美国FDA医疗器械510(k)申请深度分析系列一
如何准备电子血压计FDA 510 K 注册案例

后面根据FDA 510K Screen checklist 的要求项目对上述的报告内容要求,以电 子血压计为范例做详细描述
如何准备电子血压计510K 申请文件
FDA 510K Screen checklist 要求的申请文档或报告(1):
-
MDUFMA Cover Sheet 审核费用付款首页 相关信息链接:Medical Device User Fee Cover Sheet3 该项是本次510K提交项目的审核费用表格, 客户付款后要把paymentI 填 写在3601 FORM CDRH Premarket Review Submission Cover Sheet 提交审核信息首页 相关信息链接:CDRH Premarket Review Submission Cover Sheet4 该项主要是要求提交者填写Form FDA 3514, 是有关本次申请的基本信 息, 如sponsor, manufacturer等
如何准备电子血压计510K 申请文件
510(k)的提交报告内容简述—21 CFR第807.87部分(二)
510(k)总结或声明 510(k)必须包括下列内容之一: (1)510(k)安全性和有效性资料的“总结”,批准后公示于医疗器械与放射 卫生中心(CDRH)网站上(21CFR第807.92部分);或者 (2)510(k)‖声明“——如获要求,申请人必须在30天内提供510(k)信息 (21CFR第807.93部分) 器械用途声明 真实性和准确性声明 计划使用的标识(广告、盒标) 是否符合自愿标准(安全,电磁兼容以及专用标准) 财务证明或公告声明或者二者同时提供性能数据(实验室、动物实验、临床试 验) 灭菌、软件和硬件信息以及风险分析评估(如适用) 特别指南文件所需地址信息
510(K)

510(K)目录概述510(k)简介FDA 等价器械谁必须递交510(k)何时需要510(k)何时无需510(k)概述为了在美国上市医疗器械,制造商必须经过两个评估过程其中之一:上市前通知书[510(k)](如果没有被510(k)赦免),或者上市前批准(PMA)。
大多数在美国进行商业分销的医疗器械都是通过上市前通知书[510(k)]的形式得到批准的。
在某些情况下,在1976年5月28日之前合法上市的器械,既不要求递交510(k)也不要求递交PMA。
510(k)简介510(k)文件是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上市前批准(PMA)影响的合法上市器械同样安全有效,即为等价器械(substantially equivalent)。
申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。
合法上市器械是在1976年5月28日之前合法上市的器械(preamendment device),或者从III类器械中分入II或I类的器械,或者通过510(k)程序发现与这样的器械等价的器械,或者通过自动的III 类器械定义的评价建立的器械。
与之等价的器械被称为“predicate device(s)”。
申请者必须提交描述性的数据,必要的时候,要提交性能数据来说明器械是predicate device的等价器械。
再次说明,510(k)的数据是显示相似性的数据,即,新器械与predicate device的等价程度。
FDA 等价器械510(k)不像PMA那样要求合理的安全性和有效性的证明,而是要求等价器械的证明。
等价器械就是新的器械与predicate device一样安全有效。
与predicate device相比,如果符合下列条件,就认为器械是等价器械:—与predicate device有相同的使用目的,具有相同的技术性能;或者—与predicate device有相同的使用目的,具有不同的技术性能,但是并没有增加安全性和有效性的问题,并且证明人证明器械与合法上市器械一样安全有效。
FDA医疗器械510K申请文件介绍

FDA医疗器械510K申请文件介绍1.510(K)文件也即FDA对PMN所需的文件,因其相应FD&C Act第510章节,故通常称510(K)文件。
对510(K)文件所必须包含的信息,FDA有一个基本的要求,其内容大致如下16个方面:1)申请函,此部分应包括申请人(或联系人)和企业的基本信息、510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(K)号码;2)目录,即510(K)文件中所含全部资料的清单(包括附件);3)真实性保证声明,对此声明,FDA有一个标准的样本;4)器材名称,即产品通用名、FDA分类名、产品贸易名;5)注册号码,如企业在递交510(K)时已进行企业注册,则应给出注册信息,若未注册,也予注明;6)分类,即产品的分类组、类别、管理号和产品代码;7)性能标准,产品所满足的强制性标准或自愿性标准;8)产品标识,包括企业包装标识、使用说明书、包装附件、产品标示等;9)实质相等性比较(SE);10)510(K)摘要或声明;11)产品描述,包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等;12)产品的安全性与有效性,包括各种设计、测试资料;13)生物相容性;14)色素添加剂(如适用);15)软件验证(如适用);16)灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。
2.实质相等性比较(SE)实质相等性比较是要证明所申请上市的产品和已在美国市场上合法销售的产品在安全性和有效性方面比较是实质相等的。
选择合适的产品进行比较是510(K)申请的关键步骤。
在进行比较时应从如下方面进行考虑:企业必须提供充足的资料证明,所申请上市的器械和被比较的器械是实质相等的(SE),否则510(K)申请不会通过。
3.510(K)审查程序FDA在收到企业递交的510(K)资料后,首先检查资料是否齐全,如资料齐全,则受理并给企业发出确认性,同时给出申请受理编号(K YYXXXX),此号码也将作为正式批准后的号码;如不齐全,则要求企业在规定时间内补充齐全,否则作企业放弃处理。
心电监护产品的FDA 510(K)申请

适用范围-心律失常检测和报警器
• 心律失常检测和报警器
– 是一种监护ECG,并设计为在发生房性或室性心律失常(例如期 前收缩或室颤)时,产生视觉或听觉信号或报警的心电监护设备 – 可包括(或不包括)ST段报警功能,产品代码为MLD(Monitor, ST Segment with Alarm)
• 病人生理参数监护仪(MHX)Patient Physiological Monitor (with Arrhythmia Detector or Alarm)
健康风险-3
• 具有患者应用部分的心律失常检测器应被认为与 患者的完整皮肤有长时间的接触
– 对这些部分的材料进行生物相容性评价:依照标准 ISO10993 Biological Evaluation of Medical Devices Part 1: Evaluation and Testing – 在设计历史文件中记录评价结果并作为QS的一部分 (21 CFR 820.30) – 根据与设备接触的长短和程度来选择合适的测试方法 – 若已有同样的材料用于与患者接触具有相同类型和长 短的已上市产品,可以确认该已上市产品而代替生物 相容性试验
第四部分
软件确认活动 (Software Validation Activity)
软件确认活动-1
• 包含在医疗器械中的软件
– 按照 Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices 提供所需的软件文档 – 严重级别(level of concern)是反映软件失效的可能后 果的指标,分成较大、中等、较小三级 – 通常,FDA认为心律失常检测器属于“major level of concern” – 若申请机构相信心律失常检测器中的软件应被认为是较 小或中等严重级别,应提供一份讨论发生软件失效的可 能后果的清晰的科学辩护报告 – 鼓励利用任何认可的软件标准,并提供一致性声明, FDA网站提供被认可的软件标准搜索(包括其他标准)
美国FDA医疗器械认证详解

美国FDA医疗器械认证详解美国FDA(Food and Drug Administration)是美国联邦政府的一家机构,负责确保食品、药物、生物医学器械和医疗器械的安全和有效性。
医疗器械是指用于预防、诊断、治疗或缓解疾病的设备,包括诊断用设备(如X射线机、医学成像仪器),治疗用设备(如手术器械、人工心脏),支持用设备(如人工呼吸机、血压监测设备)等。
美国FDA对医疗器械的认证需要遵守一系列法规和规定。
首先,医疗器械制造商需要申请一个510(k)预市通知,或者一个PMA(前期市场批准)提交给FDA。
由于大多数医疗器械能通过510(k)程序获得市场准入,以下将重点介绍这一程序。
510(k)程序是一种适用于“预市”设备的快速审核方法,它要求新设备与已经获得FDA认可的“已存在”的设备相似,并且其安全性和有效性也应该和已存在设备相似。
该程序的目的是确保新设备不会对患者的健康造成不必要的风险。
在提交510(k)预市通知之前,制造商需要确定适用于他们的设备的类别。
FDA将医疗器械分为三个类别:Class I(一类)、Class II(二类)和Class III(三类)。
一类设备一般具有较低的风险,二类设备具有中等风险,而三类设备则具有较高的风险。
根据设备的类别和特征,制造商需要遵守不同的法规和审核要求。
在提交510(k)预市通知时,制造商需要提供一份详细的510(k)申请文件,其中包括以下内容:设备的描述和分类、设备的设计和原理、与已存在的类似设备的比较、设备的制造和控制、设备的安全和有效性数据等。
制造商还需要提供临床试验数据、实验室测试结果、文献资料等证据来支持设备的安全性和有效性。
一旦510(k)预市通知被提交,FDA会对申请进行审核。
审核的时间取决于设备的类别和申请文件的完整性。
在审核过程中,FDA可能要求进一步补充资料或进行其他形式的沟通和讨论。
如果FDA认为申请材料不完整或设备存在安全风险,它可能会拒绝该申请并要求制造商重新提交。
510k测试报告 可接受标准

510k测试报告可接受标准一、引言在医疗器械行业,510k测试报告是评估和验证医疗器械安全性和效能的重要依据。
该报告的准确性和合规性对于保障患者安全和满足监管要求至关重要。
本文将探讨510k测试报告的可接受标准,并对其相关要求进行详细解析。
二、背景医疗器械的研发和上市需要符合相关法规和标准,其中510k测试报告是美国食品药品监督管理局(FDA)要求的重要文件之一。
该报告旨在评估新医疗器械与现有市场上类似器械的相似性,并验证其安全性和有效性。
根据食品药品管理局,510k测试报告需满足一定的可接受标准,以确保医疗器械符合相关法规和规定。
三、可接受标准详解1. 完整性:510k测试报告应包含完整的信息,包括但不限于产品描述、材料成分、技术规格、操作说明、安全性和效能测试等。
报告应涵盖所有必要的信息,以便医疗器械行业的专业人士进行准确的评估和验证。
2. 准确性:510k测试报告所包含的信息需要准确无误,并符合实际情况。
任何虚假、误导或不准确的信息都会导致报告被拒绝或被撤销。
因此,在编制报告时,厂商必须确保所提供的信息真实、准确,并经过充分的验证和验证。
3. 透明度:510k测试报告应具有透明度,即对医疗器械的各个环节进行明确的描述和解释。
报告中应清楚说明医疗器械的设计原理、材料使用、生产工艺、安全性和效能测试的结果等。
透明度可以帮助监管部门和专业人士更好地理解医疗器械并做出准确的评估。
4. 法规符合性:510k测试报告必须符合相关的法规和标准要求。
在编制报告时,厂商需要了解并遵守FDA的规定,确保医疗器械满足美国国内的安全性和有效性要求。
此外,还需考虑国际标准和其他国家的法规要求,以确保医疗器械在全球范围内的合规性。
四、总结510k测试报告的可接受标准非常重要,对于医疗器械行业来说具有指导和约束作用。
厂商在编制报告时应重视完整性、准确性、透明度和法规符合性等要素,并进行详尽的验证和验证工作。
只有符合可接受标准的510k测试报告才能确保医疗器械的合规性和患者的安全性。
fda traditional 510k 分类

关于FDA传统510(k)分类的解析一、概述在医疗器械行业,为了保障患者的安全和权益,美国食品药品监督管理局(FDA)实施了一系列的规定和标准,其中包括510(k)分类。
在这篇文章中,我们将重点关注FDA传统510(k)分类的相关内容,对其进行深入解析。
二、FDA传统510(k)分类概述1. 510(k)分类的背景510(k)分类是FDA根据《联邦食品、药品和化妆品法》中的相关规定制定的一种医疗器械分类和审批制度。
根据该法规,对于新的医疗器械或对现有医疗器械的修改,需要进行相应的分类和审批,以确保其安全性和有效性。
2. 510(k)分类的含义510(k)分类是指医疗器械制造商通过向FDA提交510(k)申请,证明其新研发的医疗器械与FDA已经批准上市的同类医疗器械相比,具有相似的安全性和有效性。
通过这种方式,制造商可以避免重新进行临床试验,节省时间和成本。
3. 510(k)分类的适用范围510(k)分类适用于许多类型的医疗器械,包括但不限于体外诊断设备、手术器械、植入式器械、放射性医疗器械等。
对于不同类型的医疗器械,FDA制定了相应的分类标准和审批流程。
三、FDA传统510(k)分类的申请流程1. 510(k)申请材料的准备制造商在向FDA提交510(k)申请之前,需要准备充分的申请材料。
这些材料包括但不限于医疗器械的技术文件、临床试验数据、质量管理体系文件、风险分析报告等。
这些材料需要详细描述医疗器械的结构、功能、性能指标、材料成分、使用方法、适应症和禁忌症等内容。
2. 510(k)申请的提交一旦制造商完成了申请材料的准备,可以通过FDA的电子提交系统eSubmit,向FDA提交510(k)申请。
在提交申请之后,FDA将对申请材料进行初步审核,确定是否符合基本要求。
3. 510(k)申请的审核和决定一旦申请材料通过初步审核,FDA将进行全面的技术评估和风险评估。
这一过程通常包括FDA内部专家的评审、对外部专家的交流、对临床试验数据的审查等环节。
fda510k 命名规则

fda510k 命名规则FDA 510(k)命名规则是指美国食品药品监督管理局(FDA)对于医疗器械510(k)递交申请的命名规则。
这个规则的目的是为了确保递交的申请能够被准确地识别和跟踪,以便进行有效的审查和监管。
本文将详细介绍FDA 510(k)命名规则的背景、重要性以及一些常见的命名规则。
我们来了解一下什么是FDA 510(k)。
根据美国FDA的规定,对于那些已经获得FDA批准的类似医疗器械的新产品,可以通过递交510(k)申请来获得市场准入。
这个申请的名字来源于1976年颁布的美国食品、药品、化妆品法案中的第510(k)条款。
递交510(k)申请的目的是证明新产品与已经获得批准的类似产品在安全性和有效性方面没有本质差异。
为了确保对于递交的申请能够准确地进行识别和跟踪,FDA制定了一套命名规则。
这些规则包括了申请编号的格式、命名要求和命名规则的使用方法。
其中,最常见的命名规则是使用公司名称或产品名称作为申请编号的一部分。
这样一来,FDA可以根据申请编号快速找到对应的申请,进行审查和监管。
为什么FDA 510(k)命名规则如此重要呢?首先,这些规则确保了申请的唯一性。
每个申请都有一个独特的编号,避免了混淆和重复。
其次,这些规则使得对于申请的跟踪和审查变得更加高效。
FDA可以根据申请编号快速找到对应的申请,提高审查的速度和准确性。
另外,这些规则还有助于建立一个规范的申请管理系统,使得信息的记录和管理更加方便和可靠。
那么,根据FDA 510(k)命名规则,具体有哪些常见的命名规则呢?首先,公司名称可以作为申请编号的一部分。
这个规则可以方便地将不同公司的申请进行区分。
其次,产品名称也可以作为申请编号的一部分。
这个规则可以使得同一公司不同产品的申请进行区分。
另外,申请的递交时间也可以作为申请编号的一部分。
这个规则可以帮助FDA对于申请进行时序管理和跟踪。
除了上述的常见命名规则外,还有一些其他的命名规则。
510k测试报告 可接受标准

510k测试报告可接受标准
510(k)测试报告是针对美国食品和药物管理局(FDA)的510(k)递交的医疗器械产品进行的测试报告。
根据FDA的要求,医疗器械产品需要进行一系列的测试以确保其安全性和有效性,以及符合适用的标准和法规。
具体的可接受标准通常取决于所涉及的医疗器械产品类别和所在国家的法规要求。
一般来说,医疗器械的测试标准可能涉及以下方面:
安全性测试:包括机械安全性、电气安全性、生物相容性等方面的测试,通常需要符合ISO 14971风险管理标准。
性能测试:针对医疗器械产品的功能和性能进行测试,确保其设计的预期功能可以得到满足。
电磁兼容性测试:针对电气医疗器械产品,需要进行电磁兼容性测试,以确保其在电磁环境中的稳定性和安全性。
医疗设备标准:不同类型的医疗器械可能需要遵守特定的行业标准,比如对于心脏起搏器和除颤器产品,可能需要符合AAMI/IEC 60601-2-31的标准。
在测试报告中,应包含产品的具体测试项目、测试方法、测试结果以及测试依据的标准和法规。
这些测试报告需要确保符合FDA的要求,以确保产品的安全性和有效性。
如果您有具体的产品类型或测试标准,可以向测试机构或专业顾问咨询,以获取更详细的信息。
美国FDA医疗器械510(k)深度分析系列一
美国FDA医疗器械510(k)深度分析系列一中国对美国的医疗器械出口数量逐年增长,越来越多的中国企业需要面对美国食品与药品监督管理局FDA的审核。
对于高风险的医疗器械,通常需要向美国FDA提交510(k)申请(也叫上市前通知),在获得510(k)批准以后,方可销售到美国。
往往510(k)申请是企业需要面对的一大难题。
相比CE认证,美国FDA 510(k)申请显的更加灵活、往往企业遇到的困难更大。
在全面深入理解美国FDA相应法规要求的基础上,对美国FDA 510(k)历年申请情况进行全面的分析和总结是非常有必要的。
针对这种情况,美国美德思有限公司(MEDevice Services, LLC)在参考众多资料并结合其多年来积累的FDA 510(k)丰富经验,对历年510(k)进行了系统总结和归纳。
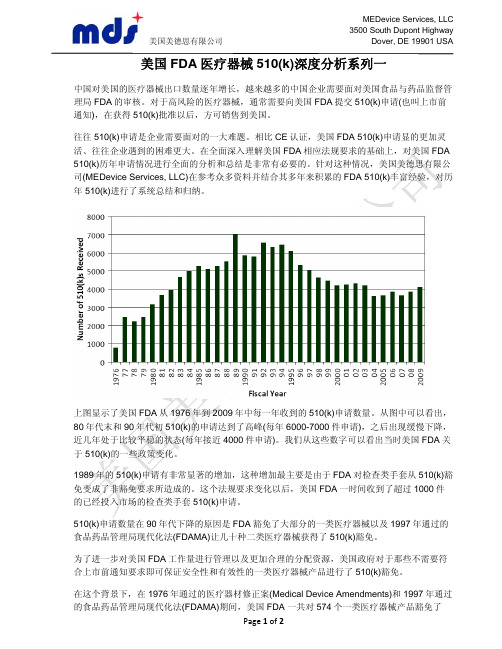
上图显示了美国FDA从1976年到2009年中每一年收到的510(k)申请数量。
从图中可以看出,80年代末和90年代初510(k)的申请达到了高峰(每年6000-7000件申请),之后出现缓慢下降,近几年处于比较平稳的状态(每年接近4000件申请)。
我们从这些数字可以看出当时美国FDA关于510(k)的一些政策变化。
1989年的510(k)申请有非常显著的增加,这种增加最主要是由于FDA对检查类手套从510(k)豁免变成了非豁免要求所造成的。
这个法规要求变化以后,美国FDA一时间收到了超过1000件的已经投入市场的检查类手套510(k)申请。
510(k)申请数量在90年代下降的原因是FDA豁免了大部分的一类医疗器械以及1997年通过的食品药品管理局现代化法(FDAMA)让几十种二类医疗器械获得了510(k)豁免。
为了进一步对美国FDA工作量进行管理以及更加合理的分配资源,美国政府对于那些不需要符合上市前通知要求即可保证安全性和有效性的一类医疗器械产品进行了510(k)豁免。
在这个背景下,在1976年通过的医疗器材修正案(Medical Device Amendments)和1997年通过的食品药品管理局现代化法(FDAMA)期间,美国FDA 一共对574个一类医疗器械产品豁免了510(k)。
关于美国FDA第三方510(k)审核项目的分析研究

综 述收稿日期:2020-11-26[15][16][17]scale Medical Equipment for Hospital[J].Chin Med Equ J,2012,33(8):91-92,115.安恒.基于移动数据终端的医疗设备智能管理系统应用研究[D].济南:山东大学,2010.李云,王涵,王作涪,等.基于综合净现值的医疗设备成本效益分析[J].中国医疗设备,2017,32(6):156-158,169.李健,王天苗,戴国强.发展数字化医疗设备产业迫在眉睫[J].中国科技产业,2003,3(1):28-29.沈玲丽,朱人杰,范璐敏,等.基于综合净现值的医院医疗设备成本效益分析及改进建议[J].医疗卫生装备,2019,40(2):71-74,108.张昊,王韬,白波.数字化医院大型医疗设备绩效评价方式的改进研究[J].中国医学装备,2014,11(10):5-7.赵翊君,孙皓月,董颢霞.基于物联网的医疗设备智能化管理应用研究[J].河北建筑工程学院学报,2014,32(2):98-101.[18][19][20]*基金项目:浙江省食品药品监督管理局科技计划项目(2018023)“我国医疗器械评审模式改革研究”①浙江省医疗器械审评中心审评一科 浙江 杭州 311121②国家药品监督管理局医疗器械注册管理司 北京 100037*通信作者:cool.wenwu@作者简介:俞卉,女,(1992- ),硕士,工程师,研究方向:医疗器械技术审评。
[文章编号] 1672-8270(2021)05-0191-05 [中图分类号] R197.39 [文献标识码] AAnalysis and research of the third party 510(k) audit project of American FDA/YU Hui, YUAN Peng, YANG Ting, et al//China Medical Equipment,2021,18(5):191-195.[Abstract] The differences of organizational structure, team construction of technician, professional background and understanding of regulations among various provincial institutes of technique evaluation of medical device were larger. And the unified standards of evaluation are the responsibility of the reform of the evaluation and approval system of medical device. The third party 510 (k) audit project of American Food and Drug Administration (FDA) was sorted out and analyzed, and the supervision on the third party audit institute was studied from its’ evaluation flow . And the operation performance and routine 510 (k) audit of this project were compared and analyzed so as to promote the reform and development of the evaluation system of China. In the reform of the domestic evaluation and approval system of medical device, the experience of FDA should be referred to implement effective evaluation system within the nationwide evaluation institutes. And the third party institute should be used to improve the works of product recording of the first kinds of medical devices, and to develop the evaluation of the third party , and to optimize the ability check of evaluation and approval of provincial level and promote the information sharing and technical exchange of evaluations, and to accelerate the development with high quality of the industry of medical device.[Key words] The 510(k) Third Party Review Program; The review and approval of the medical device; System reform; the record of class I medical devices; The availability of review information[First-author’s address] Department of Evaluation I, Evaluation center of medical device in Zhejiang Province, Hangzhou 31112,China.[摘要] 鉴于国内各省级局的医疗器械技术审评机构在组织架构、技术人员队伍建设、专业背景及法规理解等差异较大,统一审评尺度一直是医疗器械审评审批制度改革的应有之义。
美国FDA医疗器械注册(510k)

FDA 2892表格—医疗器械列名登记
4、每年递交FDA 2891a表格更新注册
医疗器械列名登记—2892表格
(21CFR第807部分)
FDA 2892表格—医疗器械列名登记 1、/cdrh/devadvice/342.html
/scripts/cdrh/cfdocs/cfRL/FDA-2892.pdf - 每份表格可登记 4 件产品; - 每种器械类型(不是每个型号、目录编号和品牌)登记一个列名; - 填写的4件产品中,每件产品均记作独立的条目;
美国FDA食品药品监督管理局 法规概览
William (Bill) M. Sutton 医疗器械与放射健康中心 小型制造商、国际及消费者协助分部 副主管
美国健康与人类服务部
美国健康与人类服务部(DHHS) 部长 公共卫生服务署 署长 国立卫生研究院 卫生资源与服务管理局 卫生保健研究与质检局 印第安人卫生服务 药物滥用与精神卫生管理局 疾病控制中心 美国FDA食品药品管理局
510(k)——上市前通知
何时需要510(k) 医疗器械初次投放美国市场 已上市医疗器械预期用途变更 已上市器械经过重大改动 /cdrh/devadvice/314.html 免于510(k)审批的器械—798 / 47% 第一类:729 / 93% 第二类:69 / 9%
510(k)——上市前通知
510(k)的内容——21 CFR第807.87部分(一) 申请人姓名、地址、电话/传真号码/、联系人、代表/顾问姓名、公司注册编号 器械分类名称、CFR编号、器械等级 、产品代码 通用名/常用名、商品名/专有名及型号 与此器械具有实质等效性的已上市器械名称 是否符合第514节特殊控制的规定 计划使用的商标、标识和广告宣传 照片、工程图纸 实质等效性声明及与已上市的类似器械之比较 与已上市的类似器械之相似性和/或差异性声明 改动的器械应附上改动数据
k注册

510(K)注册510(k)注册FDA510(K)即上市前通告(Pre-market Notification),旨在证明该产品与已经合法上市的产品实质性等同(Substantially Equivalent)。
因其相应FD&C Act第510(K)章节故通常称510(K)。
因此FDA510(K)并不是产品【信息咨询】,而是产品注册。
1、根据FDA有关规定合适需申请510(K):1)首次将一种医疗器械引入美国市场进行销售的医疗器械制造商;Manufacturers of medical devices who would introduce a kind of medical devices in . market for the first time.2)是再次向美国市场引入其改变或更新的医疗器械进行销售的制造商(这种变更或更新会影响器械的安全或有效性,这种改变或更新包括设计、材料、化学成分、驱动力、生产流程或者预期用途)。
实质性等同(SE)的含义:证明所申请上市的产品和已在美国市场上合法销售的产品在安全性和有效性方面比较是实质相等的。
1)与已上市的产品预期用途相同;产品的新特性不会对安全性或有效性产生影响,或者对安全有效性产生影响的新特性有可接收的科学方法用于评估新技术的影响以及有证据证明这些新技术不会降低安全性或有效性。
2)选择合适的产品进行比较是510(K)申请中实质性等同的关键步骤。
实质性等同代表要素件表1。
需要特别关注:510(K)申请时很少需要临床试验结果(Results from Human Clinical Studies),并且由申请方自行决定是否提交临床资料。
3)申请方必须提供充足的资料证明,所申请上市的器械和被比较的器械是实质性等同的,否则510(K)申请不会通过。
实质性等同代表要素表12、510(K)申请流程1.申请登记;2.FDA确认发布制造商序列号;3.产品分类:市场准入认可(即510(K)认可);4.委托代理:《FDA注册与通报委托协议》(法人代表签字,加盖公章);5.提供资料。
《510K注册》

510(K) 注册510(k) 注册FDA510 (K)即上市前通知( Pre-market Notification),旨在证明该产品与已经合法上市的产品本质性等同(Substantially Equivalent)。
因其相应FD&C Act第510(K)章节故往常称510 (K)。
所以 FDA510 (K)其实不是产品【信息咨询】,而是产品注册。
1 、依据FDA相关规定适合需申请510 (K):1)初次将一种医疗器材引入美国市场进行销售的医疗器材制造商;Manufacturers of medical devices who would introduce a kind of medicaldevices in U.S. market for the first time.2)是再次向美国市场引入其改变或更新的医疗器材进行销售的制造商(这类变更或更新会影响器材的安全或有效性,这类改变或更新包含设计、资料、化学成分、驱动力、生产流程或许预期用途)。
本质性等同( SE)的含义:证明所申请上市的产品和已在美国市场上合法销售的产品在安全性和有效性方面比较是本质相等的。
1)与已上市的产品预期用途同样;产品的新特征不会对安全性或有效性产生影响,或许对安全有效性产生影响的新特征有可接收的科学方法用于评估新技术的影响以及有凭证证明这些新技术不会降低安全性或有效性。
2)选择适合的产品进行比较是510 (K)申请中本质性等同的重点步骤。
本质性等同代表因素件表 1 。
需要特别关注: 510 (K)申请时极少需要临床试验结果( Results from Human Clinical Studies),并且由申请方自行决定是否提交临床资料。
3)申请方一定供给充分的资料证明,所申请上市的器材和被比较的器材是本质性等同的,不然510 (K)申请不会经过。
本质性等同代表因素表 1代表因素能否使用Indications for Use合用症Summary纲要Proposed Labeling建议标签Description of the Device器材描绘Results from Laboratory & Animal实验室和动物研究结果StudiesResults from Biocompatibility生物相容性要就结果StudiesSterilization Information灭菌信息Comparison to Predicate Device与 Predicate器材比较Manufacturing Information生产信息2 、510 (K)申请流程1.申请登记;2.FDA 确认公布制造商序列号;3.产品分类:市场准入认同(即 510 (K)认同);4.拜托代理:《 FDA 注册与通告拜托协议》(法人代表署名,加盖公章);5.供给资料。
FDA医疗器械510K申请注意事项

FDA医疗器械510K申请注意事项
向FDA申请时需注意的一些问题
1.在申请前必须明确产品是否被FDA认作医疗器械、产品类别、管理要求,明确申请工作内容;
2.对申请上市的产品查阅有否美国强制标准,产品是否符合该标准
3.在准备510(K)申请文件前,需考虑是否真正需要递交、何时递交以及递交哪一种性质的510(K)申请:常规510(K)、特殊510(K)、简化510(K);
4.对申请过程中FDA所提出的问题应及时给予书面的、及时的回答;
5.向FDA递交的所有资料纸张大小应采用Letter Size(21.5cm X 29.7cm);
6.所有递交FDA的资料企业需留有备份,因为FDA在收到申请资料后即电子扫描登录,同时销毁申请资料,并不归还企业。
7.对少部分产品,FDA将对企业进行现场GMP考核,企业需参照美国GMP管理要求,并在FDA现场审核时配备合适的、对GMP和企业有一定了解的翻译人员;
8.告知FDA的正式联系人需对FDA法规和工作程序有一定的了解,并能与FDA直接交流,以方便及时反馈,企业可明确自己或委托咨询机构负责与FDA 的日常沟通。
心电监护产品的FDA510K申请

申请者可以寻求专业意见,以了解如何改进产品或提交的 数据,以满足FDA的要求。
重新提交申请
如果认为已经解决了问题,申请者可以重新提交申请。
与FDA沟通的技巧和策略
01
充分准备
在与FDA沟通之前,应充分了解产品的特性和FDA的要求,准备好所有
必要的文件和数据。
02
清晰、准确地回答问题
在与FDA沟通时,应清晰、准确地回答他们的问题,不要提供不准确或
心电监护产品的FDA 510(k)申请
• 引言 • 心电监护产品概述 • FDA 510(k)申请流程 • 心电监护产品FDA 510(k)申请要
求 • 常见问题和解决方案 • 未来展望
目录
01
引言
目的和背景
确保心电监护产品的 安全性和有效性
促进心电监护产品的 市场准入和合规性
保障患者的生命安全 和健康权益
批准或拒绝
根据审核结果,FDA会做出是否批准 产品的决定。如果获得批准,产品可 以上市销售;如果被拒绝,企业需要 进一步改进或重新提交申请。
04
心电监护产品FDA 510(k)
申请要求
申请材料准备
提交申请表格
01
填写完整的FDA 510(k)申请表格,包括产品名称、型号、规格
等信息。
附上技术规格
FDA 510(k)简介
FDA 510(k)是美国的医疗器械上市前 审查程序,要求申请人提交申请资料, 证明其产品与已上市产品实质等同,符 合相关标准和规定,并获得FDA的批准。
心电监护产品作为医疗器械的一种,需 要进行FDA 510(k)申请,以确保其安
全性和有效性。ຫໍສະໝຸດ 通过FDA 510(k)申请,心电监护产品 可以获得市场准入,并在美国合法销售
二类医疗器械FDA认证流程——510K提交步骤

二类医疗器械FDA认证流程——510K提交步骤FDA(美国食品药品监督管理局)是负责对美国市场上销售的医疗器械进行监管和审查的机构。
在美国销售医疗器械,必须获得FDA的认证才能合法销售。
根据医疗器械的分类,FDA认证分为三个类别:一类、二类和三类。
本文将着重介绍二类医疗器械的FDA认证流程中的510(k)提交步骤。
1.确定器械的分类:首先,需要确定医疗器械的分类。
根据FDA的定义,二类医疗器械是指需要通过一份称为510(k)的材料进行市场申报的器械。
目的是证明新器械与FDA已批准上市的同类器械相似,具有相同的作用和安全性。
2.收集现有资料:在进行510(k)提交之前,需要收集一系列相关的资料,如市场调查报告、设计开发文件、生产质量计划等。
这些资料将用于证明新器械与同类已上市器械的相似性。
3.编写510(k)报告:根据FDA规定的格式,编写510(k)报告。
该报告应包括以下内容:a.适用范围:说明器械的适用范围和预期用途。
b.相似性比较:详细对比新器械与已上市同类器械的特性、注射剂、材料等,证明其具有相似性。
必要时,可以提供测试报告或数据支持。
c.临床数据:如有必要,需提供临床试验的数据,以证明新器械的安全性和有效性。
d.风险分析:分析新器械可能产生的各种风险,并提供相应的风险控制措施。
5. 提交FDA注册申请:将完成的510(k)报告和相关资料提交给FDA 进行注册申请。
申请可以通过FDA的电子提交系统(eSubmitter)或纸质方式提交。
7.通知:若FDA审核通过,批准器械上市销售,FDA将向申请人发出确认信。
若审核未通过,FDA将提供不通过的原因和建议。
需要注意的是,以上是一般的510(k)提交步骤,不同的医疗器械可能会有不同的要求和程序。
因此,在进行510(k)提交之前,确保充分了解和遵守FDA的相关规定非常重要。
总结起来,二类医疗器械的FDA认证流程中的510(k)提交步骤包括确定器械分类、收集现有资料、编写510(k)报告、审核、提交FDA注册申请、FDA审核和通知。
510(k)申请提交临床数据的情况

510(k)申请哪些情况下提交临床数据
可能存在四种情况需要提交临床数据:
适应症差异体现在:
1.设备的患者人群不同,疾病不同,申请器械和对比器械作用的部
位不同,申报器械和对比器械的通用或者特定的适应症不同,适
应症的扩展,收益和风险比未知的情况
2.技术特征的差异:材料,器械的设计,能量的来源,其他特征
3.对比器械存在新识别的风险或风险增加:无法通过非临床测试做
出实质等同的决定:这里可能会有几个原因,第一个是没有相关
的模型;第二个模型有缺陷,第三个是模型并不能很好的预测产
品在临床使用的临床结果,第四个,动物模型和人体间的差异,
解剖结构的差异,生理和病例的差异(
4.通过检索FDA的召回数据库,了解到对比器械有新的的风险,那
么针对这些风险就需要考虑,在申报产品上是不是有没有可能会
发生,可能要提供临床数据,证明这个风险是可以接受。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
美国FDA医疗器械510(k)深度分析系列一
中国对美国的医疗器械出口数量逐年增长,越来越多的中国企业需要面对美国食品与药品监督管理局FDA的审核。
对于高风险的医疗器械,通常需要向美国FDA提交510(k)申请(也叫上市前通知),在获得510(k)批准以后,方可销售到美国。
往往510(k)申请是企业需要面对的一大难题。
相比CE认证,美国FDA 510(k)认证显的更加灵活、往往企业遇到的困难更大。
在全面深入理解美国FDA相应法规要求的基础上,对美国FDA 510(k)历年申请情况进行全面的分析和总结是非常有必要的。
针对这种情况,美国美德思国际医疗器械咨询机构(MEDevice Services, LLC)在参考众多资料并结合其多年来积累的FDA 510(k)丰富经验,对历年510(k)进行了系统总结和归纳。
上图显示了美国FDA从1976年到2009年中每一年收到的510(k)申请数量。
从图中可以看出,80年代末和90年代初510(k)的申请达到了高峰(每年6000-7000件申请),之后出现缓慢下降,近几年处于比较平稳的状态(每年接近4000件申请)。
我们从这些数字可以看出当时美国FDA关于510(k)的一些政策变化。
1989年的510(k)申请有非常显著的增加,这种增加最主要是由于FDA对检查类手套从510(k)豁免变成了非豁免要求所造成的。
这个法规要求变化以后,美国FDA一时间收到了超过1000件的已经投入市场的检查类手套510(k)申请。
510(k)申请数量在90年代下降的原因是FDA豁免了大部分的一类医疗器械以及1997年通过的食品药品管理局现代化法(FDAMA)让几十种二类医疗器械获得了510(k)豁免。
为了进一步对美国FDA工作量进行管理以及更加合理的分配资源,美国政府对于那些不需要符合上市前通知要求即可保证安全性和有效性的一类医疗器械产品进行了510(k)豁免。
在这个背景下,在1976年通过的医疗器材修正案(Medical Device Amendments)和1997年通过的食品药品管理局现代化法(FDAMA)期间,美国FDA 一共对574个一类医疗器械产品豁免了
510(k)。
FDAMA的通过使绝大部分一类医疗器械产品获得了510(k)豁免,除了那些预期用途是为了防止危害人体健康方面及其重要或者具有造成生病或受伤等重大潜在风险的产品(“保留”条件)。
因此,一类医疗器械产品中只有那些符合保留条件的产品需要遵守510(k)上市前通知的要求。
同时FDAMA也授权FDA直接对某些二类医疗器械产品进行510(k)豁免。
1998年1月21日,FDA公布了二类医疗器械510k(k)豁免名单,参见法规63 FR 3142。
随着2002年医疗器械使用费现代化法(MDUFMA)的通过,510(k)申请数量再一次下降。
最近几年又有少量回升的趋势。
我们预计将来会有更多的二类医疗器械获得510(k) 豁免。
上图显示了美国FDA从1998年到2009年中每一年收到的不同类别的510(k)申请数量。
从图中可以看出,Traditional类型的传统510(k)数量有所减少,Abbreviated类型的简略510(k)数量基本维持,Special类型的特殊510(k)数量有所增加,第三方审核的510(k)数量有微小上升。
我们从这些数字可以看出510(k)申请类别仍然以Traditional和Special的方式为主,Abbreviated以及第三方审核仍然占据很少的份额。
下一系列,我们将对美国FDA医疗器械510(k)做进一步的深入分析。