欧盟-who标准比较
中国GMP 2010 与EU、WHO 及FDA 的原则比较
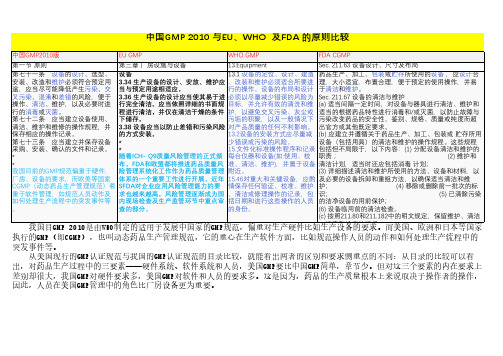
执行的GMP(即CGMP),也叫动态药品生产管理规范,它的重心在生产软件方面,比如规范操作人员的动作和如何处理生产流程中的突发事件等。
从美国现行的GMP认证规范与我国的GMP认证规范的目录比较,就能看出两者的区别和要求侧重点的不同:从目录的比较可以看出,对药品生产过程中的三要素——硬件系统、软件系统和人员,美国GMP要比中国GMP简单,章节少。
但对这三个要素的内在要求上差别却很大,我国GMP对硬件要求多,美国GMP对软件和人员的要求多。
这是因为,药品的生产质量根本上来说取决于操作者的操作,因此,人员在美国GMP管理中的角色比厂房设备更为重要。
总硬度地表水质量标准

总硬度地表水质量标准
总硬度是指水中的钙和镁离子的总含量。
地表水总硬度标准可以根据不同国家和地区的水质管理法规和标准来确定。
以下是一些国际上常见的地表水总硬度标准的范围:
1. 世界卫生组织(WHO)建议的地表水总硬度标准为:
- 极高硬度:>500 mg/L(以钙的化学计量单位mg/L计算) - 高硬度: 200-500 mg/L
- 中等硬度: 75-200 mg/L
- 低硬度: <75 mg/L
2. 欧盟饮用水标准下的总硬度标准为:
- 极高硬度:>500 mg/L
- 高硬度: 200-500 mg/L
- 适中硬度:150-200 mg/L
- 低硬度: <150 mg/L
需要注意的是,地表水总硬度标准的具体数值范围可能根据当地的法律法规、水质管理机构的要求和地质水文特征等因素而有所不同。
因此,在确定具体的总硬度标准时,应参考当地的法规和标准。
WHO和世界主要国家生活饮用水卫生标准介绍

览等章节,2006年WHO又发布了《准则》增补本资
料。
WHO决定以滚动修订的方式来推进本准则的进展。
【收稿日期]2007一髓一2l
万方数据
・354・
中国卫生监督Leabharlann 志《准则》主要根据卫生学意义提出水质指标,依 次分为微生物指标、化学物质指标、放射性指标、由 于感官可能引发消费者不满的指标等指标。该版包 括了水源性疾病病原体27项(细菌12项,病毒6 项,原虫7项,寄生虫2项),具有健康意义的化学指
2007年第14卷第5期
・353・
水单位的积极性,在推动水泥水箱改造方面起到了 积极作用。
和一些居民小区采用管道分质供水,这些采用先进 工艺的供水方式,使市民可以饮用更卫生、更安全的 饮用水。
目前北京已有部分新建、改建的项目中使用了
无负压、无吸程供水方式;部分奥运场馆、宾馆饭店
WHO和世界主要国家生活饮用水卫生标准介绍
甘日华 (广东省卫生监督所,广东广州510300) [摘要】 作者舟绍了WHO和世界主要国家生活饮用水卫生标准。各国饮用水标准均明确控制微生物污染
是极端重要的,世界喜国和相关机构纷纷修改原有的或制订新的水质标准,新标准共同特点是有机污染和农药 指标、消毒剂及其副产物项目增多,对人体危害指标要求趋严,科学求实调整放宽个别指标,增加有益的健康
何发挥个人的作用。
2.3美国饮用水水质标准(2006)
标、有机消毒副产物指标、其它有机化合物指标、农 药、饮用水中的放射性指标,总共248项,其中有些
项目未列出指标值。该标准考虑项目全面,特别是 微生物学项目分为细菌、原生动物、病毒和毒藻等几
强制性的一级饮用水标准指标98项,其中有机
物指标63项,无机物指标22项,微生物指标8项
中国、美国、日本、欧盟四国饮用水质标准比较

快适水水质卫生标准1、快适水概念水质指标达到美国、欧盟、日本和中国现行饮用水标准中最高要求,适合于婴幼儿等对水中污染物较为敏感的人群长期饮用的优质饮用水。
2、美国、日本、欧盟和中国四国或地区饮用水水质标准比较2.1分类的区别(1)美国水质标准分为国家一级饮用水水质标准和二级饮用水标准。
国家一级饮用水水质标准中又有最大污染物浓度(MCL)和最大污染物浓度目标(MCLG)两个指标,MCL为强制性的标准,MCLG是非强制性的更高目标值。
美国一级饮用水指标共有78个,分为:无机物、有机物、放射性物质、微生物学指标,其中无机物指标有15个,有机物指标54个,放射性物质指标有3个,微生物学指标有6个;二级饮用水污染物指标共有15个,没有细分。
(2)欧盟水质标准并没有特别分类,水质指标分为微生物学指标、化学物质指标和指示指标,其中微生物学指标2个(瓶桶装水是5个),化学物质指标26个,指示指标20个。
(3)日本水质标准分为水质基准项目和水质管理目标设定项目。
水质指标项目分为病原微生物、重金属、无机物、金属类、有机物、消毒剂残留及消毒副生成物、基本特性、其他类。
日本水质基准项目共50个,其中病原微生物指标有2个,重金属指标有10个,无机物指标有9个,有机物指标有11个,消毒剂和消毒副生成物10个,基本特性指标5个,其他类指标2个;水质管理目标设定项目共27个,其中重金属和金属指标有4个,无机物指标有2个,有机物指标有9个,消毒剂和消毒副生成物6个,其他类6个。
(4)中国生活饮用水卫生标准GB5749分为水质常规指标及限值、饮用水中消毒剂常规指标及要求、水质非常规指标及限值,共计106项。
饮用水标准水质常规指标分为微生物指标、毒理指标、感官性状和一般化学指标、放射性指标四类共38个,其中微生物指标有4个,毒理指标有15个,感官性状和一般化学指标有17个,放射性指标2个;饮用水中消毒剂常规指标4个;水质非常规指标及限值分为微生物指标2个、毒理指标59个、感官性状和一般化学指标3个。
食品安全标准的国际比较与我国标准的完善

食品安全标准的国际比较与我国标准的完善随着经济全球化和贸易自由化的发展,食品安全问题已越来越受到各国政府和民众的关注。
每年都有大量的食品案件发生,严重影响了人们的健康和生命安全。
为了保证食品安全,各国都采取了一系列措施,并制定了相关的食品安全标准。
本文将试图通过国际比较的方法,探讨各国的食品安全标准和我国的标准在哪些方面还需要完善。
一般来说,各国的食品安全标准主要包括以下几个方面:1.化学污染物限量标准:比如苯并芘、氯霉素、四环素、呋喃丙酸、黄曲霉毒素等。
2.微生物标准:比如大肠菌群、沙门氏菌、金黄色葡萄球菌、霉菌等。
3.营养成分标准:比如蛋白质、脂肪、碳水化合物、糖分、维生素、矿物质等。
4.残留农药标准:比如DDT、克百威、甲基对硫磷等。
5.添加剂标准:比如防腐剂、甜味剂、色素等。
现在我们来比较一下几个国家的食品安全标准:一、美国美国是全世界最严格的食品安全立法国家之一,其食品安全标准极为严格。
美国食品和药品管理局(FDA)制定了数千项化学物质、农药、兽药和食品添加剂的授权限制,对其含量和用途均作出严格规定。
举个例子,美国对于苯并芘的限量是0.5ppb,而我国仅为10ppb。
另外,美国还制定了营养成分标准,如维生素和矿物质含量的规定,以保证食品营养价值。
二、欧盟欧盟也非常重视食品安全问题,其食品安全标准颇为严格。
欧盟委员会负责为食品和农产品制定标准,欧盟食品安全局(EFSA)则负责审查和评估有关食品安全的科学证据。
举个例子,欧盟对于黄曲霉毒素的限量是4ppb,而我国仅为20ppb。
此外,欧盟还规定了动物药品残留的标准,并监测食品中残留物质的含量,以保障消费者的健康。
三、日本日本尤其注重营养成分的标准,并非常注重保护儿童和孕妇的健康。
日本制定了较为严格的食品添加剂控制标准,通过食品添加剂的分类和加工食品的标签告知等方法,保障了消费者的知情权。
举个例子,日本对于三氯甲烷的限量是10ppb,而我国仅为40ppb。
臭氧超标标准

臭氧超标标准
臭氧超标标准是指空气中臭氧浓度超过一定限值的情况。
根据不同国家和地区的环境标准,臭氧超标的限值可能会有所不同。
以下是一些国际上常用的臭氧超标标准:
1. 世界卫生组织(WHO)标准:日均最大8小时平均浓度为100微克/立方米。
2. 美国环境保护署(EPA)标准:日均最大8小时平均浓度为70-75微克/立方米。
3. 欧盟标准:日均最大8小时平均浓度为120微克/立方米。
4. 中国标准:日均最大8小时平均浓度为160微克/立方米。
需要注意的是,这些标准是根据臭氧对人体健康和环境的影响而制定的,超过臭氧超标标准可能对人体健康产生不良影响,例如刺激呼吸道、导致哮喘、增加心血管疾病风险等。
因此,控制空气中臭氧浓度对于保护公众健康和环境非常重要。
中国、美国、日本、欧盟四国饮用水质标准比较

快适水水质卫生标准1、快适水概念水质指标达到美国、欧盟、日本和中国现行饮用水标准中最高要求,适合于婴幼儿等对水中污染物较为敏感的人群长期饮用的优质饮用水。
2、美国、日本、欧盟和中国四国或地区饮用水水质标准比较2.1分类的区别(1)美国水质标准分为国家一级饮用水水质标准和二级饮用水标准。
国家一级饮用水水质标准中又有最大污染物浓度(MCL)和最大污染物浓度目标(MCLG)两个指标,MCL为强制性的标准,MCLG是非强制性的更高目标值。
美国一级饮用水指标共有78个,分为:无机物、有机物、放射性物质、微生物学指标,其中无机物指标有15个,有机物指标54个,放射性物质指标有3个,微生物学指标有6个;二级饮用水污染物指标共有15个,没有细分。
(2)欧盟水质标准并没有特别分类,水质指标分为微生物学指标、化学物质指标和指示指标,其中微生物学指标2个(瓶桶装水是5个),化学物质指标26个,指示指标20个。
(3)日本水质标准分为水质基准项目和水质管理目标设定项目。
水质指标项目分为病原微生物、重金属、无机物、金属类、有机物、消毒剂残留及消毒副生成物、基本特性、其他类。
日本水质基准项目共50个,其中病原微生物指标有2个,重金属指标有10个,无机物指标有9个,有机物指标有11个,消毒剂和消毒副生成物10个,基本特性指标5个,其他类指标2个;水质管理目标设定项目共27个,其中重金属和金属指标有4个,无机物指标有2个,有机物指标有9个,消毒剂和消毒副生成物6个,其他类6个。
(4)中国生活饮用水卫生标准GB5749分为水质常规指标及限值、饮用水中消毒剂常规指标及要求、水质非常规指标及限值,共计106项。
饮用水标准水质常规指标分为微生物指标、毒理指标、感官性状和一般化学指标、放射性指标四类共38个,其中微生物指标有4个,毒理指标有15个,感官性状和一般化学指标有17个,放射性指标2个;饮用水中消毒剂常规指标4个;水质非常规指标及限值分为微生物指标2个、毒理指标59个、感官性状和一般化学指标3个。
WHO用于国际比较的主要卫生指标
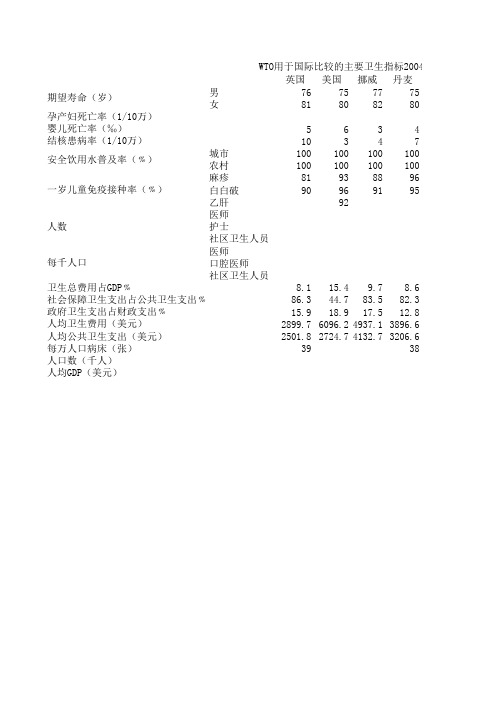
英国美国挪威丹麦
男76757775女
81808280
563410347城市100100100100农村100100100100麻疹81938896白白破90
9691
95
乙肝92
医师护士
社区卫生人员医师口腔医师社区卫生人员
8.115.49.78.686.344.783.582.315.918.917.512.82899.76096.24937.13896.62501.82724.74132.73206.6
3938
人口数(千人)期望寿命(岁)WTO用于国际比较的主要卫生指标2004年
人均GDP(美元)
卫生总费用占GDP﹪
社会保障卫生支出占公共卫生支出﹪政府卫生支出占财政支出﹪人均卫生费用(美元)人均公共卫生支出(美元)每万人口病床(张)安全饮用水普及率(﹪)一岁儿童免疫接种率(﹪)
人数
每千人口
孕产妇死亡率(1/10万)婴儿死亡率(‰)结核患病率(1/10万)
004年
瑞典新加坡香港首尔(韩国)台北东京(日本) 78777379
83828086
3253
43111334 10010097100
10071100
94949999
99948899
9392
9.1 3.7 5.57.8
84.93452.681.3
13.6 6.210.317.2
3532942.9776.92823.2 2999.8321408.52295.2。
饮用水铊含量标准

饮用水铊含量标准铊是一种有毒的金属元素,它可以引起人体中枢神经系统、心血管系统、肝脏、肾脏等多个器官的损害。
因此,饮用水中铊的含量非常重要。
为了保障公众的健康和安全,各国家和地区都有相应的标准来规定饮用水中铊含量的限制。
在本文中,我将探讨一下中国和其他国家和地区的饮用水铊含量标准。
一、中国的饮用水铊含量标准当前,中国国家标准规定饮用水中铊含量的限制为0.1毫克/升(以下简称mg/L)。
这个标准是由卫生部、发改委、环保部等多个部门联合制定的《饮用水卫生标准》(GB 5749-2006)中明确提出的。
这个标准是根据中国国情以及国际上对铊的毒性研究结果制定的。
在国际上,WHO也对饮用水中的铊含量进行了限制,其标准是0.01mg/L。
相比之下,中国的标准限制要限制松一些。
二、其他国家和地区的饮用水铊含量标准1、美国美国是世界上认为水质规定最为严格的国家之一,它针对铊的毒性还制定了非常严格的标准。
在美国的《饮用水规定》(Safe Drinking Water Act)中,铊被列为二价金属元素的一种,并规定饮用水中铊含量的限制为0.002mg/L。
这个标准非常严格,相比之下,中国的标准要宽松许多。
2、欧盟欧盟对饮用水中铊含量的限制标准为0.01mg/L,这个标准与WHO 的标准相同。
欧盟成员国在执行这一标准时还需要遵守EC 98/83/EC,也就是《关于供水质量的欧盟理事会指令》中的规定,以确保饮用水的健康和安全。
3、日本日本的饮用水标准将铊含量的限制标准设定为0.001mg/L。
这个标准非常严格,相比之下,中国和欧盟的标准都要宽松许多。
三、结论通过对比可以发现,各国和地区对饮用水中的铊含量都设置了不同的标准,但总的来说,这些标准大多都很严格。
在中国,饮用水中铊的含量限制只有0.1mg/L,仅仅相当于美国标准的五分之一。
然而,人体对铊的饮用是相当危险的,因此我们应该尽可能地避免在饮用水中受到铊的污染。
只有通过科学和合理的控制措施,才能保障饮用水中铊的含量在安全范围内,从而保证公众的健康和安全。
茶叶农残限量——欧盟400多项VS中国28项

S pecial cial专题行业聚焦
近年来,国内茶叶的消费需求量越来越大,人们对茶叶的质量安全越发关注,对茶叶的品质要求越来越高,对茶叶质量安全尤其茶叶农残的关注度更为集中,所以本文比较CAC、美国、日本、欧盟、中国几大茶叶生产、消费国对茶叶农残限量的要求,分析我国茶叶相关标准中涉及茶叶农药最大残留限量的问题,探讨我国茶叶质量安全和品质提升问题。
美国、法国、英国、德国和加拿该原则是联合国粮农组织(FAO)和世
界卫生组织(WHO)提出的安全性评价
原则,根据人体接触化合物量和对人
体或动物的毒性制定的标准。
人体可
能摄入量较大和对人体毒性大的化合
物制定农残限量标准较严,反之较宽
松。
(2)“零风险”原则。
该原则不
考虑化合物的毒性,只根据自己的考
虑点制定。
(3)不同国家、地区在茶
叶生产技术、农残检测技术等方面存
在一定的差距,有些发达国家为本国
茶叶农药最大残留标准共有
见表1。
美国茶叶农残限量
标准
美国茶叶农残限量的法规与标准
采用的是风险性评估原则,仅对三氯
杀螨醇、唑酮草酯等8项有限量要求,
见表2,未列出的采用最低检出限。
检
测方法由美国食品和药品监督管理局
(FDA)在具体执行时给出,这些检测
方法通常最灵敏。
茶叶农残限量——欧盟400多项VS中国28项
□ 刘 洋 云南出入境检验检疫局检验检疫技术中心DOI:10.16043/ki.cfs.2016.33.055
80食品安全导刊 2016年11月。
美国、欧盟、日本和中国标准比较
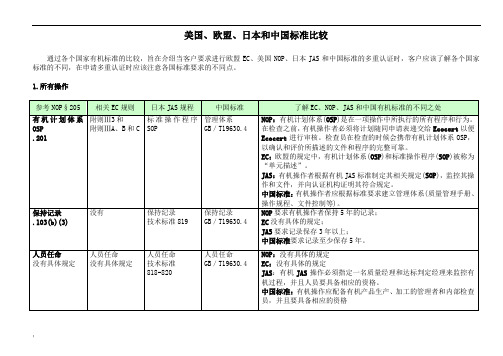
美国、欧盟、日本和中国标准比较通过各个国家有机标准的比较,旨在介绍当客户要求进行欧盟EC、美国NOP、日本JAS和中国标准的多重认证时,客户应该了解各个国家标准的不同,在申请多重认证时应该注意各国标准要求的不同点。
1.所有操作2.作物的种植3.加工和经营4.有机产品的标签条提供的方法。
-NOP规则中有机成份的计算与EC不同。
有可能出现这样的情况,一种产品用EC计算含有95%有机产品,但按照NOP规则它不属于有机产品。
JAS:计算时同样不包括盐和水。
中国标准:规则中有机成份百分比的计算:对于固体形式的有机产品,有机配料百分比按照下列算式计算:有机配料百分比(%)=产品中有机配料总质量(不包括水和食盐)÷产品总质量(不包括水和食盐)×100%对于液体形式的有机产品,其有机配料百分比按照下列算式计算:有机配料百分比(%)=产品中机配料的总体积(不包括水和食盐)÷产品总体积(不包括水和食盐)×100%对于包含固体和液体形式的有机产品,其有机配料百分比按照下列算式计算:有机配料百分比(%)=产品中有机配料总质量(不包括水和食盐)÷产品总质量(不包括水和食盐)×100%中国标准有机成份百分比的计算与NOP类似。
USDA标识.303 没有具体规定技术标准819 GB/T19630.3-2005第7条NOP:USDA标识用在标签中沙自愿的。
但是不允许其它有关认证的标识比USDA标识大。
有机的和100%有机的产品才能使用USDA标识。
EC:没有具体规定JAS:有机产品必须使用JAS标识中国标准:中国有机产品认证标志和中国有机转换产品认证标志仅用于按照有机产品国家标准生产或者加工并经认证机构认证的相应的有机产品或者有机转换产品。
WHO与欧盟对甲型H1N1流感防控评估报告的比较研究

将流感 大流 行警 戒 级 别从 3级 提 高 到 4级 , 4月 2 9 日, 界卫生 组织 将 流 感 大流 行 警 戒 级别 再 次 提 高 世 到 5级 。20 09年 4月 3 t世 界 卫生 组 织 ( O) 0E, H W 、
1 09年 甲型 H1 1 感的基本概况 20 N 流
联合 国粮食及农业组织和世界动物卫生组织经过研 究讨论 , 将这一新型流感命名为“ 甲型 H N 流感” I1 。
甲型 H N 1 1流 感 在 不 到 半 年 的 时 间 里 传 遍 全 球, 截止 到 20 年 6月 中旬 , 09 全球 7 国家 和地 区 4个 都 报告 了流 感 病 例 , 数 达 到 了 2 4 总 874例 , 亡 病 死 例 1 例 , O再 次 将 流 感 警 戒级 别 提 升 至 6级 , 4 H W 宣 布全球 进 入 流 感 大 流行 状 态 。截 止 到 21 00年 8
发公 共卫生 事件 ” 20 ,09年 4 2 月 7日, 界卫 生组 织 世
最好“ 教材” 。我们要通过危机评估 , 充分借鉴其他
国家成 功 的经 验 和优 秀 的研 究成 果 , 且 在 实践 工 并 作 中不 断探索 , 断完 善 , 现 评估 工 作 的科 学性 、 不 实 规范性 、 效性 , 有 提高我 国政 府应 对突发 公共 卫生事 件 的能力 , 在未来 的应 急 管理 中更 好 的保 障人 民生
些诸如隔离等传统措施与人权保护之 间的争议 等。而对禽流感的应对 , 则显示出了动物卫生的计
划和应 对工 作 的重要 角 色 ; 共 卫 生 机构 与 动 物 医 公
疗机 构之 间的协 调 问题 等 。
球公共健康受到重大冲击的 同时, 也让世界各 国重
噪声等级标准

噪声等级标准
噪声等级标准通常由各国政府、国际标准化组织和环保机构等制定,旨在规定不同环境下允许的噪声水平,以保护公众健康和环境。
不同国家和地区可能有不同的噪声等级标准,以下是一些常见的噪声等级标准:
1. 国际标准:
-世界卫生组织(WHO):WHO制定了《环境噪声指南》(Guidelines for Community Noise),为城市、工业和交通等不同环境下的噪声制定了指导性标准。
-国际标准化组织(ISO):ISO也发布了一些与噪声相关的标准,例如ISO 1996-1:2016《声学——建筑物噪声和室内噪声的评估》。
2. 欧洲标准:
-欧盟环境噪声指令:欧盟发布了《环境噪声指令》(Environmental Noise Directive),要求成员国制定噪声监测和管理计划。
-英国环境噪声政策:英国政府发布了噪声政策,将噪声区分为日间、夜间和深夜三个时段,并制定了相应的噪声等级标准。
3. 美国标准:
-环境保护署(EPA):美国环境保护署发布了《环境噪声控制政策》(Environmental Noise Control Policy),用于指导各州和城市管理噪声。
-联邦航空管理局(FAA):FAA制定了噪声等级标准,以规定民用航空器的噪声限制。
4. 中国标准:
-《城市居住区环境噪声标准》:中国环境保护部发布的标准,用于城市居住区环境噪声的管理和评估。
以上仅是一些常见的噪声等级标准,不同国家和地区根据其实际情况和立法要求,可能会有更为详细和具体的标准。
噪声等级标准的目的是保护居民的健康和提高生活质量,减少噪声污染对环境的影响。
欧盟-WHO标准比较

欧盟与WHO无菌药品要求比较一、章节对照表二、灭菌方式比较三、欧盟与WHO无菌药品文本的对比1、微粒表述方式不同1)欧盟以≥0.5μm及>5.0μm;2)WHO则是0.5-5.0μm和>5.0μm二种规格,(DH:由于大的微粒很少,实际测试数据可能二者可能一样)。
2、微生物控制限度不同1)欧盟对A区微生物控制(空气沉降菌及浮游菌、表面、手套)均为不得检出(小于一个菌);(DH:欧盟与FDA一样,比WHO严格)。
2)WHO则为 3个菌,B、C、D级别区控制指标与欧盟完全相同。
3、监控要求不同1)欧盟第3条洁净级别划分表的说明中提到:应对A级区的空气粒子进行连续测定,并建议对B级区也连续进行测定…。
修订版第5条提到:…除有充分理由以外,例如活病毒疫苗的灌装,A区的微粒应连续监控或频繁取样。
建议B区也采用类似的系统,尽管取样频率可适当降低。
这类体系通常需要独立的粒子计数仪;或有一个粒子计数器,这个计数器通过一个多接口的管子,与数个连续的取样点相接。
如使用远程监控系统,管子的长度及任何管的弯曲半径均应验证。
A区监控的频率应足以保证检出所有的干扰及偶发事件,一旦发生偏离正常情况时,即能启动报警系统(DH:基本均是连续取样的要求,如不是连续,如何能检出干扰?)欧盟第93条要求:容器的密封应采用经验证的方法。
熔封性容器,如玻璃或塑料安瓿,应进行100%的完好性检查。
应根据适当的方法对其它容器的样品进行密封完好性检查。
半压塞冻干的小瓶,从半压塞开始至轧盖应始终处在A级的保护之下。
无菌灌装的小瓶在完成轧盖前,尚不是一个完整的密封系统。
因此,应在A级保护下直到轧盖完成。
2)WHO无连续测试要求;无菌灌装小瓶的轧盖,无百级保护的要求。
4、对D区的要求1)欧盟附表说明(f)中提到:须根据生产操作的性质来决定此洁净区的要求和限度。
2)WHO及欧盟对动态微粒均不作规定。
5、换气次数1)欧盟第3条表下说明提到:为了达到B、C、D级区的要求,空气换气次数应根据房间的大小、室内的设备和操作人员数决定。
国内外洁净区标准等级对比

*
对平面布局影响大的条款-2
浪费了投资
增大了污染的风险
降低了标准
混淆了无菌及非无菌控制区的界限
许多设计不分无菌控制万级及非无菌控制万级
在过大、过复杂更衣室的同时
E
D
C
B
A
F
*
送风速度和换气次数
达到动态洁净度标准,须一定的条件 换气次数,单位: 次/小时
01
总送风量÷房间体积 风速,层流要求 0.45 (1±20%)米/秒 折算-比较
级别
静态
动态
最大允许粒子数/m3
最大允许粒子数/m3
0.5-5.0μm
>5.0μm
0.5-5.0μm
>5.0μm
A
3 500
0
3500
0
B
3 500
0
350 000
2 000
C
350 000
2 000
3 500 000
20 000
D
3 500 000
20 000
不作规定
不作规定
*
WHO GMP-2002 说明-1
01
03
02
*
WHO GMP 2002 微生物限度
级别b
空气样CFU/m3
沉降碟(90mm)CFU/4小时c
接触碟(55mm)CFU/碟
5指手套CFU/手套
A
<3
<3
<3
<3
B
10
5
5
5
C
100
50
25
-
D
200
100
50
-
*
国际GMP规范对洁净度要求的特点
我国GAP与欧盟GACP世界卫生组织GACP的比较

我国GAP与欧盟GACP世界卫生组织GACP的比较研究背景2002年3月18日,我国国家食品药品监督管理局颁布了《中药材生产质量管理规范(试行)》,简称中药GAP。
2016年2月3日,国务院印发《关于取消13项国务院部门行政许可事项的决定》,规定取消中药材生产质量管理规范认证。
截至到今日,约有194个中药材种植基地获得国家食药监管总局批准的GAP认证。
如同仁堂、云南白药、香雪制药、天士力、康美、东阿阿胶、以岭药业等过半A股中药上市公司都建有中药材GAP生产基地。
(参考自金羊网-羊城晚报作者:陆志霖)欧盟GACP与世界卫生组织GACP背景欧盟的GACP,提出了植物和芳香植物生产和加工的标准,这对于中国GAP的制定与实施起到了至关重要的作用。
2003年世界卫生组织发布了《药用植物种植与采集的生产质量管理规范指南》,其宗旨是为了使各国政府确保草药产品的优质,安全,可持续利用且对人体和环境没有威胁。
我国中药材GAP与国外相关标准的一致性分析(1)人员管理的一致性分析» 2-1我国中药材GAF关徳人员资厦与WHO.欧盟GACP的一致性Table2-1 Consistency of Chinese herb GAP with WHO s GACP and EUM GACP on the keypersonnel qualification-(GAP第四十五条生产企业的技术负责人应有药学或农学、畜牧学等相关专业的大专以上学历,并有药材生产实践经验。
GAP第四十六条质量管理部门负责人应有大专以上学历,并有药材质量管理经验。
)-(WHO与欧盟的GACP无)山表可知,在对关键人员资质进行具体规范方面,是我国中药材GAP在人员方面与WHO的GACP,欧盟的GACP的明显不同。
我国GAP规定,生产企业技术负责人和质量管理部门负责人都应有大专以上学历,而前者还需要是药学,农学或畜牧业等相关专业的学历。
并且还规定前者须有药材生产实践经验,后者须有药材质量管理经验,这些都是WHO和欧盟的GACP中所没有涉及到的。
欧盟与中国食品添加剂法规标准的对比分析

欧盟与中国食品添加剂法规标准的对比分析【论著】Contrast on Food Additive regulations and Standards between EU and China姚斯洁1,代汉慧2,李杏1,邹志飞3YAO Si-jie ,DAI Han-hui ,LI Xing ,ZOU Zhi-fei摘要目的为加强中国食品添加剂的管理及使用提供技术支持。
方法分别从概念、分类、编码、使用品种、范围和限量等方面对欧盟与中国食品添加剂的法规标准进行对比分析。
结果中国食品添加剂在概念、分类、编码、使用品种、范围和限量标准等方面与欧盟仍有差距。
探讨了欧盟与中国食品添加剂标准结构以及色素、甜味剂、防腐保鲜类品种的使用差异,提出了根据中国食品添加剂在生产、使用和检测技术中存在的问题,进一步完善法规标准建设,并探讨我国进出口食品及食品添加剂贸易的对策。
结论中国和欧盟的食品添加剂标准法规存在较大差异,会影响到双方的进出口贸易。
关键词食品添加剂;中国;欧盟;法规;标准;对比中国图书资料分类号:R155.5文献标识码:A文章编号:1004-1257(2011)12-1332-07Subject Contrast on Food Additive regulations and Standards between EU and ChinaAuthorsYAO Si-jie ,DAI Han-hui ,LI Xing ,ZOU Zhi-fei (Guangdong College of Pharmacy ,Guangzhou ,510310,China )Abstract [Objective ]To offer technical support for management and use of food additives in China.[Methods ]The differences in regulations and standards of food additives between EU and China from the definition ,functional categories ,coding ,variety ,service-able range ,dose limits were compared and analyzed.[Results ]There were differences in definition ,functional categories ,coding ,variety ,serviceable range ,dose limits and so on of food additives between EU and China.The paper explored the difference of ad-dictive use between EU and China on standard structure ,and the use of pigment ,sweetener ,preservation class ;put forward the problems which exist in production ,use and detection of addictive ;modified the regulations and standards ;and offered some coun-termeasures on food and food addictive which involved in imports and exports in China.[Conclusion ]There are more differences be-tween China and the EU in food additives standards and regulations ,which will affect the import and export trade.Key wordsFood additives ;China ;EU ;Standard ;Contrast基金项目:国家质检总局科技计划项目(项目编号:2009IK310);中国检科院资助项目(项目编号:2009JK011);广东省科技基础条件建设项目(粤科财字[2010]885)作者简介:姚斯洁,女,食品质量与安全专业在学。
饮用水细菌总数标准

饮用水细菌总数标准
饮用水的细菌总数是衡量水质安全和卫生的一个重要指标。
不同国家和地区对于饮用水中细菌总数的标准可能有所不同,但通常都有明确的限值以确保饮用水的安全。
以下是一些常见的指导原则和标准:
1.世界卫生组织(WHO)标准:WHO提供了饮用水质量
指南,但并没有为细菌总数设定具体的数值限制。
WHO强调,饮用水中不应检出大肠杆菌或其他病原
体,这通常被视为水质良好的标志。
2.美国环保署(EPA)标准:美国对饮用水的细菌标准
是非常严格的。
EPA规定,自来水中大肠杆菌或类
似病原体的存在应为零。
3.欧盟标准:欧盟的饮用水指令要求成员国保证供水
系统中水的微生物安全。
与EPA类似,欧盟也强调
大肠杆菌的不存在。
4.中国标准:中国《生活饮用水卫生标准》中规定,
自来水的细菌总数(以每1毫升计)不得超过100
个单位,且不能检出大肠杆菌。
5.日本标准:日本的饮用水标准要求细菌总数每1毫
升不超过100个。
以上标准强调,虽然细菌总数是一个重要的指标,但更关键的是病原性微生物(如大肠杆菌)的存在。
水中的细菌总数可能因自然原因略有波动,但病原体的存在通常表示水源受到了污染。
对于个人和家庭来说,了解当地的饮用水标准并定期检测家中水质是保证饮用水安全的重要步骤。
如果对水质有疑虑,使用过滤器或沸腾水来消毒是较好的选择。
QA法规培训材料--浅析美国、欧洲和WHO工艺验证指南类

QA法规培训材料--浅析美国、欧洲和WHO工艺验证指南类因欧洲的《工艺验证指南(草案)》文件在“立法基础”一段中提到与2001/83/EC和2001/82/EC 相关内容一道阅读,所以其目的应在于对某些问题的强调、补充说明或解释。
该文件对工艺验证的具体活动要求阐述相对较少,但将“没有使用连续工艺确证的工艺”做了定义,称之为“非标准工艺”,并对“非标准工艺”的划分做出了较为详细的解读,方便应用和识别。
在“定义”一章中,有两个新的名词——“设计空间”和“高影响模型”。
其中,“设计空间”可以理解为保证产品质量的工艺参数和物料属性等变量的相互影响和相互作用的集合。
而如果来自模型的预测是一个重要的产品质量指标则这个模型可以认为是“高影响模型”。
WHO的《药品生产质量管理规范补充指南:验证,附录7:非无菌工艺验证》(修订提案)因其仅仅适用于制剂产品,所以篇幅不长。
该指南参考了美国的《工艺验证:一般原则与规范》,在用词风格与欧洲的《工艺验证指南(草案)》近似。
整篇文件短小精悍,在“持续的工艺确证”之后,一个表格——“表2.工艺验证新途径”将工艺验证的概要汇总其中,工艺验证的各个阶段和每阶段的主要任务一目了然。
在“变更”一章中,除了提出总体要求,也列举出了需要考虑再验证的变更情况。
浅析三方工艺验证的共同理念该3篇工艺验证指南类的文件虽然篇幅长短迥异,内容上也可以列出近10条的不同之处,但是工艺验证的精髓即理念是完全一致的——即在药品的整个生命周期内,应用足够的产品和工艺知识,采用质量风险管理和质量体系管理的相关技术实施工艺验证。
从工艺设计开始,直到商业化生产后的持续工艺确证,工艺验证贯穿于整个产品的生命周期。
而ICH-Q8(《药品开发》)、ICH-Q9(《质量风险管理》)和ICH-Q10(《药品质量体系》)3个指导原则被3个工艺验证指南类的文件共同引用参考,他们的理念也潜移默化地渗透到对工艺验证的方方面面要求之中。
国内外饮用水水质对比分析

国内外饮用水水质对比分析一、国内外饮用水水质标准项目的比较世界卫生组织(WHO)、美国、欧盟、日本等饮用水水质标准代表了当今世界饮用水标准方面的最高水平。
分析其指标可以发现有机物指标的数目均超过水质指标总数的2/3左右,特别是消毒副产物项目的增加,反映了人类对控制有害有毒有机物认识的加深及其有关分析检测技术的进步。
我国的生活饮用水卫生规范与国外饮水标准项目的比较见表1。
从指标数量来看,生活饮用水卫生规范基本与世界接轨,表明我国的饮用水卫生标准已经向前迈出了一大步。
从检测项目上看,这次标准的修改,增加了大量的有机污染物的毒理学指标,这与国际上水质标准的总体发展趋势相一致。
生活饮用水卫生规范总体上克服了以前标准中有毒有害项目偏少、指标值不严、感官项目重视不够、微生物项目尤其是致病原生动物检测指标过于简单的缺点。
表1我国生活饮用水卫生规范与国外饮水标准项目比较我国目前饮用水标准的执行、实施、修订没有相关的法律法规。
这就造成了在实施过程中,其执法的力度不够,没有起到标准本该有的作用,许多标准成为“软标准",在实施中根本就没有执行,美国1974年制定了《安全饮水法》,此后又制定了《国家暂行饮用水基本规则》,以保证生活水质标准的执行和实施。
这一点是值得我国借鉴的。
另一方面,由于城市水质监测技术的落后及城市供水管网设备的陈旧,成为实施饮用水标准的障碍,这一障碍与我国水质标准的前瞻性有很大关系。
标准从颁布到实施,留给供水企业的适应时间太短,使其无法在短时期内适应人员素质提高、管网更新、引进先进监测技术等要求。
二、国内外饮用水水质标准的对比分析综合评价根据世界卫生组织(WHO)的《饮用水水质准则》、欧盟(EC)的《饮用水水质指令》和美国环保局(USEPA)的《国家饮用水水质标准、英国《生活饮用水水质标准》及我国《生活饮用水水质标准》GB5749-2006等,可能影响人体健康或可能导致“三致”的指标将近两百项,其中有一部分具有地区选择性(即有的地区存在的污染物在另一个地区并不存在),还有一部分指标各国限值相同(如细菌总数等),为了科学起见,选择其中对人体影响较大的一些指标,建立水质评价指标体系。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
欧盟与WHO无菌药品要求比较
一、章节对照表
二、灭菌方式比较
三、欧盟与WHO无菌药品文本的对比
1、微粒表述方式不同
1)欧盟以≥0.5μm及>5.0μm;
2)WHO则是0.5-5.0μm和>5.0μm二种规格,(DH:由于大的微粒很少,实际
测试数据可能二者可能一样)。
2、微生物控制限度不同
1)欧盟对A区微生物控制(空气沉降菌及浮游菌、表面、手套)均为不得
检出(小于一个菌);(DH:欧盟与FDA一样,比WHO严格)。
2)WHO则为 3个菌,B、C、D级别区控制指标与欧盟完全相同。
3、监控要求不同
1)欧盟第3条洁净级别划分表的说明中提到:应对A级区的空气粒子进行
连续测定,并建议对B级区也连续进行测定…。
修订版第5条提到:…
除有充分理由以外,例如活病毒疫苗的灌装,A区的微粒应连续监控或
频繁取样。
建议B区也采用类似的系统,尽管取样频率可适当降低。
这
类体系通常需要独立的粒子计数仪;或有一个粒子计数器,这个计数器
通过一个多接口的管子,与数个连续的取样点相接。
如使用远程监控系
统,管子的长度及任何管的弯曲半径均应验证。
A区监控的频率应足以
保证检出所有的干扰及偶发事件,一旦发生偏离正常情况时,即能启动
报警系统(DH:基本均是连续取样的要求,如不是连续,如何能检出干扰?)
欧盟第93条要求:容器的密封应采用经验证的方法。
熔封性容器,如玻
璃或塑料安瓿,应进行100%的完好性检查。
应根据适当的方法对其它容
器的样品进行密封完好性检查。
半压塞冻干的小瓶,从半压塞开始至轧
盖应始终处在A级的保护之下。
无菌灌装的小瓶在完成轧盖前,尚不是一个完整的密封系统。
因此,应
在A级保护下直到轧盖完成。
2)WHO无连续测试要求;无菌灌装小瓶的轧盖,无百级保护的要求。
4、对D区的要求
1)欧盟附表说明(f)中提到:须根据生产操作的性质来决定此洁净区的要求
和限度。
2)WHO及欧盟对动态微粒均不作规定。
5、换气次数
1)欧盟第3条表下说明提到:为了达到B、C、D级区的要求,空气换气次
数应根据房间的大小、室内的设备和操作人员数决定。
空调净化系统应
当配有适当的过滤器,如:A、B和C级区应采用高效(HEPA)过滤器。
(DH:没有要求D区安装高效过滤器,现国内基本都是高效)。
2)WHO GMP 在4.1的说明中,提到:为了达到B、C、D级区的要求,换
气次数应根据房间的大小、室内的设备和操作人员的数量来决定。
每一
洁净室换气次数不低于20次/时,通常应使用合适的高效过滤器并有良
好的气流方式。
6、温湿度控制
1)欧盟第9条提到:温度、相对湿度等其它指标取决于产品及操作的性质,
这些参数不应对规定的洁净度造成不良影响。
(DH:洁净区不规定温、湿指
标,第50条内容与WHO相同。
)
2)WHO则在4.20提到:洁净区内环境的温湿度不应使穿洁净服的操作人
员感到不适。
7、注射用水
1)欧盟35条提到;…注射用水的生产、贮存和分配方式应能防止微生物生
长,例如,在70℃以上保持循环。
第44条.提到:水源、水处理设施及
水的化学和微生物污染状况应定期监测,必要时还应监测细菌内毒素,
但没有提到中间物料。
(DH:没有冷循环的提法,当然也不排除)
2)WHO 10.6条中提到:…注射用水的生产、贮存和输送方式,应能防止
微生物生长,如在70℃以上或4℃以下连续循环。
2.3条提到:…应对注
射剂的WFI及中间物料的细菌内毒素水平进行监控。
(DH:这是防污染方
式的示例,不是强制要求)
8、气体灭菌方法
1)欧盟灭菌法中只提到环氧乙烷。
2)WHO在6.15中提到:多种气体和熏蒸剂都可用于灭菌(例如环氧乙烷、
汽化过氧化氢)。
(DH:过氧化氢是指南中首次出现的新方法。
我国滴眼剂用臭
氧消毒,情况在些类似WHO的提法,当然这要验证)。
9、培养基模拟试验
1)欧盟的标准现与FDA 2004年9月的标准相一致,第47条提到:…每一工
艺的培养基模拟试验通常每班每年进行2次。
培养基灌装容器的数量应足
以保证评价的有效性。
对批次量比较小的产品而言,培养基灌装的数量
应至少等于产品的批次量。
培养基模拟试验的目标是不长菌,应按以下
建议对无菌工艺进行评估:
i 灌封数低于5000时,不得检出污染。
ii 灌封数在5000至10000时:
--当有1瓶污染时,需进行调查,并应考虑重复培养基灌封试验
--当有2瓶污染时,需进行调查,并在调查后应进行再验证iii 灌封数超过10000时:
--当有1瓶污染时,需进行调查
--当有2瓶污染时,需进行调查,并应在调查后进行再验证对失败进行全面调查的内容,应包括上一次培养基灌装试验合格后,所
有生产批中可能影响产品无菌的因素(邓:现欧盟与FDA完全一致,要求比
WHO严格一些。
但WHO的要求,总体上与欧盟是一致的。
)。
2)WHO GMP 第42条:--培养基模拟试验的目标是不出现长菌,但可信度
为95%时,污染率应小于0.1%。
企业应建立警戒限度及纠偏限度标准。
发生任何污染时,均应进行调查。
10、生物负荷测试
1)欧盟在第57条提到:应监控灭菌前产品的微生物污染水平并确立控制标
准,此标准与所采用灭菌方法的功效相关。
无论是无菌灌装产品还是最
终灭菌产品,均应进行灭菌前微生物污染水平的检查。
采用过度杀灭程
序的最终灭菌产品,灭菌前微生物污染水平的检查也许可定期进行。
对
实施参数放行的产品而言,应将此试验视作中间控制并须每批检查。
…。
2)WHO 在4.26条也有相类似的要求,但没有作为中间控制来处理。
11、隔离操作技术吹/灌/封
1)欧盟有二节内容是WHO中没有的。
隔离操作技术:…隔离操作器所处
环境的级别取决于它们的设计及其应用。
无菌操作的隔离操作器所处环
境的级别至少应为D级;在吹-灌-封技术一节中提到:…该设备可以安
装在洁净度至少为C级的环境中。
在静态条件下,此环境微粒和微生物
均应达到标准,在动态条件下,此环境的微生物应达到标准。
用于生产
最终灭菌产品的吹/灌/封设备至少应安装在D级环境中(DH:现国内企业
已有多家采用隔离操作器,用于无菌检查;另杭州眼力健及正在研制的粉液包装,属类似工艺,因此,这二节的内容可收载入我国的标准中)
2)WHO 无此内容。
12、参数放行
1)欧盟稿中二次提到参数放行。
2)WHO 无参数放行的提法。
四、小结
1)二个文件的章节对应,欧盟多了二小节隔离操作技术吹/灌/封,这二个小
节是我国目前制药行业可采用的标准。
2)随着参数放行的实施,有关生物负荷测试要求可明确写入文件中。
3)对可最终灭菌产品而言,执行WHO标准最大的难点是生产环境的动态
监控,涉及的问题是HV AC的改造;
4)欧盟的连续监控及控制限度比WHO严格,除微粒连续监控的要求外,
欧盟新版要求冻干产品从半压塞至轧盖,均应有局部百级保护,这一原
则也适用于无菌分装。
我国有不少企业连无菌万级的设置都没有,压盖
在十万级是比较常见的做法,执行欧盟标准,不仅HV AC要改造,要增
加B区的设置,还要考虑厂房轧盖的保护问题。
新标准也不得不考虑欧
盟的合理要求,否则WHO标准一变,企业需又得重新改造一次。
具有
前瞻性的标准,才能使二次改造“合二为一”。
执行WHO的标准,当然
执行欧盟标准要轻松些,但对我国采用无菌制造工艺企业而言,执行
WHO的标准也存在很多困难。
5)草案编写总体思路:综合二个文件,基本采用欧盟文件的编写次序+
WHO的控制标准+ 适当的余地,使标准具有一定的前瞻性。
2006-12-11。