达拉滨杂质14 磷酸氟达拉滨杂质15 磷酸氟达拉滨杂质16 磷酸氟达拉滨杂质17 磷酸氟达拉滨杂质18 磷酸氟达拉滨杂质19 磷酸氟达拉滨杂质20 磷酸氟达拉滨杂质21 磷酸氟达拉滨杂质22
新药研发申报注册/ 鉴别、检查、含量测定
等
266360-74-1
新药研发申报注册/ 鉴别、检查、含量测定
等
新药研发申报注册/
N/A
鉴别、检查、含量测定
等
102783-36-8
新药研发申报注册/ 鉴别、检查、含量测定
等
新药研发申报注册/
N/A
鉴别、检查、含量测定
等
磷酸氟达拉滨杂质35
新药研发申报注册/ 2734853-80-4 鉴别、检查、含量测定
等
新药研发申报注册/
N/A
鉴别、检查、含量测定
等
新药研发申报注册/
N/A
鉴别、检查、含量测定
等
《生命科学中的微量元素》氟和碘的生物学作用与临床(可编辑)

《生命科学中的微量元素》氟和碘的生物学作用与临床生命科学中的微量元素――氟和碘的生物学作用与临床氟的生物学作用与临床――人体必需微量元素――对牙齿及骨骼的形成和结构具有重要作用――对钙磷的代谢具有重要作用氟的生物学作用与临床一、氟的含量与分布二、氟的生物学作用与临床三、氟的吸收、运输与排泄四、氟的毒性与防治氟的生物学作用与临床一、氟的含量与分布一、氟的含量与分布地壳:625μg/g 空气:小于0.01μg/m3 人体:正常成人体内共含氟2.6g,占体内微量元素的第三位,仅次于硅和铁人体几乎所有的各种器官内均含有氟,但绝大部分分布在硬组织骨骼和牙齿中,两者约占人体总含氟量的90%以上一、氟的含量与分布我国正常人骨骼中氟:200~300μg/g,最高可达800μg/g,高氟区居民骨骼氟含量甚至高达1000μg/g,地方性氟中毒病区成人骨氟达15000μg/g!人体不同部位骨骼含氟量差异很大,以长骨含氟量较高,依次是股骨>肱骨>掌骨>颅骨>腰椎骨,而且男性骨骼含氟量高于女性。
一、氟的含量与分布头发含氟量也较高,可达14~30μg/g 软组织中含氟最多的是皮肤,约3~50μg/g,其次为肌腱(膜)、主动脉、肺、肾、心、胰、脑、脾、肝等氟在生物体内还有一个重要的特性,即无生物降解作用,能在生物体内富集。
骨骼中的含氟量具有随年龄的增长而增高的趋势二、氟的生物学作用与临床 1、氟的生理作用 2、氟的毒理作用 1、氟的生理作用(1)参与骨骼代谢(2)防龋作用(3)对神经系统的作用(4)其他作用(1)参与骨骼代谢机体正常的钙、磷代谢离不开适量的氟在一定的pH条件下,氟有助于钙和磷形成羟基磷灰石,促进成骨过程。
适量的氟,羟基磷灰石的羟基可被氟取代,形成均匀一致的氟化磷灰石, 溶解度明显降低,其热力学的稳定性明显升高,增强了骨骼的强度(2)防龋作用氟可以起到防龋的作用:①直接提高牙齿的防龋能力②消除有关的致龋因素,改善口腔环境(3)对神经系统的作用氟对神经系统兴奋性的影响主要是通过对某些酶的作用而体现的氟能抑制胆碱脂酶活性,减少乙酰胆碱分解,从而使其含量增多,提高神经的兴奋性和传导作用氟还能抑制三磷酸腺苷酶,使体内ATP含量增多。
血液科常用化疗方案

药物剂量急性淋巴细胞白血病方案用法及疗程备注VDCP 长春新硷(VCR)柔红霉素(DNR)环磷酰胺(CTX)强的松(Pred)长春新硷(VCR)柔红霉素(DNR)1.5mg/m230-40mg/m2600-800mg/m240-60mg/m21.5mg/m230-40mg/m2Ⅳ,第1,2,3Ⅳ,第1-3天,第15-17天Ⅳ,第1,15天口服,第1-28天Ⅳ,第1,8,15,21天Ⅳ,第1-3和第15-17天28天为1疗程VDPA强的松(Pred)门冬酰胺酶(L-ASP)足叶乙甙(VP16)EA阿糖胞苷(Ara-C)长春新硷(VCR)柔红霉素(DNR)D-2V-P足叶乙甙(VP16)强的松(Pred)HD-MTX甲氨喋呤(MTX)柔红霉素(DNR)DA阿糖胞苷(Ara-C)HA三尖杉酯硷(HAT)阿糖胞苷(Ara-C)柔红霉素(DNR)40-60mg/m26000u/m275mg/m2100-150mg/m21.5mg/m230-40mg/m275mg/m240-60mg/m21-1.5g/m2静滴24h,急非淋方案30-40mg/m2100-150mg/m23-4mg/m2100-150mg/m230-60mg/m228天为1疗程口服1―28天ⅣGTT,第19-28天Ⅳ,第1-7天7天为1疗程Ⅳ,第1-7天Ⅳ,第1,8,15,21天Ⅳ,第1-3和第15-17天28天为1疗程Ⅳ,第8-10,21-23天口服1―28天药后12h以四氢叶酸钙解救,6-9mg/m2,Q6h,共8次停Ⅳ,第1-3天7天为1疗程Ⅳ,第1-7天Ⅳ,第1-7天7天为1疗程Ⅳ,第1-7天Ⅳ,第1-3天DAE阿糖胞苷(Ara-C)足叶乙甙(VP16)米托蒽醌(MTZ)MA100-150mg/m275mg/m25mg/m2Ⅳ,第1-7天Ⅳ,第5-7天Ⅳ,第1-3天7天为1疗程7天为1疗程阿糖胞苷(Ara-C)D H-A柔红霉素(DNR)100-150mg/m230-40mg/m2Ⅳ,第1-7天Ⅳ,第1-3天3天为1疗程阿糖胞苷(Ara-C)三尖杉酯硷(HAT)1.0g/m23-4mg/m2Ⅳ,Q12h1~3天Ⅳ,第1-7天H H-A7天为1疗程CHOP DAEP 阿糖胞苷(Ara-C)环磷酰胺(CTX)阿霉素(ADM)长春新硷(VCR)强的松(Pred)柔红霉素(DNR)阿糖胞苷(Ara-C)足叶乙甙(VP16)强的松(Pred)米托蒽醌(MTZ)长春新硷(VCR)1.0g/m2恶组化疗方案600mg/m230mg/m21.4mg/m260mg/m225mg/m2100mg/m270mg/m240mg/m25mg/m21.4mg/m2Ⅳ,Q12h1~3天IV,第1天IV,第1天IV,第1天口服1~5天IV,第1~3天皮下2/日,第1~7天IVGTT(2h),第5~7天口服第1~7天IV,第1~3天IV,第1天每2~3周重复每2~3周重复每2~3周重复MOAP阿糖胞苷(Ara-C)强的松(Pred)氮芥长春新硷(VCR)MOPP甲基苄肼强的松(Pred)阿霉素(ADM)平阳霉素ABVD长春硷氮烯咪胺氮芥MOPP/ABV长春新硷(VCR)甲基苄肼100mg/m240mg/m2霍奇金淋巴瘤主要化疗方案4mg/m21.4mg/m270mg/m240mg/m225mg/m210mg/m26mg/m2375mg/m26mg/m21.4mg/m2100mg/m2皮下2/日第1~7天口服第1~7天Ⅳ第1,8天Ⅳ第1,8天口服,第1~14天口服,第1~14天Ⅳ第1,15天Ⅳ第1,15天Ⅳ第1,15天Ⅳ第1,15天Ⅳ,第1天Ⅳ,第1天口服,第1~7天28天重复疗程每28天重复疗程28天重复疗程共6个疗程强的松(Pred)阿霉素(ADM)平阳霉素长春花硷环磷酰胺(CTX)COP长春新硷(VCR)强的松(Pred)环磷酰胺(CTX)阿霉素(ADM)CHOP(米托蒽醌长春新硷(VCR)强的松(Pred)环磷酰胺(CTX)长春新硷(VCR)COPP甲基苄肼强的松(Pred)博来霉素阿霉素(ADM)环磷酰胺(CTX)m-BACOB长春新硷(VCR)地塞米松(DXM)甲氨喋呤(MTX)亚叶酸钙(甲酰四氢叶酸钙)环磷酰胺(CTX)长春新硷(VCR)COP-BLAM强的松(Pred)博来霉素(BLAM)阿霉素(ADM)40mg/m235mg/m210mg/m26mg/m2非霍奇金淋巴瘤化疗方案400mg/m21.4mg/m2100mg/m2750mg/m250mg/m212-14mg/m21.4mg/m2100mg/m2600mg/m21.4mg/m2100mg/m240mg/m24mg/m245mg/m2600mg/m21mg/m26mg/m2200mg/m210mg/m2400mg/m21mg/m240mg/m215mg40mg/m2口服,第1~14天Ⅳ,第8天Ⅳ,第8天Ⅳ,第8天口服,第1~5天Ⅳ,第1天口服,第1~5天Ⅳ,第1天Ⅳ,第1天IV第1天)Ⅳ第1天口服。
硼替佐米联合氟达拉滨及环磷酰胺治疗复发难治性套细胞淋巴瘤完全缓解2例并文献复习

硼替佐米联合氟达拉滨及环磷酰胺治疗复发难治性套细胞淋巴瘤完全缓解2例并文献复习常智;李书苹;张亚瑞;曹晓艳;宋腾;陈海珠;王华庆【期刊名称】《癌症进展》【年(卷),期】2017(015)007【总页数】3页(P848-850)【关键词】硼替佐米;套细胞淋巴瘤;疗效【作者】常智;李书苹;张亚瑞;曹晓艳;宋腾;陈海珠;王华庆【作者单位】天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121;天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121;天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121;天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121;天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121;天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121;天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所,天津300121【正文语种】中文【中图分类】R733.4套细胞淋巴瘤(mantle cell lymphoma,MCL)是一种具有高度侵袭性的起源于淋巴结滤泡套区的B细胞非霍奇金淋巴瘤(non-Hodgkin’s lymphoma,NHL),占NHL的3%~10%,高发于老年人群,中位发病年龄为60~65岁,男女比例为3∶1[1]。
MCL具有特征性的遗传学易位特点t(11;14)(q13;q32),细胞周期蛋白D1(CyclinD1)过表达,恶性程度高,中位生存时间为3~5年[2]。
目前尚无标准的治疗方案,常以环磷酰胺+多柔比星+长春新碱+强的松(CHOP)方案为主,可获得较高的缓解率,但复发率高。
硼替佐米是一种新型的蛋白酶体抑制剂,以硼替佐米为主的联合化疗目前成为MCL治疗领域研究的热点。
天津市人民医院肿瘤诊治中心天津市中西医结合肿瘤研究所使用硼替佐米联合氟达拉滨及环磷酰胺治疗2例复发难治MCL患者并获得了完全缓解,现报道如下。
慢性淋巴细胞白血病的造血干细胞移植治疗

慢性淋巴细胞白血病的造血干细胞移植治疗杨申淼;江倩;许兰平【摘要】慢性淋巴细胞白血病(CLL)存在很大的临床异质性。
尽管免疫化疗方案的进步带来了显著的疗效,部分患者仍可在短期内发生疾病进展,或处于疾病难治耐药的状态。
由于移植物抗CLL效应的存在,异基因造血干细胞移植的根治性意义获得肯定。
年轻CLL患者如具有高危因素,包括:嘌呤类似物耐药或治疗后早期复发,以及具有17p(TP53位点)缺失和TP53突变,异基因造血干细胞移植是合理的治疗选择。
减低强度的预处理方案有效降低了患者的治疗相关死亡率。
%Chronic lymphocytic leukemia(CLL) is an indolent lymphoproliferative disorder with great clinical heterogeneity. The immunochemotherapeutic regimens have improved the outcome of majority of the patients. Nonetheless, in some patients, the disease progresses shortly after the immunochemotherapy, while in others, it becomes refractory and drug resistant. Allogeneic hematopoietic stem cell transplantation(allo-SCT) potentially cures CLL due to the graft versus CLL effect. It has been wildly accepted that allo-SCT is a good option for young CLL patients with high risk factors, including purine analogue resistance, early relapse after purine analogue-based treatment, and 17p deletion and TP53 mutation. Reduced-intensity conditioning regimens effectively decrease the treatment-related mortality.【期刊名称】《中华老年多器官疾病杂志》【年(卷),期】2013(000)008【总页数】7页(P587-593)【关键词】慢性淋巴细胞白血病;造血干细胞移植【作者】杨申淼;江倩;许兰平【作者单位】北京大学人民医院血液病研究所,北京 100044;北京大学人民医院血液病研究所,北京 100044;北京大学人民医院血液病研究所,北京 100044【正文语种】中文【中图分类】R733.72慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)是一种在临床特点上存在巨大异质性的惰性淋巴增殖性疾病。
氟达拉滨单药或联合环磷酰胺治疗慢性淋巴细胞白血病临床观察

氟达拉滨单药或联合环磷酰胺治疗慢性淋巴细胞白血病临床观察目的:观察氟达拉滨单药(F方案)或联合环磷酰胺(FC方案)治疗慢性淋巴细胞白血病的疗效及不良反应观察。
方法:2006-2012年33例慢性淋巴细胞白血病患者分为3组,F组11例,FC组11例,CHOP组11例,研究含氟达拉滨的治疗方案与传统CHOP方案对慢性淋巴细胞白血病的疗效及不良反应,同时研究F方案与FC方案在治疗慢性淋巴细胞白血病方面的疗效及不良反应。
结果:F和FC方案与CHOP方案相比,完全缓解率(CR)提高,总有效率(OR)提高,差异具有统计学意义(P<0.05)。
三种方案不良反应在血液学改变方面及在非血液学方面,差异无统计学意义(P>0.05)。
结论:含有氟达拉滨的方案治疗慢性淋巴细胞白血病疗效优于传统的CHOP一线治疗方案,氟达拉滨与环磷酰胺联合优于氟达拉滨单药治疗,不良反应无明显增加。
标签:氟达拉滨;环磷酰胺;慢性淋巴细胞白血病随着我国人口老龄化逐渐加重,慢性淋巴细胞白血病的发病率呈上升趋势。
慢性淋巴细胞白血病是老年人多发的一种血液系统肿瘤,由B淋巴细胞恶性克隆性增殖所致,临床上它的特点是产生大量不成熟的淋巴细胞,这些细胞在骨髓内聚集,抑制骨髓的正常造血;并且能够通过血液在全身扩散,导致患者出现贫血、容易出血、感染及器官浸润等。
传统治疗方案多为含烷化剂的联合化疗,完全缓解率低。
氟达拉滨能特异性作用于淋巴细胞,目前含有氟达拉滨的治疗方案已取代传统治疗方案,成为一线治疗。
本研究回顾性分析含有氟达拉滨的治疗方案与传统方案治疗慢性淋巴细胞白血病的临床疗效与副反应。
1 资料与方法1.1 一般资料2006-2012年在锦州市中心医院经临床,血常规,骨髓象和骨髓细胞免疫分型确诊的慢性淋巴细胞白血病患者33例,均符合张之南血液病诊断及疗效标准(第3 版)诊断标准[1]。
男22例,女11例,年龄45 ~77 岁,中位年龄64 岁;按Binet临床分期, A 期3例,B期18例,C期12例。
十七氟癸基三甲氧基硅烷怎么水解达到120℃水滴角

十七氟癸基三甲氧基硅烷怎么水解达到120℃水滴角
十七氟癸基三甲氧基硅烷是一种有机硅化合物,也称为FAS17。
它是一种表面活性剂,可以在水和油之间降低表面张力。
水解是指化合物在水中被分解成其组成部分。
FAS17的水解过程可以通过以下步骤实现:
首先,在120℃下将FAS17加入到水中,并将其搅拌均匀,使其充分分散。
接下来,在这个体系中添加一定量的碱性物质,例如氢氧化钠(NaOH)。
碱性物质可以促进FAS17的水解,使其分解成十七氟癸酸和三甲氧基硅醇。
随着水解的进行,表面张力逐渐降低,水滴开始变得更加平滑,水滴角也会逐渐达到120度。
水滴角是表征固体表面亲水性或疏水性的度量,120度的水滴角表明FAS17已经成功地降低了水的表面张力,使其成为疏水表面。
总之,在120℃下将FAS17加入水中,添加适量的碱性物质,可以实现FAS17的水解,并且达到120度的水滴角。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
磷酸氟达拉滨药效学研究试验和文献资料氟达拉滨(Fludarabinr)商品名Fludara IV,化学名:9-β-D-arabinofuranosyl-2-fluoroadenine 5’-monophosphate,代号NSC 321887, 分子式:C10H11N5O7PF。
静脉注射制剂内主要含2-fluoro-araadenine monophosphate (2-fluoro-ara-AMP)。
属嘌呤核苷酸类抗代谢药,临床上主要用于抗淋巴细胞性增殖性疾病。
氟达拉滨是Montgomery及同工用腺苷单核苷酸和ara-A合成的。
由于在嘌呤环上的氟原子位置,使氟达拉滨不容易去氨和溶解性比ara-A更好一些。
临床前研究证实氟达拉滨有抗多种肿瘤(血液系统和实体瘤)作用,如生长在裸鼠L1210白血病、P388白血病和LX1人肺肿瘤。
在L1210和P388模型,药物的作用依赖给药程序。
多剂量(给药)比单剂量给药的效果要好得多。
对于人肿瘤细胞株作用测定结果表明,氟达拉滨有抗非何杰金淋巴瘤和乳腺癌形成株的活性。
但对生长在裸鼠的B16黑色素瘤、Lewis 肺38colon,和MX1人乳腺肿瘤无效。
对非小细胞肺癌和卵巢肿瘤细胞株作用很小。
氟达拉滨可能通过多机制发挥抗肿瘤作用。
氟达拉滨属前药,在动物和人体内通过血清磷酸酶迅速去磷酸化形成2-fluoro-A,通过载体参与的转运机制进入细胞内。
细胞内氟达拉滨在脱氧胞啶激酶作用下磷酸化形成2-fluoro-ara-AMP,并进一步形成有抗肿瘤活性的产物2-fluoro-ara-ATP,2-fluoro-ara-ATP可掺入细胞DNA和RNA,在正常细胞和肿瘤细胞,F-ara-A的转运和磷酸化不同,F-ara-ATP的蓄积也不同,从而形成正的治疗指数的代谢基础。
因此可以认为氟达拉滨抑制DNA合成。
许多离体和在体研究表明F-ara-ATP通过DNA多聚酶与三磷酸脱氧腺苷竞争掺入扩展DNA链的A位,在掺入部位抑制DNA的合成,2-fluoro-ara-ATP有抑制核糖核苷酸还原酶,从DNA 以其3’到5’exonuclease活性,降低细胞内dATP可增强该药的作用。
DNA多聚酶αδ和ε、DNA引物合成酶和DNA连接酶的作用,体外实验证实:DNA 多聚酶δ能运动掺入的F-ara-AMP残基,F-ara-AMP终末掺入DNA导致遗传物质的删除,从而抑制DNA的合成。
部分抑制RNA多聚酶II,从而减少蛋白合成。
2F-ara-ATP 在DNA,RNA和蛋白合成的作用的某些方面作用机制,目前仍尚不清楚。
药物通过抑制DNA合成从而抑制肿瘤细胞生长。
离体研究表明慢性淋巴细胞性白血病的淋巴细胞暴露2F-ara-A后引起广泛的DNA断裂和凋亡的细胞或死亡。
阿糖胞苷抗血液系统恶性肿瘤,特别成人髓性白血病的治疗成功,自然引导人们致力于在嘌呤系列中确定能补充和扩展这种活性的阿糖核苷酸,在这些核苷酸中第一个进入临床的是ara-A(9-β-D-arabinofuranosyladenine),该药显示了边界的抗肿瘤作用和抗病毒作用。
如临床前评价所预示的,ara-A 通过普遍存在的腺苷脱氨酶对脱氨的易感性限制药物的生物利用性,虽然阿糖胞苷是相似的代谢消除途径,但那药更大的强度和易溶性已围绕这些问题,管理者致力于ara-A更易溶单磷酸形式,但主要因为缺乏这样的成分,初始的肿瘤临床试验正精力旺盛地追求着。
Ara-A对由去氨灭活的易感性提示需要结构修饰以对腺苷脱氨酶抵抗而保留理想的代谢和抑制特性,由Montgomery和Hewson提供的对2-fluoroadenosine(F-Ado)评价,证明在培养的细胞和小鼠细胞毒作用,腺苷激酶能使F-Ado有效地磷酸化,它的5’triphosphate蓄积在Ehrlich as cites cell.tetrahymena pyriformis,人红细胞、血小板和淋巴细胞。
然而通过腺核苷脱氨酶的核苷酸分解代谢的可比较研究证明F-Ado对脱氨抵抗,由F-Ado显示的代谢特征指出阿糖2-氟腺苷衍生物可能围绕ara-A的缺点。
Montgomery 和Hewson成功地合成磷酸法达拉滨(9-β-D-arabinosyl-2-fluoroadenosine,F-ara-A,fludara,iv),以后该组改进合成路线,由于法达拉滨相对不溶性5’单磷酸盐(FludaraI.V,F-ara-AMP)已用于许多实验研究和临床试验。
1、F-ara-A的代谢F-ara-A通过高亲和(Km=69μmol/L)和低亲和(Km=305μmol/L)双系统进入小鼠L1210细胞。
相反在小鼠小肠上皮crypt 细胞仅有低亲和转运F-ara-A系统(Km=301μmol/L),有趣的是这两类细胞都能抗浓度梯度,蓄积F-ara-A(Dagnino等的报告支持),转运动力学的差异,在L1210细胞蓄积F-ara-A的能力比小肠细胞大7-8倍,是核苷酸同系物在敏感肿瘤正治疗指数的基础,最近用[3H]标记F-ara-A的研究表明:在一次腹腔注射最大耐受剂量后小鼠小肠粘膜F-ara-ATP的浓度约为鼠ascitic P388细胞的2.5%.2、脱氨化初始的代谢研究表明:似F-Ado,F-ara-A不是来自小牛肠粘膜,P388或L1210细胞脱氨酶的底物,然而在以后的研究表明与小牛小肠酶稳定活性延长孵育(18小时)后,可以分离到脱氨化产物arabinosyl-2-fluorohypoxanthine,此外代谢研究确定在猴,犬和小鼠尿中有arabinosyl-2-fluorohypoxanthine,赋予腺苷脱氨酶抵抗的核苷酸同系物,就存在5’单磷酸F-ara-AMP可能是AMP脱氨酶的底物的可能性,虽然这一假设的评价尚未报告。
2-fluoroadenine的产生动物学研究已证实:在血浆、尿和脑脊液存在2-fluoroadenine自由基,这个F-ara-A催化物的产生,由于它的强细胞毒作用和缺乏治疗价值,可能是物质性关注,假定F-ade的作用是通过形成5’triphosphate,F-ATP发挥的,虽然可以接受diphosphate可以作为核糖核苷酸还原酶的底物,从而出现另一个毒性三磷酸盐,F-Datp。
F-Ade的生物起源最初是一个难题,F-ara-A不是哺乳类嘌呤核苷酸磷酸酶的底物,F-Ade也不是与S-homocysteine孵育释放出来的,该酶从脱氧腺苷和ara-A裂解腺苷。
现已明确细菌嘌呤核苷酸磷酸化酶可以利用腺核苷酸作为底物。
与此一致的是F-Ade仅可以从用法达拉滨治疗后动物体内或新鲜分离的肿瘤组织中检测到。
为此Huang 和Plunkett证实:完整的大肠杆菌和提取物可从F-ara-A催化释放F-Ade,该反应依赖与磷酸盐和被嘌呤核苷酸竞争性抑制。
提示细菌嘌呤核苷酸磷酸化酶是应答于释放F-Ade 的酶。
虽然哺乳类细胞没有产生F-Ade的能力,但它来自与细菌接触后,通过肝肠循环使之全身利用的可能性存在。
F-ara-A分泌到胆汁可能通过细菌的flora提供到达药物的路径,可能然后代谢它和排泄F-Ade,F-Ade吸收入血并转运到可转变为毒性三磷酸盐F-ATP的其他组织。
在注射[3H]F-ara-A和[3H]F-ara-AMP小鼠的P388细胞,骨髓和胃肠粘膜已经证明F-ATP的存在,F-ATP的形成依赖于F-ara-AMP的剂量,F-ATP在宿主组织代谢的进一步实验需要证明这种代谢物的细胞毒作用。
3、磷酸化与其他核苷酸同系物,F-ara-A要发挥细胞毒作用和治疗作用需要磷酸化,初始磷酸化需要去氧胞啶激酶,支持该观点的证据来自来自缺乏去氧胞啶激酶的突变细胞株,这些细胞对F-ara-A参与的细胞毒作用或治疗活性抵抗。
用牛胸腺、人慢性淋巴细胞性白血病细胞和L1210细胞提取物部分纯化的去氧胞啶激活酶的研究证明这些标本磷酸化F-ara-A的表观米氏常数(Km)分别为290μmol/L,213μmol/L和500μmol/L。
Krenitsky 等计算底物效率,它们仅为选择性底物去氧胞啶的6%。
假如F-ara-A出现超过是哦党适当时间,甚至低速率磷酸化也会导致在细胞内核苷酸同系物蓄积达到毒性水平,人髓性淋巴瘤K562细胞与F-ara-A和共同孵育,ara-C对于去氧胞啶激酶另一个不同的底物,其底物亲和效率高于F-ara-A,细胞消除F-ara-A快一些,腺苷激酶使ara-A磷酸化,因此可作为F-ara-A磷酸化的附加酶进行测试。
来自兔肝和L1210细胞纯化的腺苷激酶并不能将F-ara-A作为底物,由于缺乏腺苷激酶对ara-A抵抗的细胞保存对F-ara-A完全敏感的发现与腺苷激酶不有意义与F-ara-A磷酸化有关的结论相一致。
近来磷酸化腺苷,脱氧腺苷酸,脱氧鸟苷酸和脱氧胞啶的活性,假定各自激酶通过柱层析得到分离,以多种核苷酸为底物的所有片段的测定证明F-ara-A通过与脱氧胞啶激酶有关的活性而磷酸化,今后的研究目标应用较高纯度的人脱氧胞啶激酶明确F-ara-A底物的附加特性。
在L1210细胞,人成淋巴细胞,P388肿瘤细胞和K562细胞与F-ara-A孵育后,通过高效液相定量测定F-ara-A的蓄积程度。
小鼠注射F-ara-A后,观察到小鼠L1210细胞F-ara-A的浓度大于F-ara-ADP的浓度。
然而这不是体外培养新鲜收获的L1210细胞。
用放射标记药物的研究表明在P388细胞三磷酸盐的浓度与细胞内F-ara-A的浓度相似,但大于单磷酸盐或二磷酸盐的水平。
细胞中相对缺乏单磷酸盐或双磷酸盐脱氧胞啶激酶引起磷酸化对F-ara-ATP的形成是一限速率过程。
这些证据(不象在ara-C磷酸化中对于脱氧胞啶激酶建立相似作用那样有力)得到这样的观念,脱氧胞啶激酶活性在三磷酸盐的形成中是一限速过程。
虽然无特殊的报告,可能假设F-ara-AMP通过腺苷激酶进行磷酸化而实现F-ara-ATP的合成,通过核苷酸二磷酸激酶的作用相继产生三磷酸盐。
在腺苷脱氨酶抑制剂存在的情况下,单独用F-ara-AC处理的CEM细胞内F-ara-ATP与用ara-A孵育细胞细胞内ara-ATP比较前者F-ara-ATP蓄积。
F-ara-ATP蓄积呈浓度依赖性,但外源性F-ara-A达到300μmol/L时,在K562细胞存在速率饱和的特点。
在用F-ara-A孵育后洗去药液,再将细胞置于无药掖的条件下,CCRF-CEM细胞和K562细胞以一级动力学过程消除F-ara-ATP。
半衰期为分别为2.5和3.3小时.,荷瘤小鼠注射F-ara-A或F-ara-AMP后P388细胞内F-ara-ATP以单相动力学消除,半衰期分别为2.9 和4.1小时。