510K程序与费用
FDA与FDA认证及医疗器械FDA认证区别介绍与流程(图文word版)

美国FDA、FDA认证、医疗器械FDA认证介绍目录一、FDA (3)二、FDA认证 (4)(一)什么是FDA认证 (4)(二)哪些类型产品需要做FDA认证 (4)(三)FDA认证流程 (5)(四)FDA认证的误区 (5)三、医疗器械FDA认证 (6)(一)美国FDA对医疗器械的分类 (7)(二)医疗器械FDA认证步骤 (8)(三)医疗器械FDA认证资料 (9)(四)疗器械FDA认证注意事项 (10)(五)医疗器械FDA认证标准 (10)附件 (12)一、510K介绍 (12)二、GMP介绍 (14)一、FDAFDA”是美国食品药物管理局的英文缩写(Food and Drug Administration,FDA),是国际医疗审核权威机构,由美国国会即联邦政府授权,专门从事食品与药品管理的最高执法机关。
FDA是由医生、律师、微生物学家、药理学家、化学家和统计学家等专业人事组成,致力于保护、促进和提高国民健康的政府卫生管制的监控机构。
通过FDA认证的食品、药品、化妆品和医疗器具对人体是确保安全而有效的。
在美国等近百个国家,只有通过了FDA 认可的材料、器械和技术才能进行商业化临床应用。
FDA食品和药品管理局隶属于美国国务院保健与服务部(U.S.Department Of Health and Human Services)的公共健康服务署(Public Health Service),负责美国所有有关食品,药品,化妆品及辐射性仪器的管理,它也是美国最早的消费者保护机构,将近9,000名员工,管理着每年约1兆美元市场的制造、进口、运送和储藏,所管辖的动物、食物与药品制造业者超过12万家,其中以食品制造业者最多,约5万家。
其次便是医疗器械制造业者有三万两千余家,影响美国每个纳税义务人约3美元,可以说与社会大众的生活福利和生命安全息息相关。
FDA之中约有1,100名检查员,每年要赴海内外15,000个工厂,去确认他们的各种活动是否符台美国的法律规定;同时他们也必须搜集80,000项美国境内制造或进口的产品样品并施以检验。
心电图(ECG)设计面临的挑战及其应对措施

心电图(ECG)设计面临的挑战及其应对措施工程师们可以利用ADI解决方案来应对心电图子系统设计的重大挑战,包括安全、共模/差模干扰、输入动态范围要求、设备可靠性和保护、降噪以及EMC/RFI考虑。
心电图(ECG)是一种常见的医疗记录,在许多恶劣的环境中,它也必须清晰可读并保持精确。
无论是医院、救护车、飞机、轮船、诊所还是家里,干扰源无处不在。
新一代高度便携式ECG技术使我们能够在更多的环境条件下测量心脏的电活动。
随着ECG子系统越来越多地投入医院外应用,制造商面临着持续的降低系统成本并缩短开发时间,同时保持或提高性能水平的压力,这就给ECG设计工程师提出了相当严苛的要求:实现一种安全有效、能够应对目标使用环境挑战的ECG子系统。
本文说明通常所认为的ECG子系统设计的主要挑战,并提供关于如何应对的各种方法建议。
本文讨论的挑战包括安全、共模/差模干扰、输入动态范围要求、设备可靠性和保护、降噪以及EMC/RFI考虑。
挑战1:达到最高安全标准,确保ECG子系统安全有效安全始终是ECG设计师的头号关注对象。
设计师必须严防来自交流电源的电涌或过压,以及经过ECG电极的任何可能超过10 μA rms推荐限值的电流路径影响到病人和操作人员。
在ECG子系统本身或其它与病人或操作人员相连的医疗设备发生故障时,可能出现危险电压或电流,ECG设计的终极目标就是确保病人和操作人员安全,不会受此类电压或电流伤害。
图1. 交流电源耦合简图开始ECG设计之前,工程师必须确定其临床应用及在哪里使用和存放设备。
工程师必须评估所有可能导致电流施加于病人的设备误用情况和潜在外部连接。
当施加的电流(吸入或流出)小于10 μA rms时,即使在单一故障条件下,操作人员和病人的安全也不会有问题。
必须防止病人意外触电,并且保护ECG设备不受紧急使用心脏除颤器所产生的极端电压影响。
ECG系统必须符合联邦法律、国际标准和相关国家/地区指令的要求。
510k认证流程

510k认证流程
510(k)认证流程是美国食品药品监督管理局(FDA)对医疗器械进行的一种上市前审核程序,旨在确保该产品在市场上投放之前,其安全性、有效性及可靠性已经得到了充分的验证和评估。
以下是详细的510(k)认证流程:
1、准备申请材料:申请者需要准备并向FDA提交510(k)申请,包括产品描述、使用说明、安全性和有效性数据等。
在这一步,申请者需要对产品进行详细的描述,并收集所有必要的数据来证明产品的安全性和有效性。
2、递交申请:将所有准备好的申请材料递交至FDA。
FDA会对收到的申请进行形式审查,检查提交的材料是否齐全、是否符合法规要求。
3、确定审查方式:FDA会对申请进行分类,根据产品的风险等级确定是否需要进行实质审查。
对于高风险的医疗器械,FDA会进行实质审查,确保产品的安全性和有效性。
4、实质审查:如果需要进行实质审查,FDA会对申请材料进行详细的评估,包括产品的安全性、有效性、可靠性和上市后监控等方面的数据。
FDA会对这些数据进行严格的审查,以确保产品的安全性和有效性。
5、做出决定:基于实质审查的结果,FDA会做出是否批准或拒绝上市的决定。
如果申请被批准,FDA会颁发医疗器械许可证,允许产品在市场上销售。
如果申请被拒绝,申请者可以要求FDA做出进一步的评估或提出申诉。
6、保持监管:即使产品已经获得510(k)认证,FDA还会对产品进行持续的监管,以确保产品的安全性和有效性。
在整个510(k)认证流程中,申请者需要与FDA保持密切的沟通,及时回应FDA的疑问和要求。
同时,申请者还需要确保所提交的所有数据真实、准确、完整,以避免被拒绝或撤销认证。
医疗器械 510k证书格式 -回复

医疗器械510k证书格式-回复什么是医疗器械的510(k)证书?在医疗器械正式上市之前,通常需要进行严格的监管和评估。
在美国,食品药品监督管理局(FDA)负责监管医疗器械的上市,并要求厂商根据适当的程序获得必要的证书。
而医疗器械的510(k)证书就是其中之一。
510(k)证书是指厂商根据美国联邦法规第21节第807.92(b)项,向FDA提交的一份产品先例报告。
这份报告的目的是向FDA证明正在上市的医疗器械与已经通过FDA审批的类似产品具有相似的安全性和有效性。
换句话说,510(k)证书是通过比较已经获得批准的医疗器械和待上市的器械来证明其相似性,从而减少重复测试和评估的过程。
510(k)证书报告的内容通常包括以下几个方面:1. 产品介绍:包括厂商名称、产品型号和名称、适应症和用途等相关信息。
2. 先例产品信息:列出与待上市产品相似的已获得FDA批准的产品信息,包括正式名称、批准号、适应症和用途等。
3. 技术比较:对待上市产品和先例产品的技术参数进行比较,包括结构、材料、工作原理等。
4. 安全和有效性分析:通过比较待上市产品和先例产品的安全性和有效性数据,证明待上市产品具备与已批准产品相当的特性。
5. 测试和验证数据:提供与待上市产品相关的测试和验证数据,包括实验室测试结果、临床试验数据等。
6. 风险评估:评估待上市产品可能存在的风险,并提供相应的控制措施和风险分析。
7. 总结和结论:对比较结果和分析进行总结,并得出结论,证明待上市产品与已批准产品的相似性。
在提交510(k)证书后,FDA会对其进行审查,并根据证据和分析结果决定是否批准该产品上市。
如果审查通过并获得批准,厂商将获得510(k)准则对应的证书,并可以正式开始销售该产品。
需要注意的是,虽然510(k)证书可以简化产品上市的过程,但并不代表产品完全没有风险。
FDA仅通过比较已批准产品来判断待上市产品的相似性,因此仍然需要厂商对待上市产品进行严格的验证和测试。
传统和简略的510(k)格式

传统和简略的510(k)文件的格式该文件发布于2005年8月12日序言公共评论起草的评论和建议可在任何时间提交给FDA,5630Fisher Lane,1061房间,Rockville,MD,20852。
当提交评论时,请注明准确的文件标题。
直到该文件被修改或升级时,该评论才会被实施。
另外的副本另外的副本可从互联网中获取:/cdrh/oivd/guidance/1567.pdf 或拨打301-827-0111。
拨1进入系统,在第二声提示的时候,拨1或索要文件。
本指南是代表FDA现时在问题焦点的想法。
它没有产生或赋予任何人权利,并且没有在约束FDA和大众的情况下运行。
若该方法满足适用的条例、法规或两者的要求,则可使用该方法。
若您想讨论使用其他方法,直接联系FDA实施该指南。
若您未找到FDA,呼叫本指南中的电话。
简介本文件的主要观点是如何规范原始的510(k)文件。
本指南仅提供了一个大体的组织框架和传统或简略510(k)文件的内容。
这并不代表我们的建议对任何型式1的设备,特殊510(k)文件或其他型式文件,例如上市前许可申请(PMAs)或研究器械豁免申请。
(IDEs)FDA认为该指南中的建议性文件能够保存FDA和企业资源定期审核。
本指南补充其他FDA 指南中的510(k)程序和特殊设备类型,不是一个代替文件。
另一种方法,你可以提交协调格式的,该文件在“医疗器械安全和性能基本原理论证一致性的技术文件”中进行了描述,或在STED草案文件中找到。
找CDRH网站关于设备特殊指南,网址/scripts/cdrh/cfdocs/cfggp/search.cfm特殊510(k)文件的选项允许申请者澄清他们本国法规上市的医疗器械并且没有影响改设备预期使用的变化。
见/cdrh/ode/parad510.html。
包容不具约束力的建议FDA指南,对提议全球一致性的预上市程序进行全面评估的试点项目,对FDA试点程序和适宜型号的指南。
美国人道主义器械豁免政策分析

美国人道主义器械豁免政策分析[目的]对比FDA的人道主义器械和传统器械审批程序,为我国完善治疗罕见病医疗器械的审批提供一定的参考。
[方法]采用比较法,从申请标准、盈利限制以及上市后监察几个方面对FDA医疗器械不同的审批方式进行对比。
[结果]HDE政策的实施有力推动了罕见病医疗器械的创新和发展。
[结论]我国应采取激励政策,促进罕见病医疗器械的发展。
标签:人道主义用途器械;人道主义器械豁免;医疗器械;上市前许可D9随着科技的不断创新,医生在工作中较之以往任何时候都更依赖于医疗器械的辅助。
在过去几十年间,医疗器械产业经历了从种类、用途到复杂性的爆发式增长。
目前,全球总医疗器械市场价值已超过4,118亿美元,其中近半数的产出和消耗都发生在美国。
截至目前,美国共有超过6,500家医疗器械生产企业,人均医疗器械支出约为每年392美元,是全球最大的医疗器械市场。
美国食品药品监督管理局(FDA)按风险等级对医疗器械实行分类审批管理,审批方式主要有2种:上市前通告(Pre-market Notification,简称510K)和上市前审批(Pre-market Approval,简称PMA)。
为了满足罕见病患者的医疗需求,美国国会于1990年颁布了《医疗器械安全法案》,提出了人道主义器械豁免(Humanitarian Device Exemption,简称HDE)的审批途径。
如果某种医疗器械的目的在于治疗或诊断某种疾病或病症,且该疾病或病症每年在美国的影响人数不超过4,000名,则该器械可申请获得了人道主义用途器械(Humanitarian Use Device,简称HUD)资格,并通过HDE途径得到审评。
本文研究了FDA人道主义器械豁免途径和传统医疗器械审批途径的不同流程,着重从申请标准、盈利限制以及上市后监察等方面比较二者的差异,总结了HDE审批途径的潜在优缺点,以期为我国完善医疗器械审批方式提供一定的参考。
FDA-510K所需准备的文件

510(k)s) 10 Executive Summary 11 Device Description
财务证明或应行公告的财务事项
一致性声明和总结报告(简要510k) 执行总结 设备说明
序号 1 2
Title
Medical Device User Fee Cover Sheet(Form FDA 3601) CDRH Premarket Review Submission Cover Sheet
3 510(k) Cover Letter
4 Indications for Use Statement
5 510(k) Summary or 510(k) Statement 510k总结或510k声明
6 Truthful and Accuracy Statement
真实与准确性声明
7 Class III Summary and Certification III类总结和声明
Financial Certification or 8 Disclosure Statement
文件名 缴费证明文件
CDRH上市前审核提交表格 510k封面信件 预期用途
Related Information Medical Device User Fee Cover Sheet
CDRH Premarket Review Submission Cover Sheet Appendix A of "Guidance for Industry and FDA Staff Format for Traditional and Abbreviated 510(k)s" Indications for Use Statement
美国FDA医疗器械注册(510k)

510(k)申请类型
传统 特殊 简略
510(k)申请类型(1)——传统510(k)
包含21 CFR第807.87部分中所列的所有要素 在90天内审核
510(k)申请类型(2)——特殊510(k)
申请人对其合法销售器械进行了重大改动; 申请人认为需要进行新的510(k)申请; 这些改动不影响器械的预期用途或基本科学技术; 申请人按照21CFR第820.30部分(设计控制)对改 动进行评估; 申请人递交510(k)申请的同时,也递交符合设计控制 原则的声明; 在30天内审核。
510(k)——上市前通知
何时需要510(k) 医疗器械初次投放美国市场 已上市医疗器械预期用途变更 已上市器械经过重大改动 /cdrh/devadvice/314.html 免于510(k)审批的器械—798 / 47% 第一类:729 / 93% 第二类:69 / 9%
510(k)——上市前通知
已获批准类似器械的定义(Predicate device): 无需上市前批准(PMA)的合法销售器械,例如: - 在修正案制定前(1976年5月28日)就已合法上市的器械; - 美国FDA认定具有实质等效性的器械;或者 - 重新分类的器械。 510(k)对于实质等效性的判断: - 如果认定不具有实质等效性(NSE): 公司必须提交上市前批准(PMA)、产品开发协议(PDP)或 人道主义设备豁免(HDE)申请,或者 要求将产品依风险等级重新分类(De Novo Process) - 如果认定具有实质等效性: 公司可将器械合法投放美国市场
510(k)的内容和格式 联邦法典(CFR) 21 CFR第807.87部分—内容 21 CFR第807.90部分—格式 器械特别指南,例如:活塞式注射器 CFR在线浏览网址:/cdrh/devadvice/365.html 器械建议
医疗器械出口流程详解

XXXXXXXXXXX有限公司---------- XXXXXXXXXXXXXXXXXXXXXXXXXXXX出口技术服务CE专项服务FDA510K专项服务目录一、相关前置信息二、医疗器械产品出口销售证明书的获取三、出口产品信息四、CE-MDD (Wa)认证五、FDA510K认证六、相关报关报检手续一、相关前置信息2类以上医疗器械,出口美国应该经FDA审核,按510(k)的要求取得K号。
欧盟国家需要NB机构(即公告机构-认证机构)进行CE审核后签发CE证书。
其他国家一般要向国家监管部门申请注册如澳大利亚、泰国等。
但泰国等亚洲国家认可SFDA的证书。
二、医疗器械产品出口销售证明书的获取在SFDA网站上下载“医疗器械(体外诊断试剂)电子申报软件”,填写出口申请表,并提供:1、已取得医疗器械注册证的产品,向SFDA提交以下文件:①出口产品生产者的《医疗器械生产企业许可证》(复印件)②出口产品的医疗器械注册证(复印件)③出口企业的营业执照(复印件)④申请者的自我保证声明,保证所出口产品满足药品监督管理部门对该产品生产和注册的法规要求,所提交的材料真实合法。
所提交的证书复印件需加盖证书所属企业公章。
2、未取得医疗器械产品注册证的产品,提交以下文件:①生产企业的营业执照(复印件)②出口企业的营业执照(复印件)③ 申请者自我保证声明,保证所提交的材料真实合法。
所提交的证书复印件需加盖证书所属企业公章。
出口证明相对简单,提交相关文件后,会很快获得SFDA审批,取得《中华人民共和国医疗器械产品出口销售证明书》三、出口产品信息I、CE-MDD (Wa)认证1、CE-MDD (Illa)流程及周期五、FDA510K认证1、FDA510K申报流程和周期六、相关报关报检手续(一)出口货物的申报出口货物的发货人或者他们的代理人在货物出口时应在海关规定的期限内按海关规定的格式填写出口货物报关单随附有关的货运、商业单据同时提供批准货物出口的证件向海关申报。
FDA法规讲座之510K文件编写

1. 510(k)编写技巧 2. 医疗器械的变更和特殊510(k) 3. 第三方审核项目
510(k)编写技巧
•介绍 •报告基本原则 •报告内容要求
介绍
A 510(k) is a premarketing submission made to FDA to demonstrate that the device to be marketed is as safe and effective, that is, substantially equivalent (SE), to a legally marketed device that is not subject to premarket approval(PMA).
报告内容要求(Cont.)
软件验证 颜色添加剂要求 组合产品要求
行政文件要求
准确及可信声明 (Truthful and Accuracy Statement)
法规21 CRF 807
技术总结(510(k) Summary)
法规21 CRF 807.92
技术声明(510(k) Statement)
包装和标识 (Cont.)
v
包装和标识 (Cont.)
基本要求相关法规
21 CFR 801.1 制造商的名称地址 21 CFR 801.4 预期用途 21 CFR 801.5 指导文字 21 CFR 801.6 错误和误导标识 21 CFR 801.15 标识文字的清晰规范要求
包装和标识 (Cont.)
指导文字(21 CFR Part 801.5)
足够的使用指导信息,使至少非专业使用者能够安全有效的使用产品 说明产品所有能够正常使用条件 剂量等等 应用人群 使用频率 应用持续时间 应用的路径和方法 必要的使用前准备
510K介绍

1 of 14
FDA产品编码(Product Code)
• 产品分类数据:
/scripts/cdrh/cfdocs/cfPCD/classific ation.cfm
三个字 母的编
FDA 510(K)程序
——2017.12
什么是510(K)
• 510(K),又称上市前通知(Premarket Notification),是医疗器械 进入美国市场的主要途径之一。
• 联邦食品、药品和化妆品法案(Federal FD&C Act)的510(K)章节 • 实际上是医疗器械进入美国市场的最常用途径。 • FDA医疗器械分类:
5 of 14
510(K)的资料
• Medical Device User Fee Cover Sheet(Form FDA 3601)
• CDRH Premarket Review Submission Sheet
• 510(K) Cover Sheet • Indications for Use Statement • 510(K) Summary or Statement • Truthful and Accuracy Statement • Class III Summary and Certification • Financial Certification or Disclosure
8 of 14
谢谢观看!
• 或 技术特性的差异不会引起安全和有效性问题
3 of 14
考虑实质等同的基本思路
• 等同器械是否合法上市? • 器械的预期用途是否相同? • 器械的技术特性是否相同? • 技术特性的差异是否会引起安全和有效性问题? • 用科学的方法来证明技术特性的差异不会引起安全和有效性问题:
模板 中国注册与FDA 510k技术资料对照一览表
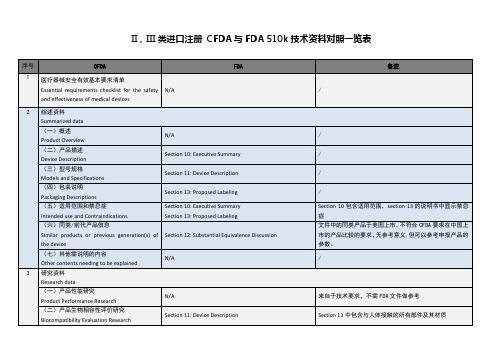
来自于技术要求,不需FDA文件做参考
(二)产品生物相容性评价研究
Biocompatibility Evaluation Resห้องสมุดไป่ตู้arch
Section 11: Device Description
Section 11中包含与人体接触的所有部件及其材质
(三)生物安全性研究
Biosafety Research
(一)概述
Product Overview
N/A
/
(二)产品描述
Device Description
Section 10: Executive Summary
/
(三)型号规格
Models and Specifications
Section 11: Device Description
/
(四)包装说明
Section 10: Executive Summary
Section 17: Electromagnetic Compatibility and Electrical Safety
Section 18: Performance Testing – Bench
Section 9包含全部标准清单及指南文件(guidance)清单,指南文件类似于CFDA的指导原则,有助于技术要求的编写
Section 20:符合FDA要求的临床试验
6
产品风险分析资料
Risk analysis data
Section 16: Software
510k只对软件风险有要求,不关注产品整体风险
7
产品技术要求
Product technical requirements (PTR)
FDA 510k注册流程

FDA 510k注册一、FDA法规要求按照美国食品药品监督管理局(FDA)相关规定,任何一种医疗器械进入美国市场前,必须厘清申请产品分类和管理要求。
根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级最高,Ⅰ类风险最低。
对I类产品实施一般控制(General Control);II类产品实施特殊控制(Special Control);对III类产品实施上市前许可。
对于任何产品,企业都需进行企业注册(Registration)和产品列名(Listing)。
对Ⅰ类产品实行的是一般控制,绝大部分产品只需进行注册、列名和实施GMP规范后产品即可进入美国市场销售,其中极少数产品连GMP也豁免,极少数保留产品则需向FDA递交510(K)申请PMN;对Ⅱ类产品实行的是特殊控制,企业在进行注册和列名后,还需实施GMP和递交510(K)申请。
FDA对大多数Ⅱ类产品均要求进行上市前通告(510K)。
生产企业须在产品上市前通过510K审查后,产品才能够上市销售;对Ⅲ类产品实施的是上市前许可,企业在进行注册和列名后,须实施GMP并向FDA递交PMA申请(部分Ⅲ类产品还是PMN)。
对于I类产品,企业向FDA递交相关资料后,FDA只进行公告,无相关证件发给企业;对于II、III类器械,企业递交PMN或PMA后,FDA在公告的同时给企业以正式的市场准入批准函件。
至于申请过程中是否进行GMP审核,则由FDA根据产品风险等级、管理要求和市场反馈等综合因素决定。
二、FDA 510K基本流程美国FDA对流入本国并在本国上市销售的医疗器械产品,实行严格的法规把控,510K产品在申报时必须通过FDA官方审查。
1、确定产品类别2、确定美国授权代表3、FDA企业注册和产品列明4、确定适用的指南3、确定适用标准和所有检测项4、确定已上市同类产品5、检测6、编写510k文件报告7、递交报告8、FDA审核。
美国 FDA课程 理解

FDA课程看完比尔萨顿所介绍的内容,我感觉到美国FDA与我国的食品和药品管理局相比,美国法律中对于医疗器械的定义对比与中国的更加广泛。
FDA规划了大约1700种医疗器械可分列为16个医疗器械专项,在联邦法规第21卷第800-1299部分。
医疗器械分为第一、二、三类医疗器械,并对其有着不同的控制。
第一类医疗器械风险度低属于普通控制;第二类医疗器械风险度中属于普通控制以及特别控制;第三类医疗器械风险度高,属于普通及特别控制和上市前批准(PMA)。
美国食品和药品管理局由五个中心及监管事务办公室构成:食品安全和应用营养中心(CFSAN)、药品评估和研究中心(CDER)、生物评估和研究中心(CBER)、医疗器械和放射健康中心(CDRH)、兽医药物中心(CVM)、管事务办公室(ORA)。
医疗器械和放射健康中心由六个办公室构成:器械评估办公室、交流、教育和放射项目办公室、监督和生物测定办公室、科学和工程实验室办公室、法规遵守办公室、体外诊断器械评估与安全办公室。
卫生与公众服务部部长直接向美国总统负责。
美国医务总监担任公共卫生局军官团团长,也是联邦政府在卫生事务上的首席发言人。
各个机构的所处位置:卫生与公众服务部-公共卫生局-NIH,HRSA,AHRQ,HIS,SAMHSA,CDC,FDA。
食品和药品管理局在美国政府部门中所处的位置。
美国国家卫生研究院(NIH)、卫生资源和服务局(HRSA)、医疗保健研究和质量署(AHRQ)、印第安人卫生服务局(HIS)、药物滥用和精神健康服务局(SAMHSA)、疾病预防和控制中心(CDC)、食品和药品管理局(FDA)隶属美国卫生与公众服务部(HHS)。
美国国立卫生研究院(NIH)是美国政府负责生物医学和保健研究的主要部门。
卫生资源和服务局(HRSA)是负责没有医疗保险、孤立无援、一级健康状况脆弱的群体改善医疗条件的主要联邦机构。
医疗保健研究和质量属(AHRQ)为改善医疗成果和质量、消减成本、解决病人安全和医疗失误问题等。
二类医疗器械FDA认证流程——510K提交步骤

二类医疗器械FDA认证流程——510K提交步骤FDA(美国食品药品监督管理局)是负责对美国市场上销售的医疗器械进行监管和审查的机构。
在美国销售医疗器械,必须获得FDA的认证才能合法销售。
根据医疗器械的分类,FDA认证分为三个类别:一类、二类和三类。
本文将着重介绍二类医疗器械的FDA认证流程中的510(k)提交步骤。
1.确定器械的分类:首先,需要确定医疗器械的分类。
根据FDA的定义,二类医疗器械是指需要通过一份称为510(k)的材料进行市场申报的器械。
目的是证明新器械与FDA已批准上市的同类器械相似,具有相同的作用和安全性。
2.收集现有资料:在进行510(k)提交之前,需要收集一系列相关的资料,如市场调查报告、设计开发文件、生产质量计划等。
这些资料将用于证明新器械与同类已上市器械的相似性。
3.编写510(k)报告:根据FDA规定的格式,编写510(k)报告。
该报告应包括以下内容:a.适用范围:说明器械的适用范围和预期用途。
b.相似性比较:详细对比新器械与已上市同类器械的特性、注射剂、材料等,证明其具有相似性。
必要时,可以提供测试报告或数据支持。
c.临床数据:如有必要,需提供临床试验的数据,以证明新器械的安全性和有效性。
d.风险分析:分析新器械可能产生的各种风险,并提供相应的风险控制措施。
5. 提交FDA注册申请:将完成的510(k)报告和相关资料提交给FDA 进行注册申请。
申请可以通过FDA的电子提交系统(eSubmitter)或纸质方式提交。
7.通知:若FDA审核通过,批准器械上市销售,FDA将向申请人发出确认信。
若审核未通过,FDA将提供不通过的原因和建议。
需要注意的是,以上是一般的510(k)提交步骤,不同的医疗器械可能会有不同的要求和程序。
因此,在进行510(k)提交之前,确保充分了解和遵守FDA的相关规定非常重要。
总结起来,二类医疗器械的FDA认证流程中的510(k)提交步骤包括确定器械分类、收集现有资料、编写510(k)报告、审核、提交FDA注册申请、FDA审核和通知。
510k申诉技巧

510k申诉技巧全文共四篇示例,供读者参考第一篇示例:在医疗器械领域,想要将新产品上市需要经过严格的审批过程。
510(k)是美国食品药品监督管理局(FDA)规定的一项重要的途径,用于加快市场准入。
有时候申请人可能会收到FDA的拒绝通知,这时就需要进行510(k)申诉。
下面我们将介绍一些有关510(k)申诉技巧,希望对需要申诉的人员有所帮助。
1. 了解拒绝通知的原因当收到FDA的拒绝通知时,申请人需要认真阅读通知并仔细分析拒绝的原因。
通常,FDA会在通知中指出产品在哪些方面不符合要求,这为申诉提供了重要的线索。
申请人需要对拒绝通知内容进行细致的剖析,找出具体的问题所在。
2. 谨慎选择申诉方式接下来,申请人需要考虑如何进行申诉。
在510(k)申诉中,有两种主要的方式,一种是向FDA提交书面申诉,另一种是请求进行面对面会议。
申请人可以根据自己的情况和需求选择合适的申诉方式。
通常情况下,如果问题比较严重或者比较复杂,建议选择面对面会议,以便更好地与FDA进行沟通和交流。
3. 准备充分的申诉材料在进行510(k)申诉之前,申请人需要准备充分的申诉材料,包括但不限于产品测试报告、技术文档、市场调研数据等。
这些材料可以帮助申请人向FDA证明产品的安全性和有效性,从而提高申诉的成功率。
申请人还需要制定申诉的详细计划和策略,确保申诉过程顺利进行。
4. 与FDA进行积极沟通在进行510(k)申诉过程中,申请人和FDA之间的沟通至关重要。
申请人需要及时回复FDA提出的问题和要求,确保信息的及时传递和沟通的畅通。
申请人还可以主动与FDA进行交流,寻求进一步的帮助和支持。
通过良好的沟通,申请人可以更好地理解FDA的要求和期望,从而更好地应对申诉过程中的挑战。
5. 寻求专业支持申请人可以寻求专业的支持和帮助,以提高申诉的成功率。
可以请律师或专业顾问协助处理申诉事务,确保申诉的合理性和可行性。
申请人还可以参加相关的培训和研讨会,以提升自身在医疗器械领域的专业知识和技能。
510k

510(k)简介为了在美国上市医疗器械,制造商必须经过两个评估过程其中之一:上市前通知书[510(k)](如果没有被510(k)赦免),或者上市前批准 (PMA)。
大多数在美国进行商业分销的医疗器械都是通过上市前通知书[510(k)]的形式得到批准的。
在某些情况下,在1976年5月28日之前合法上市的器械,既不要求递交510(k)也不要求递交PMA。
510(k)文件是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上市前批准 (PMA)影响的合法上市器械同样安全有效,即为等价器械(substantially equivalent)。
申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。
合法上市器械是在1976年 5月28日之前合法上市的器械(preamendment device),或者从III类器械中分入II或I类的器械,或者通过510(k)程序发现与这样的器械等价的器械,或者通过自动的III 类器械定义的评价建立的器械。
与之等价的器械被称为“predicate device(s)”。
申请者必须提交描述性的数据,必要的时候,要提交性能数据来说明器械是predicate device的等价器械。
再次说明,510(k)的数据是显示相似性的数据,即,新器械与predicate device的等价程度。
FDA 等价器械510(k)不像PMA那样要求合理的安全性和有效性的证明,而是要求等价器械的证明。
等价器械就是新的器械与predicate device一样安全有效。
与predicate device相比,如果符合下列条件,就认为器械是等价器械:—与predicate device有相同的使用目的,具有相同的技术性能;或者—与predicate device有相同的使用目的,具有不同的技术性能,但是并没有增加安全性和有效性的问题,并且证明人证明器械与合法上市器械一样安全有效。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
PROFESSIONAL SERVICE PROPOSAL
BETWEEN
REGULATORY COMPLIANCE SOLUTIONS, LLC
And
HuBei Weil Kang Protective Products Co, LTD
Regulatory Compliance Solutions, LLC (RCS) is pleased to be able to contribute to HuBei Weil Kang Protective Products Co, LTD planned medical device product commercialization efforts and has made a genuine attempt to provide meaningful and realistic cost estimates.
Services:
Regulatory Guidance and Project Management
RCS will provide routine correspondences as to the status of the project via teleconference or email.
1. 510K Review and Advisory Assessment
A) Review existing 510K submission and all FDA correspondences.
i.FDA Interaction – Limit to 1 or 2 calls
1.Identify the next steps.
B) Provide information to client and discuss next steps.
2. Revision of the Present 510K Submission and Interaction with FDA.
A) Prepare revised 510K.
3. Prepare a New 510K Submission and Interaction with FDA.
A) Prepare 510K.
4. FDA Site Readiness Assessment
1
A) Evaluate and Prepare Documentation and Instructions to existing client staff for FDA Site
visit or audit.
Timelines: The timelines and hours will be agreed upon by the HuBei Weil Kang Protective Products Co, LTD and RCS and depend on the final submission of data and reports from the HuBei Weil Kang Protective Products Co, LTD and/or the HuBei Weil Kang Protective Products Co, LTD’s contractors. TERMS:
RETAINER FEE: $15,000.00 USD IS REQUIRED PRIOR TO STARTING. Retainer Fee will be applied to balance of due.
PAYMENT; DEFAULT: Payment in full for invoices shall be due within Pre-paid or Net 15 days from invoice date, at Regulatory Compliance Solutions, L.L.C. - 12 Holly Drive – Suite 100 Upper Saddle River, NJ 07458. Invoices that are more than (7) days past due the amount of the past due are subject to a late charge of one percent (3 %) per month on the amount of the past due balance. Late charges shall be calculated using the U.S. Method, therefore interest will not be compounded on the past due balance. If the
Client’s account is past due and REGULATORY COMPLIANCE SOLUTIONS, L.L.C. has notified Client
2
verbally or in writing of the past due balance, REGULATORY COMPLIANCE SOLUTIONS, L.L.C. may, without advance notice, immediately cease providing any and all further Contract Employee services without any liability to Client for interruption of pending work.
EXPENSES: Client shall reimburse REGULATORY COMPLIANCE SOLUTIONS, L.L.C. for all ordinary, necessary, and reasonable travel expenses incurred by Contract Employee(s) while performing services on behalf of Client. This estimate is based on the work being completed at Regulatory Compliance Solutions, L.L.C. offices.
COLLECTION:If the Client’s account, after default, is referred to an attorney or col lection agency for collection, Client shall pay all of REGULATORY COMPLIANCE SOLUTIONS, L.L.C. expenses incurred in such collection efforts including, but not limited to, court costs and reasonable attorneys’ fees. Notwithstanding the above and the terms of this Agreement, REGULATORY COMPLIANCE SOLUTIONS, L.L.C. may institute proceedings to seek a default judgment in any court of competent jurisdiction in the United States.
All payments to RCS are pre-paid or Net 15.
Client will give a Retainer Fee of $15,000.00 towards this project. This retainer fee will be for a maximum of 20 hours.
Total Estimated Costs of the Project is: $ 57,000.00 - $75,000 U.S. Dollars Disclaimer:
Regulatory Compliance Solutions, L.L.C. does not in any way or expressed the views of the FDA during the approval process. Regulatory Compliance Solutions, L.L.C. can not in any way guarantee FDA approvals.
Effective Date. The Effective Date of this PROFESSIONAL SERVICES Agreement is May 20, 2011. This proposal is valid for 14 days from today.
IN WITNESS WHEREOF, the parties have so agreed as of the date written above. REGULATORY COMPLIANCE SOLUTIONS, LLC 12 Holly Drive – Suite 100 Upper Saddle River, New Jersey 07458
3
Ms. Deborah Cenci
CEO and President
By: Its:
Date: 05/20/2011
Spenser (full name required and address) V.P. Product Development
HuBei Weil Kang Protective Products Co, LTD Address:
Address:
By: Its: ____________________________________ Date:
4。