配合物的离子吸收光谱
理解金属离子配位化合物的光谱性质与应用

第1章 理论基础 第2章 紫外-可见吸收光谱 第3章 荧光光谱 第4章 红外光谱 第5章 核磁共振光谱 第6章 应用与展望
目录
● 01
第1章 理论基础
金属离子配位化合物的定 义
金属离子配位化合物由中心金属离子与周围配体 形成,配体通过配位键与金属离子结合形成配合 物。这种化合物具有独特的结构和性质,在化学 研究中具有重要意义。
推断配合物 的结构和配
位环境
关键步骤
不同金属离 子和配体的 吸收光谱特
征
多样性
吸收峰的位 置和强度的
观察
信息丰富
紫外-可见吸收光谱的实验操作
样品制备
关键步骤
数据处理
精准分析
仪器调试
确保准确性
总结
紫外-可见吸收光谱在研究金属离子配位化合物 的光谱性质方面具有重要意义。深入了解其基本 原理、应用、解读和实验操作,有助于提升对配 位化合物的认识和分析能力。
紫外-可见吸收光 谱的基本原理
紫外-可见吸收光谱 是研究配位化合物色 彩的重要手段。金属 离子在特定波长下吸 收光子,使电子跃迁 至高能级轨道。
紫外-可见吸收光谱的应用
确定金属离 子的氧化态 和配位数
重要性不可忽视
研究金属离 子与配体之 间的相互作
用
深入了解配合物 性质
紫外-可见吸收光谱的解读
确定结构
通过特定吸收峰 判断
推断配体配 位方式
红外吸收峰呈现 不同特性
识别功能团
各功能团有特定 特征
红外光谱的解读
观察红外光谱的吸收峰位置和强度,可以推断配 位化合物的结构和键合情况。不同的功能团和金 属离子在不同波数下具有独特的红外吸收特征, 对照库片段可以帮助解读。
配合物吸收光波长怎么比较

配合物吸收光波长怎么比较
在化学中,配合物的吸收光波长与其分子结构和配位离子有关。
常用的比较方法包括:
1. 吸收光谱比较:通过测量配合物溶液在紫外可见光谱仪中的吸收光谱,比较吸收峰的强度和位置。
吸收光谱的位置对应着吸收光波长,较短的波长表示配合物能够吸收更高能量的光。
2. 摩尔消光系数比较:摩尔消光系数(molar absorption coefficient)是描述物质吸收光强度的指标。
在吸收峰处,摩尔消光系数越大,表示更多的光被吸收,即配合物的吸收强度越高。
通过比较不同配合物的摩尔消光系数,可以得出它们吸收光的强度差异。
3. 光谱带的宽度比较:光谱带的宽度代表了配合物能级的分布范围,也间接反映了配合物的电子结构。
带宽较宽的配合物吸收范围较广,说明其能级分布比较宽泛,可能存在多个能级和吸收能力。
以上方法可以在实验室中进行,以帮助确定不同配合物的吸收光波长。
高中化学竞赛 中级无机化学 第四章 配位场理论和配合物的电子光谱

ML= 0 MS =0 (2S+1)(2L+1)=1
15
能量相同的微状态归为一组,得到自由离子的5个光谱项:
L=4, ML= 4, 3, 2, 1 0, S=0 MS= 0
1G
L=3, ML= 3, 2, 1 0,
S=1 MS= 1, 0 3F
25
基本性质:
为什么可以把d1、d4、d6、d9组 态放到一张图中?
这是因为: (1) d0、d5、d10 在八面体弱场和
四面体场中都是球形对称的, 稳定化能 均为0, 其静电行为相同;
( 2 ) 而 d6 可 认 为 是 在 d5 上 增 加 1 个 电子, 尤如从d0上增加1个电子成d1一 样, 因而d1和d6的静电行为应该相同;
d2与d8, d3与d7的能级分布情况可用同一张Orgel 图定性描述
28
28
由图可以发现: ① F谱项在配位场中分裂为T1、
T2和A2, 而P谱项不分裂但变成T1, 基 态F谱项与P谱项有能量差。
② 相同类型的线, 如T1(P)和 T1(F)(图的左边)是禁止相交的, 他们 发生弯曲, 互相回避, 其弯曲的程度以 C表示, 称为弯曲系数。(不相交规 则)
颜色: 吸收绿黄光 显紫红色
31
Orgel图
优点:简单方便
缺点: ① 不能用于强场配合物, 它只适用于弱场、高自
旋的情况, 而在强场情况下的谱项能量的变化在图上未反映。 ② Orgel图缺乏定量准确性, 那怕是同一电子组态
的离子, 不同的配体就要用不同的Orgel图, 因而特别不方 便。这是因为它的谱项能量和分裂能都是以绝对单位表示的, 不同的中心离子和不同的配体有不同的谱项能量和分裂能。
分光光度法测定配合物的分裂能的原理和方法

1.1962×10-2 kJ ·mol-1相当于1 cm-1,则得hc = 1。
配合物在最大吸收波长 λmax 处吸收的能量即为分裂能。当 波长的单位为 nm ,波数单位为 cm-1 时,分裂能为:
ES
hc
hc
max
1
107 (cm1)
二、实验原理
本实验采用一定浓度的 [Ti(H2O)6]3+ 溶液,用分光光度计 测定不同波长 λ 时的吸光度A ,再做A-λ 曲线,即得 [Ti(H2O)6]3+ 的吸收光谱曲线。பைடு நூலகம்出曲线最大吸收峰对应的波
长λmax,按上式即可求算 [Ti(H2O)6]3+ 的分裂能。
三、仪器与试剂
1. 仪器:
分光光度计,吸量管(5 mL),容量瓶(25 mL)3个,洗耳球,擦镜纸。
2. 试剂:
TiCl3溶液(150 ~200 g·L-1),HCl溶液(2 mol·L-1)
四、实验步骤
1. 计算150 ~200 g·L-1 TiCl3 溶液的浓度。 2. 移取5 mL、3 mL、2 mL上述溶液,置于25 mL容量瓶中,用 2mol·L-1 HCl溶液稀释至刻度,即得 [Ti(H2O)6]3+ 测量液。 3. 用分光光度计测出上述 [Ti(H2O)6]3+ 溶液在不同波长时的吸光度。 4. 绘制 A-λ曲线。以 λ 为横坐标,A为纵坐标作图,在吸收曲线上找 出 [Ti(H2O)6]3+配离子最大吸收峰所对应的波长 λmax。
五、实验数据记录与处理
1. 170 g·L-1 TiCl3 溶液的浓度c =_________。 2. [Ti(H2O)6]3+ 测量液的浓度c =________,________,_________。 3. 不同波长下,三种不同浓度的 [Ti(H2O)6]3+ 配离子溶液的吸光度A:
第四章 配合物的电子光谱

t2g、eg*主要为金属离子轨道成份,而t2g*主要为配体轨道。 例:[Co(CN)6]3-, M→L跃迁, ν1=49500cm-1
3、金属对金属荷移 普鲁士兰KFeIII[FeII(CN)6] 钼蓝中的MoV 和 MoVI 黑金化合物CS2AuIAuIIICl6 [AuCl4]-和[AuCl2]-基团之间产生电荷迁移
3.旋-轨偶合 旋-轨偶合使谱项进一步裂分为光谱 支项,导致谱带加宽。
二、 配合物的电荷迁移光谱 1、配位体对金属L→M的荷移(LMCT,还原迁移)
荷移迁移的实质是氧化还原过程,
即中心离子是氧化剂,它本身被还原,
而配体是还原剂,它本身被氧化。
σ* d π σ M MO L π*
π
σ
[MX6]n-的简化分子轨道能级图
3、d-d 光谱带的理论分析
d-d 跃迁光谱的四个特征
谱带的数目 谱带的位置 谱带的强度 谱带的宽度
(一)光谱带强度的分析
I = ∫ε(v)dv
0 1.光谱选律 ΔE = hv (1)自旋选律 多重性选律 即多重度(2S+1)相同谱项间的跃 迁是允许的;而多重度不同的谱项间的跃 迁是禁止的。
∞
按选律,ΔS=0,电子跃迁前后自旋状 态必须相同;当ΔS≠0时,电子跃迁要改变 自旋状态,需要更高的能量因而这种自旋禁 止的跃迁摩尔吸光系数很小 。 用公式表示如下:
[CoIII(NH3)5X-]2+→[CoII(NH3)5X0 Nhomakorabea2+
VO43->CrO42->MnO4白 黄 紫红
2、金属对配位体荷移(MLCT,氧化跃迁))
t2g* eg*
Δo ν1
eg *
ν2
10 晶体场稳定化能及其应用,配离子的电子吸收光谱

PPT编号13-4-3-11
从 Ca2+ 到 Zn2+ 离子的水合 热却呈现出“双 驼峰”的变化规 律。如何解释这 一规律?
PPT编号13-4-3-12
解释: 为什么实验数据与理论分析不一致呢?
原来在分析问题时将这些离子与水形成的八 面体弱场配合物的 CFSE 忽略了!如果将弱场配 合物的 CFSE 曲线叠加到水合热曲线上,就完全 吻合了。
eg + 6Dq
Es 0 t2g – 4Dq
弱场
分裂前
eg
t2g
强场
在考虑了电子成对引起的能量升高,即考虑电子成对能 (P)的影响后,CFSE分别为:
CFSE(d6 强场)= 6×(– 4Dq) + 2P = – 24 Dq + 2P CFSE(d6 弱场)= 4×(– 4Dq) + 2×(+ 6Dq) = – 4Dq
四、晶体场稳定化能
PPT编号13-4-3-1
晶体场稳定化能的定义
在晶体场中,中心离子的 d 电子进入分裂后的轨道中, 与在球形场的未分裂的轨道(设其能量 Es = 0)相比,系统 的总能量有所下降,该下降的能量叫做晶体场稳定化能 (CFSE, crystal field stability energy)。
局限:晶体场理论只考虑中心离子与配体之间的静电作用, 着重考虑了配位体对中心离子的d轨道的影响。而没有考虑 它们之间在一定程度上的共价结合,因此不能解释像 [Ni(CO)4] 这一类羰基配合物及一些 π 配合物的形成,也不 能从本质上说明“光谱化学序列”。
在此基础上,诞生了新的配合物化学键理论d1
d2
d3
d5
d6
d7
d8
d9 d10
第五章 配合物的电子光谱

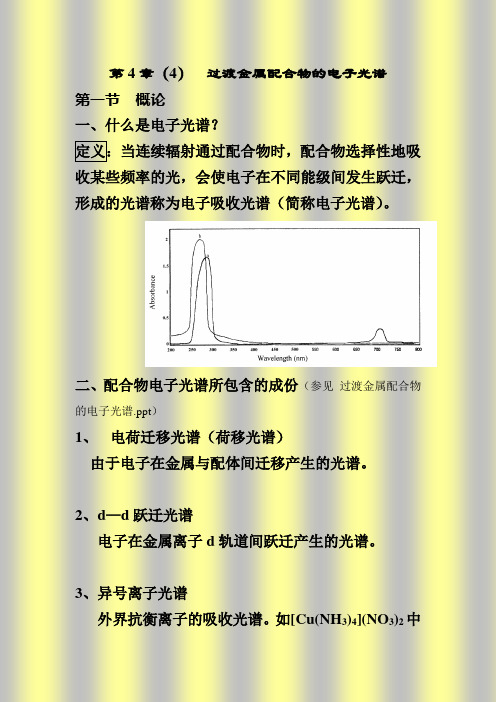
第五章过渡金属配合物的电子光谱第一节概论一、什么是电子光谱?定义:当连续辐射通过配合物时,配合物选择性地吸收某些频率的光,会使电子在不同能级间发生跃迁,形成的光谱称为电子吸收光谱(简称电子光谱)。
2004006008A b s o r b a n c eWavelength(nm)配体配合物二、配合物电子光谱所包含的成份 1、 电荷迁移光谱(荷移光谱) 由于电子在金属与配体间迁移产生的光谱。
2、d —d 跃迁光谱电子在金属离子d 轨道间跃迁产生的光谱。
3、异号离子光谱外界抗衡离子的吸收光谱。
如[Cu(NH3)4](NO3)2中NO3-的吸收。
4、配体光谱配体本身的吸收光谱。
如[Ti(H2O)6]3+中H2O的吸收。
第二节电荷迁移光谱、异号离子光谱及配体光谱一、电荷迁移光谱不同对称类型的轨道间跃迁是允许的(u → g、u → g)1、无π分子轨道的配体(NH3、CH3−)L →M 跃迁:跃迁类型 Lσ(t 1u )→ M(t 2g ) = ν1 Lσ(t 1u )→ M(eg *) = ν2弱场(Δ<P )t 2g e g *e g V 1V 2NH 3、CH 3-作配体t 1ut 1u *a 1g *a 1gd0d1 d2ν1<ν2ν1<ν2 ν1<ν2d3d4 d5ν2<ν1 ν2<ν1ν1<ν2d6d7 d8ν1<ν2 ν1<ν2 无ν1d9d10无ν1 无ν1和ν2强场(Δ>P)d0d1 d2ν1<ν2ν1<ν2 ν1<ν2d3d4 d5ν1<ν2ν1<ν2 ν1<ν2d6d7 d8无ν1 无ν1 无ν1d9d10无ν1 无ν1和ν22、具有低能充满π轨道的配体(Cl −、F −)L →M 跃迁:跃迁类型Lπ(t 1u 和t 2u ) → M(t 2g *) = ν1Lπ(t 1u 和t 2u ) → M(e g *) = ν2e g *t 2gt 2g *e g *t 2g e gt 2g +t 1g +t 2u +t 1ut 2u +t 1ut 1gv 1v 2v 3v 4a 1g *t 1u *L σ(t1u)→ M(t2g*)= ν3L σ(t1u)→ M(e g*) = ν4ν3和ν4跃迁能量太高,观测不到。
等离子吸收光谱

等离子吸收光谱
等离子吸收光谱(Plasma absorption spectroscopy)是一种常用的光谱分析方法,它利用等离子体作为光源来产生特定波长的光,然后通过测量这些光的吸收情况来分析物质。
等离子体是由高温或高能量激励下,从原子和分子中释放出电子,并且产生了原子和分子间的运动而形成的电离状态,是一种带电体系。
通俗地说,等离子体就是带电气体。
等离子体光谱是等离子体发射光谱和吸收光谱的总称。
等离子体光谱的产生涉及到等离子体的能级结构、能量转移和辐射过程。
物理学家通过对等离子体光谱的光谱线、光谱强度和光谱形状等特性进行分析,可以了解物质的性质和结构。
等离子吸收光谱技术具有许多独特的优点,例如高灵敏度、高精度和高分辨率,因此在许多领域都有广泛的应用。
例如,它可以用于环境监测、化学分析、医学诊断、材料科学和天文学等领域。
在环境监测领域,等离子吸收光谱可以用于分析空气、水和土壤中的有害物质,例如重金属、有机物和有毒气体等。
在化学分析领域,它可以用于测定化合物的结构和组成,例如有机物、无机物和合金等。
在医学诊断领域,它可以用于检测生物样品中的药物和代谢物,例如毒品、酒精和血糖等。
在材料科学领域,它可以用于研究材料的结构和性质,例如半导体、陶瓷和聚合物等。
在天文学领域,它可以用于分析星际气体和星云中的元素。
等离子吸收光谱是一种非常有用的技术,可以帮助科学家和工程师们更好地了解物质的性质和结构,为科学研究和技术发展做出贡献。
3.1-配合物的UV-vis光谱

过度金属配合物的紫外-可见吸收光谱主要是由于配体与金属离子间 的结合而引起的电子跃迁,因此也称为电子光谱(electronic spectrum)
在紫外-可见吸收光谱中,根据吸收带来源的 不同划分:
紫外-可见吸收光谱
配位场吸收带Leabharlann 电荷跃迁吸收带配体内的电子跃迁吸收带
定性定量分析
金属向配体的跃迁 MLCT
n
等跃迁,研究配体间的作用方式和关系。波长范围大多在近紫外-可见光区。
生色团:分子结构的某些基团吸收某种波长的光,而不吸收另 外波长的光,从而使人觉得好像这一物质“发出颜色”似的, 因此把这些基团称为“发色基团/发色团”。例如>C=C<, >C=O,-N=N-,-C C-,-C N-等。 助色团:分子中本身不吸收辐射而能使分子中生色基团的吸收峰 向长波长移动并增强其强度的基团,如羟基、胺基和卤素等。助 色团可以分为两类: 酸性助色团: -COOH, -OH, -SO3H 碱性助色团:-NHR,-NR2,-NH2
包括d-d跃迁和f-f跃迁,对于过渡金属配合物而言也称为d-d跃 迁吸收带,其位置变化和裂分可跟踪考察配合物的反应和形式, 波长范围大多在可见光区。下面为d-d跃迁。
中心离子所带电量越大,周期数越高,分裂能越大。不同的配位场, 平面四方>八面体>四面体。
配体向金属的跃迁 LMCT 电荷跃迁吸收带
吸收的光与透过的光互为补色。
定性分析
定量分析
第4章(4)过渡金属配合物的电子光谱
二、配合物电子光谱所包含的成份(参见过渡金属配合物.ppt)电荷迁移光谱(荷移光谱)由于电子在金属与配体间迁移产生的光谱。
—轨道角量子数注*矢量用黑体字母表示。
*角动量:就是质量乘以角速度(单位角度/秒)。
自旋角动量:角动量是由物体自旋产生的,而不是外力给它的。
轨道角动量:角动量是由轨道运动产生的2、电子间相互作用在多电子体系中,l i与l j主要是通过电性相互作用;而s i与l i或s j之间则主要通过磁性作用。
s i s jl i l j对轻元素(原子序数<30),电子间偶合强于电子内偶合,即:l i——l js i——s j的作用要大于s i——l i的作用。
此时电子间相互作用,可用L—S偶合方案处理:(参见L—S偶合方案.pdf)Σl L (总轨道角动量)Σs S (总自旋角动量)即可用L、S描述多电子体系的状态。
│S│=[S(S+1)]1/2(h/2π) │L│=[L(L+1)] 1/2(h/2π)S——总自旋角量子数L——总轨道角量子数如何求S、L见“物质结构”。
3、d n组态金属离子的谱项多电子体系的能量状态可用谱项符号表示:2S+1L L 0 1 2 3 4 5符号S P D F G H(2S+1)为谱项的自旋多重度。
如S=1/2,L=2时,为2D谱项。
组态谱项d1 d92Dd2 d83F,3P,1G,1D,1Sd3 d74F,4P,2H,2G,2F,2x2D,2Pd4d65D,3H,3G,2x3F,3D,2x3P,1I,2x1G,1F,2x1D,2x1Sd56S,4G,4F,4D,4P,2I,2H,2x2G,2x2G,2x2F,3x2D,2P,2S* d n体系,不考虑电子间作用时,只有一种能量状态。
考虑电子间作用后,产生多个能量状态(谱项)。
d1体系除外,因其只含一个电子。
4、基态谱项1)定义:能量最低的谱项称为基态谱项(基谱项)。
1)如何确定基谱项?A、同一电子组态的各谱项中,自旋多重度最大者能量最低。
实验一八五配合物的光谱化学序列的测定

实验一八五配合物的光谱化学序列的测定一、实验目的1、通过测定若干个铬配合物的吸收光谱,计算晶体场分裂能;了解不同配体对配合物中心金属离子d轨道能级分裂的影响。
2、通过实验进一步掌握光谱化学序列及其应用。
二、实验原理在过渡金属配合物中,由于配体场的影响使中心离子原来简并的d轨道发生分裂。
配体的对称性不同,d轨道的分裂形式和分裂轨道间的能级差也不同。
如图185-1所示:dx2-y2dx2-y2Δdyz dxz dxy dxydΔΔdx2-y2dz2 dz2dxy dxz dyzdxz dyzTd(四面体场) O h(八面体场) D4h(四边形场)图185-1 d轨道在不同配体场中的分裂电子在分裂后的d轨道间的跃迁称为d-d跃迁。
这种跃迁的能量相当于可见光区的能量范围,这就是过渡金属配合物呈现颜色的原因。
图185-1中所示△为两个不同能级d轨道之间的能量差,称为分裂能。
△值的大小受中心离子的电荷、周期数、d电子数和配体性质等因素的影响。
对于同一中心离子和相同构型的配合物,△值随配体场强度的增强而增大。
按照△值相对大小排列的配位体顺序称为“光谱化学序列”,它反映了配体所产生的配位场强度的相对大小。
分裂能△可以通过测定配合物的吸收光谱来求得。
过渡金属配合物的吸收光谱通常包括d-d跃迁,电荷迁移和配体内电子迁移等三种类型的吸收带,其中最重要的是d-d跃迁吸收带。
研究配合物的吸收光谱必须同时考虑电子间的排斥作用和配位场的作用。
根据研究离子的电子光谱的弱场方法,首先考虑d电子间相互作用引起的能级改变,获得d n组态的光谱项。
然后考虑各光谱项在配位场中的分裂情况。
以各光谱项在配位场中分裂后的能级能量对分裂能△作图,就可得到d n组态的奥格尔(Orgel)能级图。
各电子组态的奥格尔能级图可通过量子力学计算得到。
图185-2是Cr 3+(d 3)离子在八面体体场(O h )中的简化奥格尔能级图。
)(14P T g P 4)(14F T g E F 4g T 24g A 24Δ(cm )图185-2 Cr 3+ 离子在八面体场中的简化奥格尔能级图图中纵坐标表示光谱项能量, 是Cr F 43+的基态光谱项,P 4是与基态光谱项具有相同多重态的激发态光谱项。
配合物的离子吸收光谱
轨道角动量的偶合(l-l偶合)
单个电子的轨道运动的角量子数l按向量加和起来,就 可以得到一组电子的总轨道角量子数L。由于向量l相 对的可能取向有几个,故可得几个向量和,即几个L值。 即 L = ∑ li
L在Z轴上的分量为(令磁场方向与Z轴重合) │ML │≤ L,即ML = L,L-1,L-2,……-L
+ 3/2
4
P1/2
2
mJ= + 1/2
2
J=5/2 d1 L=2 S=1/2 J=3/2
D5/2 mJ= + 1/2 + 3/2 + 5/2 D3/2 mJ= + 1/2 + 3/2
2
由电子组态确定自由离子光谱项
非等价电子
组态 2P13P1 l1 = 1, l2 = 1 L = 2, 1, 0 S = 1, 0 6个光谱项 : 1S、1D、3P、 3S、3D、1P
D2
D1
D2
3
P2
3 3
P1
P0 P1 S1 S0
1
3
1
由电子组态确定自由离子光谱项
等价电子 —— 考虑Pauli原理 某一电子组态的微观状态数为
其中m是未充满轨道最多可容纳的电子数,n是实际填入
的电子数
以np2组态为例:
p2组态有15种排布方式
mL +1 0 -1 ML=ΣmL Ms=Σms
跃迁能量较小, 一般出现在可见区, 所以许多过渡
金属配合物都有颜色
自由离子的光谱项
d-d跃迁 能级分裂 一个d电子的金属离子的配合物, 如Ti3+的水合离子 多个d电子的金属离子的配合物 d电子间相互作用 弱场处理
无电子相 互作用
Cr(Ⅲ)配合物的合成与分光序的测定(1)

配合物的分光化学序的测定摘 要 本文中介绍了几种Cr 3+的配合物的合成方法并测定了它们的电子光谱,根据吸收光谱曲线中吸收峰的位置, 研究了不同的配体的分裂能和光化学序,分析了各配体能力强弱的原因。
关键词 Cr 3+;配离子;吸收光谱;分裂能;光谱化学序晶体场理论的认为:配位体与中心离子间的化学键是电价键,也就是他们之间的相互作用是纯粹的静电作用,依靠带正电荷的金属离子吸引带负电荷的配位体而组成配合物。
中心离子d 轨道具有5种伸展方向不同,而能量相同的简并轨道,在配位体电场影响下,5个d 轨道会分裂成两组以上能量不同的轨道。
分裂的情况主要决定于配位体的空间分布。
轨道分裂后,最高d 轨道的能量与最低d 轨道的能量差,称为分裂能(∆)。
由于d 电子没有充满,电子可吸收光子后在分裂后的d 轨道之间跃迁(d-d 跃迁)。
所吸收的能量恰好等于d 轨道分裂能。
对于同一中心离子和相同构型的配合物,∆值的大小取决于配体的强弱。
按分裂能的相对大小来排列的配体顺序称为,分光化学序。
配合物的分光化学序可以通过测定它的电子光谱,计算∆值得到。
()71110cm λ-∆=⨯1、实验部分1.1实验仪器及试剂仪器:烧杯、容量瓶、玻璃棒、酒精灯、研钵、蒸发皿、PH 试纸、分光光度计。
试剂:草酸钾、重铬酸钾、丙酮、EDTA 、硫酸溶液、氯化铬、硫酸铬钾、硫氰酸钾、乙醇。
1.2实验步骤1.2.1配合物的制备 [1] K 3[Cr(C 2O 4)3]·3H 2O称3 g 草酸钾和7 g 草酸于蒸发皿中,加入50 mL 水溶解。
完全溶解后,再慢慢加入2.5 g 研细(玻璃研钵)的重铬酸钾,不断搅拌(一定要缓慢加入注意实验现象写出化学方程式)。
蒸发浓缩溶液使晶体析出,冷却、抽滤并用丙酮(少量)洗涤沉淀。
[2][Cr-EDTA]-称0.5 g EDTA 于烧杯中,加入50 mL 水,加热使其溶解,用1M 的硫酸溶液(先用试纸测溶液的pH ,加1-2滴)调节溶液pH 在3-5之间,然后加入0.5 g 氯化铬,加热得到[Cr-EDTA]-配合物溶液。
铜离子的紫外可见吸收光谱的原理

铜离子的紫外可见吸收光谱的原理
铜离子的紫外可见吸收光谱学习理解已经成为分析化学研究中不可或缺的领域。
铜离子的紫外可见吸收光谱是指在更长的光谱范围内,金属—有机配合物的受紫外线照射后,产生的光谱现象,这一现象由铜离子决定,是一种非常有价值的分析技术。
首先要了解的是铜离子的紫外可见吸收光谱的基本原理。
所谓吸收光谱,是指
特定成分受紫外线照射后,能吸收特定波长的光,反应在紫外和可见光谱范围内,显示出特定的光谱线和吸收率曲线。
铜离子的紫外可见吸收光谱的原理,是指铜离子以各种配位形式接受到紫外线
的能量,在局部发生四价铜络合能级担体在激发状态间过渡,其过渡涉及到它的紫外紫外可见光谱以及可见光谱,以及可见光谱吸收曲线的形成,由于其能量过渡的不同,会吸收在紫外紫外光谱和可见光谱展示不同的吸收曲线,从而鉴定特定的有机物质。
此外,铜离子的紫外可见吸收光谱也能够揭示其与金属配合合物之间的关系。
金属与各种有机配体之间,会形成键类似取向络合,有些金属会影响物质的吸收能力,从而改变物质在紫外紫外可见光谱以及可见光谱的吸收曲线。
铜离子的紫外可见吸收光谱学习可以说是分析化学新的一大突破,它可以帮助
我们迅速辨认有机物质的性质,推测合物的结构,并可帮助更精确的探明金属有机配合物的详细情况。
它的发展及应用,在化学分析上大有裨益,对于物质的结构分析、物质性质的分析等有着重要的价值。
配位离子的光谱性质

配位离子的光谱性质在化学中,配位离子是指由一个或多个配体与中心金属离子形成稳定的配合物的离子。
配位离子的光谱性质是指通过光谱分析方法研究配位离子的特性和行为。
本文将从分子结构、电子转移与吸收以及荧光发射等方面探讨配位离子的光谱性质。
一、分子结构对光谱性质的影响配位离子的分子结构对其光谱性质具有重要影响。
通常情况下,配合物的分子结构可以通过X射线晶体学、核磁共振等方法确定。
配位离子的分子结构会影响到其电子态的能级分布情况、自旋-轨道耦合以及电子跃迁等过程。
例如,八配位的配合物往往具有比较复杂的分子结构,其光谱性质也较为复杂。
这是因为八配位的配合物中,配体通常以正八面体或扭曲八面体的方式周围环绕着中心金属离子。
这种结构对光的吸收和发射过程产生了多条可能的跃迁路径,导致光谱呈现出多个峰值。
相反,六配位的配合物通常具有更简洁的结构,其光谱性质也较为简单。
这是因为六配位的配合物中,配体通常以八面体或六面体的方式周围环绕着中心金属离子。
这种结构对光的吸收和发射过程产生了较为明确的跃迁路径,光谱呈现出单一的峰值。
由此可见,配位离子的分子结构对其光谱性质有着直接的影响,而不同的结构特点也使得光谱分析成为了研究配位离子的重要手段之一。
二、电子转移与吸收对光谱性质的影响在配位离子的光谱性质中,电子转移和吸收过程发挥着关键作用。
当配位离子吸收和发射光线时,电子会在不同的能级之间跃迁,这一过程产生了光谱图。
具体来说,当光线照射到配位离子上时,电子会从基态跃迁至激发态,此时会发生吸收过程。
吸收光的波长与电子跃迁的能级差有关,因此可以通过测量吸收光谱确定配合物的电子能级结构。
另一方面,在激发态的配位离子中,电子受到分子振动和旋转等过程的影响,可能会产生不同的电子跃迁路径,从而产生不同波长的发射光。
通过测量发射光谱,可以了解激发态电子的能级分布情况,进而研究配合物的特性和行为。
三、荧光发射对光谱性质的影响荧光发射是配位离子光谱性质中的重要现象之一。
配位化合物 紫外可见吸收

配位化合物紫外可见吸收配位化合物的紫外可见吸收配位化合物是一种含有中心金属离子和配体的化合物。
当光照射到配位化合物时,如果光的能量足够高,化合物中的电子就会被激发到更高的能级。
这种电子跃迁会导致紫外可见(UV-Vis)吸收。
UV-Vis吸收光谱可以提供有关配位化合物中金属离子和配体的有用信息,包括:d-d跃迁:这是中心金属离子的d轨道之间的电子跃迁。
d-d跃迁的能量取决于金属离子的电子构型、配体的场强和配位化合物的几何形状。
电荷转移跃迁:发生在金属离子和配体之间或配体之间的电子转移。
电荷转移跃迁的能量受金属-配体键强度的影响。
π-π跃迁:这是配体中π键的电子跃迁到π反键轨道。
π-π跃迁的能量取决于配体的共轭度。
n-π跃迁:这是配体中非键电子跃迁到π反键轨道的跃迁。
n-π跃迁的能量受配体的杂化程度的影响。
UV-Vis吸收带的特征:吸收最大值(λmax):吸收带的最大强度出现的光波长。
吸收强度(ε):吸收带的面积,表示吸收光的量。
半高宽(Δν1/2):在吸收最大值处吸收强度降至一半所需的光波长范围。
影响UV-Vis吸收的因素:金属离子:不同金属离子的电子构型不同,导致d-d跃迁的能量不同。
配体:配体的场强和几何形状会影响d-d跃迁的能量。
溶剂:溶剂可以与配位化合物相互作用,影响配体的场强和配位化合物的构型。
温度:温度会影响配位化合物的构型和稳定性,从而影响UV-Vis吸收光谱。
UV-Vis吸收光谱的应用:金属离子鉴定:通过分析d-d跃迁的能量和模式,可以确定配位化合物中的金属离子。
配体场分析:UV-Vis吸收光谱可以用于研究配体的场强和配位化合物的几何形状。
反应监测:UV-Vis吸收光谱可以用来监测配位化合物的反应,例如配体交换或氧化还原反应。
颜色预测:UV-Vis吸收光谱可以用来预测配位化合物的颜色,因为吸收光的互补色会被化合物反射。
结论:UV-Vis吸收光谱是配位化合物的重要表征技术,可提供有关中心金属离子、配体和配位化合物的结构和电子性质的信息。
配合物显色原理

配合物显色原理
配合物显色原理是指在某些过渡金属离子与配体形成配位化合物时,由于电子跃迁和分子结构改变等因素的影响,使配合物在可见光范围内吸收光线,产生显色现象。
通常情况下,过渡金属离子的d轨道中的电子有较大的能级差,当配体与金属离子结合形成配合物时,这些能级差会被扩展或改变,导致电子的吸收能力发生变化。
当配合物处于基态时,部分或全部d轨道上的电子被激发到高能级的空d轨道上,吸收了特定波长的光线。
其吸收光谱往往位于可见光区域,使配合物呈现出不同的颜色。
这种显色现象主要取决于配合物中的金属离子、配体的性质以及配合物的空间构型等因素。
通过调整配体的种类、浓度和金属离子的性质,可以实现对配合物显色的控制和调控。
配合物显色原理在化学分析、催化反应、材料科学等领域有着广泛的应用。
通过分析和研究配合物的显色特点,可以实现对金属离子的检测和分离,也可以用于制备颜料、染料等各种应用材料。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
有自旋轨 道偶合
光谱支项
自由离子的能态
组态:无磁场作用下的电子状态,用n, l, m表示
微观状态:原子在磁场作用下的运动状态,用n, l,
m, s, ms描述——价电子轨道上电子的排布方式 原子的能态——光谱项:各个电子所处的轨道和 自旋状态的总和 + 电子间的相互作用,轨道运动 和自旋运动相互作用,由原子的量子数L, S, J描述
收带。称为紫外-可见(UV-vis)光谱
ቤተ መጻሕፍቲ ባይዱ
过渡金属配合物电子光谱的分类
根据电子跃迁的机理分为三种
d 轨道在配位场中的能级分裂即配位场光谱(电 子光谱的基础和来源)
配位体至金属离子或金属离子至配位体之间的电
荷迁移光谱(charge transfer, CT光谱) 配体内部的电子转移光谱
过渡金属配合物的电子光谱
两个显著的特点
30,000 40,000
10,000
20,000
a)
为带状光谱。这是因为电子跃迁时伴 随有不同振动精细结构能级间的跃迁
CT
b)
CT 之故
在可见光区有吸收, 但强度不大。但在
c) 紫外区 , 常有强度很大的配位体内部吸
迁移到一个主要具有金属性
质的轨道中去,相当于金属 被还原,配体被氧化
L→M 跃迁
金属离子越容易被还原(或金属的氧化性 越强)和配体越容易被氧化(或配体的还原 性越强), 则这种跃迁的能量越低, 跃迁越 容易, 产生的荷移光谱的波长越长, 观察 到的颜色越深
L→M 跃迁
F-、Cl-、Br-、I-所形成的
分子光谱的产生
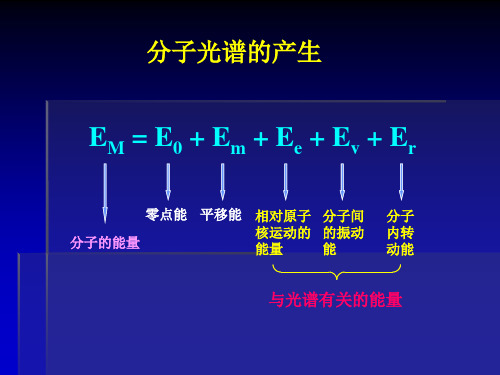
EM = E0 + Em + Ee + Ev + Er
零点能 平移能 相对原子 分子间 核运动的 的振动 分子的能量 能量 能 分子 内转 动能
与光谱有关的能量
电子-振动-转动光谱
分子光谱
分 子 光 谱
电子光谱(紫外光谱):远紫外、紫外及可见区,Ec、 Ev和Er都改变 振动光谱(近红外光谱):近红外区,Ev和Er改变 转动光谱 (远红外光谱):在远红外区,Er改变 (微波谱):微波区,Er改变
跃迁能量较小, 一般出现在可见区, 所以许多过渡
金属配合物都有颜色
自由离子的光谱项
d-d跃迁 能级分裂 一个d电子的金属离子的配合物, 如Ti3+的水合离子 多个d电子的金属离子的配合物 d电子间相互作用 弱场处理
无电子相 互作用
组态
配位场 强场处理
有配体 存在
配位场谱项
有电子相 互作用
光谱项
配体内部的电子光谱
配位体内部的光谱包括 以下三种类型
n→* 跃迁。水、醇、 胺、卤化物等 n→* 跃迁。含羰基的 醛和酮类分子
*
E
K
E, B
R
*
n
→* 跃迁。含双键、 叁键的有机分子 出现在紫外区, 吸收强度大
电荷迁移光谱
电荷迁移光谱也常简称为荷移光谱, 特点是
电子自旋偶合(s-s偶合)
一组N个电子的总自旋量子数S是由每一个电子的自旋量子
数s的向量和得到的,即
S = ∑ si (si向量值为±1/2,S取值为n/2, n/2-1, n/2-2, …, 0) S在Z轴上的分量为(令磁场方向与Z轴重合) │MS│≤ S,即MS = S,S-1,S-2,…… -S (2S+1)个
轨道角动量的偶合(l-l偶合)
单个电子的轨道运动的角量子数l按向量加和起来,就 可以得到一组电子的总轨道角量子数L。由于向量l相 对的可能取向有几个,故可得几个向量和,即几个L值。 即 L = ∑ li
L在Z轴上的分量为(令磁场方向与Z轴重合) │ML │≤ L,即ML = L,L-1,L-2,……-L
配合物中, 配体还原性依次 增强,碘化物颜色最深
金属含氧酸的颜色 VO43– CrO42– 显示 吸收 无色 紫外 黄色 紫色
MnO4– 紫色 黄色
NH3
金属还原谱带, 电荷
d的能量
20000
30000
40000 cm-1
[Co(NH3)5X]2+离子的光谱
M→L 跃迁
这类光谱发生在金属很
自由离子的能态
mJ=1 np2
3
P
3
P1
mJ=0 mJ=-1
微观状态
电子组态
谱项
光谱支项
微观能态
轨道和自旋 分开考虑
轨道和自旋 一起考虑 L-S耦合
磁场作用 下的轨道 分裂
多电子原子的能态——光谱项的推求
L-S偶合:适用于电子之间的轨道角动量和自旋角动量相 互作用强于每个电子自身轨道角动量与自旋角动量相互作 用的情况。这种偶合方式一般适用于原子序数小于30的轻 元素
容易被氧化, 配体又容易 被还原(低氧化态金属离 子和不饱和配体)的配合 物中——金属离子具有 充满的或接近充满t2g轨 道, 而配体具有最低空轨
道, 例如: py, phen,
bipy, CN-, CO, NO等配 体
M→M’ 跃迁
这种光谱出现在一种金属离子以不同价态同时存 在于一个体系的时候, 这时在不同价态之间常发 生电荷的迁移。
Mm+→Mn+ (n>m)
如 , 普鲁氏蓝 KFe(Ⅲ)[Fe(Ⅱ)(CN)6] 的 Fe(Ⅱ)→Fe(Ⅲ) 的电 荷迁移, 钼蓝中的Mo(Ⅳ)→Mo(Ⅴ)的迁移。
配位场光谱
配位场光谱是指配合物中心离子的电子光谱。由d电
子在d电子组态衍生出来的能级间跃迁产生的, 所以
又称为d-d跃迁光谱或电子光谱 这种光谱有以下三个特点 一般包含一个或多个吸收 强度比较弱, 这是因为d-d跃迁是光谱选律所禁阻
S = ∑si L = ∑li J = L + S
j-j 偶合:适用于每个电子自身的轨道角动量和自旋角动量 相互作用强于电子之间轨道角动量和自旋角动量相互作用 的情况。这种偶合方式一般用于原子序数大于30的较重的 元素。这种情况下,应首先将每个电子的l和s偶合起来求 出j,然后把每个电子的j偶合起来得J j = l + s,l + s -1,……│ l - s│,J=∑j
吸收强度大 跃迁能量高, 常出现在紫外区
电荷迁移光谱
配体到金属的荷移(
还原迁移)(L→M)
金属对配体的荷移(氧 化迁移) (M→L) 金属到金属的荷移 (M→M’)
L→M 跃迁
[ML6]n-为例,
1=→*(t2g) 2=→*(eg) 3=→*(t2g) 4=→*(eg) 每一个跃迁都表明电荷由一 个主要具有配体性质的轨道