Q3C残留溶剂中英文
usp 溶剂残留 中英文版

<467>溶剂残留简介:INTRODUCTIONThis general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.本章节适用于现有的原料药,辅料和制剂。
应对原料药或制剂产品中可能存在溶剂的所有原料及制剂产品进行控制。
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs, because the solvents employed may vary from one manufacturer to another.当限值与下面提供的数值相符合,残留溶剂的测试方法一般不会在专论中特别,因为不同制造商所使用的溶剂不同。
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
Q3C残留溶剂中英文

杂质:残留溶剂的指导原则1.介绍本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。
在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。
纯度和溶解度。
因此.有时溶剂是合成中非常关键的因素。
本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。
出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。
制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。
一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。
使用低毒溶剂(第三类,表3)较为理想。
附录1中列出了指导原则中的全部溶剂。
表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。
第一、二类溶剂的建议限度或溶剂的分类会随着。
新的安全性资料的获得而调整。
含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。
2. 指导原则的范围指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。
应进行残留溶剂的检验。
也只有在上述情况下,才有必要作溶剂的检查。
虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。
如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。
EP2.4.24残留溶剂的鉴定与控制中英文对照版(带图)

残留溶剂的鉴定及控制该方法适用于: .1.在活性物质、赋形剂或药品中的未知的一类或二类溶剂残留量的鉴定;2.在活性物质、赋形剂或药品中的一类或二类溶剂残留量的限量试验;3.当二类溶剂的限度大于1000ppm(0.1%)或要求检测三类溶剂时的限量试验。
—类溶剂、二类溶剂、三类溶剂的分类见5.4以下给出了三种样品溶液的稀释方法,以及气相色谱顶空进样的系统条件。
还给出了两种气相色谱的系统条件,系统A为首选方法,同时系统B适用于一般的鉴别试验。
样品溶液的制备方法由样品的溶解性和待测的溶剂种类决定。
下列溶剂不适于用顶空进样法测定:甲酰胺、2—乙氧基乙醇、2—甲氧基乙醇、乙二醇、N-甲基吡咯烷酮、环丁砜(四氢噻吩砜),但可采用其他的适当的方法测定。
当采用其他的方法定量测定有机残留量时,必须进行方法验证采用静态顶空进样法测定样品溶液制备1:适用于在水中易溶的物质的残留溶剂的测定样品溶液(1):取0.200g待测物质,用水溶解并稀释至20m1样品溶液制备2:适用于在水中不溶的物质的残留溶剂的测定样品溶液(2):取0.200g待测物质,用N,N--二甲基甲酰胺(DMF)溶解并稀释至20ml样品溶液制备3:适用于测定N,N---二甲基乙酰胺或N,N--二甲基甲酰胺的残留量,当怀疑他们存在时。
样品溶液(3):取0.200g待测物质,用1,3—二甲基-2-咪唑啉酮(DMI)溶解并稀释至20ml如果上述方法均不适宜,那么所用稀释方法及静态顶空进样条件均必须验证其合理性。
溶剂溶液(a):吸取一类残留溶剂标准溶液1.0ml,用水稀释至100.0ml,再吸取该溶液1.0ml,用水稀释至10.0ml。
溶剂溶液(b):吸取适量二类溶剂溶于二甲基亚砜,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格二)。
溶剂溶液(c):称取1.00g溶剂或待测物质中存在的,用二甲基亚砜或水溶解,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格一或二)。
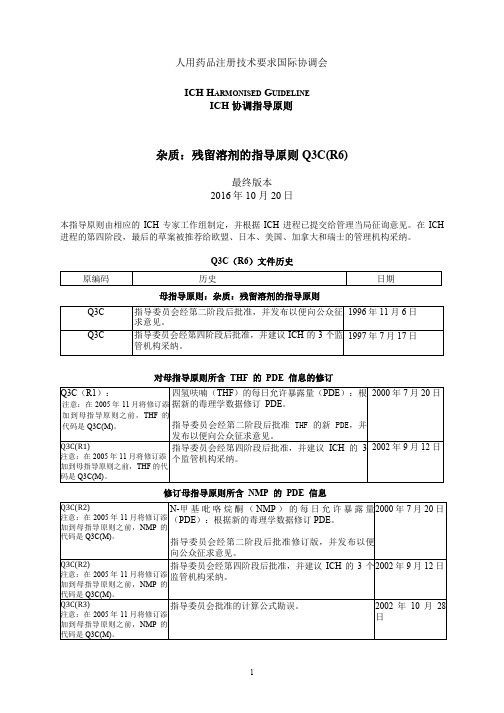
ICH 杂质:残留溶剂的指导原则 Q3C(R6)

人用药品注册技术要求国际协调会ICH协调指导原则杂质:残留溶剂的指导原则Q3C(R6)现行第四阶段版本2016年10月20日本指导原则由相应的ICH专家工作组制定,并根据ICH进程已提交给管理当局征询意见。
在ICH进程的第四阶段,最后的草案被推荐给欧盟、日本、美国、加拿大和瑞士的管理机构采纳。
Q3C(R5)文件历史母指导原则:杂质:残留溶剂的指导原则对母指导原则所含THF的PDE信息的修订修订母指导原则所含NMP的PDE信息母指导原则:杂质:残留溶剂的指导原则对母指导原则所含异丙基苯的PDE信息的修订修订母指导原则所含甲基异丁基酮的PDE信息,并纳入三乙胺的PDE杂质:残留溶剂的指导原则ICH协调指导原则目录第一部分:1. 引言 (1)2. 指导原则的适用范围 (1)3. 通则 (2)3.1 基于风险评估的残留溶剂的分类 (2)3.2 建立暴露限度的方法 (2)3.3 2类溶剂限度的表示方法 (2)3.4 分析方法 (4)3.5 残留溶剂的报告水平 (4)4. 残留溶剂的限度 (4)4.1 应避免的溶剂 (4)4.2 应限制的溶剂 (5)4.3 低潜在毒性的溶剂 (6)4.4 没有足够毒理学数据的溶剂 (7)词汇表 (8)附录1:指导原则中包括的溶剂列表 (9)附录2:其他背景 (13)A2.1 环境领域对有机挥发性溶剂的监管 (13)A2.2 药物中的残留溶剂 (13)附录3:建立暴露限度的方法 (14)第二部分:四氢呋喃的PDE (17)第三部分:N-甲基吡咯烷酮(NMP)的PDE (19)第四部分:异丙基苯的PDE (21)第五部分:三乙胺的PDE和甲基异丁基酮的PDE (24)第一部分:杂质:残留溶剂的指导原则在1997年7月17日的ICH指导委员会会议上进入ICH进程第四阶段,并建议ICH的三方监管机构采纳该指导原则1. 引言本指导原则旨在建议为保证患者安全而应规定的药物中残留溶剂的可接受量。
残留溶剂指导原则ICHQ3CR5

August 2011EMA/CHMP/ICH/82260/2006ICH guideline Q3C (R5) on impurities: guideline for residual solventsStep 5Part I (Parent guideline)Transmission to CHMP November 1996 Adoption by CHMP for release for consultation November 1996 End of consultation (deadline for comments) May 1997 Final adoption by CHMP September 1997 Date for coming into effect March 1998 Part II and part III (PDE for Tetrahydrofuran and N-Methylpyrrolidone)Transmission to CHMP July 2000 Adoption by CHMP for release for consultation July 2000 End of consultation (deadline for comments) September 2000 Final adoption by CHMP September 2002 Corrigendum to calculation formula for NMP November 2002 Transmission to CHMP March 2003February 2009 Update of table 2, table 3 and appendix 1 to reflect therevision of the PDEs for N-Methylpyrrolidone andTetrahydrofuran Q3C(R4)Part IV (PDE for cumene)Transmission to CHMP June 2010 Adoption by CHMP for release for consultation June 20107 Westferry Circus ● Canary Wharf ● London E14 4HB ● United KingdomEnd of consultation (deadline for comments) September 2010 Final adoption by CHMP March 2011 Date for coming into effect August 2011Q3C (R5) on impurities: guideline for residual solventsTable of contentsPart I (4)Impurities: Residual solvents - Parent guideline (4)1. Introduction (4)2. Scope of the guideline (4)3. General principles (5)3.1. Classification of residual solvents by risk assessment (5)3.2. Methods for establishing exposure limits (5)3.3. Options for describing limits of class 2 solvents (6)3.4. Analytical procedures (7)3.5. Reporting levels of residual solvents (7)4. Limits of residual solvents (8)4.1. Solvents to be avoided (8)4.2. Solvents to be limited (8)4.3. Solvents with low toxic potential (9)4.4. Solvents for which no adequate toxicological data was found (10)Glossary (11)Appendix 1: List of solvents included in the guideline (12)Appendix 2: Additional background (16)Appendix 3: Methods for establishing exposure limits (17)PART II: (20)PDE for Tetrahydrofuran (20)PART III: (22)PDE for N-Methylpyrrolidone (NMP) (22)PART IV (24)PDE for cumene (24)Part IImpurities: Residual solvents - Parent guideline1. IntroductionThe objective of this guideline is to recommend acceptable amounts for residual solvents in pharmaceuticals for the safety of the patient. The guideline recommends use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents. Residual solvents in pharmaceuticals are defined here as organic volatile chemicals that are used or produced in the manufacture of drug substances or excipients, or in the preparation of drug products. The solvents are not completely removed by practical manufacturing techniques. Appropriate selection of the solvent for the synthesis of drug substance may enhance the yield, or determine characteristics such as crystal form, purity, and solubility. Therefore, the solvent may sometimes be a critical parameter in the synthetic process. This guideline does not address solvents deliberately used as excipients nor does it address solvates. However, the content of solvents in such products should be evaluated and justified.Since there is no therapeutic benefit from residual solvents, all residual solvents should be removed to the extent possible to meet product specifications, good manufacturing practices, or other quality-based requirements. Drug products should contain no higher levels of residual solvents than can be supported by safety data. Some solvents that are known to cause unacceptable toxicities (Class 1, Table 1) should be avoided in the production of drug substances, excipients, or drug products unless their use can be strongly justified in a risk-benefit assessment. Some solvents associated with less severe toxicity (Class 2, Table 2) should be limited in order to protect patients from potential adverse effects. Ideally, less toxic solvents (Class 3, Table 3) should be used where practical. The complete list of solvents included in this guideline is given in Appendix 1.The lists are not exhaustive and other solvents can be used and later added to the lists. Recommended limits of Class 1 and 2 solvents or classification of solvents may change as new safety data becomes available. Supporting safety data in a marketing application for a new drug product containing a new solvent may be based on concepts in this guideline or the concept of qualification of impurities as expressed in the guideline for drug substance (Q3A, Impurities in New Drug Substances) or drug product (Q3B, Impurities in New Drug Products), or all three guidelines.2. Scope of the guidelineResidual solvents in drug substances, excipients, and in drug products are within the scope of this guideline. Therefore, testing should be performed for residual solvents when production or purification processes are known to result in the presence of such solvents. It is only necessary to test for solvents that are used or produced in the manufacture or purification of drug substances, excipients, or drug product. Although manufacturers may choose to test the drug product, a cumulative method may be used to calculate the residual solvent levels in the drug product from the levels in the ingredients used to produce the drug product. If the calculation results in a level equal to or below that recommended in this guideline, no testing of the drug product for residual solvents need be considered. If, however, the calculated level is above the recommended level, the drug product should be tested to ascertain whether the formulation process has reduced therelevant solvent level to within the acceptable amount. Drug product should also be tested if a solvent is used during its manufacture.This guideline does not apply to potential new drug substances, excipients, or drug products used during the clinical research stages of development, nor does it apply to existing marketed drug products.The guideline applies to all dosage forms and routes of administration. Higher levels of residual solvents may be acceptable in certain cases such as short term (30 days or less) or topical application. Justification for these levels should be made on a case by case basis.See Appendix 2 for additional background information related to residual solvents.3. General principles3.1. Classification of residual solvents by risk assessmentThe term "tolerable daily intake" (TDI) is used by the International Program on Chemical Safety (IPCS) to describe exposure limits of toxic chemicals and "acceptable daily intake" (ADI) is used by the World Health Organization (WHO) and other national and international health authorities and institutes. The new term "permitted daily exposure" (PDE) is defined in the present guideline as a pharmaceutically acceptable intake of residual solvents to avoid confusion of differing values for ADI's of the same substance.Residual solvents assessed in this guideline are listed in Appendix 1 by common names and structures. They were evaluated for their possible risk to human health and placed into one of three classes as follows:Class 1 solvents: Solvents to be avoidedKnown human carcinogens, strongly suspected human carcinogens, and environmental hazards. Class 2 solvents: Solvents to be limitedNon-genotoxic animal carcinogens or possible causative agents of other irreversible toxicity such as neurotoxicity or teratogenicity.Solvents suspected of other significant but reversible toxicities.Class 3 solvents: Solvents with low toxic potentialSolvents with low toxic potential to man; no health-based exposure limit is needed. Class 3 solvents have PDEs of 50 mg or more per day.3.2. Methods for establishing exposure limitsThe method used to establish permitted daily exposures for residual solvents is presented in Appendix 3. Summaries of the toxicity data that were used to establish limits are published in Pharmeuropa, Vol. 9, No. 1, Supplement, April 1997.3.3. Options for describing limits of class 2 solventsTwo options are available when setting limits for Class 2 solvents.Option 1: The concentration limits in ppm stated in Table 2 can be used. They were calculated using equation (1) below by assuming a product mass of 10 g administered daily. Concentration (ppm) = 1000 x PDE(1)Here, PDE is given in terms of mg/day and dose is given in g/day.These limits are considered acceptable for all substances, excipients, or products. Therefore this option may be applied if the daily dose is not known or fixed. If all excipients and drug substances in a formulation meet the limits given in Option 1, then these components may be used in any proportion. No further calculation is necessary provided the daily dose does not exceed 10 g. Products that are administered in doses greater than 10 g per day should be considered under Option 2.Option 2: It is not considered necessary for each component of the drug product to comply with the limits given in Option 1. The PDE in terms of mg/day as stated in Table 2 can be used with the known maximum daily dose and equation (1) above to determine the concentration of residual solvent allowed in drug product. Such limits are considered acceptable provided that it has been demonstrated that the residual solvent has been reduced to the practical minimum. The limits should be realistic in relation to analytical precision, manufacturing capability, reasonable variation in the manufacturing process, and the limits should reflect contemporary manufacturing standards. Option 2 may be applied by adding the amounts of a residual solvent present in each of the components of the drug product. The sum of the amounts of solvent per day should be less than that given by the PDE.Consider an example of the use of Option 1 and Option 2 applied to acetonitrile in a drug product. The permitted daily exposure to acetonitrile is 4.1 mg per day; thus, the Option 1 limit is 410 ppm. The maximum administered daily mass of a drug product is 5.0 g, and the drug product contains two excipients. The composition of the drug product and the calculated maximum content of residual acetonitrile are given in the following table.Acetonitrile content Daily exposure Component Amount informulationDrug substance 0.3 g 800 ppm 0.24 mgExcipient 1 0.9 g 400 ppm 0.36 mgExcipient 2 3.8 g 800 ppm 3.04 mgDrug Product 5.0 g 728 ppm 3.64 mgExcipient 1 meets the Option 1 limit, but the drug substance, excipient 2, and drug product do not meet the Option 1 limit. Nevertheless, the product meets the Option 2 limit of 4.1 mg per day and thus conforms to the recommendations in this guideline.Consider another example using acetonitrile as residual solvent. The maximum administered daily mass of a drug product is 5.0 g, and the drug product contains two excipients. The composition of the drug product and the calculated maximum content of residual acetonitrile is given in the following table.Acetonitrile content Daily exposure Component Amount informulationDrug substance 0.3 g 800 ppm 0.24 mgExcipient 1 0.9 g 2000 ppm 1.80 mgExcipient 2 3.8 g 800 ppm 3.04 mgDrug Product 5.0 g 1016 ppm 5.08 mgIn this example, the product meets neither the Option 1 nor the Option 2 limit according to this summation. The manufacturer could test the drug product to determine if the formulation process reduced the level of acetonitrile. If the level of acetonitrile was not reduced during formulation to the allowed limit, then the manufacturer of the drug product should take other steps to reduce the amount of acetonitrile in the drug product. If all of these steps fail to reduce the level of residual solvent, in exceptional cases the manufacturer could provide a summary of efforts made to reduce the solvent level to meet the guideline value, and provide a risk-benefit analysis to support allowing the product to be utilised with residual solvent at a higher level.3.4. Analytical proceduresResidual solvents are typically determined using chromatographic techniques such as gas chromatography. Any harmonised procedures for determining levels of residual solvents as described in the pharmacopoeias should be used, if feasible. Otherwise, manufacturers would be free to select the most appropriate validated analytical procedure for a particular application. If only Class 3 solvents are present, a non-specific method such as loss on drying may be used. Validation of methods for residual solvents should conform to ICH guidelines Text on Validation of Analytical Procedures and Extension of the ICH Text on Validation of Analytical Procedures.3.5. Reporting levels of residual solventsManufacturers of pharmaceutical products need certain information about the content of residual solvents in excipients or drug substances in order to meet the criteria of this guideline. The following statements are given as acceptable examples of the information that could be provided from a supplier of excipients or drug substances to a pharmaceutical manufacturer. The supplier might choose one of the following as appropriate:Only Class 3 solvents are likely to be present. Loss on drying is less than 0.5%.Only Class 2 solvents X, Y, ... are likely to be present. All are below the Option 1 limit. (Here the supplier would name the Class 2 solvents represented by X, Y, ...)Only Class 2 solvents X, Y, ... and Class 3 solvents are likely to be present. Residual Class 2 solvents are below the Option 1 limit and residual Class 3 solvents are below 0.5%.If Class 1 solvents are likely to be present, they should be identified and quantified."Likely to be present" refers to the solvent used in the final manufacturing step and to solvents that are used in earlier manufacturing steps and not removed consistently by a validated process.If solvents of Class 2 or Class 3 are present at greater than their Option 1 limits or 0.5%, respectively, they should be identified and quantified.4. Limits of residual solvents4.1. Solvents to be avoidedSolvents in Class 1 should not be employed in the manufacture of drug substances, excipients, and drug products because of their unacceptable toxicity or their deleterious environmental effect. However, if their use is unavoidable in order to produce a drug product with a significant therapeutic advance, then their levels should be restricted as shown in Table 1, unless otherwise justified. 1,1,1-Trichloroethane is included in Table 1 because it is an environmental hazard. The stated limit of 1500 ppm is based on a review of the safety data.TABLE 1. Class 1 solvents in pharmaceutical products (solvents that should be avoided).Solvent Concentration limitConcern(ppm)Benzene 2 CarcinogenCarbon tetrachloride 4 Toxic and environmental hazard1,2-Dichloroethane 5 Toxic1,1-Dichloroethene 8 Toxic1,1,1-Trichloroethane 1500 Environmental hazard4.2. Solvents to be limitedSolvents in Table 2 should be limited in pharmaceutical products because of their inherent toxicity. PDEs are given to the nearest 0.1 mg/day, and concentrations are given to the nearest 10 ppm. The stated values do not reflect the necessary analytical precision of determination. Precision should be determined as part of the validation of the method.TABLE 2. Class 2 solvents in pharmaceutical products.Solvent PDE (mg/day) Concentration limit(ppm)Acetonitrile 4.1 410Chlorobenzene 3.6 360Chloroform 0.6 60Cyclohexane 38.8 38801,2-Dichloroethene 18.7 1870Dichloromethane 6.0 6001,2-Dimethoxyethane 1.0 100N,N-Dimethylacetamide 10.9 1090N,N-Dimethylformamide 8.8 8801,4-Dioxane 3.8 3802-Ethoxyethanol 1.6 160Ethyleneglycol 6.2 620Formamide 2.2 220Hexane 2.9 290Methanol 30.0 30002-Methoxyethanol 0.5 50Methylbutyl ketone 0.5 50Methylcyclohexane 11.8 1180N-Methylpyrrolidone1 5.3 530Nitromethane 0.5 50Pyridine 2.0 200Sulfolane 1.6 160Tetrahydrofuran27.2 720Tetralin 1.0 100Toluene 8.9 8901,1,2-Trichloroethene 0.8 80Xylene* 21.7 2170*usually 60% m-xylene, 14% p-xylene, 9% o-xylene with 17% ethyl benzene4.3. Solvents with low toxic potentialSolvents in Class 3 (shown in Table 3) may be regarded as less toxic and of lower risk to human health. Class 3 includes no solvent known as a human health hazard at levels normally accepted in pharmaceuticals. However, there are no long-term toxicity or carcinogenicity studies for many of the solvents in Class 3. Available data indicate that they are less toxic in acute or short-term studies and negative in genotoxicity studies. It is considered that amounts of these residual solvents of 50 mg per day or less (corresponding to 5000 ppm or 0.5% under Option 1) would be acceptable without justification. Higher amounts may also be acceptable provided they are realistic in relation to manufacturing capability and good manufacturing practice.Table 3: Class 3 solvents which should be limited by GMP or other quality-based requirements. Acetic acid HeptaneAcetone Isobutyl acetateAnisole Isopropyl acetate1 The information included for N-Methylpyrrolidone reflects that included in the Revision of PDE Information for NMP which reached Step 4 in September 2002 (two mistyping corrections made in October 2002), and was incorporated into the core guideline in November 2005. See Part III (pages 20-21).2 The information included for Tetrahydrofuran reflects that included in the Revision of PDE Information for THF which reached Step 4 in September 2002, and was incorporated into the core guideline in November 2005. See Part II (pages 18-19).1-Butanol Methyl acetate2-Butanol 3-Methyl-1-butanolButyl acetate Methylethyl ketonetert-Butylmethyl ether Methylisobutyl ketoneCumene 2-Methyl-1-propanolDimethyl sulfoxide PentaneEthanol 1-PentanolEthyl acetate 1-PropanolEthyl ether 2-PropanolEthyl formate Propyl acetateFormic acid4.4. Solvents for which no adequate toxicological data was foundThe following solvents (Table 4) may also be of interest to manufacturers of excipients, drug substances, or drug products. However, no adequate toxicological data on which to base a PDE was found. Manufacturers should supply justification for residual levels of these solvents in pharmaceutical products.Table 4 Solvents for which no adequate toxicological data was found.1,1-Diethoxypropane Methylisopropyl ketone1,1-Dimethoxymethane Methyltetrahydrofuran2,2-Dimethoxypropane Petroleum etherIsooctane Trichloroacetic acidIsopropyl ether Trifluoroacetic acidGlossaryGenotoxic Carcinogens:Carcinogens which produce cancer by affecting genes or chromosomes.LOEL:Abbreviation for lowest-observed effect level.Lowest-Observed Effect Level:The lowest dose of substance in a study or group of studies that produces biologically significant increases in frequency or severity of any effects in the exposed humans or animals.Modifying Factor:A factor determined by professional judgment of a toxicologist and applied to bioassay data to relate that data safely to humans.Neurotoxicity:The ability of a substance to cause adverse effects on the nervous system.NOEL:Abbreviation for no-observed-effect level.No-Observed-Effect Level:The highest dose of substance at which there are no biologically significant increases in frequency or severity of any effects in the exposed humans or animals.PDE:Abbreviation for permitted daily exposure.Permitted Daily Exposure:The maximum acceptable intake per day of residual solvent in pharmaceutical products. Reversible Toxicity:The occurrence of harmful effects that are caused by a substance and which disappear after exposure to the substance ends.Strongly Suspected Human Carcinogen:A substance for which there is no epidemiological evidence of carcinogenesis but there are positive genotoxicity data and clear evidence of carcinogenesis in rodents.Teratogenicity:The occurrence of structural malformations in a developing fetus when a substance is administered during pregnancy.Appendix 1: List of solvents included in the guideline Solvent Other Names Structure Class Acetic acid Ethanoic acid CH3COOH Class 3Acetone 2-PropanoneCH3COCH3 Class 3Propan-2-oneAcetonitrile CH3CN Class 2Anisole Methoxybenzene OCHClass 33Benzene Benzol Class 11-Butanol n-Butyl alcoholCH3(CH2)3OH Class 3Butan-1-olCH3CH2CH(OH)CH3 Class 3 2-Butanol sec-Butyl alcoholButan-2-olButyl acetate Acetic acid butyl ester CH3COO(CH2)3CH3 Class 3tert-Butylmethyl ether 2-Methoxy-2-methyl- propane (CH3)3COCH3 Class 3Carbon tetrachloride Tetrachloromethane CCl4 Class 1Chlorobenzene Cl Class 2Chloroform Trichloromethane CHCl3 Class 2Cumene IsopropylbenzeneCH(CH3)2Class 3(1-Methyl)ethylbenzeneCyclohexane Hexamethylene Class 2CH2ClCH2Cl Class 1 1,2-Dichloroethane sym-DichloroethaneEthylene dichlorideEthylene chloride1,1-Dichloroethene 1,1-DichloroethyleneH2C=CCl2 Class 1Vinylidene chloride1,2-Dichloroethene 1,2-DichloroethyleneClHC=CHCl Class 2Acetylene dichlorideDichloromethane Methylene chloride CH2Cl2 Class 2H3COCH2CH2OCH3 Class 2 1,2-Dimethoxyethane Ethyleneglycol dimethyl etherMonoglymeDimethyl CellosolveN,N-Dimethylacetamide DMA CH3CON(CH3)2 Class 2 N,N-Dimethylformamide DMF HCON(CH3)2 Class 2(CH3)2SO Class 3 Dimethyl sulfoxide MethylsulfinylmethaneMethyl sulfoxideDMSO1,4-Dioxane p-DioxaneO O Class 2[1,4]DioxaneEthanol Ethyl alcohol CH3CH2OH Class 3 2-Ethoxyethanol Cellosolve CH3CH2OCH2CH2OH Class 2 Ethyl acetate Acetic acid ethyl ester CH3COOCH2CH3 Class 3HOCH2CH2OH Class 2 Ethyleneglycol 1,2-Dihydroxyethane1,2-EthanediolCH3CH2OCH2CH3 Class 3 Ethyl ether Diethyl etherEthoxyethane1,1’-OxybisethaneEthyl formate Formic acid ethyl ester HCOOCH2CH3 Class 3 Formamide Methanamide HCONH2 Class 2 Formic acid HCOOH Class 3 Heptane n-Heptane CH3(CH2)5CH3 Class 3Hexane n-Hexane CH3(CH2)4CH3 Class 2Isobutyl acetate Acetic acid isobutyl ester CH3COOCH2CH(CH3)2 Class 3 Isopropyl acetate Acetic acid isopropyl ester CH3COOCH(CH3)2 Class 3 Methanol Methyl alcohol CH3OH Class 2 2-Methoxyethanol Methyl Cellosolve CH3OCH2CH2OH Class 2 Methyl acetate Acetic acid methyl ester CH3COOCH3 Class 33-Methyl-1-butanol Isoamyl alcoholIsopentyl alcohol3-Methylbutan-1-ol(CH3)2CHCH2CH2OH Class 3Methylbutyl ketone 2-HexanoneHexan-2-oneCH3(CH2)3COCH3 Class 2Methylcyclohexane Cyclohexylmethane CH3Class 2 Methylethyl ketone 2-ButanoneMEKButan-2-oneCH3CH2COCH3 Class 3Methylisobutyl ketone 4-Methylpentan-2-one4-Methyl-2-pentanoneMIBKCH3COCH2CH(CH3)2 Class 32-Methyl-1-propanol Isobutyl alcohol2-Methylpropan-1-ol(CH3)2CHCH2OH Class 3 N-Methylpyrrolidone 1-Methylpyrrolidin-2-one1-Methyl-2-pyrrolidinone NCH3OClass 2Nitromethane CH3NO2 Class 2 Pentane n-Pentane CH3(CH2)3CH3 Class 3 1-Pentanol Amyl alcohol CH3(CH2)3CH2OH Class 3Pentan-1-olPentyl alcohol1-Propanol Propan-1-olPropyl alcoholCH3CH2CH2OH Class 32-Propanol Propan-2-olIsopropyl alcohol(CH3)2CHOH Class 3 Propyl acetate Acetic acid propyl ester CH3COOCH2CH2CH3 Class 3PyridineNClass 2Sulfolane Tetrahydrothiophene 1,1-dioxideSO OClass 2Tetrahydrofuran1Tetramethylene oxideOxacyclopentane OClass 2Tetralin 1,2,3,4-Tetrahydro-naphthalene Class 2Toluene Methylbenzene CH3Class 2 1,1,1-Trichloroethane Methylchloroform CH3CCl3 Class 1 1,1,2-Trichloroethene Trichloroethene HClC=CCl2 Class 2Xylene* DimethybenzeneXylolCH3CH3Class 2*usually 60% m-xylene, 14% p-xylene, 9% o-xylene with 17% ethyl benzene1 The information included for Tetrahydrofuran reflects that included in the Revision of PDE Information for THF which reached Step 4 in September 2002, and was incorporated into the core guideline in November 2005. See Part II (pages 18-19).Appendix 2: Additional backgroundA2.1 Environmental Regulation of Organic Volatile SolventsSeveral of the residual solvents frequently used in the production of pharmaceuticals are listed as toxic chemicals in Environmental Health Criteria (EHC) monographs and the Integrated Risk Information System (IRIS). The objectives of such groups as the International Programme on Chemical Safety (IPCS), the United States Environmental Protection Agency (USEPA), and the United States Food and Drug Administration (USFDA) include the determination of acceptable exposure levels. The goal is protection of human health and maintenance of environmental integrity against the possible deleterious effects of chemicals resulting from long-term environmental exposure. The methods involved in the estimation of maximum safe exposure limits are usually based on long-term studies. When long-term study data are unavailable, shorter term study data can be used with modification of the approach such as use of larger safety factors. The approach described therein relates primarily to long-term or life-time exposure of the general population in the ambient environment, i.e. ambient air, food, drinking water and other media.A2.2 Residual Solvents in PharmaceuticalsExposure limits in this guideline are established by referring to methodologies and toxicity data described in EHC and IRIS monographs. However, some specific assumptions about residual solvents to be used in the synthesis and formulation of pharmaceutical products should be taken into account in establishing exposure limits. They are:1) Patients (not the general population) use pharmaceuticals to treat their diseases or forprophylaxis to prevent infection or disease.2) The assumption of life-time patient exposure is not necessary for most pharmaceuticalproducts but may be appropriate as a working hypothesis to reduce risk to human health.3) Residual solvents are unavoidable components in pharmaceutical production and will oftenbe a part of drug products.4) Residual solvents should not exceed recommended levels except in exceptionalcircumstances.5) Data from toxicological studies that are used to determine acceptable levels for residualsolvents should have been generated using appropriate protocols such as those describedfor example by OECD, EPA, and the FDA Red Book.Appendix 3: Methods for establishing exposure limitsThe Gaylor-Kodell method of risk assessment (Gaylor, D. W. and Kodell, R. L.: Linear Interpolation algorithm for low dose assessment of toxic substance. J Environ. Pathology, 4, 305, 1980) is appropriate for Class 1 carcinogenic solvents. Only in cases where reliable carcinogenicity data are available should extrapolation by the use of mathematical models be applied to setting exposure limits. Exposure limits for Class 1 solvents could be determined with the use of a large safety factor (i.e., 10,000to 100,000) with respect to the no-observed-effect level (NOEL). Detection and quantitation of these solvents should be by state-of-the-art analytical techniques.Acceptable exposure levels in this guideline for Class 2 solvents were established by calculation of PDE values according to the procedures for setting exposure limits in pharmaceuticals (Pharmacopeial Forum, Nov-Dec 1989), and the method adopted by IPCS for Assessing Human Health Risk of Chemicals (Environmental Health Criteria 170, WHO, 1994). These methods are similar to those used by the USEPA (IRIS) and the USFDA (Red Book) and others. The method is outlined here to give a better understanding of the origin of the PDE values. It is not necessary to perform these calculations in order to use the PDE values tabulated in Section 4 of this document. PDE is derived from the no-observed-effect level (NOEL), or the lowest-observed effect level (LOEL) in the most relevant animal study as follows:PDE =NOEL x Weight Adjustment(1)F1 x F2 x F3 x F4 x F5The PDE is derived preferably from a NOEL. If no NOEL is obtained, the LOEL may be used. Modifying factors proposed here, for relating the data to humans, are the same kind of "uncertainty factors" used in Environmental Health Criteria (Environmental Health Criteria 170, World Health Organization, Geneva, 1994), and "modifying factors" or "safety factors" in Pharmacopeial Forum. The assumption of 100% systemic exposure is used in all calculations regardless of route of administration.The modifying factors are as follows:F1 = A factor to account for extrapolation between speciesF1 = 5 for extrapolation from rats to humansF1 = 12 for extrapolation from mice to humansF1 = 2 for extrapolation from dogs to humansF1 = 2.5 for extrapolation from rabbits to humansF1 = 3 for extrapolation from monkeys to humansF1 = 10 for extrapolation from other animals to humans。
关于国际药品注册翻译说明

国际药品注册翻译医药翻译网的国际药品注册翻译译员多毕业于国内外著名医科大学,并在各自的国际药品注册翻译领域有过丰富翻译经验。
国际药品注册翻译人员都经过严格测试,大多有国外留学、工作经历,具有良好的国际药品注册翻译能力。
国际药品注册翻译网项目组成员对国际药品注册翻译的文化背景、语言习惯、专业术语等有深入的把握。
医药翻译网鼎力提供每位国际药品注册翻译客户质量最高、速度最快的国际药品注册翻译。
医药翻译网凭借严格的质量控制体系、规范化的运作流程和独特的审核标准已为各组织机构及来自全球的医药公司提供了高水准的国际药品注册翻译,不少的医药公司还跟我们签定了长期合作协议。
国际药品注册翻译的质量和速度质量是企业生存和发展的根本,为确保国际药品注册翻译的准确性,项目的全过程如下:一、庞大国际药品注册翻译团队保证各类国际药品注册翻译稿件均由专业人士担任。
二、规范化的国际药品注册翻译流程。
从获得资料的开始到交稿全过程进行质量的全面控制,并同时做到高效率,快速度的原则。
三、及时组建若干翻译小组,分析各项要求,统一专业词汇,确定语言风格,译文格式要求。
四、国际药品注册翻译均有严格的语言和专业技术双重校对。
从初稿的完成到统稿,从校对到最终审核定稿,甚至词汇间的细微差别也力求精确。
五、不间断的进行招聘,充足的人力资源不断汇集国际药品注册翻译界的精英和高手。
不断对内部及外聘国际药品注册翻译人员进行系统的再培训工程。
六、曾6 小时翻译4.5 万字的速度客户所需。
七、有效沟通。
国际药品注册翻译大项目组协调各方面工作:高级项目经理项目经理(Project Manager)翻译(Translation)编辑(Editing)校对(Profreading)质量控制(Quality Assurance)国际药品注册翻译技术配备一、制作部配备有先进的计算机处理设备,多台扫描仪、打印机、光盘刻录机、宽带网络接入、公司拥有独立的服务器,各项领先技术确保所有文件系统化处理和全球同步传输。
Q3C(R6)-20191004-S-Step 4杂质:残留溶剂指导原则
人用药品注册技术要求国际协调会ICH H ARMONISED G UIDELINEICH 协调指导原则杂质:残留溶剂的指导原则 Q3C(R6)最终版本2016年 10 月 20日本指导原则由相应的ICH 专家工作组制定,并根据ICH 进程已提交给管理当局征询意见。
在ICH 进程的第四阶段,最后的草案被推荐给欧盟、日本、美国、加拿大和瑞士的管理机构采纳。
Q3C(R6)文件历史母指导原则:杂质:残留溶剂的指导原则对母指导原则所含THF的PDE信息的修订修订母指导原则所含NMP的PDE信息母指导原则:杂质:残留溶剂的指导原则对母指导原则所含异丙基苯的PDE信息的修订修订母指导原则所含甲基异丁基酮的PDE信息,并纳入三乙胺的PDE乙二醇PDE修正I MPURITIES: G UIDELINE FOR R ESIDUAL S OLVENTS杂质:残留溶剂的指导原则TABLE OF CONTENTS目录PART I:IMPURITIES: GUIDELINE FOR RESIDUAL SOLVENTS (4)第一部分:杂质:残留溶剂的指导原则 (4)1.INTRODUCTION引言 (4)2.SCOPE OF THE GUIDELINE 指导原则的适用范围 (5)3.GENERAL PRINCIPLES通则 (6)3.1Classification of Residual Solvents by Risk Assessment基于风险评估的残留溶剂的分类 (6)3.2 Methods for Establishing Exposure Limits 建立暴露限度的方法 (6)3.3 Options for Describing Limits of Class 2 Solvents 2 类溶剂限度的表示方法 (7)3.4 Analytical Procedures分析方法 (9)3.5 Reporting levels of residual solvents残留溶剂的报告水平 (9)4.LIMITS OF RESIDUAL SOLVENTS 残留溶剂的限度 (10)4.1 Solvents to Be Avoided应避免的溶剂 (10)4.2 Solvents to Be Limited应限制的溶剂 (10)4.3 Solvents with Low Toxic Potential低潜在毒性的溶剂 (12)4.4 Solvents for which No Adequate Toxicological Data was Found 没有足够毒理学数据的溶剂 (13)GLOSSARY术语 (14)APPENDIX 1. LIST OF SOLVENTS INCLUDED IN THE GUIDELINE附录 1:指导原则中包括的溶剂列表 (15)APPENDIX 2. ADDITIONAL BACKGROUND附录 2:其他背景 (18)APPENDIX 3. METHODS FOR ESTABLISHING EXPOSURE LIMITS附录 3:建立暴露限度的方法 (19)PART II:IMPURITIES: RESIDUAL SOLVENTS (MAINTENANCE)PDE FOR TETRAHYDROFURAN第二部分:杂质:残留溶剂(修订)四氢呋喃的 PDE (23)PART III:IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE) PDE FOR N-METHYLPYRROLIDONE (NMP) 第三部分:杂质:残留溶剂(修订)N-甲基吡咯烷酮(NMP)的 PDE25 PART IV:IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE) PDE FOR CUMENEICH Harmonised Tripartite Guideline第四部分:杂质:残留溶剂(修订)异丙基苯的 PDE (27)PART V:IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE)PDE FOR TRIETHYLAMINE AND PDE OF METHYLISOBUTYLKETONE第五部分:杂质:残留溶剂(修订)三乙胺的 PDE 和甲基异丁基酮的 PDE (31)PART I:IMPURITIES: GUIDELINE FOR RESIDUAL SOLVENTS第一部分:杂质:残留溶剂的指导原则Having reached Step 4 of the ICH Process at the ICH Steering Committee meeting on 17 July 1997, this Guideline is recommended for adoption to the three regulatory parties to ICH在 1997 年 7 月 17 日的 ICH 指导委员会会议上进入 ICH 进程第四阶段,并建议 ICH 的三方监管机构采纳该指导原则1.INTRODUCTION引言The objective of this guideline is to recommend acceptable amounts for residual solvents in pharmaceuticals for the safety of the patient. The guideline recommends use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本指导原则旨在建议为保证患者安全而应规定的药物中残留溶剂的可接受量。
药物中的残留溶剂测定

有机溶剂的分类
分类依据: 允许日暴露量(permitted daily exposure PDE) 定义: 是指某一有机溶剂被允许摄入而不产生毒性的日平 均最大剂量,单位为mg/天
有机溶剂的分类
类别 第一类溶剂 毒性 人体致癌物、疑为人 体致癌物或环境危害 物 有非遗传致癌毒性或 其他不可逆毒性、或 其他严重的可逆毒性 对人体低毒 没有足够毒性资料 PDE( mg/天) 0.1以下(1,1,1) 三氯乙烷除外 0.5-50
PDE : mg/天
剂量: g/天
有机溶剂的引入
根据研究对象具体情况 分析有机溶剂的引入 有机溶剂毒性
确定何种有机溶剂需要进行研究
研究方法的建立及方法学验证
研究结果的分析及质量标准的建立
有机溶剂的引入
原料药/辅料:合成过程中引入 包括: 作为合成原料或反应溶剂引入 作为反应副产物引入 由其他合成原料或其他溶剂带入 制剂 各种成份(原料药、辅料)带入 制剂制备过程中引入
有机溶剂的引入
研究集中在:原料药的第一种情况 影响因素: 有机溶剂在合成过程中使用的步骤 后续步骤中使用的有机溶剂对之前使用的溶剂的影 响 中间体的影响(中间体的纯化方法、干燥条件) 终产品精制方法和条件等等
有机溶剂的引入
制剂 控制原料药的残留溶剂,最终目的是控制制剂的残留 溶剂,使之符合规定。有时候根据制剂的一些特点, 可能对原料药残留溶剂的研究和限度要求进行特殊性 的考虑。
药物中的残留溶剂测定
浙江省药品检验所 高素英
前 言
定义:药物中的残留溶剂( Residual Solvent )系指 在原料药或辅料的生产中,以及在制剂制备过程中使 用的,但在工艺过程中未能完全去除的有机溶剂。 研究的性质:杂质研究的范畴 研究的目的:控制药物质量,保障病人用药安全。 在原料药合成中,溶剂的选择是合成中非常关键的因 素,选择适当的溶剂可提高得率或决定药物的性质,如 晶型、纯度和溶解度,从而影响疗效。同时,在某些 特定的制剂生产中,其工艺也要求使用特定的溶剂。 但由于溶剂没有疗效,故所有残留溶剂均应尽可能除 去,以使产品符合其规范、GMP或其他基本的质量要 求 。
Q3C 溶剂残留和分类

摘自Q3C-残留溶剂的指导选择一类溶剂:应避免致癌物;备受怀疑的致癌物;环境危害物二类溶剂:设定残余量,限量使用非基因性动物致癌物;可能导致不可逆中毒,比如神经性中毒,畸形;可能导致其他可逆性中毒三类溶剂: 低毒对人体有潜在毒性,可以接触,但不超过50mg/day。
If Class 1 solvents are likely to be present, they should be identified and quantified."Likely to be present" refers to the solvent used in the final manufacturing step and to solvents that are used in earlier manufacturing steps and not removed consistently by a validated process.ICH规定3类只要干失小于0.5, 但1,2类并不是不能出现。
按照毒性大小和对环境的危害程度,该指导原则将溶剂分成三类(所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进行评估):第一类溶剂是指已知可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应避免使用这类溶剂。
如果在生产治疗价值较大的药品时不可避免地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的范围内,如:苯(2ppm)、四氯化碳(4ppm)、1,2-二氯乙烷(5ppm)、1,1-二氯乙烷(8ppm)、1,1,1-三氯乙烷(1500ppm)。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
按每日用药10克计算的每日允许接触量如下:2-甲氧基乙醇(50ppm)、氯仿(60ppm)、1,1,2-三氯乙烯(80ppm)、1,2-二甲氧基乙烷(100ppm)、1,2,3,4-四氢化萘(100ppm)、2-乙氧基乙醇(160ppm)、环丁砜(160ppm)、嘧啶(200ppm)、甲酰胺(220ppm)、正己烷(290ppm)、氯苯(360ppm)、二氧杂环己烷(380ppm)、乙腈(410ppm)、二氯甲烷(600ppm)、乙烯基乙二醇(620ppm)、N,N-二甲基甲酰胺(880ppm)、甲苯(890ppm)、N,N-二甲基乙酰胺(1090ppm)、甲基环己烷(1180ppm)、1,2-二氯乙烯(1870ppm)、二甲苯(2170ppm)、甲醇(3000ppm)、环己烷(3880ppm)、N-甲基吡咯烷酮(4840ppm)、。
Q3A(R2)新原料药中的杂质(中英文)

INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USEICH H ARMONISED T RIPARTITE G UIDELINEI MPURITIES I N N EW D RUG S UBSTANCESQ3A(R2)Current Step 4 versiondated 25 October 2006This Guideline has been developed by the appropriate ICH Expert Working Group and has been subject to consultation by the regulatory parties, in accordance with the ICH Process. At Step 4 of the Process the final draft is recommended for adoption to the regulatory bodies of the European Union, Japan and USA.Q3A(R2) Document HistoryCurrent Step 4 versionI MPURITIES I N N EW D RUG S UBSTANCESICH Harmonised Tripartite GuidelineHaving reached Step 4 of the ICH Process at the ICH Steering Committee meeting on 7 February 2002, this guideline is recommended foradoption to the three regulatory parties to ICH.Attachment 2 has been revised on 25 October 2006.TABLE OF CONTENTSNo table of contents entries found.I MPURITIES I N N EW D RUG S UBSTANCES新原料药中的杂质1. PREAMBLE 序言This document is intended to provide guidance for registration applications on the content and qualification of impurities in new drug substances produced by chemical syntheses and not previously registered in a region or member state. It is not intended to apply to new drug substances used during the clinical research stage of development. The following types of drug substances are not covered in this guideline: biological/biotechnological, peptide, oligonucleotide, radiopharmaceutical, fermentation product and semi-synthetic products derived therefrom, herbal products, and crude products of animal or plant origin.本文件旨在为化学合成的新原料药(这些新原料药尚未在任何地区或成员国注册)在注册申请时,对其杂质的含量和界定的申报提供指导。
Q3C(R7)杂质:残留溶剂指导原则(中文翻译公开征求意见稿)

注:2005 年 11 月 机构采纳。
修订前本版本命
名为 Q3C(M):
THF
对母指导原则所含 NMP 的 PDE 信息的修订
Q3C(R2)
N-甲基吡咯烷酮(NMP)的每日允许暴露量(PDE):根据2000 年 7 月 20 日
注:2005 年 11 月新的毒理学数据修订 PDE。
修 订 前 本 版 本 命指导委员会经第二阶段后批准修订版,并发布以便向公众征
药物中的残留溶剂在此定义为在原料药或辅料的生产中以及制剂制备过程中使用 或产生的有机挥发性化合物。这些溶剂在实际生产技术中不能完全除去。选择适当的溶 剂合成原料药可提高收率或决定药物的性质,如晶型、纯度和溶解度。因此,溶剂有时 可能是合成工艺的关键因素。本指导原则并不针对特意用作辅料的溶剂,也不针对溶剂 化物。然而这些制剂中的溶剂也应进行评价,并论证其合理性。
异丙基苯文档的 PDE 已经作为第 IV 部分整合在核心 Q3C (R4)指导原则中,指导原则更名为 Q3C(R5)。
已对表 2、表 3 和附录 1 进行更新,以反映对异丙基苯的 PDE 进行的修订。
修订母指导原则所含甲基异丁基酮的 PDE 信息,并纳入三乙胺的 PDE
Q3C(R6) Q3C(ห้องสมุดไป่ตู้6)
名为 Q3C(M):求意见。
NMP
Q3C(R2)
指导委员会经第四阶段后批准,并建议 ICH 的 3 个监管机2002 年 9 月 12 日
注:2005 年 11 月 构采纳。
修订前本版本命
名为 Q3C(M):
NMP
Q3C(R3)
指导委员会批准的计算公式勘误。
注:2005 年 11 月
修订前本版本命
2002 年 10 月 28 日
ICH Q3c 中文版)

药、赋形剂及制剂生产中不应该使用。但是,为了生产一种有特殊疗
效的药品而不得不使用时,除非经过其他论证,否则应按表 1 控制,
1,1,1-三氯乙烷因会造成环境公害列人表 1,其限度 1500ppm 是
基于安全性数据而定的。
杂质:残留溶剂的指导原则
杂质:残留溶剂的指导原则
1.介绍 本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的
可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理 学上的可接受水平。
药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以 及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中 不能完全除尽。在合成原料药中选择适当的溶剂可提高产量或决定药 物的性质,如结晶型。纯度和溶解度。因此.有时溶剂是合成中非常 关键的因素。本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂, 也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作 出合理的判断。
四氢呋喃
4.4 没有足够毒性资料的溶剂
以下溶剂(表4)在赋形剂、原料药和制剂生产中也许会被生产
商采用,但尚无足够的毒理学数据,故无PDE值,生产厂在使用时
应提供这些溶剂在制剂中残留水平的合理性论证报告。
表4 无足够毒理学数据的溶剂
1,1-二乙氧基丙烷 1,1-二甲基甲烷 2,2-二甲丙烷 异辛烷 异丙醚
表 1 药物制剂中含第一类溶剂的限度(应避免使用)
溶剂
浓度限度(ppm)
备注
苯
2
致癌物
四氯化碳
4
毒性及环境公害
1,2-二氯乙烷
5
毒性
1,1-二氯乙烯
8
毒性
Q3C-残留溶剂的指导选择

European Medicines Agency7 Westferry Circus, Canary Wharf, London, E14 4HB, UKTel. (44-20) 74 18 85 75 Fax (44-20) 75 23 70 40E-mail: mail@emea.eu.int http://www.emea.eu.intMarch 1998 CPMP/ICH/283/95ICH Topic Q 3 C (R3)Impurities: Residual SolventsStep 5NOTE FOR GUIDANCE ONIMPURITIES: RESIDUAL SOLVENTS(CPMP/ICH/283/95)TRANSMISSION TO CPMP November 1996 TRANSMISSION TO INTERESTED PARTIES November 1996 COMMENTS REQUESTED BEFORE May 1997 FINAL APPROVAL BY CPMP September 1997 DATE FOR COMING INTO OPERATION March 1998ICH Harmonised Tripartite GuidelineTable of Contents1. INTRODUCTION (3)2. SCOPE OF THE GUIDELINE (3)3. GENERAL PRINCIPLES (4)3.1 Classification of Residual Solvents by Risk Assessment (4)3.2 Methods for Establishing Exposure Limits (4)3.3 Options for Describing Limits of Class 2 Solvents (4)3.4 Analytical Procedures (6)3.5 Reporting levels of residual solvents (6)4. LIMITS OF RESIDUAL SOLVENTS (7)4.1 Solvents to Be Avoided (7)4.2 Solvents to Be Limited (7)4.3 Solvents with Low Toxic Potential (8)4.4 Solvents for which No Adequate Toxicological Data was Found (9)GLOSSARY (10)APPENDIX 1. LIST OF SOLVENTS INCLUDED IN THE GUIDELINE (11)APPENDIX 2. ADDITIONAL BACKGROUND (16)A2.1 Environmental Regulation of Organic Volatile Solvents (16)A2.2 Residual Solvents in Pharmaceuticals (16)APPENDIX 3. METHODS FOR ESTABLISHING EXPOSURE LIMITS (16)PART II: (21)IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE) (21)PART III: (23)IMPURITIES : RESIDUAL SOLVENTS (MAINTENANCE) (23)PART I:1. INTRODUCTIONThe objective of this guideline is to recommend acceptable amounts for residual solvents in pharmaceuticals for the safety of the patient. The guideline recommends use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.Residual solvents in pharmaceuticals are defined here as organic volatile chemicals that are used or produced in the manufacture of drug substances or excipients, or in the preparation of drug products. The solvents are not completely removed by practical manufacturing techniques. Appropriate selection of the solvent for the synthesis of drug substance may enhance the yield, or determine characteristics such as crystal form, purity, and solubility. Therefore, the solvent may sometimes be a critical parameter in the synthetic process. This guideline does not address solvents deliberately used as excipients nor does it address solvates. However, the content of solvents in such products should be evaluated and justified.Since there is no therapeutic benefit from residual solvents, all residual solvents should be removed to the extent possible to meet product specifications, good manufacturing practices, or other quality-based requirements. Drug products should contain no higher levels of residual solvents than can be supported by safety data. Some solvents that are known to cause unacceptable toxicities (Class 1, Table 1) should be avoided in the production of drug substances, excipients, or drug products unless their use can be strongly justified in a risk-benefit assessment. Some solvents associated with less severe toxicity (Class 2, Table 2) should be limited in order to protect patients from potential adverse effects. Ideally, less toxic solvents (Class 3, Table 3) should be used where practical. The complete list of solvents included in this guideline is given in Appendix 1The lists are not exhaustive and other solvents can be used and later added to the lists. Recommended limits of Class 1 and 2 solvents or classification of solvents may change as new safety data becomes available. Supporting safety data in a marketing application for a new drug product containing a new solvent may be based on concepts in this guideline or the concept of qualification of impurities as expressed in the guideline for drug substance (Q3A, Impurities in New Drug Substances) or drug product (Q3B, Impurities in New Drug Products), or all three guidelines.2. SCOPE OF THE GUIDELINEResidual solvents in drug substances, excipients, and in drug products are within the scope of this guideline. Therefore, testing should be performed for residual solvents when production or purification processes are known to result in the presence of such solvents. It is only necessary to test for solvents that are used or produced in the manufacture or purification of drug substances, excipients, or drug product. Although manufacturers may choose to test the drug product, a cumulative method may be used to calculate the residual solvent levels in the drug product from the levels in the ingredients used to produce the drug product. If the calculation results in a level equal to or below that recommended in this guideline, no testing of the drug product for residual solvents need be considered. If, however, the calculated level is above the recommended level, the drug product should be tested to ascertain whether the formulation process has reduced the relevant solvent level to within the acceptable amount. Drug product should also be tested if a solvent is used during its manufacture.This guideline does not apply to potential new drug substances, excipients, or drug products used during the clinical research stages of development, nor does it apply to existing marketeddrug products. The guideline applies to all dosage forms and routes of administration. Higher levels ofresidual solvents may be acceptable in certain cases such as short term (30 days or less) or topical application. Justification for these levels should be made on a case by case basis.See Appendix 2 for additional background information related to residual solvents.3. GENERAL PRINCIPLES3.1 Classification of Residual Solvents by Risk AssessmentThe term "tolerable daily intake" (TDI) is used by the International Program on Chemical Safety (IPCS) to describe exposure limits of toxic chemicals and "acceptable daily intake" (ADI) is used by the World Health Organization (WHO) and other national and internationalhealth authorities and institutes. The new term "permitted daily exposure" (PDE) is defined in the present guideline as a pharmaceutically acceptable intake of residual solvents to avoidconfusion of differingvalues for ADI's of the same substance.Residual solvents assessed in this guideline are listed in Appendix 1 by common names and structures. They were evaluated for their possible risk to human health and placed into one ofthree classes as follows:Class 1 solvents: Solvents to be avoided Known human carcinogens, strongly suspected human carcinogens, and environmentalhazards.Class 2 solvents: Solvents to be limited Non-genotoxic animal carcinogens or possible causative agents of other irreversible toxicitysuch as neurotoxicity or teratogenicity. Solvents suspected of other significant but reversible toxicities.Class 3 solvents: Solvents with low toxic potential Solvents with low toxic potential to man; no health-based exposure limit is needed. Class 3solvents have PDEs of 50 mg or more per day.3.2 Methods for Establishing Exposure LimitsThe method used to establish permitted daily exposures for residual solvents is presented inAppendix 3. Summaries of the toxicity data that were used to establish limits are published in Pharmeuropa, Vol. 9, No. 1, Supplement, April 1997.3.3 Options for Describing Limits of Class 2 SolventsTwo options are available when setting limits for Class 2 solvents.Option 1: The concentration limits in ppm stated in Table 2 can be used. They were calculated using equation (1) below by assuming a product mass of 10 g administered daily.dose PDE x 1000(ppm)ion Concentrat :(1) =Here, PDE is given in terms of mg/day and dose is given in g/day.These limits are considered acceptable for all substances, excipients, or products. Therefore this option may be applied if the daily dose is not known or fixed. If all excipients and drugsubstances in a formulation meet the limits given in Option 1, then these components may be used in any proportion. No further calculation is necessary provided the daily dose does notexceed 10 g. Products that are administered in doses greater than 10 g per day should be considered under Option 2. Option 2: It is not considered necessary for each component of the drug product to complywith the limits given in Option 1. The PDE in terms of mg/day as stated in Table 2 can be used with the known maximum daily dose and equation (1) above to determine theconcentration of residual solvent allowed in drug product. Such limits are considered acceptable provided that it has been demonstrated that the residual solvent has been reducedto the practical minimum. The limits should be realistic in relation to analytical precision, manufacturing capability, reasonable variation in the manufacturing process, and the limitsshould reflect contemporary manufacturing standards.Option 2 may be applied by adding the amounts of a residual solvent present in each of the components of the drug product. The sum of the amounts of solvent per day should be lessthan that given by the PDE.Consider an example of the use of Option 1 and Option 2 applied to acetonitrile in a drug product. The permitted daily exposure to acetonitrile is 4.1 mg per day; thus, the Option 1limit is 410 ppm. The maximum administered daily mass of a drug product is 5.0 g, and the drug product contains two excipients. The composition of the drug product and the calculated maximum content of residual acetonitrile are given in the following table.Component Amount in formulation Acetonitrile contentDaily exposure Active substance0.3 g 800 ppm 0.24 mg Excipient 10.9 g 400 ppm 0.36 mg Excipient 23.8 g 800 ppm 3.04 mg Medicinal product 5.0 g 728 ppm 3.64 mgExcipient 1 meets the Option 1 limit, but the drug substance, excipient 2, and drug product do not meet the Option 1 limit. Nevertheless, the product meets the Option 2 limit of 4.1 mg per day and thus conforms to the recommendations in this guideline.Consider another example using acetonitrile as residual solvent. The maximumadministered daily mass of a drug product is 5.0 g, and the drug product contains two excipients. Thecomposition of the drug product and the calculated maximum content of residual acetonitrile is given in the following table.Component Amount in formulation AcetonitrilecontentDaily exposure Active substance0.3 g 800 ppm 0.24 mg Excipient 10.9 g 2000 ppm 1.80 mg Excipient 23.8 g 800 ppm 3.04 mg Medicinal product 5.0 g 1016 ppm 5.08 mgIn this example, the product meets neither the Option 1 nor the Option 2 limit according to this summation. The manufacturer could test the drug product to determine if the formulationprocess reduced the level of acetonitrile. If the level of acetonitrile was not reduced during formulation to the allowed limit, then the manufacturer of the drug product should take othersteps to reduce the amount of acetonitrile in the drug product. If all of these steps fail to reduce the level of residual solvent, in exceptional cases the manufacturer could provide asummary of efforts made to reduce the solvent level to meet the guideline value, and provide a riskbenefit analysis to support allowing the product to be utilised with residual solvent atahigher level.3.4 Analytical ProceduresResidual solvents are typically determined using chromatographic techniques such as gas chromatography. Any harmonised procedures for determining levels of residual solvents as described in the pharmacopoeias should be used, if feasible. Otherwise, manufacturers wouldbe free to select the most appropriate validated analytical procedure for a particular application. If only Class 3 solvents are present, a nonspecific method such as loss on dryingmay be used.Validation of methods for residual solvents should conform to ICH guidelines Text on Validation of Analytical Procedures and Extension of the ICH Text on Validation ofAnalytical Procedures .3.5 Reporting levels of residual solventsManufacturers of pharmaceutical products need certain information about the content of residual solvents in excipients or drug substances in order to meet the criteria of thisguideline. The following statements are given as acceptable examples of the information that could be provided from a supplier of excipients or drug substances to a pharmaceuticalmanufacturer. The supplier might choose one of the following as appropriate:• Only Class 3 solvents are likely to be present. Loss on drying is less than 0.5%.• Only Class 2 solvents X, Y, ... are likely to be present. All are below the Option 1 limit. (Here the supplier would name the Class 2 solvents represented by X, Y, ...)• Only Class 2 solvents X, Y, ... and Class 3 solvents are likely to be present. Residual Class 2 solvents are below the Option 1 limit and residual Class 3 solvents are below0.5%.If Class 1 solvents are likely to be present, they should be identified and quantified."Likely to be present" refers to the solvent used in the final manufacturing step and tosolvents that are used in earlier manufacturing steps and not removed consistently by avalidated process.If solvents of Class 2 or Class 3 are present at greater than their Option 1 limits or 0.5%, respectively, they should be identified and quantified.4. LIMITS OF RESIDUAL SOLVENTS4.1 Solvents to Be AvoidedSolvents in Class 1 should not be employed in the manufacture of drug substances, excipients,and drug products because of their unacceptable toxicity or their deleterious environmentaleffect. However, if their use is unavoidable in order to produce a drug product with asignificant therapeutic advance, then their levels should be restricted as shown in Table 1,unless otherwise justified. 1,1,1- Trichloroethane is included in Table 1 because it is an environmental hazard. The stated limit of 1500 ppm is based on a review of the safety data.ConcernLimitSolvent Concentration(ppm)Benzene 2 Carcinogen Carbon tetrachloride 4 Toxic and environmentalhazard1,2-Dichloroethane 5 Toxic1,1-Dichloroethene 8 Toxic1,1,1-Trichloroethane 1500 Environmentalhazard4.2 Solvents to Be LimitedSolvents in Table 2 should be limited in pharmaceutical products because of their inherenttoxicity. PDEs are given to the nearest 0.1 mg/day, and concentrations are given to the nearest10 ppm. The stated values do not reflect the necessary analytical precision of determination.Precision should be determined as part of the validation of the method.TABLE 2. Class 2 solvents in pharmaceutical products.Solvent PDE (mg/day) Concentration Limit(ppm)Acetonitrile 4.1 410 Chlorobenzene 3.6 360 Chloroform 0.6 60 Cyclohexane 38.8 3880 1,2-Dichloroethene 18.7 1870 Dichloromethane 6.0 600 1,2-Dimethoxyethane 1.0100 N,N-Dimethylacetamide 10.91090 N,N-Dimethylformamide 8.8 8801,4-Dioxane 3.8 380 2-Ethoxyethanol 1.6 160 Ethylene glycol 6.2 620Formamide 2.2 220 Hexane 2.9 290 Methanol 30.0 3000 2-Methoxyethanol 0.5 50 Methylbutylketone 0.5 50 Methylcyclohexane 11.8 1180 N-Methylpyrrolidone 48.4 4840Nitromethane 0.5 50Pyridine 2.0 200 Sulfolane 1.6 160 Tetralin 1.0 100 Toluene 8.9 8901,1,2-Trichloroethene 0.8 80 Xylene* 21.7 2170 * usually 60% m-xylene, 14% p-xylene, 9% o-xylene with 17% ethyl benzene.4.3 Solvents with Low Toxic PotentialSolvents in Class 3 (shown in Table 3) may be regarded as less toxic and of lower risk to human health. Class 3 includes no solvent known as a human health hazard at levels normallyaccepted in pharmaceuticals. However, there are no long-term toxicity or carcinogenicity studies for many of the solvents in Class 3. Available data indicate that they are less toxic inacute or short-term studies and negative in genotoxicity studies. It is considered that amounts of these residual solvents of 50 mg per day or less (corresponding to 5000 ppm or 0.5% underOption 1) would be acceptable without justification. Higher amounts may also be acceptable provided they are realistic in relation to manufacturing capability and good manufacturingpractice.Table 3. Class 3 solvents which should be limited by GMP or other qualitybased requirements.Acetic acid Heptaneacetate Acetone IsobutylAnisole Isopropylacetateacetate1-Butanol Methyl2-Butanol 3-Methyl-1-butanol Butyl acetate Methylethyl ketonetert-Butylmethyl ether Methylisobutyl ketoneCumene 2-Methyl-1-propanol Dimethylsulfoxide PentaneEthanol 1-PentanolEthyl acetate 1-PropanolEthyl ether 2-PropanolEthyl formate Propyl acetateFormic acid Tetrahydrofuran4.4 Solvents for which No Adequate Toxicological Data was FoundThe following solvents (Table 4) may also be of interest to manufacturers of excipients, drug substances, or drug products. However, no adequate toxicological data on which to base aPDE was found. Manufacturers should supply justification for residual levels of these solvents in pharmaceutical products.Table 4. Solvents for which no adequate toxicological data was found.1,1-Diethoxypropane Methylisopropylketone1,1-Dimethoxymethane Methyltetrahydrofuranether2,2-Dimethoxypropane PetroleumIsooctane Trichloroaceticacid Isopropyl ether Trifluoroacetic acidGLOSSARYGenotoxic Carcinogens:Carcinogens which produce cancer by affecting genes or chromosomes.LOEL:Abbreviation for lowest-observed effect level.Lowest-Observed Effect Level:The lowest dose of substance in a study or group of studies that produces biologically significant increases in frequency or severity of any effects in the exposed humans or animals.Modifying Factor:A factor determined by professional judgment of a toxicologist and applied to bioassay data to relate that data safely to humans.Neurotoxicity:The ability of a substance to cause adverse effects on the nervous system.NOEL:Abbreviation for no-observed-effect level.No-Observed-Effect Level:The highest dose of substance at which there are no biologically significant increases in frequency or severity of any effects in the exposed humans or animals.PDE:Abbreviation for permitted daily exposure.Permitted Daily Exposure:The maximum acceptable intake per day of residual solvent in pharmaceutical products.Reversible Toxicity:The occurrence of harmful effects that are caused by a substance and which disappear after exposure to the substance ends.Strongly Suspected Human Carcinogen:A substance for which there is no epidemiological evidence of carcinogenesis but there are positive genotoxicity data and clear evidence of carcinogenesis in rodents.Teratogenicity:The occurrence of structural malformations in a developing fetus when a substance is administered during pregnancy.APPENDIX 1. LIST OF SOLVENTS INCLUDED IN THE GUIDELINESolvent OtherNames Structure Class Acetic acid Ethanoic acid CH3COOH Class3Acetone 2-Propanone CH3COCH3 Class 3Propan-2-oneAcetonitrile CH3CN Class2Anisole Methoxybenzene OCH3Class3Benzene Benzol Class11-Butanol n-Butyl alcohol CH3(CH2)3OH Class3Butan-1-ol2-Butanol sec-Butyl alcohol CH3CH2CH(OH)CH3 Class 3Butan-2-olButyl acetate Acetic acid butyl ester CH3COO(CH2)3CH3 Class3tert-Butylmethyl ether 2-Methoxy-2-methyl-propane (CH3)3COCH3 Class3Carbon tetrachloride Tetrachloromethane CCl4 Class1Chlorobenzene ClClass2Chloroform Trichloromethane CHCl3 Class 2Cumene IsopropylbenzeneCH(CH 3)2Class 3(1-Methyl)ethylbenzeneCyclohexane Hexamethylene Class 21,2-Dichloroethane sym -Dichloroethane CH 2ClCH 2Cl Class 1Ethylene dichloride Ethylene chloride 1,1-Dichloroethene 1,1-Dichloroethylene H 2C=CCl 2 Class 1 Vinylidene chloride 1,2-Dichloroethene 1,2-Dichloroethylene ClHC =CHCl Class 2Acetylene dichloride Dichloromethane Methylene chlorideCH 2Cl 2Class 21,2-Dimethoxyethane Ethyleneglycol dimethyl ether H 3COCH 2CH 2OCH 3 Class 2 Monoglyme Dimethyl Cellosolve N,N-Dimethylacetamide DMACH 3CON(CH 3)2 Class 2 N,N-Dimethylformamide DMFHCON(CH 3)2 Class 2Dimethyl sulfoxide Methylsulfinylmethane (CH 3)2SO Class 3 Methyl sulfoxide DMSO1,4-Dioxane p-DioxaneOO Class 2[1,4]Dioxane Ethanol Ethyl alcoholCH 3CH 2OH Class 32-Ethoxyethanol Cellosolve CH3CH2OCH2CH2OH Class 23Ethyl acetate Acetic acid ethyl ester CH3COOCH2CH3 Class2 Ethyleneglycol 1,2-Dihydroxyethane HOCH2CH2OH Class1,2-Ethanediol3Ethyl ether Diethyl ether CH3CH2OCH2CH3 ClassEthoxyethane1,1’-Oxybisethane3Ethyl formate Formic acid ethyl ester HCOOCH2CH3 Class2 Formamide Methanamide HCONH2 Class Formic acid H C O O H Class 33 Heptane n-Heptane CH3(CH2)5CH3 Class2 Hexane n-Hexane CH3(CH2)4CH3 Class Isobutyl acetate Acetic acid isobutyl ester CH3COOCH2CH(CH3)2 Class 33Isopropyl acetate Acetic acid isopropyl ester CH3COOCH(CH3)2 Class2alcohol CH3OH Class Methanol MethylCellosolve CH3OCH2CH2OH Class22-Methoxyethanol Methyl3 Methyl acetate Acetic acid methyl ester CH3COOCH3 Classalcohol (CH3)2CHCH2CH2OH Class 33-Methyl-1-butanol IsoamylalcoholIsopentyl3-Methylbutan-1-ol2 Methylbutyl ketone 2-Hexanone CH3(CH2)3COCH3 ClassHexan-2-oneMethylcyclohexane Cyclohexylmethane CH3Class2Methylethyl ketone 2-Butanone CH3CH2COCH3 Class3 MEKButan-2-oneMethylisobutyl ketone 4-Methylpentan-2-one CH3COCH2CH(CH3)2 Class 34-Methyl-2-pentanoneMIBK2-Methyl-1-propanol Isobutyl alcohol (CH3)2CHCH2OH Class3 2-Methylpropan-1-olN-Methylpyrrolidone 1-Methylpyrrolidin-2-one N OCH3 Class21-Methyl-2-pyrrolidinoneNitromethane CH3NO2 Class2 Pentane n-Pentane CH3(CH2)3CH3 Class31-Pentanol Amylalcohol CH3(CH2)3CH2OH Class3Pentan-1-olPentylalcohol1-Propanol Propan-1-ol CH3CH2CH2OH Class3Propylalcohol2-Propanol Propan-2-ol (CH3)2CHOH Class3IsopropylalcoholPropyl acetate Acetic acid propyl ester CH3COOCH2CH2CH3 Class 3Pyridine NClass2Sulfonane Tetrahydrothiophene1,1-dioxideSO OClass2Tetrahydrofuran Tetramethyleneoxide O Class3 OxacyclopentaneTetralin 1,2,3,4-Tetrahydro-naphthalene Class2Toluene Methylbenzene CH3Class21,1,1-Trichloroethane Methylchlororoform CH3CCl3 Class1 1,1,2-Trichloroethene Trichloroethene HClC=CCl2 Class2Xylene* Dimethybenzene CH3CH3Class2Xylol* usually 60 % m-xylene, 14 % p-xylene, 9 % o-xylene with 17 % ethyl benzeneAPPENDIX 2. ADDITIONAL BACKGROUNDA2.1 Environmental Regulation of Organic Volatile SolventsSeveral of the residual solvents frequently used in the production of pharmaceuticalsare listed as toxic chemicals in Environmental Health Criteria (EHC) monographs andthe Integrated Risk Information System (IRIS). The objectives of such groups as theInternational Programme on Chemical Safety (IPCS), the United States EnvironmentalProtection Agency (USEPA), and the United States Food and Drug Administration(USFDA) include the determination of acceptable exposure levels. The goal is protectionof human health and maintenance of environmental integrity against the possible deleterious effects of chemicals resulting from long-term environmental exposure. The methods involved in the estimation of maximum safe exposure limits are usually based on long-term studies. When long-term study data are unavailable, shorter term study data can be used with modification of the approach such as use of larger safety factors. The approach described therein relates primarily to long-term or life-time exposure of the general population in the ambient environment, i.e. ambient air, food, drinking water and other media.A2.2 Residual Solvents in PharmaceuticalsExposure limits in this guideline are established by referring to methodologies and toxicity data described in EHC and IRIS monographs. However, some specific assumptions about residual solvents to be used in the synthesis and formulation of pharmaceutical products should be taken into account in establishing exposure limits. They are:1) Patients (not the general population) use pharmaceuticals to treat their diseases or forprophylaxis to prevent infection or disease.2) The assumption of life-time patient exposure is not necessary for mostpharmaceuticalproducts but may be appropriate as a working hypothesis to reduce risk to human health3) Residual solvents are unavoidable components in pharmaceutical production and willoften be a part of drug products.4) Residual solvents should not exceed recommended levels except in exceptionalcircumstances.5) Data from toxicological studies that are used to determine acceptable levels forresidual solvents should have been generated using appropriate protocols such as those described for example by OECD, EPA, and the FDA Red Book.APPENDIX 3. METHODS FOR ESTABLISHING EXPOSURE LIMITSThe Gaylor-Kodell method of risk assessment (Gaylor, D. W. and Kodell, R. L.: Linear Interpolation algorithm for low dose assessment of toxic substance. J Environ. Pathology, 4, 305, 1980) is appropriate for Class 1 carcinogenic solvents. Only in cases where reliable carcinogenicity data are available should extrapolation by the use of mathematical models be applied to setting exposure limits. Exposure limits for Class 1 solvents could be determined with the use of a large safety factor (i.e., 10,000 to 100,000) with respect to the no-observed-effect level (NOEL). Detection and quantitation of these solvents should be by state-of-the-art analytical techniques.Acceptable exposure levels in this guideline for Class 2 solvents were established by calculation of PDE values according to the procedures for setting exposure limits in pharmaceuticals (Pharmacopeial Forum, Nov-Dec 1989), and the method adopted by IPCS for Assessing Human Health Risk of Chemicals (Environmental Health Criteria 170, WHO, 1994). These methods are similar to those used by the USEPA (IRIS) and the USFDA (Red Book) and others. The method is outlined here to give a better understanding of the origin of the PDE values. It is not necessary to perform these calculations in order to use the PDE values tabulated in Section 4 of this document.PDE is derived from the no-observed-effect level (NOEL), or the lowest-observed effect level (LOEL) in the most relevant animal study as follows:PDE =NOEL x Weight Adjustment F1x F2x F3x F4x F5The PDE is derived preferably from a NOEL. If no NOEL is obtained, the LOEL may be used. Modifying factors proposed here, for relating the data to humans, are the same kind of "uncertainty factors" used in Environmental Health Criteria (Environmental Health Criteria 170, World Health Organization, Geneva, 1994), and "modifying factors" or "safety factors" in Pharmacopeial Forum. The assumption of 100% systemic exposure is used in all calculations regardless of route of administration.The modifying factors are as follows:F1 = A factor to account for extrapolation between speciesF1 = 5 for extrapolation from rats to humansF1 = 12 for extrapolation from mice to humansF1 = 2 for extrapolation from dogs to humansF1 = 2.5 for extrapolation from rabbits to humansF1 = 3 for extrapolation from monkeys to humansF1 = 10 for extrapolation from other animals to humansF1 takes into account the comparative surface area:body weight ratios for the species concerned and for man. Surface area (S) is calculated as:S = kM0.67in which M = body mass, and the constant k has been taken to be 10. The body weights used in the equation are those shown below in Table A3.1.F2 = A factor of 10 to account for variability between individuals.A factor of 10 is generally given for all organic solvents, and 10 is used consistently in this guideline.F3 = A variable factor to account for toxicity studies of short-term exposureF3 = 1 for studies that last at least one half lifetime (1 year for rodents or rabbits; 7 years for cats, dogs and monkeys).F3 = 1 for reproductive studies in which the whole period of organogenesis is covered.F3 = 2 for a 6-month study in rodents, or a 3.5-year study in non-rodents.F3 = 5 for a 3-month study in rodents, or a 2-year study in non-rodents.F3 = 10 for studies of a shorter duration.。
FDA,GMP,ICH临床实验专业英语词汇互译

FDA,GMP,ICH临床实验专业英语词汇互译FDA,GMP,ICH临床实验专业英语词汇互译FDA常用词中英对照FDA(food and drug adminisration)美国)食品药品监督管理局NDA(new drug application):新药申请ANDA(abbreviated new drug application):简化新药申请EP(export application):出口药申请(申请出口不被批准在美国销售的药品)treatment IND:研究中的新药用于治疗abbreviated(new)drug:简化申请的新药DMF(drug master file):药物主文件(持有者为谨慎起见而准备的保密资料,可以包括一个或多个人用药物在制备,加工,包装和贮存过程中所涉及的设备,生产过程或物品.只有在DMF 持有者或授权代表以授权书的形式授权给FDA,FDA在审查IND, NDA,ANDA时才能参考其内容)holderMF持有者CFR(code of federal regulation)美国)联邦法规PANEL:专家小组batch production:批量生产;分批生产batch production records:生产批号记录post or pre-market surveillance:销售前或销售后监督informed consent:知情同意(患者对治疗或受试者对医疗试验了解后表示同意接受治疗或试验)prescription drug:处方药OTC drug(over—the—counter drug):非处方药U.S. public health service:美国卫生福利部NIH(national institute of health)美国)全国卫生研究所animal trail:动物试验accelerated approval:加速批准standard drug:标准药物investigator :研究人员;调研人员preparing and submitting:起草和申报submission:申报;递交benefit(s):受益risk(s):受害drug product:药物产品drug substance:原料药established name:确定的名称generic name:非专利名称proprietary name:专有名称;INN(international nonproprietary name):国际非专有名称narrative summary: 记叙体概要adverse effect:副作用adverse reaction:不良反应protocol:方案archival copy:存档用副本review copy:审查用副本official compendium:法定药典(主要指USP, NF).USP(the united state pharmacopeia):美国药典(现已和NF合并一起出版)NF(national formulary)美国)国家药品集official=pharmacopeial = compendial:药典的;法定的;官方的agency:审理部门(指FDA)sponsor:主办者(指负责并着手临床研究者)identity:真伪;鉴别;特性strength:规格;规格含量(每一剂量单位所含有效成分的量)labeled amount:标示量regulatory specification:质量管理规格标准(NDA提供)regulatory methodology:质量管理方法(FDA用于考核原料药或药物产品是否符合批准了的质量管理规格标准的整套步骤)regulatory methods validation:管理用分析方法的验证(FDA对NDA提供的方法进行验证)Dietary supplement:食用补充品ICH(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use)人用药物注册技术要求国际协调会议ICHuality-质量Q1A(R2): Stability Testing of New Drug Substances and Products (Second Revision)新原料药和制剂的稳定性试验(第二版)Q1B: Photostability Testing of New Drug Substances and Products新原料药和制剂的光稳定性试验Q1C: Stability Testing for New Dosage Forms新制剂的稳定性试验Q1D: Bracketing and Matrixing Designs for Stability Testing of Drug Substances and Drug Products原料药和制剂稳定性试验的交叉和矩阵设计Q1E: Evaluation of Stability Data对稳定性数据的评估处理Q1F: Stability Data Package for Registration Applications in Climatic Zones III and IV在气候带III和IV,药物注册申请所提供的稳定性数据Q2A: Text on Validation of Analytical Procedures分析程序的验证Q2B: Validation of Analytical Procedures: Methodology分析程序的验证:方法学Q3A(R): Impurities in New Drug Substances (Revised Guideline)新原料药中的杂质(修订版)Q3B(R): Impurities in New Drug Products (Revised Guideline)新制剂中的杂质(修订版)Q3C: Impurities: Guideline for Residual Solvents杂质:残留溶剂指南Q3C(M): Impurities: Guideline for Residual Solvents (Maintenance)杂质:残留溶剂指南(修改内容)Q4: Pharmacopoeias药典Q4A: Pharmacopoeial Harmonisation 药典的协调Q4B: Regulatory Acceptance of Pharmacopoeial Interchangeability药典互替在法规上的可接受性Q5A: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin来源于人或者动物细胞系的生物技术产品的病毒安全性评估Q5B: Quality of Biotechnological Products: Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products生物技术产品的质量:源于重组DNA的蛋白质产品的生产中所用的细胞中的表达构建分析Q5C: Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products生物技术产品的质量:生物技术/生物产品的稳定性试验Q5D: Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products用于生产生物技术/生物产品的细胞底物的起源和特征描述Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process基于不同生产工艺的生物技术产品/生物产品的可比较性Q6: Specifications for New Drug Substances and Products新原料药和制剂的质量规格Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances质量规格:新原料药和新制剂的检验程序和可接收标准:化学物质Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products质量规格:生物技术/生物产品的检验程序和可接收标准-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:34:00--Q7: Good Manufacturing Practices for Pharmaceutical Ingredients活性药物成份的GMPQ7A: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients活性药物成份的GMP指南Q8: Pharmaceutical Development药物研发Q9: Quality Risk Management质量风险管理ICH:Safety-安全S1A: Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals药物致癌性研究需要的指南S1B: Testing for Carcinogenicity of Pharmaceuticals药物致癌性的检验S1C: Dose Selection for Carcinogenicity Studies of Pharmaceuticals药物致癌性研究之剂量选择S1C(R): Addendum: Addition of a Limit Dose and Related Notes附录:极限剂量和有关注释的的补充S2A: Guidance on Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals受法规管辖的药物基因毒性检验的特定方面的指南S2B: Genotoxicity: A Standard Battery for Genotoxicity Testing for Pharmaceuticals 基因毒性:药物基因毒性检验的标准S3A: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies毒物代谢动力学指南的注释:毒性研究中的全身性暴露量的评估S3B: Pharmacokinetics: Guidance for Repeated Dose Tissue Distribution Studies药物代谢动力学:重复剂量的组织分布研究指南S4: Single Dose Toxicity Tests单剂量毒性检验S4A: Duration of Chronic Toxicity Testing in Animals (Rodent and Non-Rodent Toxicity Testing)动物体内慢性毒性持续时间的检验(啮齿动物和非啮齿动物毒性检验)S5A: Detection of Toxicity to Reproduction for Medicinal Products药物对生殖发育的毒性的检验S5B(M): Maintenance of the ICH Guideline on Toxicity to Male Fertility: An Addendum to the Guideline on Detection of Toxicity to Reproduction for Medicinal Products 对男性生殖能力的毒性的指南的变动:药物对生殖发育的毒性的检验指南增加了一个附录S6: Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals生物技术生产的药物的临床前安全评价S7A: Safety Pharmacology Studies for Human Pharmaceuticals人用药的安全药理学研究S7B: The Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization(QT Interval Prolongation) By Human Pharmaceuticals药物延迟心室复极化(QT间期)潜在作用的非临床评价S8: Immunotoxicology Studies for Human Pharmaceuticals人用药免疫毒理学研究M3(M): Maintenance of the ICH Guideline on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals药物的对人临床试验的非临床安全研究指南的变动E-Efficacy(有效)E1: The Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life-Threatening Conditions对用于无生命危险情况下长期治疗的药物进行临床安全评估的族群暴露量范围E2A: Clinical Safety Data Management: Definitions and Standards for Expedited Reporting临床安全数据管理:速报制度的定义和标准E2B(R): Revision of the E2B(M) ICH Guideline on Clinical Safety Data Management Data Elements for Transmission of Individual Case Safety Reports个案安全报告送交的临床安全数据管理的数据要素指南(E2B(M))的修订版E2B (M): Maintenance of the Clinical Safety Data Management including: Data Elements for Transmission of Individual Case Safety Reports临床安全数据管理的变动包括:个案安全报告送交的数据要素E2B(M): Maintenance of the Clinical Safety Data Management including Questions and Answers临床安全数据管理的变动,包括问答E2C: Clinical Safety Data Management: Periodic Safety Update Reports for Marketed Drugs临床安全数据管理:已上市药品的周期性安全数据更新报告Addendum to E2C: Periodic Safety Update Reports for Marketed DrugsE2C的附录:已上市药品的周期性安全数据更新报告E2D: Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting批准后的安全数据管理:速报制度的定义和标准E2E: Pharmacovigilance Planning药物警戒计划E3: Structure and Content of Clinical Study Reports临床研究报告的结构和内容E4: Dose-Response Information to Support Drug Registration支持药品注册的剂量-效应资料E5: Ethnic Factors in the Acceptability of Foreign Clinical Data引入海外临床数据时要考虑的人种因素E6: Good Clinical Practice: Consolidated GuidelineGCP:良好的临床规范:统一的指南E7: Studies in Support of Special Populations: Geriatrics对特定族群的支持的研究:老人病学E8: General Considerations for Clinical Trials对临床试验的总的考虑E9: Statistical Principles for Clinical Trials临床试验的统计原则E10: Choice of Control Group and Related Issues in Clinical Trials临床试验中控制组和有关课题的选择E11: Clinical Investigation of Medicinal Products in the Pediatric Population小儿科药物的临床调查E12A: Principles for Clinical Evaluation of New Antihypertensive Drugs新抗高血压药物的临床评价原则E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs非抗心率失常药物的QT/QTc 间期和致心率失常潜在作用的临床评价Multidisciplinary Guidelines 多学科兼容的指南M1: Medical Terminology医学术语M2: Electronic Standards for Transmission of Regulatory Information (ESTRI)药政信息传递之电子标准M3: Timing of Pre-clinical Studies in Relation to Clinical Trials (See Safety Topics)有关临床试验的临床前研究的时间安排M4: The Common Technical Document (See CTD section for complete Status of the guidelines)通用技术文件(见有关CTD章节)M5: Data Elements and Standards for Drug Dictionaries药物词典的数据要素和标准临床试验常用的英文缩略语TTP: time-to-progression 疾病进展时间SAE: severity Adverse Event 严重不良事件AE: Adverse Event 不良事件-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:34:00--SOP: Standard Operating Procedure 标准操作规程CRF: Case Report form 病例报告表DLT: 剂量限制毒性MTD: 最大耐受剂量KPS: Karnofsky Performance Status行为状态评分CR: complete response完全缓解PR: partial response部分缓解SD: 病情稳定PD: progressive disease病情进展CTC: 常用药物毒性标准IEC: independent ethics committee 独立伦理委员会IRB : institutional review board 伦理委员会CRA: 临床研究助理CRO: Contract Research Organization 合同研究组织DFS: Disease Free Survival 无病生存期OS: (Overall Survival) 总生存时间IC: Informed consent 知情同意ADR: Adverse Drug Reaction 不良反应GAP:Good Agricultural Practice 中药材种植管理规范GCP:Good Clinical Practice 药物临床试验质量管理规范GLP:Good Laboratory Practice 药品实验室管理规范GMP:Good Manufacturing Practice 药品生产质量管理规范GSP:Good Supply Practice 药品经营质量管理规范GUP:Good Use Practice 药品使用质量管理规范PI rincipal investigator 主要研究者CI: Co-inveatigator 合作研究者SI :Sub-investigator 助理研究者COI :Coordinating investigtor 协调研究者DGMP: 医疗器械生产质量管理规范ICF: Informed consent form 知情同意书RCT : randomized controlled trial, 随机对照试验NRCCT: non-randomized concurrent controlled trial, 非随机同期对照试验EBM: evidence-based medicine 循证医学RCD: randomized cross-over disgn 随机交叉对照试验HCT: historial control trial, 历史对照研究RECIST: Response Evaluation Criteria In Solid Tumors. 实体瘤疗效反应的评价标准QC: Quality Control质量控制UADR: Unexpected Adverse Drug Reaction,非预期药物不良反应-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:34:00--GMP英语PIC/S的全称为harmaceutical Inspection Convention/Pharmaceutical Inspection Cooperation Scheme, PIC/S(制药检查草案), 药品检查协会(PIC/S) ,也有人称PIC/S为医药审查会议/合作计划(PIC/S)PIC的权威翻译:药品生产检查相互承认公约API(Active Pharmaceutical Ingrediet) 原料药又称:活性药物组分AirLock 气闸Authorized Person 授权人Batch/Lot 批次Batch Number/Lot-Number 批号;Batch Numbering System 批次编码系统;Batch Records 批记录;Bulk Product 待包装品;Calibration 校正;Clean area洁净区;Consignmecnt(Delivery)托销药品.ABPI Association of the British Pharmaceutical IndustryADR Adverse Drug ReactionAE Adverse EventAIM Active Ingredient ManufacturerANDA Abbreviated New Drug ApplicationANOVA Analysis of VarianceASM: Active Substance ManufacturerATC Anatomical Therapeutic ChemicalATX Animal Test Exemption CertificateBANBritish Approved NameBIRABritish Institute of Regulatory AffairsBNF British National FormularyBP British PharmacopoeiaC of A Certificate of AnalysisC of S Certificate of SuitabilityCENTRE FOR DRUG EVALUATION (CDE)Centre for Pharmaceutical Administration (CPA)CMS Concerned Member StateCMS每个成员国COS Certificate of SuitabilityCPMP Committee for Proprietary Medicinal ProductsCRA Clinical Research AssociateCRF Case Report FormCRO Contract Research OrganisationCTA Clinical Trial ApplicationCTC Clinical Trial CertificateCTD Common Technical DocumentCTX Clinical Trials ExemptionDDD Defined Daily DoseDGC Daily Global ComparisonDIA Drug Information AssociationDMF Drug Master FileDrug Registration Branch (DR, Product Evaluation & Registration Division, CPA EDQM (European Directorate for the Quality of Medicines) 欧洲联盟药品质量指导委员会EEA 欧洲经济地区EGMA European Generics Medicine AssociationELA Established Licence ApplicationEMEA European Medicines Evaluation AgencyEMEA (European Agency for the Evaluation of Medicinal Products) 欧洲联盟药品评价机构EP European PharmacopoeiaEPAR European Public Assessment ReportsESRA European Society of Regulatory AffairsEuropean Pharmacopoeia Commission 欧洲药典委员会FDA Food and Drug Administrationfinal evaluation report (FER)free sale certificates (FSCs)Health Sciences Authority (HSA)HSA's Medicines Advisory Committee (MAC)IB Investigators BrochureICH International Conference for HarmonisationIDMC Independent Data-Monitoring CommitteeIEC Independent Ethics CommitteeIND Investigational New DrugINN International Non-proprietary NameInternational Conference on Harmonisation (ICH)IPC In Process ControlIRB Institutional Review BoardLICENCE HOLDERMA Marketing AuthorisationMAA Marketing Authorisation ApplicationMAA上市申请MAH Marketing Authorisation HolderMAH 销售许可持有者MCA Medicines Control AgencyMHW Ministry of Health and Welfare (Japan)MR Mutual RecognitionMRA 美国与欧盟的互认协议MRAs (Mutual Recognition Agreements) 互相认证同意MRFG Mutual Recognition Facilitation Group MRPMutual Recognition ProcedureNASNew Active SubstanceNCENew Chemical EntityNDANew Drug Applicationnew chemical entities (NCEs)new drug applications (NDAs)NSAID Non Steroidal Anti Inflammatory DrugNTA Notice To ApplicantsOOS Out of SpecificationOTC Over The CounterPAGB Proprietary Association of Great BritainPh Eur European PharmacopoeiaPIL Patient Information LeafletPL Product LicencePOM Prescription Only MedicinePRODUCT OWNERPSU Periodic Safety UpdatesQA Quality AssuranceQC Quality ControlRAJ Regulatory Affairs JournalRMS Reference Member StateRMS相互认可另一成员国RSD Relative Standard DeviationRx Prescription OnlySAE Serious Adverse EventSMF Site Master FileSOP Standard Operating ProcedureSOP (STANDARD OPERATION PROCEDURE) 标准运作程序SPC/SmPC Summary of Product Characteristics summary of product characteristics(SPC)Therapeutic Goods Administration (TGA)USP US PharmacopoeiaVMF Veterinary Master FileVPC Veterinary Products CommitteeA.A.A Addition and Amendments 增补和修订AC Air Conditioner 空调器ADR Adverse Drug Reaction 药物不良反应AFDO Association of Food and Drug Officials 食品与药品官员协会(美国) ACC Accept 接受AQL Acceptable Quality Level 合格质量标准ADNA Abbreviated New Drug Application 简化的新药申请BOM Bill of Material 物料清单BPC Bulk pharmaceutical Chemiclls 原料药CBER Center for Biologics Evaluation Research 生物制品评价与研究中心CFU Colony Forming Unet 菌落形成单位DMF Drug Master File 药品管理档案CDER Cemter for Drug Evaluation amd Research 药物评价与研究中心CI Corporate Identity (Image) 企业识别(形象)CIP Cleaning in Place 在线清洗CSI Consumer Safety Insepctor 消费者安全调查员CLP Cleaning Line Procedure 在线清洗程序DAL Defect Action Level 缺陷作用水平DEA Drug Enforcement Adminestration 管制药品管理DS Documentation Systim 文件系统FDA Food and Drug Administration 食品与药品管理局(美国)GATT General Agreemernt on Tariffs and Trade 关贸总协会GMP Good Manufacturing Practice Gvp 药品生质量管理规范GCP Good Clinical Practice 药品临床实验管理规范GLP Good Laboratory Practice 实验室管理规范GSP Good Supply Practice 药品商业质量规范GRP Gook RaTAIL Practice 药品零业质量管理规范GAP Good Agriculture Practice 药材生产管理规范GVP Gook Validation Prctice 验证管理规范GUP Gook Use Practice 药品重用规范HVAC Heating Ventilation Air Conditioning 空调净化系统ISO Intematonal Organization for Standardization 车际标准化组织MOU Memorandum of Understanding 谅解备忘录PF Porduction File 生产记录用表格OTC Over the Counter (Drug) 非处方药品PLA Product License Application 产品许可申请QA Quality Assurance 质量保证QC Quality Control 质量控制QMP Quality Management Procedure 质量管理程序SDA State Drug Administration 国家药品监督管理局SMP Standard Managmert Procedure 标准管理程序SOP Standard Operating Procedure 标准操作程序TQC Tatal Quality Control 全面质量管理USA Uneted States Pharmacopeia 美国药典-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:35:00--ICH 安全性领域常用专业术语中英文对照表Dead offspring at birth 出生时死亡的子代Degradation 降解 Delay of parturition 分娩延迟Deletion 缺失 Descriptive statistics 描述性统计 Distribution 分布Detection of bacterial mutagen 细菌诱变剂检测 Detection of clastogen 染色体断裂剂检测Determination of metabolites 测定代谢产物 Development of the offspring 子代发育Developmental toxicity 发育毒性 Diminution of the background lawn 背景减少Direct genetic damage 直接遗传损伤DNA adduct DNA加合物 DNA damage DNA损伤DNA repair DNA修复 DNA strand breaks DNA链断裂Dose escalation 剂量递增 Dose dependence 剂量依赖关系 Dose level 剂量水平Dose-limiting toxicity 剂量限制性毒性 Dose-raging studies 剂量范围研究Dose-relatived mutagenicity 剂量相关性诱变性 Dose-related 剂量相关Dose-relatived cytotoxicity 剂量相关性细胞毒性Dose-relatived genotoxic activity 剂量相关性遗传毒性Dose-response curve 剂量-反应曲线 Dosing route 给药途径Duration 周期 Duration of pregnancy 妊娠周期Eaning 断奶 Earlier physical malformation 早期躯体畸形Early embryonic development 早期胚胎发育Early embryonic development to implantation 着床早期的胚胎发育Electro ejaculation 电射精Elimination 清除Embryofetal deaths 胚胎和胎仔死亡 Embryo-fetal development 胚胎-胎仔发育Embryo-fetal toxicity 胚胎-胎仔毒性 Embryonic death 胚胎死亡Embryonic development 胚胎发育 Embryonic period 胚胎期Embryos 胚胎 Embryotoxicity 胚胎毒性Enantiomer 对映异构体End of pregnancy 怀孕终止 Endocytic 内吞噬(胞饮)Endocytic activity 内吞噬活性 Endogenous proteins 内源性蛋白Endogenous components 内源性物质 Endogenous gene 内源性基因Endonuclease 核酸内切酶 Emdpmiclease release from lysosomes 溶酶体释放核酸内切酶End-point 终点Epididymal sperm maturation 附睾精子成熟性 Epitope 抗原决定部位Error prone repair 易错性修复 Escalation 递增Escherichia coli strain 大肠杆菌菌株 Escherichia coli 大肠杆菌Evaluation of test result 试验结果评价Exaggerated pharmacological response 超常增强的药理作用Excretion 排泄(清除) Exposure assessment 接触剂量评价Exposure period 接解期 External metabolizing system 体外代谢系统F1-animals 子一代动物False positive result 假阳性结果Fecundity 多产 Feed-back 反馈 Fertilisation 受精 Fertility 生育力Fertility studies 生育力研究 Fetal abnormalities 胎仔异常Fetal and neonatal parameters 胎仔和仔鼠的生长发育参数Fetal development and growth 肿仔发育和生长 Fetal period 胎仔期 Fetotoxicity 胎仔毒性False negative result 假阴性结果First pass testing 一期试验Fluorescence in situ hybridization(FISH) 原位荧光分子杂交-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:35:00--average deviation 平均差Bbar chart 直条图,条图bias 偏性binomial distribution 二项分布biometrics 生物统计学bivariate normal population 双变量正态总体Ccartogram 统计图case fatality rate(or case mortality) 病死率census 普查chi-sguare(X2) test 卡方检验central tendency 集中趋势class interval 组距classification 分组,分类cluster sampling 整群抽样coefficient of correlation 相关系数coefficient of regression 回归系数coefficient of variability(or coefficieut of variation) 变异系数collection of data 收集资料column 列(栏)combinative table 组合表combined standard deviation 合并标准差combined variance(or poolled variance) 合并方差complete survey 全面调查completely correlation 完全相关completely random design 完全随机设计confidence level 可信水平,置信水平confidence limit 可信限,置信限constituent ratio 构成比,结构相对数continuity 连续性control 对照control group 对照组coordinate 坐标correction for continuity 连续性校正correction for grouping 归组校正correction number 校正数correction value 校正值correlation 相关,联系correlation analysis 相关分析correlation coefficient 相关系数critical value 临界值cumulative frequency 累积频率Ddata 资料degree of dispersion 离散程度degree of freedom 自由度degree of variation 变异度dependent variable 应变量design of experiment 实验设计deviation from the mean 离均差diagnose accordance rate 诊断符合率difference with significance 差别不显著difference with significance 差别显著discrete variable 离散变量dispersion tendency 离中趋势distribution 分布,分配-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:35:00--Eeffective rate 有效率eigenvalue 特征值enumeration data 计数资料equation of linear regression 线性回归方程error 误差error of replication 重复误差estimate value 估计值event 事件experiment design 实验设计experiment error 实验误差experimental group 实验组extreme value 极值Ffatality rate 病死率field survey 现场调查fourfold table 四格表freguency 频数freguency distribution 频数分布GGaussian curve 高斯曲线geometric mean 几何均数grouped data 分组资料Hhistogram 直方图homogeneity of variance 方差齐性homogeneity test of variances 方差齐性检验hypothesis test 假设检验hypothetical universe 假设总体Iincidence rate 发病率incomplete survey 非全面调检indepindent variable 自变量indivedual difference 个体差异infection rate 感染率inferior limit 下限initial data 原始数据inspection of data 检查资料intercept 截距interpolation method 内插法interval estimation 区间估计inverse correlation 负相关Kkurtosis coefficient 峰度系数Llatin sguare design 拉丁方设计least significant difference 最小显著差数least square method 最小平方法,最小乘法leptokurtic distribution 尖峭态分布leptokurtosis 峰态,峭度linear chart 线图linear correlation 直线相关linear regression 直线回归linear regression eguation 直线回归方程link relative 环比logarithmic normal distribution 对数正态分布logarithmic scale 对数尺度lognormal distribution 对数正态分布lower limit 下限Mmatched pair design 配对设计mathematical statistics 数理统计(学) maximum value 极大值mean 均值mean of population 总体均数mean square 均方mean variance 均方,方差measurement data 讲量资料median 中位数medical statistics 医学统计学mesokurtosis 正态峰method of least squares 最小平方法,最小乘法method of grouping 分组法method of percentiles 百分位数法mid-value of class 组中值minimum value 极小值mode 众数moment 动差,矩morbidity 患病率mortality 死亡率Nnatality 出生率natural logarithm 自然对数negative correlation 负相关negative skewness 负偏志no correlation 无相关non-linear correlation 非线性相关non-parametric statistics 非参数统计normal curve 正态曲线normal deviate 正态离差normal distribution 正态分布normal population 正态总体normal probability curve 正态概率曲线normal range 正常范围normal value 正常值normal kurtosis 正态峰normality test 正态性检验nosometry 患病率-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:35:00--Oobserved unit 观察单位observed value 观察值one-sided test 单测检验one-tailed test 单尾检验order statistic 顺序统计量ordinal number 秩号ordinate 纵坐标Ppairing data 配对资料parameter 参数percent 百分率percentage 百分数,百分率percentage bar chart 百分条图percentile 百分位数pie diagram 园图placebo 安慰剂planning of survey 调查计划point estimation 点估计population 总体,人口population mean 总体均数population rate 总体率population variance 总体方差positive correlation 正相关positive skewness 正偏态prevalence rate 患病率probability 概率,机率probability error 偶然误差proportion 比,比率prospective study 前瞻研究prospective survey 前瞻调查public health statistics 卫生统计学Qquality eontrol 质量控制quartile 四分位数Rrandom 随机random digits 随机数字random numbers table 随机数目表random sample 随机样本random sampling 随机抽样random variable 随机变量randomization 随机化randomized blocks 随机区组,随机单位组randomized blocks analysis of variance 随机单位组方差分析randomized blocks design 随机单位组设计randomness 随机性range 极差,全距range of normal values 正常值范围rank 秩,秩次,等级rank correlation 等级相关rank correlation coefficent 等级相关系数rank-sum test 秩和检验ranked data 等级资料rate 率ratio 比recovery rate 治愈率registration 登记regression 回归regression analysis 回归分析regression coefficient 回归系数regression eguation 回归方程relative number 相对数relative ratio 比较相对数relative ratio with fixed base 定基比remainder error 剩余误差replication 重复retrospective survey 回顾调查Ridit analysis 参照单位分析Ridit value 参照单位值Ssample 样本sample average 样本均数sample size 样本含量sampling 抽样sampling error 抽样误差sampling statistics 样本统计量sampling survay 抽样调查scaller diagram 散点图schedule of survey 调查表semi-logarithmic chart 半对数线图semi-measursement data 半计量资料semi-guartile range 四分位数间距sensitivity 灵敏度sex ratio 性比例sign test 符号检验significance 显著性,意义significance level 显著性水平significance test 显著性检验significant difference 差别显著simple random sampling 单纯随机抽样simple table 简单表size of sample 样本含量skewness 偏态slope 斜率sorting data 整理资料sorting table 整理表sources of variation 变异来源square deviation 方差standard deviation(SD) 标准差standard error (SE) 标准误standard error of estimate 标准估计误差standard error of the mean 均数的标准误standardization 标准化standardized rate 标化率standardized normal distribution 标准正态分布statistic 统计量statistics 统计学statistical induction 统计图statistical inference 统计归纳statistical map 统计推断statistical method 统计地图statistical survey 统计方法statistical table 统计调查statistical test 统计表statistical treatment 统计检验stratified sampling 统计处理stochastic variable 分层抽样sum of cross products of 随机变量deviation from mean 离均差积和sum of ranks 秩和sum of sguares of deviation from mean 离均差平方和superior limit 上限survival rate 生存率symmetry 对称(性)systematic error 系统误差systematic sampling 机械抽样-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:35:00--Tt-distribution t分布t-test t检验tabulation method 划记法test of normality 正态性检验test of one-sided 单侧检验test of one-tailed 单尾检验test of significance 显著性检验test of two-sided 双侧检验test of two-tailed 双尾检验theoretical frequency 理论频数theoretical number 理论数treatment 处理treatment factor 处理因素treatment of date 数据处理two-factor analysis of variance 双因素方差分析two-sided test 双侧检验two-tailed test 双尾检验type I error 第一类误差type II error 第二类误差typical survey 典型调查Uu test u检验universe 总体,全域ungrouped data 未分组资料upper limit 上限Vvariable 变量variance 方差,均方variance analysis 方差分析variance ratio 方差比variate 变量variation coefficient 变异系数velocity of development 发展速度velocity of increase 增长速度Wweight 权数weighted mean 加权均数Zzero correlation 零相关-- 作者:月萝兰魂-- 发布时间:2006-12-22 13:36:00--世界500强制药企业名称中英对照排名公司名称中文名称总部收入百万美元77 Pfizer 辉瑞美国 45950.092 Johnson & Johnson 强生美国 41862.0114 GlaxoSmithKline 葛兰素史克英国 35050.9193 Novartis 诺华瑞士 24864.0205 Roche Group 罗氏瑞士 23212.9222 Merck 默克美国 22485.9239 Bristol-Myers Squibb 百时美施贵宝美国 20894.0 248 Aventis 安万特法国 20162.4254 Abbott Laboratories 雅培美国 19680.6269 AstraZeneca 阿斯利康英国 18849.0330 Wyeth 惠氏美国 15850.6433 Eli Lilly 礼来大药厂美国 12582.5100 BASF 巴斯夫德国 37757.0125 Dow Chemical 道化学美国 32632.0129 Bayer 拜耳德国 32331.1365 Akzo Nobel 阿克苏诺贝尔荷兰 14770.7。
EP2.4.24残留溶剂的鉴定与控制中英文对照版(带图)
残留溶剂的鉴定及控制该方法适用于: .1.在活性物质、赋形剂或药品中的未知的一类或二类溶剂残留量的鉴定;2.在活性物质、赋形剂或药品中的一类或二类溶剂残留量的限量试验;3.当二类溶剂的限度大于1000ppm(0.1%)或要求检测三类溶剂时的限量试验。
—类溶剂、二类溶剂、三类溶剂的分类见5.4以下给出了三种样品溶液的稀释方法,以及气相色谱顶空进样的系统条件。
还给出了两种气相色谱的系统条件,系统A为首选方法,同时系统B适用于一般的鉴别试验。
样品溶液的制备方法由样品的溶解性和待测的溶剂种类决定。
下列溶剂不适于用顶空进样法测定:甲酰胺、2—乙氧基乙醇、2—甲氧基乙醇、乙二醇、N-甲基吡咯烷酮、环丁砜(四氢噻吩砜),但可采用其他的适当的方法测定。
当采用其他的方法定量测定有机残留量时,必须进行方法验证采用静态顶空进样法测定样品溶液制备1:适用于在水中易溶的物质的残留溶剂的测定样品溶液(1):取0.200g待测物质,用水溶解并稀释至20m1样品溶液制备2:适用于在水中不溶的物质的残留溶剂的测定样品溶液(2):取0.200g待测物质,用N,N--二甲基甲酰胺(DMF)溶解并稀释至20ml样品溶液制备3:适用于测定N,N---二甲基乙酰胺或N,N--二甲基甲酰胺的残留量,当怀疑他们存在时。
样品溶液(3):取0.200g待测物质,用1,3—二甲基-2-咪唑啉酮(DMI)溶解并稀释至20ml如果上述方法均不适宜,那么所用稀释方法及静态顶空进样条件均必须验证其合理性。
溶剂溶液(a):吸取一类残留溶剂标准溶液1.0ml,用水稀释至100.0ml,再吸取该溶液1.0ml,用水稀释至10.0ml。
溶剂溶液(b):吸取适量二类溶剂溶于二甲基亚砜,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格二)。
溶剂溶液(c):称取1.00g溶剂或待测物质中存在的,用二甲基亚砜或水溶解,用水稀释至100.0ml,再用水将该溶液稀释至限量的1/20(限量见5.4表格一或二)。
ICH Q3C一类二类三类溶剂

一类溶剂:已知可致癌且对人和环境有害的溶剂,尽可能避免使用(事实上很多大公司如罗氏等已将这些甚至包括某些二类溶剂打入“黑名单”),如果实在无法避免,残留量必须控制在规定限度:苯(2ppm)四氯化碳(4ppm)1,2-二氯乙烷(5ppm)(我注意到隆莱还有使用)1,1-二氯乙烯(8ppm)1,1,1-三氯乙烷(1500ppm)二类溶剂:有动物致癌性的溶剂,按每日允许接触量计算的规定限度如下:乙腈(410ppm)氯苯(360ppm)氯仿(60ppm)(罗氏禁用)环己烷(3880ppm)1,2-二氯乙烯(1870ppm)二氯甲烷(600ppm)1,2-二甲氧乙烷(100ppm)N,N-二甲基乙酰胺(1090ppm)DMF(880ppm)二氧六环(380ppm)2-乙氧基乙醇(160ppm)乙二醇(620ppm)甲酰胺(220ppm)正己烷(290ppm)甲醇(3000ppm)乙二醇甲醚(50ppm)甲丁酮(50ppm)甲基环己烷(1180ppm)N-甲基吡咯烷酮(4840ppm)硝基甲烷(50ppm)吡啶(200ppm)环丁砜(160ppm)1,2,3,4-四氢化萘(100ppm)甲苯(890ppm)1,1,2-三氯乙烯(80ppm)二甲苯(2170ppm)三类溶剂:低毒溶剂,限度为≤%,即5000ppm,如我们常用的乙醇、EA、TBME、丙酮、THF、庚烷、异丙醇等除上述三类溶剂外,还有一些溶剂如我们用过的石油醚、甲基四氢呋喃等尚无毒理资料,必须证明其残留量的合理性。
尤其石油醚,建议今后避免使用,尤其在后道反应最好避免,由于其成分复杂,无法测定准确残留量。
总之,在工艺开发阶段应优先选择三类溶剂,控制二类溶剂,尽量避免一类溶剂的使用。
医药中常用有机溶剂分类和残留限度

医药中常用有机溶剂分类与残留限度医药中常用有机溶剂分类与残留限度药品的残留溶剂无治疗作用并可能对人体的健康和环境造成危害,本文对国际协调大会〔ICH〕制订的指导原如此与各国执行情况作了较为详尽的介绍。
药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。
药品还可被来自包装、运输、仓储中的有机溶剂污染。
药品生产商有责任确保终产品中的任何一种残留溶剂对人体无害。
各国药监部门曾使用不同的药品残留溶剂指导原如此,为此国际组织展开了协调工作。
经相关程序讨论和审查后,国际协调大会的指导原如此于1997年7月17日获得通过,被推荐至国际协调大会〔ICH〕的指导委员会采用。
该指导原如此要求,如果某个药品的生产或纯化过程可导致溶剂残留,就应对这个药品进展检测,并且只检测生产过程或纯化中使用或产生的那种溶剂。
根据使用量的多少,可采用累加的方法计算药品中残留溶剂的量。
如果累加量低于或等于指导原如此中的推荐量,如此该药品无需进展残留溶剂检测;如果累加量高于推荐量,如此必须对该药品进展残留溶剂检测。
该指导原如此适用于颁布以后上市的所有剂型和给药途径,但不适用于在临床研究阶段使用的潜在新药和新辅料,也不适用于已上市的现有药物。
在某些情况如短期〔小于30天〕或局部应用下,视具体情况,溶剂的高残留量也可承受。
按照毒性大小和对环境的危害程度,该指导原如此将溶剂分成三类〔所列举的溶剂并不完全,应对合成和生产过程所有可能的残留溶剂进展评估〕:第一类溶剂是指可以致癌并被强烈怀疑对人和环境有害的溶剂。
在可能的情况下,应防止使用这类溶剂。
如果在生产治疗价值较大的药品时不可防止地使用了这类溶剂,除非能证明其合理性,残留量必须控制在规定的X围内,如:苯〔2ppm〕、四氯化碳〔4ppm〕、1,2-二氯乙烷〔5ppm〕、1,1-二氯乙烷〔8ppm〕、1,1,1-三氯乙烷〔1500ppm〕。
第二类溶剂是指无基因毒性但有动物致癌性的溶剂。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
杂质:残留溶剂的指导原则1.介绍本指导原则旨在介绍药物中残留溶剂在保证人体安全条件下的可接受量,指导原则建议使用低毒的溶剂,提出了一些残留溶剂毒理学上的可接受水平。
药物中的残留溶剂在此定义为在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,它们在工艺中不能完全除尽。
在合成原料药中选择适当的溶剂可提高产量或决定药物的性质,如结晶型。
纯度和溶解度。
因此.有时溶剂是合成中非常关键的因素。
本指导原则所指的溶剂不是谨慎地用作赋形剂的溶剂,也不是溶剂化物,然而在这些制剂中的溶剂含量也应进行测定,并作出合理的判断。
出于残留溶剂没有疗效,故所有残留溶剂均应尽可能.去,以符合产品规范、GMP或其他基本的质量要求。
制剂所含残留溶剂的水平不能高于安全值,已知一些溶剂可导致不接受的毒性(第一类,表1),除非被证明特别合理,在原药、赋形剂及制剂生产中应避免使用。
一些溶剂毒性不太大(第二类,表2)应限制使用,以防止病人潜在的不良反应。
使用低毒溶剂(第三类,表3)较为理想。
附录1中列出了指导原则中的全部溶剂。
表中所列溶剂并非详尽无遗,其他可能使用的溶剂有待日后补充列人。
第一、二类溶剂的建议限度或溶剂的分类会随着。
新的安全性资料的获得而调整。
含有新溶剂的新药制剂、其上市申请的安全性资料应符合本指导原则或原料药指导原则(Q3A新原料药中的杂质)或新药制剂(Q3B新药制剂中的杂质)中所述的杂质控制原则,或者符合上述三者。
2. 指导原则的范围指导原则范围包括原料药、赋形剂或制剂中所含残留溶剂.因此,当生产或纯化过程中会出现这些溶剂时。
应进行残留溶剂的检验。
也只有在上述情况下,才有必要作溶剂的检查。
虽然生产商可以选择性地测定制剂,但也可以从制剂中各成分的残留溶液水平来累积计算制剂中的残留溶剂。
如果计算结果等于或低于本原则的建议水平,该制剂可考虑不检查残留溶剂,但如果计算结果高于建议水平则应进行检测,以确定制剂制备过程中是否降低了有关溶剂的量以达到可接受水平。
果制剂生产中用到某种溶剂,也应进行测定。
本指导原则不适用于临床研究阶段的准新原料药、准赋形剂和准制剂。
也不适用于已上市的药品。
本指导原则适用于所有剂型和给药途径。
短期(如30天或更短)使用或局部使用时,允许存在的残留溶剂水平可以较高。
应根据不同的情况评判这些溶剂水平。
有关残留溶剂的背景附加说明见附录2。
3.通则3.1 根据危害程度对残留溶剂分类“可耐受的日摄人量”(TDI)是国际化学品安全纲要(IPCS)用于描述毒性化合物接触限度的术语。
“可接受的日摄人量”(ADI)是WHO及一些国家和国际卫生组织所用的术语。
新术语“允许的日接触量”(PDE)是本指导原则中用于定义药物中可接受的有机溶剂摄人量,以避免与同一物质的ADI混淆。
本原则中残留溶剂的评价以通用名和结构列于附录1,根据它们对人体可能造成的危害分为以下三类;(1)第一类溶剂:应避兔的溶剂为人体致癌物、疑为人体致癌物或环境危害物。
(2)第二类溶剂。
应限制的溶剂非遗传毒性动物致癌或可能导致其他不可逆毒性测神经毒性或致畸性)的试剂。
可能具其他严重的但可逆毒性的溶剂。
(3)第三类溶剂:低毒性溶剂对人体低毒的溶剂,无须制定接触限度;第三类溶剂的PDE为每天50mg或50mg以上。
3.2 建立接触限度的方法用于建立残留溶剂的PDE方法见附录3。
用于建立限度的毒理数据的总结见Pharmeuropa,V ol . 9,No . l,Suplement,April 1997.3.3 第二类溶剂限度的选择方法制定第二类溶剂的限度时有两种选择。
方法1: 使用表2中以 ppm为单位的浓度限度,假定日给药量为10g,以方程(1)计算。
方程(1) C(ppm)=PDE:mg/天 剂量:g/天这些限度对所有原料药、赋形剂和制剂均适用。
因此,这一方法可用于日剂量未知或未定的情况、只要在处方中所有的赋形剂和原料药都符合方法1给定的限度,就可以以任何比例用于制剂。
只要日剂量不超过10g,就无须进一步计算。
服用剂量超过 10g/天,应考虑用方法2。
方法2:制剂中的每一种成分不必符合方法1的限度。
药物中允许的残留溶剂限度水平,可根据表2中 PDE mg/天及已知最大日剂量,用方程(1)来计算。
只要证明已降低至实际最低水平,便可以认为这种限度是可接受的、该限度能说明分析方法的精度、生产能力和生产工艺的合理变异,并能反映当前生产的标准水平。
应用方法2时可将药物制剂的每种成分中残留溶剂叠加起来,每天的总溶剂量应低于PDE给定的值。
下面举例说明如何用方法l和2来考虑制剂中的乙睛限度。
乙睛的允许日接触量是4.1 mg/天,因此由方法1算出限度是410PPm;如现在日最大给药量是5.0g,制剂中含两种赋形剂,制剂中的成分和计算得到的最大残留乙睛量见下表:成分处方量乙睛量 日(摄人)量原料药 0.3g 800ppm0.24mg辅料一 0.9g 400ppm 0.36mg辅料二 3.8g 800PPm 3.04mg药物制剂 5.09 728ppm 3.64mg 辅料1符合方法1限度,但原料、辅料2和药物制剂不符合方法1限度,而制剂符合方法2规定的4.1mg/天,故符合本指导原则的建议值。
乙睛作为残留溶剂的另一例子,曰最大给药量5刀g,制剂中含两种赋形剂,各组分及计算得到的最大残留的乙睛最见下表:成分 处方量 乙睛量 日(摄人)量原料药 0.3g 800ppm 0.24mg辅料1 0.9g 2000ppm 1.80mg辅料 3.8g 800ppm 3.04mg药物制剂 5.0g 1016ppm 5.08mg此例制剂中乙睛限度总量既不符合方法1也不符合方法2。
生产厂可先测定制剂,以确定在处方工艺中能否降低已睛水平,如果不能将乙腈水平降至允许范围,生产厂应采取措施降低制剂中的乙腈量;若所有措施均不能降低残留溶剂的水平,厂方应提供其尝试降低残留溶剂以符合指导原则所做工作的总结报告,并以利弊分析报告证明允许该制剂存在的较高水平的残留溶剂。
3.4 分析方法残留溶剂通常用色谱技术,如用GC法测定,如可能,对药典上规定要检测的残留溶剂,应采用统一了的测定方法。
生产厂也可选用更合适的、经论证的方法来测定。
若仅存在第三类溶剂;可用非专属性的方法如干燥失重来检查。
残留溶剂的方法论证应遵循ICH指导原则:“分析方法论证:定义和术语”及“分析方法论证:方法学”。
3.5 残留溶剂的报告水平制剂生产商需要了解有关赋形剂或原料药中残留溶剂量的信息,以符合本指导原则的标准。
以下阐述了赋形剂或原料药供应商应提供给制剂牛产商的信息的~些例子。
供应商应选择以下一项:·仅可能存在第三类溶剂,干燥失重小于0.5%。
·仅可能存在第M类溶剂,X、Y……全部应低于方法1的限度。
(这里供应商应将第二类溶剂用X、Y……来表示)·仅可能存在第二类溶剂X、Y……和第三类溶剂,残留的第三类溶剂低于方法1的限度,残留的第三类溶剂低于0.5%。
如果可能存在第一类溶剂,应进行鉴定并定量。
“可能存在”系指用于工艺最后一步的溶剂和用于较前几步工艺的溶剂经论证不能全部除尽。
如果第二类溶剂高于方法1的限度或第三类溶剂高于0.5%,应鉴定并定量。
4. 残留溶剂的限度4.1应避免的溶剂因其具有不可接受的毒性或对环境造成公害,第一类溶剂在原料药、赋形剂及制剂生产中不应该使用。
但是,为了生产一种有特殊疗效的药品而不得不使用时,除非经过其他论证,否则应按表1控制,1,1,1-三氯乙烷因会造成环境公害列人表1,其限度1500ppm是基于安全性数据而定的。
表1 药物制剂中含第一类溶剂的限度(应避免使用)溶剂浓度限度(ppm)备注苯 2 致癌物四氯化碳 4 毒性及环境公害1,2-二氯乙烷 5毒性1,1-二氯乙烷 8毒性1,1,1-三氯乙烷 1500环境公害4.2 应限制的溶剂列于表2的溶剂,由于其具毒性,在制剂中应予限制,规定 PDE约 0.1mg/天,浓度约10ppm。
所列值不能反映测定所必需的分析精度,精度应为方法论证的一部分。
表2 药品中第二类溶剂溶剂 PDE(mg/天) 浓度限度(ppm)乙晴 4.1 410氯苯 3.6 360氯仿 0.6 60环氧乙烷 38.8 38801,2-二氯乙烯 18.7 1870二氯甲烷 6.0 6001,2-二甲亚砜 1.0 100N,N-二甲乙酰胺 10.9 1090N,N-二甲基甲酰胺 8.8 8801,4-二恶烷 3.8 3802-乙氧基乙醇 1.6 160乙二醇 6.2 620甲酰胺 2.2 220正己烷 2.9 290甲醇 30.0 30002-甲氧基乙醇 0.5 50甲基丁酮 0.5 50甲基环己烷 11.8 1180N-甲基吡咯烷酮 48.4 4840硝基甲烷 0.5 50吡啶 2.0 200二氧噻吩烷 1.6 160四氢萘 1.0 100甲苯 8.9 8901,1,2-三氯乙烯 0.8 80二甲苯* 21.7 2170 *通常为60% m-二甲苯,14% p-二甲苯,9% o-二甲苯和17%乙基苯。
4.3低毒溶剂第三类溶剂(见表3)可能低毒,对人体危害很小。
第三类溶剂包括人们认为在药物中以一般量存在时对人体无害的溶剂,但该类溶剂中许多尚未进行长期毒性或致癌研究。
急性毒性或短期毒性试验表明这类溶剂几乎无毒、无遗传毒性。
每日50mg或更少量无须论证即可接受(用方法1计算。
即5000ppm或0.5%)。
如果能够反映生产能力和GMP的实际情况,更大的量也可接受。
表3在GMP或其他质量要求中应限制的第三类溶剂醋酸 乙醇 甲乙酮丙酮 醋酸乙酯 甲基异丁酮苯甲醚 乙醚 2-甲基-1-丙醇1-丁醇 甲酸乙酯 戊烷2-丁醇 甲酸 正丙醇醋酸丁酯 正庚烷 正戊醇叔丁基甲基醚 醋酸异丙酯 醋酸异丁酯醋酸甲酯 2-丙醇 异丙基苯3-甲基-1-丁醇 醋酸丙酯 二甲亚砜四氢呋喃4.4 没有足够毒性资料的溶剂以下溶剂(表4)在赋形剂、原料药和制剂生产中也许会被生产商采用,但尚无足够的毒理学数据,故无PDE值,生产厂在使用时应提供这些溶剂在制剂中残留水平的合理性论证报告。
表4无足够毒理学数据的溶剂1,1-二乙氧基丙烷 甲基异丙酮1,1-二甲基甲烷 甲基四氢呋喃2,2-二甲丙烷 石油醚异辛烷 三氯乙酸异丙醚 三氟乙酸术语遗传毒性致癌指通过影响基因或染色体而致癌。
LOEL:lowest-observed effect level的缩写。
能观察到反应的最低量(lowest-obserued effect leuel)是在研究人体或动物接触某种物质时产生任何反应的频率或严重性在生物学上显著增加的最低剂量。
修正因子是由毒理学家评定的、由生物测定的结果转换成与人体安全性相关的系数。