鲁索替尼杂质总结分享
新药Ruxolitinib(鲁索替尼)合成检索总结报告

新药Ruxolitinib(鲁索替尼)合成检索总结报告一、Ruxolitinib(鲁索替尼)简介Ruxolitinib(鲁索替尼)适应症于成人真性红细胞增多症。
2020年4月2日宣布,计划与INCEL合作启动三期临床试验,以评估Ruxolitinib用于治疗COVID-19患者的危及生命的呼吸道并发症。
Ruxolitinib(鲁索替尼)分子结构式如下:英文名称:Ruxolitinib中文名称:鲁索替尼本文主要对Ruxolitinib(鲁索替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Ruxolitinib(鲁索替尼)合成路线一三、Ruxolitinib(鲁索替尼)合成路线二四、Ruxolitinib (鲁索替尼)路线一合成检索总结报告(一)Ruxolitinib (鲁索替尼)中间体3的合成(路线一)合成方法实验步骤参考文献操作方法一With stirring in an ice bath,38.4g (250.4mmol,1.0eq.).4-chloro-7H-pyrrole [2,3-d]pyrimidine 1is dissolved in 200mL of dry DMF solution.13g (305mmol,1.2eq)of 57%NaH was added.The reaction was stirred at room temperature for 1hour,then 50.9g of SEMCl 2(305mmol,1.2eq.).After the addition,the reaction was stirred in an ice bath for 1hour.quenched with water,extracted with ethyl acetate.the title compound 3(71g,yield =100%)was obtained.CN109867676;(2019);(A)Chinese;CN109867675;(2019);(A)Chinese操作方法二Under cooling with an ice-salt bath,sodium hydride(340mg,60%)was added in two portions to a solution of4-chloropyrrolopyrimidine(1)(1.0g,6.5mmol)in DMF(15mL)while keeping the temperature of the reactants no higherthan10°C,and the reaction was stirred under nitrogenatmosphere protection for1h.SEMCl2(1.4g,8.5mmol)was slowly added via a syringe while keeping thetemperature no higher than10°C.The reaction was warmedto room temperature,and stirred overnight.The reaction wasquenched with water,extracted with EA,dried overanhydrous sodium sulfate,and the organic phase wasconcentrated,and purified by preparative flashchromatography(PE:EA=19:1),to afford4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine(3)(1.834g,oil product),yield:97%.EP2018/3360878;(2018);(A1)English操作方法三Potassium t-butoxide(328.81g,2.93mol)andtetrahydrofuran were added to the reaction flask at-30°C to-20°C,a solution of4-chloro-7H-pyrrolo[2,3-d]pyrimidine1(300g,1.95mol)in tetrahydrofuran was added dropwiseto the reaction flask.after completion of the dropwiseaddition,the mixture was stirred at room temperature for4to6hours to control the reaction flask the temperature is nothigher than-15°C-5°C,2-(trimethylsilyl)ethoxymethylchloride2(390.80g,2.34mol)Is added to the reaction flask,and after completion of the dropwise addition,thetemperature is raised to room temperature and the reaction isstirred at that temperature for3to5hours.After completionof the reaction,the reaction was quenched to neutral withdilute hydrochloric acid,concentrated in tetrahydrofuran inthe reverse solution,stirred with ethyl acetate and theaqueous phase was separated.The aqueous phase wasextracted with ethyl acetate once,and the reaction solutionwas cooled and filtered to obtain a wet product.The wetproduct was crystallized from n-hexane and dried to obtain534.50g of product3,purity:99.70%,and the residue waspurified.Yield:96.40%.CN107226814;(2017);(A)Chinese操作方法四A solution of4-chloropyrrolo[2,3-d]pyrimidine1(200g,1.3mol,1.0eq.)in N,N-dimethylformamide was added to60%NaH(62.4g,1.56mol,1.2eq.)in an ice bath.After thecompletion of the addition,the resulting mixture was stirredto react at room temperature for1hour.2-(Trimethylsilyl)-ethoxymethyl chloride2(SEMCl,260g,1.56mol,1.2eq.)was slowly added dropwise under cooling in an ice bath.After the completion of the addition,the resulting mixtureEP2019/3473626;(2019);(A1)English;was stirred to react in an ice bath for1hour,and the reaction was quenched with water.The resulting mixture was extracted with ethyl acetate.The organic phases were combined,washed with saturated salt solution,dried with anhydrous sodium sulfate,and filtered.The filtrate was concentrated under reduced pressure to obtain a residue, which was purified by silica gel column chromatography to obtain4-chloro-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine3(312.2g,91.8%yield).US2019/211021; (2019);(A1) English操作方法五To a solution of4-chloro-7H-pyrrolo[2,3-d]pyrimidine1(20.0g,130.4mmol,1.0eq.)in dry DMF was added NaH(6.6g,57%content,156.8mmol,1.2eq.),under stirring inan ice bath.After the reactants were stirred for1hr at room temperature,SEMCl2(26.1g,156.5mmol,1.2eq.)wasadded dropwise under the cooling of an ice bath.After theaddition was completed,the reactants were stirred for1hr inan ice bath,then the reaction was quenched by adding water,and the resulting mixture was extracted with ethyl acetate.The combined organic phase was washed with brine,driedover sodium sulfate,filtered and concentrated in vacuo.Theresulting residue was separated by column chromatographyon silica gel column to give4-chloro-7-{[2-(trimethylsilyl)-ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine3(33.43g,90.4%yield).EP2017/3235819;(2017);(A1)English操作方法六To a flask equipped with a nitrogen inlet,an addition funnel,a thermowell,and the mechanical stirrer was added4-chloro-7H-pyrrolo[2,3-d]pyrimidine1(600g,3.91mol)andN,N-dimethylacetimide(DMAC,9.6L)at roomtemperature.The mixture was cooled to0-5°C.in anice/brine bath before solid sodium hydride(NaH,60wt%,174g,4.35mol,1.1equiv)was added in portions at0-5°C.The reaction mixture went to a dark solution during15minutes.Trimethylsilylethoxymethyl chloride2(SEM-Cl,763mL,4.31mol,1.1equiv)was then added slowly via anaddition funnel at a rate that the internal reaction temperaturedid not exceed5°C.The reaction mixture was then stirred at0-5°C.for30minutes.When the reaction was deemedcomplete determined by TLC and HPLC,the reactionmixture was quenched by water(1L).The mixture was thendiluted with water(12L)and MTBE(8L).The two layerswere separated and the aqueous layer was extracted withMTBE(8L).The combined organic layers were washedwith water(2×4L)and brine(4L)and dried over sodiumsulfate(Na2SO4).The solvents were removed under reducedUS2009/233903;(2009);(A1)English;US2010/190981;(2010);(A1)English;WO2013/36611;(2013);(A1)Englishpressure.The residue was then dissolved inheptane (2L),filtered and loaded onto a silica gel (SiO 2,3.5Kg)column eluding with heptane (6L),95%heptane/ethyl acetate (12L),90%heptane/ethyl acetate (10L),and finally 80%heptane/ethyl acetate (10L).The fractions containing the pure desired product were combined and concentrated under reduced pressure to give 4-chloro-7-((2-(trimethylsilyl)-ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine 3(987g,1109.8g theoretical,88.9%yield)as a pale yellow oil which partially solidified to an oily solid on standing at room temperature.操作方法七To a mixture of sodium hydride (60%dispersion in mineral oil;276mg;6.89mmol)in DMF (10mL)at 0°C was added drop-wise a solution of 4-chloro-2-methylsulfanyl-7H-pyrrolo[2,3-d]pyrimidine 1(1.145g;5.74mmol)in anhydrous DMF (20mL).When the addition was complete,2-(trimethylsilyl)ethoxymethyl chloride 2(1.32ml;7.46mmol)was added drop-wise and the reaction mixture was stirred at 0°C for 1.5h then allowed to warm to ambient temperature.The reaction mixture was partitioned between water (100mL)and ethyl acetate (100mL).The organic phase was separated,dried over Na 2SO 4and then filtered and the filtrate solvents evaporated in vacuo.The crude product was purified by flash chromatography on silica gel (70g)eluting with a solvent gradient of 05%ethyl acetate in hexane to afford the title compound 3(1.73g,91%)as a colourless oil.Bioorganic and Medicinal Chemistry ;vol.20;nb.22;(2012);p.6770-6789.(二)Ruxolitinib (鲁索替尼)中间体5的合成(路线一)合成方法实验步骤参考文献To a flask equipped with a reflux condenser,a nitrogen inlet,mechanical stirrer,and a thermowell was added 4-chloro-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine 3(817g,2.88mol)and dioxane (8L).To this solution was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxa-borolan-2-yl)-1H-pyrazole 4(728g,3.75mol,1.30equiv)followed by a solution of potassium carbonate (K 2CO 3,1196g,8.67mol,3.0equiv)in water (4L).The solution was degassed by passing a stream of nitrogen through the。
鲁索利替尼

安全性
安全性
在Ⅰ/Ⅱ期临床试验中,非血液不良反应如瘀斑、眩晕和头痛的发生率较低( 10% ),主要血液学毒性是贫 血和血小板减少( > 20% )。
治疗期间应评估和监测患者发生严重的细菌、结核分枝杆菌、真菌和病毒感染的风险。开始用鲁索利替尼治 疗前应解决活动性严重感染。中断或终止鲁索利替尼治疗后,一般约 1周左右骨髓纤维化症状恢复至治疗前水平。 某些病例在终止鲁索利替尼治疗后其临床病程继续恶化,但不能确定终止治疗是否影响疾病进展。一般认为,除 了血小板计数减低需要终止治疗,一般考虑减少鲁索利替尼的剂量。
临床评价
临床评价
本品适应证包括中度或高危 MF、原发性 MF、红细胞增加症后 MF和原发性血小板增加症后 MF。
在一项来自安德森癌症中心( MDACC)和 Mayo Clinic-Rochester纳入 153例 MF患者的Ⅰ/Ⅱ期临床试验 中,鲁索利替尼的疗效得到肯定,支持了其在临床上的应用。经过 3个月的治疗,客观指标得到改善(脾脏体积 减少≥50% )的患者有 44%。Mayo Clinic-Rochester分析表明,脾脏体积、贫血和临床症状长期改善的比例分 别为 29%,21%和 63%。由MADCC发布的长期数据表明, 97例脾肿大的患者在持续 166周治疗后,有 61例脾脏 体积减小超过50%。与鲁索利替尼治疗相关的症状改善超过 2年者约 60%。
在体外,鲁索利替尼的浓度在低于 1 nmol·L时即可对 JAK2产生抑制作用,且其对 JAK2的选择性是对其 他激酶的 500倍。本品抑制 JAK2V617F突变型 FDCP和 BaF/3细胞增生的 IC50为 100 ~ 130 nmol·L,但对 活化突变型 BCR/ABL和 c-Kit细胞的增生无抑制作用。本品对细胞增生的抑制作用与降低 BaF/3细胞模型中的 JAK2和 STAT5磷酸化水平有良好的相关性,表明此抑制作用由阻断 JAKSTAT通路而介导。
最新进展:鲁索替尼治疗白血病有效性显著

最新进展:鲁索替尼治疗白血病有效性显著目的在慢性中性粒细胞白血病中,集落刺激因子3受体(CSF3R)—T618I是一种复发激活突变,在不典型慢性髓系白血病中程度较小,且导致组成性JAK-STAT信号转导。
我们的评估对象是,在慢性中性粒细胞白血病和不典型慢性髓系白血病患者中,不管CSF3R突变状态如何,JAK1 / 2抑制剂鲁索替尼的安全性和有效性。
方法针对接受鲁索替尼治疗的44例患者(21例为慢性中性粒细胞白血病和23例为不典型慢性髓系白血病),我们进行了一项II期研究。
主要终点是在6个连续28天周期结束时,先入组的25例患者的血液学总反应率。
我们将反应分为部分缓解或完全缓解。
且将应计对象扩大为44例患者,以更好地评估二级终点:包括≥3级的不良事件、脾脏体积、症状评估、反应的遗传相关性和2年生存率。
结果先入组的25例患者的血液学总反应率为32%(部分缓解8例[慢性中性粒细胞白血病7例和不典型慢性髓系白血病1例])。
有反应的患者为35%(部分缓解[慢性中性粒细胞白血病9例,不典型慢性髓系白血病2例]和完全缓解[慢性中性粒细胞白血病4例]),具有致癌性CSF3R突变的患者为50%。
与部分缓解组和无反应组相比,6个周期后,完全缓解组CSF3R—T618I的平均绝对等位基因负荷减少最大。
最常见的死亡原因是疾病进展。
分别在34%和14%的患者中观察到≥3级贫血和血小板减少症。
没有观察到与鲁索替尼相关的严重不良事件。
结论鲁索替尼具有良好的耐受性,估计缓解率显示为32%。
对诊断为慢性中性粒细胞白血病和/或带有CSF3R—T618I的患者有反应的可能性最大。
讨论我们观察了3例CSF3R -S799T 患者。
该变体发生在细胞质区域,并具有不确定的致癌潜力。
在变体3 CSF3R转录物中,该位点对应于S772,它是调节受体内吞作用的基序的一部分。
S772A取代可导致细胞表面表达增加35,但尚不清楚保守取代S772T如何改变功能。
依鲁替尼杂质-(最新结构)列表
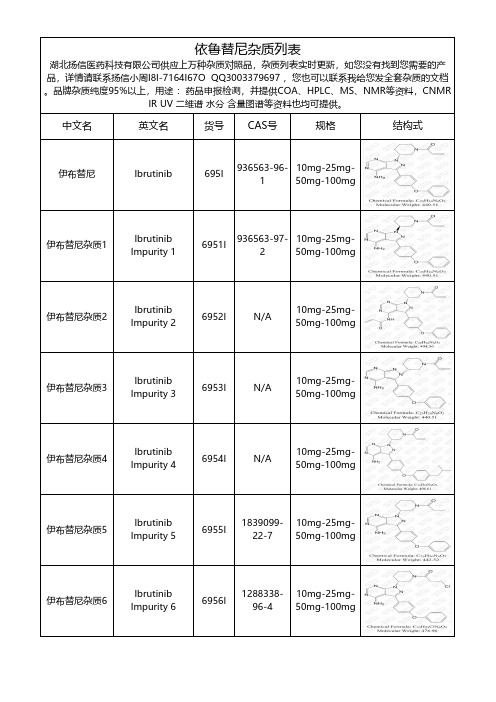
10mg-25mg50mg-100mg
伊布替尼杂质51
Ibrutinib Impurity 51
69551I
N/A
10mg-25mg50mg-100mg
伊布替尼杂质52
Ibrutinib Impurity 52
69552I
N/A
10mg-25mg50mg-100mg
伊布替尼杂质53
Ibrutinib Impurity 53
330793-492
10mg-25mg50mg-100mg
伊布替尼杂质38
Ibrutinib Impurity 38
69538I
N/A
10mg-25mg50mg-100mg
伊布替尼杂质39
Ibrutinib Impurity 39
69539I
N/A
10mg-25mg50mg-100mg
伊布替尼杂质40
Ibrutinib Impurity 15
69515I
N/A
10mg-25mg50mg-100mg
伊布替尼杂质16
Ibrutinib Impurity 16
69516I
N/A
10mg-25mg50mg-100mg
伊布替尼杂质17
Ibrutinib Impurity 17
69517I
N/A
10mg-25mg50mg-100mg
Ibrutinib Impurity 35
69535I
151266-238
10mg-25mg50mg-100mg
伊布替尼杂质36
Ibrutinib Impurity 36
69536I
51067-38-0
10mg-25mg50mg-100mg
鲁索替尼(Ruxolitinib, Jakafi)的合成

鲁索替尼(Ruxolitinib, Jakafi)的合成2014年12月4日,美国FDA批准鲁索替尼(Ruxolitinib;商品名Jakafi)用于治疗真性红细胞增多症患者,这是一种慢性骨髓疾病。
在美国鲁索替尼是获批用于这一疾病的首款药物。
2011年,FDA批准鲁索替尼用于患有另一种骨髓疾病-中或高风险骨髓纤维化患者的治疗,包括原发性骨髓纤维化、真性红细胞增多性骨髓纤维化及原发性血小板增多性骨髓纤维化。
鲁索替尼由特拉华州威尔明顿的Incyte公司上市销售。
合成路线:化合物吡唑S-1经过NBS溴代得到4-溴吡唑S-2,随后用乙基乙烯基醚保护得到S-3,S-3和iPrMgCl·LiCl(TurboGrignards,德国慕尼黑大学Paul Knochel等发展的一类较为温和的金属试剂)通过卤-镁交换后用甲基频哪醇硼酸酯S-4淬灭得到S-5,随后在酸性条件下脱保护得到频哪醇硼酸酯S-6。
Ruxolitinib的另一个片段合成如下,4,6-二羟基嘧啶在POCl3作用下将羟基氯代以及甲酰化得到化合物S-8,经过芳香亲核取代反应得到氨基嘧啶化合物S-9,接下来通过wittig反应引入烯基醚,酸性条件下水解/关环得到化合物S-11,NaH拔掉质子,SEM保护,得到S-12。
环戊基烯基腈也通过wittig 反应得到。
得到两个片段以后,通过钯催化的Suzuki反应将S-12和S-6拼接,得到化合物S-15,随后在碱性条件下,NH对烯基腈进行共轭加成,得到化合物S-16,经过拆分得到光学活性的化合物S-17,最后TFA脱出SEM保护基,即可得到鲁索替尼Ruxolitinib(Jakafi)。
合成路线参考:WO2013023119A1/US2014256941A1/US2010190981A1。
Jakavi(磷酸鲁索替尼)说明书

Jakavi (磷酸xx 替xx)---蛋白酶抑制剂有效成分:xx 替xx 磷酸盐辅料:微晶纤维素、硬脂酸镁、无水胶态二氧化硅、羧甲淀粉钠A、羟基丙基纤维素、聚乙烯吡咯烷酮规格:5mg/15mg/20mg适应症:骨髓纤维化(MF),包括原发性MF、继发于PV或ET的MF。
注意事项:1、血细胞计数:在用本药之前必须做全血细胞计数,用药初始必须每2-4 周检测一次全血细胞计数,根据临床及实验室指标调整剂量。
2、起始剂量:①Pit 100-200 X 109/L ,推荐起始剂量为15mg bid po;②Plt>200 X 109/L,推荐起始剂量为20mg bid po;③关于Plt 在50-100 X 109/L 的患者的起始剂量,尚无足够信息。
这些病人的最大推荐起始剂量为5mg bid po,且须慎重应用。
剂量调整剂量应根据用药疗效和安全性调整。
在Plt<50 X 109/L 或Neut#<0.5 X 109/L时须停药;在Plt恢复至50 X 109/L以上L或Neut#恢复至0.5 X 109/L以上时,需从5mg bid开始应用,在严密监测全血细胞计数的情况下逐渐加量。
在Plt<100X109/L 时,为避免因血小板严重减少而停药,须减量使用。
如果疗效不足,在Plt和Neut#足够的情况下,可最大按5mg bid加量。
在以起始剂量治疗的最初4 周内,不能够加量。
4周以后,加量间隔时间应至少大于 2 周。
本药推荐最大用量为25mg bid。
如果用药过程中漏服,不必补服,继续常规剂量应用即可。
如果患者受益大于所获风险,那么应坚持持续应用本药。
与强CYP3A4抑制剂合用时的剂量调整强CYP3A4抑制剂与本药同时使用,可增加本药相关的血液学毒性和临床副作用的发生率,所以如果本药和CY P3A4抑制剂合用,则日剂量应减半。
例:原先用药为15mg bid,合并应用CYP3A4抑制剂时,应减量为7.5mg bid或15mgqd。
鲁索替尼(Ruxolitinib, Jakafi)的合成
鲁索替尼(Ruxolitinib, Jakafi)的合成2014年12月4日,美国FDA批准鲁索替尼(Ruxolitinib;商品名Jakafi)用于治疗真性红细胞增多症患者,这是一种慢性骨髓疾病。
在美国鲁索替尼是获批用于这一疾病的首款药物。
2011年,FDA批准鲁索替尼用于患有另一种骨髓疾病-中或高风险骨髓纤维化患者的治疗,包括原发性骨髓纤维化、真性红细胞增多性骨髓纤维化及原发性血小板增多性骨髓纤维化。
鲁索替尼由特拉华州威尔明顿的Incyte公司上市销售。
合成路线:化合物吡唑S-1经过NBS溴代得到4-溴吡唑S-2,随后用乙基乙烯基醚保护得到S-3,S-3和iPrMgCl·LiCl(TurboGrignards,德国慕尼黑大学Paul Knochel等发展的一类较为温和的金属试剂)通过卤-镁交换后用甲基频哪醇硼酸酯S-4淬灭得到S-5,随后在酸性条件下脱保护得到频哪醇硼酸酯S-6。
Ruxolitinib的另一个片段合成如下,4,6-二羟基嘧啶在POCl3作用下将羟基氯代以及甲酰化得到化合物S-8,经过芳香亲核取代反应得到氨基嘧啶化合物S-9,接下来通过wittig反应引入烯基醚,酸性条件下水解/关环得到化合物S-11,NaH拔掉质子,SEM保护,得到S-12。
环戊基烯基腈也通过wittig 反应得到。
得到两个片段以后,通过钯催化的Suzuki反应将S-12和S-6拼接,得到化合物S-15,随后在碱性条件下,NH对烯基腈进行共轭加成,得到化合物S-16,经过拆分得到光学活性的化合物S-17,最后TFA脱出SEM保护基,即可得到鲁索替尼Ruxolitinib(Jakafi)。
合成路线参考:WO2013023119A1/US2014256941A1/US2010190981A1。
磷酸鲁索替尼分解温度

磷酸鲁索替尼分解温度然而,磷酸鲁索替尼也有其一定的不足之处,其中之一就是它的分解温度。
磷酸鲁索替尼的分解温度是指在受热作用下,该药物会发生分解的最低温度。
当药物受热至分解温度时,分子结构发生改变,导致其药效降低甚至失效,从而影响治疗效果。
因此,了解和控制磷酸鲁索替尼的分解温度对于确保该药物疗效的稳定性至关重要。
磷酸鲁索替尼的分解温度与多种因素相关,包括化学成分、结构特点、环境条件等。
通过合理设计药物分子结构、加工工艺和储存条件等措施,可以有效控制磷酸鲁索替尼的分解温度,延长其有效期,确保药效的稳定性。
磷酸鲁索替尼的分解温度与其分子结构紧密相关。
磷酸鲁索替尼的分子式为C17H21N7O4P,分子量为425.37。
它是一种含氮杂环与磷酸酯键共存的分子,具有较强的生物活性。
磷酸鲁索替尼在受热时,由于分子内部键合的不稳定性,容易发生断裂和重组,导致其结构发生改变。
磷酸鲁索替尼的环境条件也会对其分解温度造成影响。
在常见的温度下,磷酸鲁索替尼相对稳定,不易发生分解。
但是在高温或潮湿条件下,磷酸鲁索替尼的热敏性增加,分解反应加速,药效降低。
因此在生产、运输和储存过程中,需要注意控制环境条件,避免药物暴露在过高温度或湿度的环境中。
除了分子结构和环境条件外,磷酸鲁索替尼的制备工艺也会对其分解温度产生影响。
在合成和提纯过程中,可能存在杂质残留、溶剂残留等问题,加速磷酸鲁索替尼的分解反应。
因此在生产工艺中,需要严格控制反应条件、提纯工艺和溶剂选择,确保磷酸鲁索替尼的质量和稳定性。
在储存和使用过程中,也需要注意控制磷酸鲁索替尼的分解温度。
通常建议将药物存放在干燥、阴凉的环境中,避免暴露在阳光下或高温环境中。
在使用时,需要按照说明书中的用药方法和剂量进行,避免过量或不当使用导致药物分解和失效。
总的来说,了解和控制磷酸鲁索替尼的分解温度是确保药效稳定性和有效性的关键因素。
通过合理设计药物结构、制备工艺和环境条件,可以有效控制药物的分解反应,延长其有效期,提高治疗效果。
白血病的靶向药物研究关注靶向药物在白血病治疗中的最新研究成果

每个白血病患者的基因突变和表达情况都有所不同,因此 需要开发针对个体的定制化治疗策略,以实现最佳治疗效 果。
未来发展方向
联合用药
针对白血病细胞的多种突变和信 号通路,未来可能采用多种靶向 药物联合使用的方法,以提高治
疗效果和降低耐药性。
精准医学
随着基因组学和蛋白质组学等技 术的发展,未来有望实现更精准 的白血病分型,从而为每位患者
靶向药物联合治疗的研究
1 2
BTK抑制剂与化疗药物联用
通过结合BTK抑制剂和传统化疗药物,实现对白 血病细胞的多靶点攻击,降低耐药性的产生。
免疫疗法与靶向药物联用
结合靶向药物和免疫检查点抑制剂,激活患者免 疫系统,提高对白血病细胞的清除能力。
3
多靶点靶向药物联合应用
同时针对多个关键基因或信号通路的靶向药物联 合应用,以实现对白血病细胞的全面打击。
跨学科合作与创新
倡导医学、生物学、化学、药学等多学科之间的紧密合作与交流,共同推动白血病靶向药物研究的创新 与发展,为战胜白血病汇聚更多智慧与力量。
THANKS
感谢观看
临床试验成果及数据分析
01ห้องสมุดไป่ตู้
02
03
总体生存率提升
通过大规模临床试验数据 证明,靶向药物单用或联 合治疗可显著提高白血病 患者的总体生存率。
副作用降低
与传统化疗相比,靶向药 物治疗的副作用明显减少 ,患者生活质量得到显著 改善。
精准医疗策略优化
通过生物信息学分析,为 不同白血病亚型患者制定 个性化靶向药物治疗方案 ,提高治疗效果。
要点二
总结
随着对白血病发病机制研究的深入,越来越多的靶向药物 被开发并应用于临床。这些靶向药物针对特定的分子靶点 ,能够精准地抑制白血病细胞的增殖和扩散,提高治疗效 果和患者生存率。然而,靶向药物的应用仍面临一些挑战 ,如耐药性的产生、毒副作用等,未来仍需要继续优化药 物设计和治疗方案,以更好地满足白血病患者的临床需求 。
【综述】鲁索替尼乳膏治疗白癜风的Ⅱ期随机、对照试验(三)

【综述】鲁索替尼乳膏治疗白癜风的Ⅱ期随机、对照试验(三)我们将为大家提供四期连载内容,本文为第三期结论2017年6月7日至2018年3月21日,对205例患者进行了筛查,排除了48例患者,且157例患者随机接受鲁索替尼乳膏或安慰剂治疗(图1)。
32例患者接受每天两次安慰剂治疗,125例患者接受鲁索替尼乳膏治疗(31例患者每天一次0.15%鲁索替尼乳膏治疗,31例患者每天一次0.5%鲁索替尼乳膏治疗,30例患者每天一次1.5%鲁索替尼乳膏治疗,33例患者每天两次1.5%鲁索替尼乳膏治疗)。
到第24周时,157例患者中有18例(11%)患者停止了治疗研究,直至第52周,停药率一直很低。
直到第24周停药的主要原因是10例(6%)患者退出治疗,三例(2%)患者出现不良事件,两例(1%)患者失访,两例患者(1%)出现方案偏差,1例(1%)患者不配合药物研究。
患者平均年龄为48.3岁(SD 12.9),中位年龄为49.0岁(范围18-73),其中157例患者中73例(46%)为男性,84例(54%)为女性,132例(84%)患者为白种人。
各治疗组基线疾病特征分布相似(表1)。
大多数患者(93%)患有非节段性白癜风和II-III型皮肤病(64%)。
中位病程为14.0年(范围0.3-67.9)。
基线时T-BSA 受累平均百分比为22.05%(标准差18·38%),F-BSA平均百分比为1.48%(0.86%)。
基线平均T-VASI评分为17.96(SD 15·45),平均F-VASI评分为1.26(0.82)。
图1,研究表1,基线特征第24周,接受2种高剂量鲁索替尼乳膏治疗(每天两次1.5%鲁索替尼乳膏治疗组中,33例患者中,有15例(45%)患者,优势比[OR]24.7, 95% CI 3.3–1121.4; p=0.0001;每天一次1.5%鲁索替尼乳膏治疗组中,30例患者中有15例,[OR]28.5,95% CI 3.7–1305.2; p<0.0001)和接受两种最低剂量鲁索替尼乳膏(每天1次0.5%鲁索替尼乳膏治疗组中 ,31例患者中存在8例 [26%];每天一次0.15% 鲁索替尼乳膏治疗组中,31例患者中存在10例)治疗患者达到F-VASI50的主要终点指标的比例显著多于安慰剂组(32例患者中存在1例[3%]; 图2A)。