神经节苷脂沉积病
婴儿型GM1神经节苷脂沉积病1例

患儿,男,9个月16 d ,因“发育落后4个月,咳嗽1周伴气喘4 d ”入院。
患儿系第3胎第3产,足月顺产,出生时无异常。
5月龄时发现患儿竖头不稳,不会侧翻,7月龄于外院行头颅MRI 未见异常,目前仍竖头不稳,不会翻身、抓物,不能独坐,仅会咿呀学语。
既往3月龄时行双侧睾丸鞘膜积液术;3月龄后已患3次喘息性支气管炎;房间隔缺损1.3 mm 。
父母体健,非近亲结婚,否认家族中类似情况及其他遗传病史。
未能按时预防接种。
患儿1周前出现咳嗽,近4 d 出现气喘,遂就诊于空军军医大学第一附属医院儿科。
入院时体格检查:体温37 ℃,脉搏129次/min ,呼吸44次/min ,体重9.9 kg ,营养好,下腹部左右两侧可见长约1.5 cm 的手术瘢痕,双肺闻及痰鸣音及喘鸣音,肝脏肋下2 cm ,双侧阴囊明显增大,右侧阴囊内可见一液性包块,透光试验阳性。
四肢活动少,肌力、肌张力减低,深反射可引出,双侧巴宾斯基征阳性。
入院后辅助检查:头颅MRI 示双侧弥漫性脑萎缩,丘脑T1呈高信号、T2呈低信号;肺部CT 示双肺野通气不均,双肺下叶见斑片状高密度影,气管支气管通畅;肝肾功能、血糖、血氨、血乳酸、血尿代谢筛查、染色体核型分析均正常。
入院后为明确患儿肌张力低下原因,充分告知患儿家长并签署知情同意书后对患儿及其父母各抽取3 ml 全血,置乙二胺四乙酸抗凝管,送北京海思特医学检验实验室,采用安捷伦外显子芯片捕获+高通量测序法,进行医学外显子5 000 种疾病筛查。
采用 Mutation Taster (http://www.mutationtaster. org/)、SIFT婴儿型GM1神经节苷脂沉积病1例陶东英 牛焕红 南亚萍 乔红玉 成胜权(空军军医大学第一附属医院 儿科,陕西 西安 710032)·病例报告·DOI :10.3969/j.issn.2095-5340.2021.02.011基金项目:国家自然科学基金(81700314) 通信作者:成胜权(Email :***************.cn )(/www/ SIFT_chr_coords_submit.html )等在线软件,预测突变基因的致病性。
上海市普陀区2023届高三下学期二模试题 生物含答案
2023学年第二学期普陀区高三质量调研生物试卷(答案在最后)说明:1.本试卷满分100分,考试时间为60分钟。
2.请将答案写在答题纸上,答案写在试卷上一律不得分。
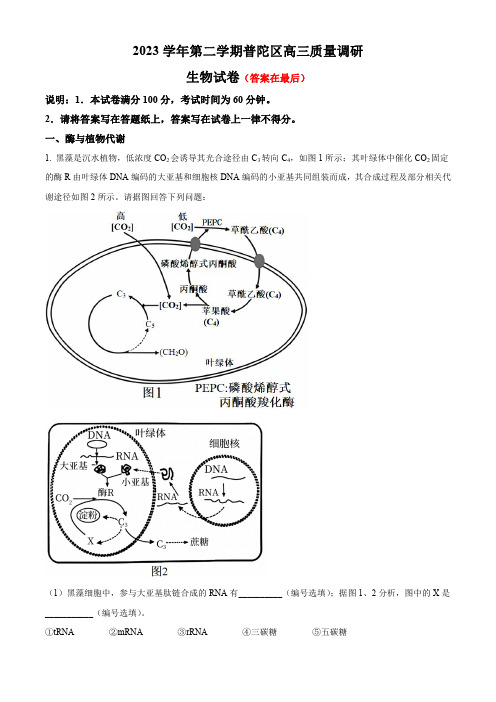
一、酶与植物代谢1.黑藻是沉水植物,低浓度CO2会诱导其光合途径由C3转向C4,如图1所示;其叶绿体中催化CO2固定的酶R由叶绿体DNA编码的大亚基和细胞核DNA编码的小亚基共同组装而成,其合成过程及部分相关代谢途径如图2所示。
请据图回答下列问题:(1)黑藻细胞中,参与大亚基肽链合成的RNA有__________(编号选填);据图1、2分析,图中的X是___________(编号选填)。
①tRNA②mRNA③rRNA④三碳糖⑤五碳糖(2)下列环境条件和物质代谢过程,短时间变化对图2中X浓度有影响的是____。
A.外界环境的CO2浓度B.叶绿体接受的光照强度C.三碳糖输出叶绿体的速度D.酶R的活性(3)结合题干信息和所学知识,下列描述中不正确的是____。
A.叶绿体中可以进行转录,也能进行翻译B.酶R的小亚基通过自由扩散进入叶绿体C.如果叶绿体中大量累积可溶性糖,可能导致叶绿体吸水涨破D.叶绿体DNA上出现碱基对的替换不一定会导致蛋白质功能改变(4)请简述黑藻细胞在低浓度CO2条件下的暗反应合成途径:___________。
甘蔗光合作用产物蔗糖是重要的糖浆原料。
某工厂寻求改进工艺,尝试使用蔗糖酶催化蔗糖水解的方法生产转化糖浆。
现有不同来源的三种只含有蔗糖酶的样品溶液A、B、C,为了选出酶活性最高的样品,现取含有等量酶的三种样品进行测试,试验条件及结果如下:实验甲:向样品中加入等量的蔗糖溶液,pH=7.4,在不同温度下重复若干次实验,结果记录如图3。
实验乙:向样品中加入等量的蔗糖溶液,温度为36℃,在不同pH下重复若干次实验,结果记录如图4。
(5)根据实验甲、乙的结果,工厂应选择蔗糖酶___________(选填“A”、“B”或“C”)催化蔗糖水解生产糖浆;催化生产中最佳反应条件是___________(编号选填)。
2020年赣州四中高三生物二模试题及答案解析

2020年赣州四中高三生物二模试题及答案解析一、选择题:本题共15小题,每小题2分,共30分。
每小题只有一个选项符合题目要求。
1. 糖酵解(EMP)属于细胞呼吸中的一个阶段,是从葡萄糖开始到生成丙酮酸的系列过程,下列有关叙述正确的是()A.EMP过程中需要氧气和多种酶的参与B.有氧呼吸和无氧呼吸中均有EMP过程C.EMP发生于细胞质基质和线粒体中D.EMP过程中会产生大量的NADH2. 甘氨酸是一种抑制性神经递质,能使处于静息状态的突触后膜上的Cl-通道开放而使Cl-顺浓度梯度进入细胞,从而抑制神经元兴奋。
下列有关叙述正确的是()A.神经元处于静息状态时,没有离子进出细胞膜B.Cl-进入突触后膜的方式为主动运输C.甘氨酸的释放与高尔基体有关,不消耗能量D.甘氨酸与突触后膜上受体结合后会使突触后膜两侧的电位差发生改变3. 某DNA分子双链均为14N,含有3000个碱基,腺嘌呤占35%。
若该DNA分子以15N标记的四种游离脱氧核苷酸为原料复制3次,将全部复制产物进行密度梯度离心,得到如图甲所示结果;若将全部复制产物加入解旋酶处理后再离心,则得到如图乙所示结果。
下列分析正确的是()A.X层全部是仅含14N的DNA分子B.Y层中含有的氢键数是X层的3倍C.W层中含15N标记的胞嘧啶有6300个D.Z层与W层的核苷酸数之比是1:44. 下图是探究某绿色植物光合速率的实验装置图,装置中的NaHCO3溶液可维持瓶内的CO2浓度相对稳定,将该装置放在20℃、一定光照条件下。
实验开始时,针筒的读数是0.2 mL。
毛细管内的有色液滴在A处。
30 min后,针筒的读数需要调至0.6 mL,才能使有色液滴维持在A处。
下列有关叙述错误的是()A.该实验过程中光合速率大于呼吸速率B.若测定植物的实际光合速率,还需要在暗处测定呼吸速率C.若将小烧杯中的NaHCO3溶液换成清水,则一段时间后光合作用会停止D.若用该装置探究该植物生长的最适温度,NaHCO3溶液浓度和光照强度等都是无关变量5. 下列有关肺炎双球菌体外转化实验和噬菌体侵染细菌实验的叙述,正确的是()A.T2噬菌体是寄生在所有细菌体内的DNA病毒B.实验都利用了放射性同位素标记法C.都能证明DNA是遗传物质,蛋白质不是遗传物质D.实验设计思路都是设法将蛋白质和DNA分开病毒6. 图是由3个圆所构成的类别关系图,其中℃为大圆,℃和℃分别为大圆之内的小圆。
神经节苷脂沉积病(黑蒙性白痴,假性Hurler病,全身性神经节苷脂病,神经节苷脂沉积症)

神经节苷脂沉积病神经节苷脂沉积病,又称黑蒙性白痴、假性Hurler病、全身性神经节苷脂病,是一种罕见的遗传性疾病。
本疾病主要由于神经节苷脂代谢的紊乱,导致该类脂质在细胞内积聚,最终影响身体各系统的功能。
本文将对神经节苷脂沉积病的病因、症状、诊断、治疗等方面进行探讨。
病因神经节苷脂沉积病主要是由遗传因素引起的。
患者通常是由一对同等和正常的亲生父母生下的子代,但他们也携带有一个异常基因。
通常,父母都是携带者,他们没有显示出明显的症状。
如果患者从父亲继承了一个异常基因,那么他们会得到本疾病。
但如果患者继承了两个异常基因,则取决于两个基因的性质,他们可能会表现出更严重的症状。
症状神经节苷脂沉积病的症状通常是多样化和复杂的。
在婴儿期,患者可能表现出发育迟缓、肝脏肉样增生、全身骨骼畸形等症状。
在稍后的发育阶段,患者可能会遇到智力落后、视力问题、听力问题、神经系统问题等。
诊断诊断神经节苷脂沉积病通常需要多种检查手段的综合运用。
例如,神经节苷脂沉积病的诊断可以通过检查尿液样本、皮肤活检、血液检测等多种方式进行。
此外,临床医生也可能会进行详细的基因检测以了解是否患者携带了与该疾病相关的特定基因突变。
治疗目前,神经节苷脂沉积病的治疗主要是针对症状的缓解和辅助支持。
对于一些患者,可能需要进行骨髓移植、酶替代疗法等治疗手段。
对于智力落后的患者,可能需要康复训练等辅助措施以提高生活质量。
预防由于神经节苷脂沉积病是一种遗传性疾病,预防方面主要是通过基因咨询和孕前诊断。
对于家庭中有神经节苷脂沉积病患者的情况,家族成员可以通过遗传咨询的方式了解自己的遗传风险,以便做出更加明智的生育决策。
结语总的来说,神经节苷脂沉积病是一种罕见但严重的遗传性疾病,对患者的生活造成了极大的影响。
希望通过对这类疾病的更深入了解,可以为未来的诊断和治疗提供更加有效的指导和支持。
在日常生活中,我们也应该关注遗传性疾病的预防和控制,为建设更加健康的社会贡献自己的一份力量。
患上神经节苷脂沉积病怎么办

患上神经节苷脂沉积病怎么办
一、概述
闺蜜的女儿在六个月的时候还没懂得爬行和对外界的反应极差,有时还会出现全身抽搐。
所以闺蜜带了她的女儿到医院去检查。
到了医院做了X线检查后,医生诊断闺蜜的女儿患上了神经节苷脂沉积病。
医生说,这是一种遗传性疾病,目前还没有办法根治。
二、步骤/方法:
1、所以医生只是开了一些药给闺蜜的女儿吃。
但是医生告诉闺蜜,即使是治疗,也只能是延长她女儿的生命而不能根治。
通常这种病是活不过10岁的。
而且还要在平常生活中要特别注意她的饮食和生活作息规律。
2、虽然闺蜜很伤心,但是为了能让自己的女儿活的更久一些,所以她很细心地在照料着她女儿的起居。
除了每天都按时按量喂医生开的药给她吃外,还特别注意给她补充一些身体内的维生素和营养物质。
3、闺蜜每天都会自制一杯果汁给女儿喝,并且有时还会煮些清汤等食物给女儿喝。
但是要注意用纱布隔渣,不然就容易是婴儿噎着。
4、同时,闺蜜每天都会帮她女儿按摩一下手脚。
并且会用些玩具锻炼着她的注意力和反应能力。
现在,闺蜜的女儿已经2岁了,虽然闺蜜知道她活不久,但也一直积极地带她到医院去治疗,希望能够尽量延长她的生命。
神经节苷脂沉积病尸检2例报告_黄润

神经节苷脂沉积病尸检2例报告黄 润(昆明医学院,昆明650031)关键词 神经节苷脂沉积病,病理学中图分类号 R 321.6 神经节苷脂沉积病(gangiosidosis GM 1),又名婴儿迟发性黑朦性痴呆,为单涎脑酰胺四已糖苷(gangliosidosis GM 1)含量过多症.1964年由Landing 等人首次报道,故又名Landing 氏病.由于半乳糖苷脂酶的ABC 异构酶缺乏所致.因为不能使神经苷脂水解,故表现为神经组织内有过多的GM 1沉积.此外,内脏细胞亦有GM 1沉积,故属神经内脏脂质沉积症,多见于2岁以下的小儿.例1:患儿男,10个月.5月时即有发育停滞,不会坐、垂头、多发性骨发育障碍,以后瘫痪、失明.早期曾出现快速自发性眼震,且一度体温不稳,常有低热,忽又突然升高,曾诊断为败血症.临床最后诊断为神经—脂质沉积症(Neuro -lipidosis ),死于小叶性肺炎.尸检肉眼观察:脑部肉眼病变不明显.肝居右肋下三横指,明显增大,重480g .脾肿大,包膜紧张,重47g .四肢长骨发育不良.双肺各叶小叶性肺炎.镜下观察:(1)大脑各叶神经细胞及胶质细胞中均有神经节苷脂沉积,两种细胞均明显肿大,可10倍于正常神经细胞大小,胞浆内有极细的泡沫状物(附图).两种肿大的细胞区别在于神经细胞中可见尼氏体,常被推挤至细胞边缘部,而胶质细胞中无尼氏体.这些细胞在常规染色中呈伊红色,PAS 染色呈紫红色颗粒.特染无异染性.本病含脂细胞与Krabbe 氏病含脂细胞的区别在于后者系存在于白质内的小胶质细胞和血管外膜细胞而非皮质的神经细胞中.与先天性黑朦性痴呆的区别在于后者罕见,且组织学上表现为白质的海绵样变性.(2)脑岛叶、壳核、苍白球及小脑分子层和浦肯野细胞内均有含量不等的上述物质沉积.(3)其它如视网膜及动眼神经核内神经细胞、骨髓白细胞及纤维细胞、肝细胞及肝脾内巨噬细胞中均有GM 1沉积.(4)双肺各叶小叶性肺炎.例2:患儿女,11月.6月时发育停滞,肝、脾肿大,逐渐瘫痪、失明、痴呆.临床诊断为神经—脂质沉积症,最后由于腹泻而亡.尸检肉眼观察:脑部各叶略显肿胀,视交叉处脑膜轻度增厚.肝居肋下三横指,重495g .脾大,重48g .肠道充血,有粘液稀便.镜下观察:(1)大脑各叶、小脑、脑干帽盖及延髓神经细胞、星形胶质细胞内有含量不等的GM 1沉积,细胞形态同例1所见.(2)中央灰质、黑质内神经细胞及脑干、延髓神经细胞内均有GM 1超载现象.(3)肝细胞及肝、脾巨噬细胞内亦有GM 1超载.(4)肠道急性炎症.附图 神经节苷脂沉积导致神经细胞明显肿大,H E ×200(1997-10-20收稿)第19卷 第2期 昆明医学院学报 Vo1.19 N o .2 1998年6月 ACADEM IC JO U RN AL OF K U NM I NG M EDICAL CO LL EGE Jun .1998。
CM1神经节苷脂贮积症科普宣传PPT

谢谢您的观赏 聆听
介绍神经节苷脂贮积症
分类和症状:神经节苷脂贮积症可分为 不同的亚型,每个亚型具有不同的症状 和影响范围。常见症状包括运动障碍、 智力退化、器官衰竭等。
神经节苷脂贮积症的诊断和 治疗
神经节苷脂贮积症的诊断和治疗
诊断方法:神经节苷脂贮积症的诊断通 常通过临床表现、遗传学检测和生物化 学检验来确定。关键是了解家族病史和 注意早期症状的出现。
CM1神经节苷脂贮积症 科普宣传PPT
目录 介绍神经节苷脂贮积症 神经节苷脂贮积症的诊断和治疗 预防和关注
介绍神经节苷脂贮积症
介绍神经节苷脂贮积症
什么是神经节苷脂贮积症:神经节苷脂 贮积症是一种遗传性疾病,会导致神经 系统和其他器官中的脂质积累。
病因和发病机制:神经节苷脂贮积症是 由特定酶的缺陷引起的,这些酶负责分 解脂质分子。当这些酶缺乏或功能异常 时,脂质在细胞内无法正常分解,逐渐 积累。
神经节苷脂贮积症的诊断和治疗
治疗方法:目前尚无治愈神经节苷脂贮 积症的方法,但可以通过辅助治疗来缓 解症状和改善生活质量。这包括药物治 疗、康复训练和支持性护理等。
预防和关注
预防和关注
预防措施:由于神经节苷脂贮积症是一 种遗传性疾病,无法完全预防。但对于 有家族病史的人来说,进行遗传咨询, 尽早进行基因检测和筛查是重要的预防 措施。
细胞生物学 相关的病

细胞生物学相关的病1、Normal human eye requires Pax6 that corresponds to eyeless.Abnormal function of Pax6 causes genetic disease --- aniridia.先天性虹膜缺失2、Tay-Sachs病,黑蒙性白痴。
神经节苷脂沉积平或脑黄斑变性3、动脉粥样硬化4、磷脂的运动:侧向扩散,旋转,摆动,翻转5、因为载体蛋白异常引发的疾病:胱氨酸尿症:肾小管上皮细胞的胱氨酸载体蛋白异常对肾小球滤出的原尿中四种氨基酸重吸收发生障碍。
肾性糖尿病:肾小管上皮细胞膜转运葡萄糖载体蛋白缺陷。
6、因为离子通道异常引发的疾病:囊性纤维化CF:CFTR(囊性纤维跨膜转到调节子)的缺失,缺乏508位苯丙氨酸的CFTR多肽不能在内质网中正常加工,不能到达上皮细胞质膜表面,患者质膜表面完全缺乏CFTR离子通道。
氯离子7、因为膜受体异常引发的疾病:家族性高胆固醇血症:LDL受体数目少,或者LDL不能和受体正常识别结合。
重症肌无力:产生抗乙酰胆碱抗体(N-Ach),阻碍乙酰胆碱与受体结合。
8、脂褐质,衰老的神经细胞、心肌细胞和肝细胞中多见;髓样小体,肿瘤细胞以及病毒感多见;含铁小体,肾肝9、溶酶体与疾病:(老师课件上说,要了解这些。
尤其是溶酶体的)黏脂病:在粘脂病病人细胞的溶酶体中发现有未被磷酸化的水解酶, 推测这些酶是通过非M6P依赖性的分选途径进入溶酶体的。
溶酶体膜失常与疾病:矽肺、石棉沉着病。
致病物质使溶酶体膜破裂,发生细胞自溶,致病物再被吸收,如此反复。
巨噬细胞释放‘致纤维化因子’,激活成纤维细胞,导致胶原纤维沉积,肺纤维化.先天性溶酶体病:1.糖原贮积病Ⅱ型:基因缺陷→α-葡萄糖苷酶↓→糖原不能降解为葡萄糖→肝脏和肌肉中糖原蓄积.进行性心衰.2.台-萨综合症:溶酶体内氨基己糖苷脂酶A↓→神经节苷脂GM2积累而影响细胞功能,造成精神痴呆3.脑苷脂沉积病: β-葡萄糖苷酶↓→溶酶体内葡萄糖脑苷脂沉积→巨噬细胞变成Gaucher细胞→肝,脾肿大.4.细胞包涵体病:N-乙酰葡萄糖胺磷酸转移酶单基因突变所致→成纤维细胞溶酶体中无水解酶→底物在溶酶体内蓄积形成‘包涵体’.溶酶体与类风湿性关节炎:类风湿因子可促使水解酶外逸溶酶体与休克:休克后缺血缺氧降低溶酶体膜的稳定性。
病理学理论指导:类脂质沉积症

类脂质沉积症(lipoidosis)是先天缺陷性脂质代谢障碍所致的组织内类脂质增多并沉积。
主要有糖脂、磷脂及胆固醇等沉积。
其发生机制,大都是由于作用于脂质分解代谢某些环节上的酶类的遗传性缺失,使其相应的底物(脂质)分解代谢不能进行而沉积在组织内。
1.糖脂沉积症糖脂是指不含磷酸的脑苷脂及神经苷脂等脂类。
它们的分解代谢障碍可分别引起脑苷脂沉积症(如高雪病)和神经节苷脂沉积症。
高雪(Gaucher)病,也称脑苷脂沉积症,是由于常染色体隐性遗传所致体内β-葡萄糖苷酶缺乏而引起的脑苷脂分解代谢障碍。
主要累及肝、脾、淋巴结及骨髓等单核吞噬细胞系统。
常发生在婴儿,为致命性疾病。
主要病变为肝、脾肿大,脾大尤为明显,可达正常脾重的20倍。
镜下,肝内聚集大量高度胀大的载脂巨噬细胞,有的胞浆呈泡沫状,有的胞浆出现红染条纹,后者排列成皱纹纸样外观,胞核小,圆形或椭圆形居于细胞中央,称为高雪细胞。
这些细胞主要分布于小叶中央静脉附近的肝窦内和汇管区。
偶见发生肝纤维化和肝硬化。
2.磷脂沉积症主要为不含甘油成分的神经磷脂的增多、蓄积,又称尼曼-皮克病。
尼曼-皮克(Niemann-Pick)病又称神经磷脂沉积症。
系由于常染色体隐性遗传所致的神经磷脂酶缺乏,使神经磷脂不能被水解而沉积于组织内所致。
另外还可伴有其它脂质贮积。
本病主要累及肝、脾、骨髓及淋巴结等器官,在儿童也侵犯神经系统。
肝病变肉眼观,肝肿大。
镜下,在肝窦内和汇管区有大量Kupffer细胞和巨噬细胞聚集,细胞体积肿大,胞浆呈泡沫状,核小居中,称为Pick细胞。
肝细胞内也可见有脂肪,主要为中性脂肪及胆固醇。
电镜下见Pick细胞内充满多数年轮样层状排列的球形包涵体。
本病常发生于幼儿,预后不佳。
GM_1和GM_2神经节苷脂沉积症

G M1和G M2神经节苷脂沉积症肖 波 肖 岚 谢光洁 彭隆祥 李 萍 谭爱玲 梁静慧目的 探讨G M1和G M2神经节苷脂沉积症的诊断。
方法 对2例婴儿型C M1神经节苷脂沉积症和6例G M2神经节苷脂沉积症进行了研究,每例患者均取右额叶脑组织做病理观察。
结果 婴儿型G M1神经节苷脂沉积症临床表现为出生后喂养困难、发育迟缓、面容丑陋、肝脾肿大,X线照片显示多发性骨发育不良。
G M2神经节苷脂沉积症婴儿型以智力差、易惊跳、眼底视网膜黄斑部樱桃红点为特点;少年型则表现为智力进行性下降、共济失调、视力减退、锥体束征。
光镜下神经元呈气球样肿胀,电镜观察见神经元胞浆内充满膜性胞浆体,夹杂少量斑马体,两种沉积症之间的沉积物形态完全一致。
结论 对临床上疑为神经节苷脂沉积症的病人,脑活检电镜观察有助于明确诊断。
关键词 神经节苷脂沉积症 超微结构G M1and G M2gangliosidosis X iao B o,X iao L an,X ie Guangj ie,et a l.Institu te of N eu rology,X i2 ang Y a H osp ita l,H unan M ed ica l U n iversity,Chang sha410008Objective T he clin ical and neu ropatho logic findings of2patien ts w ith G M1gangli o sido sis and6 patien ts w ith G M2gangli o sido sis w ere repo rted.M ethod A b i op sy of the righ t fron tal lobe w as per2 fo rm ed fo r each case.T he m ain clin icalm an ifestati on s con sisted of m en tal retardati on,facial gri m ac2 ing and viscerom egaly fo r G M1gangli o sido sis,and in tellect i m pairm en t,hyperacu sis,retinal m acu lar cherry2red spo ts fo r infan tile G M2gangli o sido sis and p rogressive visual deteri o rati on,m en tal diso r2 der,atax ia,hyperactive tendon reflexes,clonu s,ex ten so r p lan ter respon ses fo r juven ile G M2gan-gli o sido sis.Results W ith the u se of m icro scopy,gro ssly ballooned neu ron s w ere found and u ltra2 structu rally,the cytop las m s of the neu ron s w ere filled w ith m em b ranou s cytop las m ic bodies together w ith zeb ra bodies.Conclusion N o difference in the sto rage fo rm cou ld be iden tified betw een G M1 and G M2gangli o sido ses.T he resu lts suggested that electron m icro scopy w as of u se to help estab lish a diagno sis of gangli o sido sis.Key words Gangli o sido sis U ltrastructu re 神经节苷脂沉积症根据酶缺乏的不同可分为G M1神经节苷脂沉积症和G M2神经节苷脂沉积症。
克雅氏病人传人例子

克雅氏病人传人例子克雅氏病,又称神经节苷脂沉积症,是一种罕见的遗传性代谢性疾病。
以下是关于克雅氏病人的传人例子:1. 小明是一名10岁的男孩,他的父母在他很小的时候就发现他的运动能力较差。
经过多次检查,最终确定他患有克雅氏病。
虽然小明的父母都不患此病,但是他们了解到克雅氏病是一种遗传疾病,存在传人的风险。
2. 王女士是一位40岁的女性,她的哥哥在年轻时因为克雅氏病去世。
为了避免同样的悲剧再次发生,王女士决定进行基因检测,结果显示她携带了克雅氏病相关基因。
虽然她自己目前没有明显症状,但她在生育前经过咨询决定不再生育,以防止病基因传给下一代。
3. 张先生是一位50岁的男性,他的父亲在他年轻时因克雅氏病去世。
由于家族中多人患有克雅氏病,张先生决定参与一项研究,希望能为科学家提供更多关于克雅氏病传人机制的信息。
通过他的参与,研究人员发现了一种新的克雅氏病相关基因,从而为研究和治疗提供了新的线索。
4. 小红是一名15岁的女孩,她的妈妈在她出生后不久被诊断出患有克雅氏病。
尽管她的父亲没有此病,但小红遗传了母亲的病基因。
她从小就接受康复治疗,以减轻症状并延缓病情进展。
5. 李先生是一位30岁的男性,他的妻子怀孕期间被确诊为患有克雅氏病。
他们决定进行产前基因检测,结果显示他们未出生的孩子也携带克雅氏病相关基因。
李先生和他的妻子经过深思熟虑,决定终止妊娠,以避免孩子未来可能面临的困境。
6. 小华是一名12岁的男孩,他的姐姐在他出生前因克雅氏病去世。
虽然小华的父母携带克雅氏病相关基因,但他们决定继续生育,希望通过基因咨询和监测,尽可能降低下一个孩子患病的风险。
7. 王先生是一位60岁的男性,他的父亲和祖父都因克雅氏病去世。
虽然他自己没有克雅氏病的明显症状,但他决定参与一项研究,希望为科学家提供关于晚发型克雅氏病的进一步了解。
8. 郑女士是一位35岁的女性,她的丈夫患有克雅氏病。
尽管她自己没有病症,但她了解到她的孩子有可能会遗传到克雅氏病。
江苏省盐城市2025届高三生物上学期期中试题
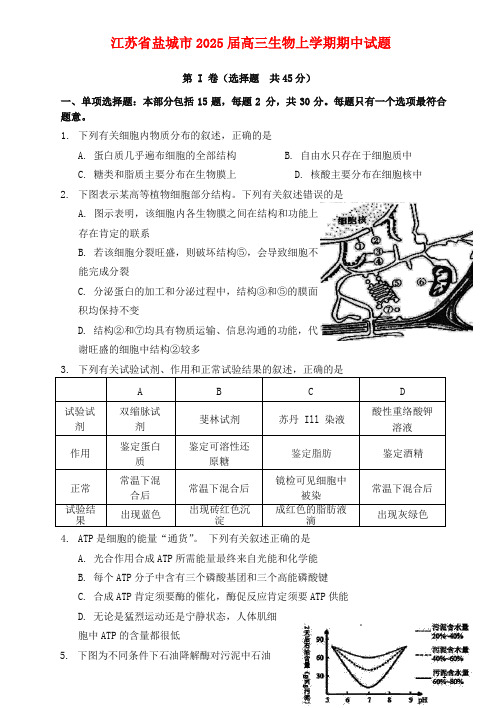
江苏省盐城市2025届高三生物上学期期中试题第 I 卷(选择题共45分)一、单项选择题:本部分包括15题,每题2 分,共30分。
每题只有一个选项最符合题意。
1.下列有关细胞内物质分布的叙述,正确的是A. 蛋白质几乎遍布细胞的全部结构B. 自由水只存在于细胞质中C. 糖类和脂质主要分布在生物膜上D. 核酸主要分布在细胞核中2.下图表示某高等植物细胞部分结构。
下列有关叙述错误的是A. 图示表明,该细胞内各生物膜之间在结构和功能上存在肯定的联系B. 若该细胞分裂旺盛,则破坏结构⑤,会导致细胞不能完成分裂C. 分泌蛋白的加工和分泌过程中,结构③和⑤的膜面积均保持不变D. 结构②和⑦均具有物质运输、信息沟通的功能,代谢旺盛的细胞中结构②较多3.下列有关试验试剂、作用和正常试验结果的叙述,正确的是A B C D试验试剂双缩脉试剂斐林试剂苏丹 Ill 染液酸性重络酸钾溶液作用鉴定蛋白质鉴定可溶性还原糖鉴定脂肪鉴定酒精正常常温下混合后常温下混合后镜检可见细胞中被染常温下混合后试验结果出现蓝色出现砖红色沉淀成红色的脂肪液滴出现灰绿色4.ATP是细胞的能量“通货”。
下列有关叙述正确的是A. 光合作用合成ATP所需能量最终来自光能和化学能B. 每个ATP分子中含有三个磷酸基团和三个高能磷酸键C. 合成ATP肯定须要酶的催化,酶促反应肯定须要ATP供能D. 无论是猛烈运动还是宁静状态,人体肌细胞中ATP的含量都很低5.下图为不同条件下石油降解酶对污泥中石油分解实力的试验测定结果。
下列有关叙述正确的是A. 该试验的自变量是pH,因变量是酶活性B. 污泥含水量对该酶的最适pH无明显影响C. 污泥含水量越高,该酶的催化实力越强D. 污泥含水量过低会使该酶的空间结构遭到破坏j-t6.某同学学习了赫尔希和蔡斯的“噬菌体侵染细菌试验”后,设计了如下试验:一组用35S标记大肠杆菌后,用未标记的噬菌体侵染;另一组用32P标记噬菌体后,侵染未标记的大肠杆菌;两组都经过相宜的培育、搅拌和离心。
脑活检诊断GM2神经节苷脂沉积症_邹婷

论著 脑活检诊断GM2神经节苷脂沉积症邹婷1 肖波2 彭隆祥3 张贺1(1.中南大学湘雅二医院神经内科,湖南 长沙 410011;2.中南大学湘雅医院神经内科,湖南 长沙 410008;3.中南大学湘雅医学院电镜室,湖南 长沙 410078)[摘要] 目的 探讨GM2神经节苷脂沉积症的临床、病理及诊断。
方法 研究8例GM2神经节苷脂沉积症患者的临床表现及诊断分型,并取右额叶脑组织行病理检查。
结果 婴儿型3例以智力低下、癫痫、锥体束征及眼底樱桃红点为特征;迟发型5例以智力倒退、锥体束征、共济失调且无癫痫及眼底樱桃红点为特征。
电镜下见神经细胞内均有膜性胞浆小体及少量斑马体沉积。
结论 临床表现结合脑活检电镜检查有助于诊断GM2神经节苷脂沉积症。
[关键词] 脂质累积病/诊断; 活组织检查Eight C ases with GM2Gangliosidosis Diagnosed by Biopsy of Brain ZO U T ing, XI A OBo, PEN G Long-x iang,et al(Dep ar tment of N eur ology,Second X iangy a H osp ital,Centr al S outhUniver sity,Changsha410008,China)[Abstract] Objective T o probe into t he clinical feature,patholog ical chang e and diagnosis o f G M2g angl-io sido sis. M ethods A biopsy of the r ight fro nta l lobe was per formed for each case.T he clinical manifestat ionsand diagnosis o f8patients wit h G M2gang lio sido sis w ere studied. Results T he main clinical manifestat ionsco nsisted of intellect im pairment,epilepsy,pyramidal t ract signs,retinal macular cherr y-r ed spots for3pa-t ients w ith infantile G M2gang lio sido sis,and mental rer ardatio n,py ramidal tract signs and atax ia for5patientswith late-onset GM2ganglio sido sis,and no epilepsy and retinal macular cherr y-red spo ts for late-onset cases.U ltr astructur ally,the deposits in neur ons w ere memberanous cy toplasmic bo dies(M CB)tog ether w ith a fewzebra bodies(ZB)in each case. Conclusion T he r esults sugg ested that clinical manifestatio ns and electro n m-icr oscopy in taking br ain biopsy contribute for establishing a diagno sis o f GM2gang lio sido sis.[Key words] lipoidosis/DI; bio psy[中图分类号] R742.89 [文献标识码] A [文章编号] 1671-7171(2007)11-1878-03GM2神经节苷脂沉积症是溶酶体贮积症的一种类型,由于遗传性酶缺陷致过量GM2神经节苷脂沉积于脑及周围脏器而发病。
神经节苷脂沉积病是怎么回事?

神经节苷脂沉积病是怎么回事?
*导读:本文向您详细介绍神经节苷脂沉积病的病理病因,神经节苷脂沉积病主要是由什么原因引起的。
*一、神经节苷脂沉积病病因
*一、发病原因
神经节苷脂沉积病(gangliosidosis)为一组常染色体隐性
遗传性疾病。
*二、发病机制
神经节苷脂水解代谢中不同酶的缺乏引起不同物质在神经
组织中的沉积而致病。
神经节苷脂为脑酰胺与一个低聚糖(oligosaccharide)分子和涎酸(sialic acid)结合而组成的葡
萄糖脂,分布于神经组织的神经细胞膜上。
酸性β-半乳糖苷酶的原发性缺乏引起上述分解过程的第一步不能进行和单涎脑酰胺四己糖苷在神经元中的沉积,产生婴儿性家族性黑矇性痴呆,称为GM1沉积病。
此型患者小脑损害较重,视网膜变性,脊髓和周围神经均有不同程度的髓鞘脱失。
氨基己糖酶的缺乏引起上述第2步分解不能和单涎脑酰胺三己糖苷在
神经组织的沉积,产生GM2沉积病。
其中Ⅰ型为婴儿型,称为Tay-Sachs病,Ⅱ型为急性早期婴儿型,称为Sandhoff病。
主
要病理改变为大脑皮质中神经细胞内有大量类脂沉积,细胞变性、消失,晚期有髓鞘脱失和胶质细胞增生。
电镜检查可见沉积物为圆形分层结构,称为膜状胞质小体。
除大脑受累外,小脑和脑干均有普遍萎缩,脑室扩大。
*温馨提示:以上就是对于神经节苷脂沉积病病因,神经节
苷脂沉积病是由什么原因引起的相关内容叙述,更多有关神经节苷脂沉积病方面的知识,请继续关注疾病库,或者在站内搜索“神经节苷脂沉积病”找到更多扩展内容,希望以上内容可以帮助到您!。
婴儿sandhoff病临床特点及基因型分析

基因缺失
部分婴儿Sandhoff病患者存在基 因大段缺失,导致编码区不完整, 影响蛋白质合成。
插入突变
少数婴儿Sandhoff病患者存在基因 插入突变,导致阅读框移位或提前 终止密码子。
基因突变与表型关系
突变位置
基因突变的位置与疾病严重程度 有一定关联,如近端或远端突变
为中度至重度表型。
婴儿Sandhoff病临床 特点及基因型分析
2023-11-11
目录
• 婴儿Sandhoff病概述 • 婴儿Sandhoff病临床特点 • 婴儿Sandhoff病基因型分析 • 婴儿Sandhoff病治疗和预后 • 相关研究和展望
01
婴儿Sandhoff病概述
定义和背景
定义
婴儿Sandhoff病是一种罕见的遗传性疾病,也称为GM2神经节苷脂沉积症, 由于β-Hexosaminidase A和B两种酶缺乏所致。
背景
Sandhoff病是一种常染色体隐性遗传病,主要影响神经系统,导致神经元和轴 突的损害。
流行病学和发病率
流行病学
婴儿Sandhoff病在新生儿中的发病率约为1/50000至1/100000。
发病率
该病的发病率因地区和种族而异,但总体来说较为罕见。
临床表现和诊断标准
临床表现
婴儿Sandhoff病的临床表现多样, 包括运动发育落后、肌张力低下、惊 厥、视力障碍、听力丧失等。
腹泻
部分患儿可能会出现腹泻 症状,可能与肠道感染或 消化不良有关。
呼吸系统异常
呼吸困难
部分患儿可能会出现呼吸困难,特别是睡觉时更为明显。
咳嗽
部分患儿可能会出现咳嗽症状,可能与呼吸道感染有关。
03
神经节苷脂沉积病怎样治疗?

神经节苷脂沉积病怎样治疗?
*导读:本文向您详细介绍神经节苷脂沉积病的治疗方法,治疗神经节苷脂沉积病常用的西医疗法和中医疗法。
神经节苷脂沉积病应该吃什么药。
*神经节苷脂沉积病怎么治疗?
*一、西医
*1、治疗
本病无特殊治疗方法,临床可对症处理,延长患儿存活期。
酶的补充疗法尚在研究之中。
*2、预后
本病预后极差。
GM1神经节苷脂沉积病Ⅰ型极少活过2周岁。
GM2神经节苷脂沉积病Ⅰ型平均病程2年左右,Ⅱ型患者起病晚,可活至10~15岁不等。
*温馨提示:上面就是对于神经节苷脂沉积病怎么治疗,神经节苷脂沉积病中西医治疗方法的相关内容介绍,更多更详尽的有关神经节苷脂沉积病方面的知识,请关注疾病库,也可以在站内搜索“神经节苷脂沉积病”找到更多扩展资料,希望以上内容对大家有帮助!。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
神经节苷脂沉积病神经节苷脂沉积病是一组先天性脂类代谢病。
神经节苷脂广泛存在于人体各种细胞内,而以脑和神经组织中含量最高。
人脑内至少含有10种不同结构的节苷脂,GM1是最主要的一种。
每一种神经节苷脂必需经一系列水解酶的作用逐步降解,其中任何一种酶的缺陷都将造成相应脂类底物的节苷脂累积在组织中沉积,进而破坏细胞和脏器,即为神经节苷沉积病。
临床表现以中枢神经系统症状为主,其共同特点是喂养困难、生长发育迟缓、智力进行性降低、特殊面容等。
目前临床上使用的注射用单涎酸四己糖神经节苷脂钠,是外源性单涎酸四己糖神经节苷脂(GM1)能以稳定的方式与神经细胞膜结合,引起膜的功能变化。
能促进由于各种原因引起的中枢神经系统损伤的功能恢复,通过改善细胞膜酶的活性减轻神经细胞水肿,对脑血流动力学参数以及因损伤后导致脑水肿有积极的作用。
其作用机理是促进“神经重构”(包括神经细胞的生存、轴突生长和突触生长),对损伤后继发性神经退化有保护作用。
动物实验显示单唾液酸四己糖神经节苷脂可改善帕金森病所致的行为障碍。
但其禁忌症是遗传性糖脂代谢异常,如家族性黑蒙性痴呆、视网膜变性病等。
根据沉积的神经苷脂种类本组疾病可分为GM1节苷脂沉积病、GM2节苷脂沉积病和GM3节苷脂沉积病几类。
其中以GM2神经节苷脂沉积病为代表。
【发病机制】单唾液酸四己糖神经节苷脂的发病机制:神经节苷脂为神经酰胺与一个低聚糖分子和涎酸结合而组成的葡萄糖脂。
其分解途径见图1图1 神经节苷脂分解途径GM1单涎酸糖酰胺四己糖苷单涎酸糖酰胺三己糖苷GM1- β-半乳糖苷酶氨基己糖苷酶GM2GM3神经酰胺糖苷脂神经酰胺二己糖苷(乳糖苷脂)GM3- β-半乳糖苷酶Farber病Ⅴ葡萄糖酸苷神经酰胺+葡萄糖图2 不同神经节苷脂沉积病分类亚型生化缺陷酶亚型缺陷酶临床特点GM1Ⅰ型婴儿型GM1-β-半乳糖苷酶,有A、B和C 3种同工酶A、B、C均缺乏6个月内起病,进展快,有特殊外貌,喂养困难,发育迟缓,早期严重惊厥,1/2有樱桃红斑,肝脾明显肿大,2岁内夭折。
Ⅱ型幼年型B、C缺乏7~16个月起病,听觉过敏,智能发育落后并倒退,共济失调,无特殊外貌,无樱桃红斑,肝脾肿大不明显。
Ⅲ型成人型6~20岁出现症状,主要为锥体外系表现如共济失调等,构音困难,进行性智力减退,无樱桃红斑及肝脾肿大。
病情进展缓慢。
GM2Ⅰ型婴儿型氨基己糖苷酶(Hex),有A、B和S 3种同工酶Hex A缺乏,Hex B增加3~5个月内起病,进展快,智能进行性衰退,伴有失明,无特殊外貌,声光刺激敏感,惊跳,发育迟缓,早期惊厥,有樱桃红斑,无肝脾肿大。
Ⅱ型Sandhoff病Hex A、Hex B缺乏,Hex S增加急性婴儿早期型,与GM2-Ⅰ型相似,但有肝脾肿大。
少年型Hex A不完全缺乏2~6岁起病,锥体外系表现+智能进行性衰退。
成年型Hex AB变异型酶发病年龄不一,共济失调,肌张力不全,眼底正常GM3GM3-β-半乳糖苷酶Farber病神经酰胺糖苷脂酶一、GM1节苷脂沉积病GM1节苷脂沉积病是常染色体隐性遗传病。
其生化特点是由于β-半乳糖苷酶缺乏,阻断了单涎酸糖酰胺四己糖节苷脂(GM1)降解过程,致使GM1在神经细胞、内脏器官、骨组织的的溶酶体中沉积,进而破坏细胞和脏器,即为GM1神经节苷脂沉积病。
其病理过程为GM1及其衍生物蓄积于脑灰质神经元的溶酶体内,导致神经元脱失,严重脱髓鞘现象。
肝、肾细胞内也有大量GM1沉积。
其临床表现以中枢神经系统症状为主,伴有肝脾肿大。
GM1-β-半乳糖苷酶,有A、B和C 3种同工酶。
依照同工酶缺乏程度将本病分三型:Ⅰ型(婴儿型)为A、B和C均缺乏,所以病情重,进展快。
Ⅱ型(幼年型)为B和C均缺乏,其脑中β-半乳糖苷酶缺乏与Ⅰ型相似,而肝和纤维细胞中酶缺乏较Ⅰ型轻。
Ⅲ型(少年型)不明,可能为不完全酶缺乏。
【临床表现】1、G M1-Ⅰ型(婴儿型)是全身性GM1沉积病,脑和内脏都有GM1沉积物。
婴儿在出生即可有异常,病情进展迅速。
患儿常在出生后不久即发病,初起表现为全身肌张低下,喂养困难,对外界反应差。
早期即可见肝脾肿大。
外貌特殊,与粘多糖Ⅰ型相似,前额突出,鼻梁凹陷,耳位低,舌体大,人中长,面部多毛。
新生儿期即哺乳困难,反应迟钝,生长发育迟缓。
病儿不能注视,有眼球震颤,听觉过敏,拥抱反射加强。
早期即出现严重惊厥,约1/2病儿有樱桃红斑。
6个月后出现肝脾肿大,脊柱后弯,关节挛缩,爪形手。
晚期肌张力增高,去大脑强直状态,对外界反应消失。
晚期可有巨颅,但不如Tay-Sachs病严重。
多在2岁以内因继发感染死亡。
2、G M1-Ⅱ型(幼年型、晚发婴儿型)一般于7~16个月间起病,首发症状常是站姿异常,听觉过敏,惊跳反射增强,生长发育落后。
继而肌张力进行性低下,腱反射亢进,上肢运动不稳、不能独坐、独站、言语不清,走路不稳。
逐渐发展至痴呆、失语、惊厥、痉挛性四肢瘫痪。
病情进展较慢,多死于继发感染。
本型与GM1-Ⅰ型不同之处:无特殊容貌,肝、脾肿大不明显,无樱桃红斑,骨骼改变不明显。
常于3-10岁内死于继发感染。
3、G M1-Ⅲ型(少年型)少年型发病一般在4岁以后。
自6~20岁间开始出现进行性智力减退,共济失调,痉挛性瘫痪。
有的智力障碍不明显,而以不自主运动为主要表现。
其病变在基底节最重。
患者常以构音障碍和肌张力改变为初始症状,病情进展缓慢,可长达数10年,智能轻度受损,多无共济失调、肌阵挛或癫癎发作;不伴面容异常、肝脾肿大;无视网膜、角膜病变。
骨骼X片大多无特殊发现。
病情进展缓慢,一般于20-30岁左右死亡。
【诊断】本病患儿尿中可见硫酸角质素排出,外周血淋巴细胞常有空泡形成,骨骼X片有特征性改变等均有助于诊断。
确诊根据在白细胞、成纤维细胞内β-半乳糖苷酶的酶活性测定。
鉴别诊断:婴儿型应与粘多糖病、Gaucher病、Niemann-Pick病、Tay-Sachs病鉴别。
【治疗】本病无特殊治疗。
二、G M2节苷脂沉积病GM2神经节苷脂沉积病是常染色体隐性遗传病,伴完全外显率。
生化缺陷是氨基已糖胺酶(hexosaminidase,Hex)缺乏,导致GM2分子所结合的N-乙酰半乳糖(NANA)不能被水解脱离,造成GM2降解障碍而沉积在体内,出现一系列临床症状。
临床特点以进行性智力和运动功能障碍伴异常惊跳反应、眼底樱桃红斑、视力减退等。
GM2神经节苷脂沉积病婴儿型最初由英国眼科学者Tay和美国Sachs所描述,故又称Tay-Sachs病,即GM2—婴儿型,也曾经称为家族性黑朦痴呆。
晚发婴儿型患儿通常在生后第2年起病,临床表现类似婴儿型。
【病理和发病机制】Hex酶有A、B和S三种同工酶。
Hex-A有α和β两个亚基,Hex-B为ββ二聚体亚基。
Hex-A和Hex-B均能水解糖蛋白和糖脂,GM2只能被Hex-A水解,且需依赖GM2激活蛋白(GM2A基因)参与。
因此,Hex-A、Hex-B、GM2A任一种基因突变,均可导致GM2神经节苷脂沉积病。
依据其不同酶缺陷,GM2神经节苷脂沉积病分为四型。
GM2—Ⅰ型为Hex-A缺乏,Hex-B代偿性增加,犹太人多见。
GM2—Ⅰ型酶基因定位于15q22-25,携带频率约3%;GM2—Ⅱ型为Hex-A和Hex-B均缺乏,Hex-S 增加。
GM2—Ⅱ型基因定位于5q13,无人种差异;GM2—Ⅲ型为Hex-A部分缺乏。
【参考文献:侯琳,Ohno Kousaku,等.GM2神经节苷脂沉积病发病的分子机理研究[J].中华医学遗传学杂志,2008.23(7):540—541】主要病理改变见于中枢神经系统,显微镜下见广泛的神经元脂质沉积,脑神经元呈球形肿胀和脂肪变性,神经元脱失,广泛脱髓鞘;电镜下可见膜状胞浆小体(MCB),呈圆形膜样分层结构,包围着颗粒状的核心,内容物为GM2、磷脂、胆固醇。
还可见指纹状脂褐质小体。
脑体积正常或增大,晚期脑组织萎缩。
眼底黄斑周围神经节细胞内有脂质沉积,出现樱桃红斑。
【参考文献:1、实用儿科学。
2、马秀伟,蒲利华,张月华,等.GM2神经节苷脂沉积症的临床特征及诊断.实用儿科临床杂志,2008.7(23):539-540】【临床表现】Ⅰ型,又称婴儿型、Tay-Sachs病、家族性黑朦性痴呆(一)GM2—本型为GM神经节苷脂沉积病最多见类型,起病早,病情发展快,但出生时可表现正常,常于3-5个月发病。
1、运动行为发育倒退:往往以对声光刺激敏感出现惊跳,易激惹及肌肉跳动症状为首发症状。
6个月后出现肌张力降低,抬头无力,躯干控制能力倒退,痉挛性瘫痪,甚至角弓反张。
高声调啼哭,不易安抚,语言能力倒退甚至不会说话。
对外界刺激从反应过度到无反应。
2、抽搐及脑电图:6个月到1岁就可以出现各型癫痫发作,2岁后抽搐发作频度和强度逐渐增加。
无抽搐发作前ECG往往正常,晚期出现阵发性高幅慢波伴有多相棘波暴发,末期仅有低幅慢波活动。
3、视力和听力:小儿病后3~4个月病情进展迅速,眼底出现樱桃红斑,视力减退,有不协调的眼球震颤,注意力减弱,常在1岁左右发展为全盲或皮质盲。
进行性听力减退至耳聋。
4、其他:头围早期增大,晚期不增。
肝脾不大。
易继发感染。
往往5岁前夭折。
(二)GM2—Ⅱ型,又称Sandhoff病GM2—Ⅱ型为急性婴儿早期型,发病年龄和病情进展与GM2—Ⅰ型相似,主要不同点为GM2—Ⅰ型表现伴有肝脾肿大。
因为Hex-A有α和β两个亚基,Hex-B为ββ二聚体亚基。
α亚基只影响Hex-A活性,β亚基对Hex-A和Hex-B均有影响。
Hex-A酶缺陷导致的90%的GM2沉积,仅有少量的GM2亚型,主要沉积在神经系统组织中。
Hex-B酶缺陷导致的GM2和GM2亚型(红细胞糖苷脂)均有沉积,以神经系统为重,在肝、脾、肾组织中均有沉积。
(三)GM2-Ⅲ型,少年型GM2-Ⅲ型生化缺陷可能为不完全性酶缺陷,起病较迟,多在2~6岁发病,病情进展较缓慢。
以锥体外系症状开始,运动共济失调、舞蹈样动作、手足徐动、肌张力不全。
逐渐出现痉挛性瘫痪、吞咽困难、言语不清、构音障碍、动眼神经麻痹、去大脑强直麻痹、视力丧失等;智力发展迟滞且倒退;很少有樱桃红斑,但有视网膜色素沉着;抽搐发作少见或轻度惊厥,巨脑少见。
多于15岁前死亡。
(四)成年型GM2神经节苷脂沉积病本型起病年龄或早至儿童期,或晚至成年期,均有共济失调表现,眼底正常,肌张力低下,痉挛性瘫痪等。
起病早者可伴有轻度智力低下,往往无抽搐。
起病晚者可有精神症状,眼震颤,眼肌麻痹等症状。
辅助检查:脑电图(EEG):婴儿型发病后常有癫痫波及高峰节律紊乱;少年型偶有癫痫波,常表现为弥漫异常;成年型可为正常或弥漫慢波。
脑干听觉诱发电位(BAEP):各型均有形态和潜伏期异常。
头颅CT/MRI:晚期可有脑萎缩。