临床实验标准操作规程SOP
临床实验标准操作规程SOP
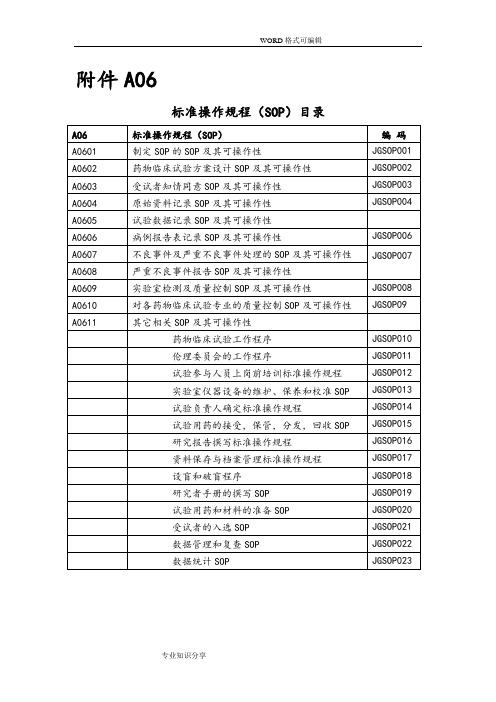
附件A06标准操作规程(SOP)目录SOP(Standard Operating Procedure):为有效地实施和完成某一临床试验中每项工作所拟定的标准和详细的书面规程。
药物临床试验机构标准操作规程制订、修订及编码的操作规程1、药物临床试验机构办公室指定人员起草或修订机构总的SOP。
2、起草人或修订人按照GCP的要求,根据医院的实际情况起草或修订SOP。
3、药物临床试验机构办公室主任审核。
4、药物临床试验机构对SOP实行统一编码。
5、编码格式为:“JGSOP×××”,“JG”代表“机构”;“×××”为顺序号。
例如:“标准操作规程的制订,修订及编码操作规程”的编码为:JGSOP001。
6、SOP经专家小组讨论通过,由药物临床试验机构主任审核批准后生效。
7、药物临床试验机构办公室对通过的SOP归档保存。
8、新的SOP通过后,旧的SOP同时废除,并统一由办公室回收。
10、药物临床试验机构办公室组织相关人员学习SOP。
由药物临床试验机构办公室监督SOP的实施。
各专业标准操作规程的制订、修订及编码的操作规程1、药物临床试验机构专业科室指定人员起草或修订本专业的SOP。
2、起草人或修订人按照GCP的要求,根据本专业的实际情况起草或修订SOP。
3、对SOP实行统一编码。
4、编码格式为:“YYSOP×××”,“YY”为本专业前两个字的汉语拼音的第一个字母(大写);“×××”为本专业SOP的顺序号。
例如:心血管专业的第一个SOP的编号为:“XXSOP001”。
5、SOP经本专业专家小组讨论通过,专业负责人审核,上报药物临床试验机构主任批准后生效6、归档保存(一份存本专业办公室,一份存医院机构办公室)。
7、新的SOP通过后,旧的SOP同时废除。
8、本专业组织相关人员学习SOP并组织实施。
1、申办者制定初步试验方案。
临床试验sop范文

今天看临床试验的文档有个sop,具体是什么意思啊SOP ,英文全称Standard Operation Procedure,标准操作规程,是临床试验中实施各个环节所拟定的标准的、详细的、书面的指导规程。
定义所谓标准规范,在这里有最优化的概念,即不是随便写出来的操作程序都可以称作SOP,而一定是经过不断实践总结出来的在当前条件下可以实现的最优化的操作程序设计。
说得更通俗一些,所谓的标准,就是尽可能地将相关操作步骤进行细化,量化和优化,细化,量化和优化的度就是在正常条件下大家都能理解又不会产生歧义。
怎么写sop标准操作规程(SOP)基础知识标准操作规程(SOP)是各种标准化管理认证和产品认证的重要内容,各行业都有SOP的要求。
什么是SOP?简单的讲,SOP就是一套包罗万象的操作说明书大全。
一套好SOP是确保产品或服务质量的必要条件。
SOP不仅仅是一套技术性范本,它更重要的涵盖了管理思想、管理理念和管理手段。
由于在成熟的行业,都有明确的管理规范和认证体系,因此其SOP的标准化和成熟性都比较高,编写SOP也有依据难度较低。
由于目前还没有成熟的实验室管理和认证体系,因此,在检验工作中编写SOP会有些盲然。
首先,SOP具有行业特点,不同的行业都有不同的SOP。
就检验工作而言,仪器有仪器的SOP,试剂有试剂的SOP,各个项目有各自不同的SOP,别说是细菌、生化免疫这些学科不同的有不同的SOP,就是同一学科内不同项目也有不同的SOP。
所以检验SOP不是一个,而一套。
第二,SOP事无巨细,也就是说只要与项目有关,要详细全面,要包括所有的可能出现的细节。
以飞行员操作规程为例,第一条竞然是“坐下”,由此可以看出,SOP涵盖细节程度。
SOP不是简单的操作说明,而应该是实用操作大全,应该成为工具书性质的东西。
一套理想的SOP应该让一个不懂的学了后就能成为专家。
以药品生产SOP为例,其要求是GMP认证所要求的,根据GMP,其SOP的重点见附。
临床试验方案设计的标准操作程序

临床试验方案设计的标准操作程序1. 试验目的临床试验方案设计的目的是确保研究按照科学原则进行,同时符合伦理和法规要求。
本标准操作程序(SOP)旨在为临床试验方案设计提供一套系统的、可重复的操作流程。
2. 适用范围本SOP适用于临床试验的方案设计,包括但不仅限于新药临床试验、生物制品临床试验和医疗器械临床试验。
3. 试验设计3.1. 试验类型根据研究目的,确定试验类型(如随机对照试验、非随机对照试验等)。
3.2. 研究对象确定研究对象的选择标准、排除和纳入标准,包括年龄、性别、病史等。
3.3. 干预措施明确干预措施,包括试验药物、对照药物或干预手段、剂量、给药方式和疗程等。
3.4. 结局指标确定主要结局指标和次要结局指标,包括终点事件、持续时间、测量方法和评价时间点等。
3.5. 样本量根据统计学原理和试验目的,计算最小样本量,确保试验结果具有统计学意义和临床意义。
4. 伦理和法规4.1. 伦理审查提交伦理委员会审查,确保研究符合伦理要求,包括知情同意、隐私保护等。
4.2. 法规遵守确保研究符合相关法律法规,如《药品管理法》、《医疗器械监督管理条例》等。
5. 数据管理和统计分析5.1. 数据管理建立严格的数据管理体系,包括数据收集、存储、核对和备份等。
5.2. 统计分析明确统计分析方法,包括数据分析软件、假设检验方法、效应量估计等。
6. 试验实施和监控6.1. 试验实施制定试验实施计划,包括试验进度、人员分工、质量控制等。
6.2. 试验监控建立试验监控机制,包括定期检查、数据审核、异常情况处理等。
7. 文件和记录确保试验过程中产生的所有文件和记录真实、完整、可追溯。
8. 修订历史记录SOP的修订历史,包括修订版本、修订日期和修订人员等信息。
9. 附录提供与临床试验方案设计相关的参考资料、工具和模板。
请注意,本SOP仅供参考,具体操作可能根据实际研究需求和法规要求进行调整。
在实施过程中,请密切关注国内外相关指南和法规的更新,确保试验方案设计始终符合最新要求。
临床试验SOP

临床试验SOP临床试验是评估新药物安全性和疗效的关键步骤之一,为了确保试验的可靠性和有效性,临床试验需遵循一套标准操作规程(SOP)。
以下是一个包括病例要求计算的临床试验SOP的范例,共计1200字以上:一、试验目的本试验旨在评估新药物X的安全性和疗效,以确定其在治疗疾病Y中的适应症和最佳剂量。
二、试验设计1.分组设计:本试验采用随机分组、双盲设计,将患者随机分配至药物X组或对照组。
2.样本容量计算:根据统计学原理和先前研究结果,选择适当的样本容量以确保试验结果具有统计学意义。
3.试验阶段:本试验分为I、II、III三个阶段,每个阶段有特定的目的和终点。
三、患者招募和入选标准1.患者招募:招募符合特定入选标准的患者进入试验。
2.入选标准:-年龄:年满18至65岁。
-确诊疾病Y,并根据国际标准明确其诊断要求。
-具有符合试验要求的病情严重度。
-没有同时进行其他临床试验。
-具备适当的知情同意能力。
四、试验操作要求1.药物配制:药物X和相应的对照物按照国际质量标准进行制备,并确保质量控制合格。
2.药物给予:根据试验设计随机分组,按照相应剂量给予药物X组和对照组的患者。
3.终点测定:定期对患者进行目标终点治疗效应(如疾病缓解、生存率等)的测定。
4.安全性监测:定期监测患者的不良事件和副作用,并根据临床试验安全管理团队的要求进行记录和处理。
五、数据分析和统计方法1.数据收集:根据试验方案和数据采集表收集完整的试验数据。
2.数据处理:对收集到的数据进行整理、校验和清洗,确保数据的准确性和完整性。
3.统计分析:根据试验目的,采用适当的统计方法和软件对试验数据进行分析,包括描述性统计、假设检验等。
4.结果解释:根据统计分析的结果,结合目标终点的变化,对试验结果进行解释和讨论。
六、质量管理和监督1.试验工作计划:制定试验的工作计划,并定期评估和更新。
2.遵守伦理原则:确保试验过程中遵守伦理原则和相关法规,并通过伦理委员会审查和批准试验方案。
临床试验标准操作规程

临床试验标准操作规程临床试验标准操作规程(Standard Operating Procedures,简称SOPs)是指在临床试验中,为确保试验的科学性、可靠性和伦理性,制定的行为规范和操作指南。
SOPs的编写是基于国家法律法规、国际指南和国内外实践经验,并经相关部门审核批准的一种规范性文件。
下面是一份关于临床试验标准操作规程的参考文档,旨在提供一种范例,仅供参考。
一、目的本SOP旨在规范临床试验过程中的相关操作,确保试验进程的科学性、可靠性和伦理性。
二、适用范围本SOP适用于本机构开展的所有临床试验项目。
三、术语和定义(根据情况修改或增加术语和定义)四、试验人员1. 主试验者:负责试验设计、管理、监督和数据分析的主要责任人。
2. 协助试验者:负责试验操作、数据采集和纪录的人员。
3. 受试者:符合试验入选标准并同意参与试验的个体。
4. 访视者:负责对受试者进行回访和记录访视纪录。
五、试验设计1. 试验目标:明确试验的科学目的和预期结果。
2. 试验方案:编写完整的试验方案,包括试验设计、入选标准、排斥标准、分组方式、随机化方法等。
3. 试验流程:绘制试验流程图,明确试验的各个阶段和相关操作。
六、试验操作1. 受试者入选:按照试验方案的入选标准和排斥标准,进行受试者的筛选和登记。
2. 受试者知情同意:确保受试者在试验前完全理解试验内容和可能风险,并签署知情同意书。
3. 随机分组:根据试验方案,使用适当的随机化方法对受试者进行分组。
4. 试验操作:按照试验方案的要求进行试验操作和数据采集,并记录相关信息。
5. 安全监测:密切监测受试者的安全情况,如出现不良反应及时处理并记录。
6. 数据管理:确保试验数据的准确性、完整性和保密性,并按照规定进行数据录入和校核。
7. 访视回访:定期对受试者进行访视并记录相关纪录。
8. 不良事件处理:对试验中的不良事件按照相关规定进行记录、报告和处理。
9. 试验中断:若试验发生重大问题或伦理审查委员会要求中断试验,应及时执行并记录。
临床试验项目标准操作规程(SOP)

临床试验项目标准操作规程(SOP)-CAL-FENGHAL-(YICAI)-Company One 1临床试验项目标准操作规程(SOP)第一部分总则第一条:为了保证新药临床试验过程中遵循科学和伦理道徳的原则,使数据的采集、录入和报告做到及时、完整、准确和一致,使受试者的权益和健康得到保护,并保障其安全,保证临床试验遵循己批准的方案、药物临床试验质量管理规范(GCP)和有关法规,使试验结论科学、可靠,根据《中华人民共和国药品管理法》、《药物临床试验质量管理规范》、《药品注册管理办法》、《赫尔辛基宣言》及ICH《人体生物医学研究国际道德指南》等相关法规文件精神,制定本标准操作程序。
第二条:药品临床试验依其流程、内容和进程不同,将其划分为临床试验前的准备、启动临床试验、临床试验过程、中期协调会和结束临床试验等五个阶段。
第三条:本标准操作规程是根据药品][期临床试验设计要求确立,临床进行的1【[、W期临床试验包括部分生物等效性试验均参照本程序执行。
第二部分临床试验询的准备第四条:申办者对临床试验中心的遴选。
⑴申办者在上报药物的临床前研究资料后,根据所申请药物的性质、作用特点、功能主治以及疾病的流行病学.样本量的大小和药品临床试验基地的专业特长等,初步遴选临床试验参加单位和确定参加单位的数量。
⑵对初选单位的专业特长、研究资质、人员组成结构、任职行医资格、相关临床试验检査和检测设备以及参研人员参加GCP培训等1W况进行现场考察,确认其资质、资源、能力和承担任务量的大小。
⑶根据现场考査结果,首先确定临床试验组长单位,经与之协商确立临床试验参加单位,并据此草拟临床试验的《多中心临床试验协调委员会联络表》和《临床试验参加单位初选报告》。
⑷国家食品药品监督管理局临床试验批文下达后,申办者根据批文精神,与临床试验组长单位一道最终确定临床试验参加单位。
第五条:申办者起草临床试验文件。
⑴申办者与研究者共同商定起草并签署试验方案、CRF和知悄同意书等临床试验文件。
实验室临床试验方案SOP

实验室临床试验方案SOP实验室临床试验方案标准操作流程(SOP)1. 引言本标准操作流程(SOP)旨在为实验室临床试验提供详细、一致的操作步骤,确保试验的准确性和可靠性。
通过遵循本SOP,研究人员可以确保试验过程的质量,提高数据的可信度,同时遵守相关法规和伦理要求。
2. 试验目的实验室临床试验的目的是通过测试和评估新的药物、治疗方法或医疗设备,以确定其在人体中的安全性和有效性。
3. 试验设计实验室临床试验应根据研究目的、试验类型(如安慰剂对照、平行组等)和样本大小进行设计。
试验设计应充分考虑伦理原则、患者安全和数据可靠性。
4. 受试者招募和筛选受试者的招募和筛选应遵循伦理原则和相关法规。
研究人员需向受试者充分告知试验的目的、过程、潜在风险和收益,并取得其知情同意。
5. 试验药物或物质的准备试验药物或物质应按照试验方案的要求进行准备。
研究人员需确保药物或物质的质量和纯度,并对其进行适当的储存和处理。
6. 样本收集和处理样本收集和处理应严格按照试验方案进行。
研究人员需确保样本的质量和完整性,避免交叉污染。
7. 数据记录和分析试验过程中产生的所有数据应进行详细记录,并按照试验方案进行数据分析。
研究人员需确保数据的真实性、准确性和完整性。
8. 结果报告和发布试验结果应以清晰、准确的方式进行报告和发布。
研究人员需确保结果报告符合相关法规和伦理要求。
9. 质量控制和监督实验室临床试验的过程应受到严格的质量控制和监督。
研究人员需定期检查试验过程和数据,以确保试验的质量和可靠性。
10. 不良事件报告和处理在试验过程中,如发现不良事件,研究人员应立即进行报告和处理。
研究人员需确保不良事件的记录和报告符合相关法规和伦理要求。
11. 试验结束和总结实验室临床试验结束后,研究人员应对试验过程和结果进行总结,并撰写试验报告。
试验报告应详细描述试验的设计、过程、结果和结论。
12. 伦理审查和批准实验室临床试验应事先提交给伦理委员会进行审查和批准。
临床试验项目标准操作规程(SOP)

临床试验项⽬标准操作规程(SOP)临床试验项⽬标准操作规程Ⅰ. ⽬的:建⽴临床试验的流程图,临床试验的SOP按此图制定。
Ⅱ. 范围:适⽤于所有临床试验SOP。
Ⅲ. 规程临床研究流程图Ⅰ. ⽬的:为使项⽬管理⼈员有所参考,提⾼项⽬管理质量和效率,特撰写此总纲。
Ⅱ. 范围:医学部。
Ⅲ. 规程1、项⽬管理的定义:项⽬的管理者,在有限的资源约束下,运⽤系统的观点、⽅法和理论,对项⽬涉及的全部⼯作进⾏有效地管理。
即对项⽬的全过程进⾏计划、组织、指挥、协调、控制和评价,以实现项⽬的⽬标。
2、项⽬管理的内容包括以下9个部分:1、项⽬范围管理是为了实现项⽬的⽬标,对项⽬的⼯作内容进⾏控制的管理过程。
它包括范围的界定,范围的规划,范围的调整等。
2、项⽬时间管理是为了确保项⽬最终的按时完成的⼀系列管理过程。
它包括具体活动的界定,如:活动排序、时间估计、进度安排及时间控制等项⼯作。
3、项⽬成本管理是为了保证完成项⽬的实际成本、费⽤不超过预算成本、费⽤的管理过程。
它包括资源的配置,成本、费⽤的预算以及费⽤的控制等项⼯作。
4、项⽬质量管理是为了确保项⽬达到客户所规定的质量要求所实施的⼀系列管理过程。
它包括质量规划,质量控制和质量保证等。
5、项⽬⼈⼒资源管理是为了保证所有项⽬关系⼈的能⼒和积极性都得到最有效地发挥和利⽤所做的⼀系列管理措施。
它包括组织的规划、团队的建设、⼈员的选聘和项⽬的班⼦建设等⼀系列⼯作。
6、项⽬沟通管理是为了确保项⽬的信息的合理收集和传输所需要实施的⼀系列措施,它包括沟通规划,信息传输和进度报告等。
7、项⽬风险管理涉及项⽬可能遇到各种不确定因素。
它包括风险识别,风险量化,制订对策和风险控制等。
8、项⽬采购管理是为了从项⽬实施组织之外获得所需资源或服务所采取的⼀系列管理措施。
它包括采购计划,采购与征购,资源的选择以及合同的管理等项⽬⼯作。
9、项⽬集成管理是指为确保项⽬各项⼯作能够有机地协调和配合所展开的综合性和全局性的项⽬管理⼯作和过程。
患者临床试验参与SOP

患者临床试验参与SOP患者临床试验参与标准化操作流程(SOP)1. 目的为确保患者临床试验的顺利进行,保障患者权益,提高临床试验质量,制定本SOP。
本SOP旨在规范患者临床试验的参与过程,包括患者筛选、入组、随访、数据收集和隐私保护等方面。
2. 适用范围本SOP适用于本机构开展的所有患者临床试验。
3. 术语和定义- 患者临床试验:指在医学研究过程中,以患者为研究对象的实验。
- 入组:指符合试验纳入标准的患者参与试验的过程。
- 随访:指在试验过程中,对入组患者进行定期或不定期的观察、评估和数据收集。
- 数据收集:指通过各种方式(如问卷、体检、实验室检测等)获取患者试验过程中的相关信息。
- 隐私保护:指在试验过程中,对患者的个人信息、医疗记录等进行保密,避免泄露。
4. 患者筛选4.1 研究人员应根据试验设计,制定明确的纳入和排除标准。
4.2 研究人员应对拟入组患者进行详细的信息收集,包括病史、体检、实验室检查等。
4.3 研究人员应根据纳入和排除标准,筛选合适的患者入组。
5. 患者入组5.1 研究人员应对入组患者进行充分的信息告知,包括试验目的、过程、可能的风险和收益等。
5.2 研究人员应获取患者的知情同意,并签订知情同意书。
5.3 研究人员应对入组患者进行编号,并建立相应的病例资料。
6. 随访管理6.1 研究人员应按照试验方案,对入组患者进行定期或不定期的随访。
6.2 研究人员应对患者的试验数据进行详细记录,并确保数据的准确性和完整性。
6.3 研究人员应密切关注患者在试验过程中的病情变化,并及时处理可能出现的问题。
7. 数据收集与隐私保护7.1 研究人员应采用标准化的数据收集工具,确保数据的准确性和一致性。
7.2 研究人员应严格执行隐私保护规定,确保患者的个人信息安全。
7.3 研究人员应对患者的医疗记录进行保密,避免泄露。
8. 质量控制与监督8.1 研究人员应定期对试验过程进行质量控制,确保试验的顺利进行。
临床试验SOP

临床试验标准操作规程(SOP)1.药品临床试验标准操作规程的制定1.1.进行药品临床试验必须符合以下原则①准备在人体进行一项药品临床试验,必须有充分理由,即已有充分的科学依据,经权衡利弊后确认有进行临床试验的必要性,并符合正当的道德原则。
②符合《赫尔辛基宣言》和《人体生物医学研究国际道德指南》规定的原则。
③有科学的、详细的并经伦理委员会批准的临床试验方案。
④临床试验应在有条件的医疗机构中进行。
我国规定临床试验由卫生部审批、国家药品监督管理局认可的临床研究基地负责进行。
⑤药品临床试验要有有资格的现职临床医师、能履行申办者职责的机构或个人和符合要求的监查员参加。
⑥有受试者自愿签署的知情同意书,受试者的权益和个人隐私权应受到充分保护。
⑦临床试验中所有数据资料及其记录、处理和保存必须有可靠的质量控制和质量保证系统。
⑧试验用药的制造、处理、贮存均应符合GMP规定,并与试验方案中的规定一致。
1.2. 药品I期临床试验标准操作规程药品I期临床试验的目的是:①研究人对新药的耐受程度;②提供安全有效的给药方案,并按下列顺序进行。
(1)准备阶段①有药政管理部门(国家药品监督管理局)批文,药检部门签发的新药质量检验报告。
②申办者提供研究者手册(Investigator'S Brochure)及其他有关资料。
③经申办者与研究者讨论并签字的临床试验方案。
研究者与申办者签订合同。
④有关文件(临床试验批文、药品质量检验报告、临床试验方案等)送伦理委员会审批,要有书面批准书。
⑤挑选参加试验的研究人员。
⑥筛选正常志愿者。
对初筛合格者进行体格检查及其他有关检查(包括实验室检查人)。
⑦经上述检查合格的正常志愿者签署知情同意书。
(2)耐受性试验①试验开始前一日住院,住院时间根据需要而定。
②根据临床前研究资料,或参考同类品种的耐受剂量范围确定最小起始剂量。
③估计最大给药剂量。
④分组通常从最小剂量至最大剂量间设3~5组,每组6~8人。
临床试验标准化操作规程

标准操作规程(SOP)Standard Operation Practice临床试验标准化操作规程I期临床试验操作规程II/III期临床试验操作规程临床试验观察表格(CRF)制定的规程临床试验方案设计规程I期临床试验方案设计规程II期临床试验方案设计规程III期临床试验方案设计规程IV期临床试验方案设计规程临床试验结束阶段操作规程临床试验总结报告设计规程I期临床耐受性试验总结报告设计规程II/III期临床试验总结报告设计规程通过新药审评会议的工作程序临床试验随机化方法及随机信封的应用规程临床试验中常用统计分析方法受试者知情同意规程试验药品供应、管理使用及处理的操作规程不良事件和严重不良事件的记录判定处理快速报告规程I期临床试验病房管理规程档案管理规程原始资料及病例报告表记录规程数据管理和输入的规程临床试验实施质量保证与控制规程《临床试验总结或小结》基地盖章操作规程临床药物基地管理委员会职责医学伦理委员会职责临床药物基地负责人职责各级临床试验研究者职责基地秘书工作职责基地统计人员工作职责研究护士工作职责临床试验标准化操作规程进行药品临床试验必须符合的原则1.准备在人体进行一项药品临床试验,必须有充分理由,即已有充分的科学依据,经权衡利弊后确认有进行临床试验的必要性,并符合正当的道德原则.2。
符合《赫尔辛基宣言》和《人体生物医学研究国际道德指南》规定的原则.3.有科学的、详细的并经伦理委员会批准的临床试验方案.4。
临床试验应在有条件的医疗机构中进行。
我国规定临床试验由卫生部审批、国家药品监督管理局认可的临床研究基地负责进行.5.药品临床试验要有有资格的现职临床医师、能履行申办者职责的机构或个人和符合要求的监查员参加。
6。
有受试者自愿签署的知情同意书,受试者的权益和个人隐私权应受到充分保护。
7.临床试验中所有数据资料及其记录、处理和保存必须有可靠的质量控制和质量保证系统。
8.试验用药的制造、处理、贮存均应符合GMP规定,并与试验方案中的规定一致。
TZRY-JG-SOP--0药物临床试验运行标准操作规程

药物临床试验运行标准操作规程目的:建立药物临床试验运行管理制度,保证试验运行规范,确保试验质量。
适用范围:适用于本机构内所有药物临床研究项目。
概述:药物临床试验机构下设机构办公室,任命主任和秘书,负责药物临床试验的承接和管理。
机构办公室对质控小组、药物临床试验机构中心药房、资料档案室及各专业科室实施统一管理。
严格按照国家GCP有关规定,对机构所承担的药物临床试验任务实行规范化管理,确保试验过程规范、结果科学可靠,最大程度的保护受试者权益并保障其安全。
(一)立项准备1.申办者/CRO(合同研究组织)与机构共同商定主要研究者。
2.PI提出研究小组成员,根据项目的具体情况并参照如下人员组成组建研究团队:临床医师、研究护士、药品管理人员、专业质量控制人员、相关科室人员(如必要)等3.研究人员的资质:①研究团队成员必须经《药物临床试验质量管理规范》培训并获取证书;②临床医务人员必须为本中心在职在岗人员。
4.准备申请临床试验的相关材料,交机构办公室秘书进行形式审查,正式受理后通知PI。
(二)立项审核机构对送审材料进行审核、立项具体事项可参考《立项审核的SOP》。
(三)伦理审核1.申办者按伦理委员会的要求准备材料,将申报材料交伦理委员会进行伦理审评。
2.最终的“伦理委员会审批件”交机构办公室档案管理员存档。
(四)合同审核1.申办者/CRO与PI拟定合同/经费预算,按《临床试验合同签订SOP)的要求,递交机构办公室完成在线审核流程。
2.协议正式签署后,方能开始临床试验。
(五)项目实施1.申办者/CRO应尽快将临床试验相关物资交研究中心。
2.申办者/CRO按照《药物管理SOP》和《药房管理SOP》将药物交予机构中心药房管理。
3.申办者/CRO协助PI主持项目启动会,具体事宜可参照《药物临床试验项目启动的SOP》4.项目管理实行PI负责制,PI对受试者安全、研究质量、进度负全责。
5.研究者遵照《药物临床试验质量管理规范》及ICH-GCP、试验方案及相关SOP,实施临床试验。
一期临床试验标准操作流程

一期临床试验标准操作流程英文回答:Clinical trial standard operating procedures (SOPs) are essential for ensuring the quality, consistency, and integrity of clinical trial data. These SOPs provide astep-by-step guide for conducting clinical trials and help ensure that all trial activities are performed in accordance with ethical and regulatory requirements.The first step in a clinical trial is the development of a protocol, which outlines the objectives, design, methodology, and statistical considerations of the trial. This protocol serves as a blueprint for the entire trial and provides detailed instructions on how the trial should be conducted.Once the protocol is finalized, the next step is to obtain regulatory and ethical approvals. This involves submitting the protocol, along with supporting documents,to the relevant regulatory authorities and ethics committees for review. These approvals are necessary to ensure that the trial meets all necessary legal and ethical requirements.After obtaining approvals, the trial can proceed to the recruitment and enrollment phase. This involves identifying and screening potential participants, obtaining informed consent, and enrolling eligible participants into the trial. The recruitment and enrollment process should be conductedin a fair and unbiased manner to ensure the integrity ofthe trial results.Once participants are enrolled, the trial interventions can be administered according to the protocol. This may involve the administration of investigational drugs,medical devices, or other interventions. It is important to follow the protocol closely and document all interventions and observations accurately to maintain data integrity.During the trial, regular monitoring and datacollection are essential. This involves conducting regularsite visits to ensure compliance with the protocol, monitoring participant safety and well-being, andcollecting and recording trial data. Any deviations from the protocol should be documented and addressed promptly to ensure the validity of the trial results.At the end of the trial, the data collected should be analyzed and interpreted. Statistical analysis should be performed to determine the efficacy and safety of the intervention being tested. The results should be reportedin a clear and concise manner, following the guidelines set forth by regulatory authorities and scientific journals.中文回答:临床试验标准操作流程(SOP)对于确保临床试验数据的质量、一致性和完整性至关重要。
临床试验sop书写模板

临床试验sop书写模板
临床试验SOP(Standard Operating Procedure,标准操作规程)是指对临床试验中各项操作流程进行规范和标准化的文件,以
确保试验过程的科学性、规范性和可靠性。
编写临床试验SOP模板
需要考虑多个方面,包括试验设计、受试者招募、试验操作、数据
收集和分析、安全监测等内容。
试验设计方面,SOP模板需要包括试验目的、研究对象、研究
方案、纳入排除标准、样本大小计算等内容。
受试者招募方面,SOP
模板应包括受试者招募的具体流程、招募标准、知情同意书签署程
序等内容。
试验操作方面,SOP模板需要详细描述试验操作的步骤、实施人员的培训和资质要求、设备使用和维护等内容。
数据收集和
分析方面,SOP模板应包括数据收集的时间点和方法、数据管理和
存储、数据分析的步骤和方法等内容。
安全监测方面,SOP模板需
要包括不良事件的报告和处理程序、安全监测委员会的设置和职责
等内容。
除了以上内容,临床试验SOP模板还需要考虑伦理审查、质量
管理、文件管理、变更控制、试验终止和报告等方面的内容。
在编
写SOP模板时,需要遵循相关的法律法规和伦理要求,确保试验过
程的合法性和道德性,并且需要经过临床试验负责单位和专业人员的审查和批准。
总之,临床试验SOP模板需要全面、清晰、具体地描述试验的各个环节,确保试验过程的科学性、规范性和可靠性,以保障试验结果的准确性和可信度。
设计临床试验方案的SOP

设计临床试验方案的SOP设计临床试验方案的标准操作流程(SOP)1. 引言本标准操作流程(SOP)旨在为临床试验方案的设计提供一个统一的框架,确保方案设计的科学性、合规性和有效性。
本SOP适用于所有临床试验项目,参与人员需严格遵守本SOP规定。
2. 试验方案设计目标试验方案设计需明确以下目标:- 确定试验类型(如随机对照试验、队列研究等);- 明确研究对象(如患者、健康志愿者等)及其纳入和排除标准;- 设定研究终点(如有效性、安全性等);- 制定样本量计算方法;- 确定试验周期、随访时间和数据收集方法;- 考虑伦理、法律和道德问题;- 制定质量控制和数据管理计划。
3. 试验方案设计流程3.1 初步设计- 收集和分析相关文献,了解同类研究的最新进展;- 确定研究问题和研究假设;- 初步确定试验类型和研究对象;- 制定初步研究终点和样本量计算方法。
3.2 详细设计- 完善研究对象纳入和排除标准;- 细化研究终点和评价指标;- 确定试验周期、随访时间和数据收集方法;- 考虑伦理、法律和道德问题,制定相应的解决方案;- 制定质量控制和数据管理计划;- 形成试验方案初稿。
3.3 方案评审- 组织专家对试验方案进行评审;- 针对评审意见进行修改和完善;- 形成试验方案定稿。
4. 试验方案文档管理- 试验方案应采用规范的文档格式进行撰写;- 确保文档清晰、简洁、易于理解;- 试验方案文档应包括所有相关表格、图表和附录;- 试验方案文档应进行编号和版本控制;- 确保试验方案文档的保密性和完整性。
5. 试验方案的实施与监督- 试验方案实施过程中,应严格按照方案规定进行;- 定期对试验进展进行监督和评估,确保试验方案的执行质量;- 如有必要,可对试验方案进行调整和优化;- 试验结束后,对试验结果进行总结和分析,为后续研究提供参考。
6. 附录- 参考文献:列出本SOP编写过程中参考的相关文献;- 相关法规和指南:列出适用于临床试验方案设计的法规和指南;- 试验方案模板:提供一份试验方案的模板,以便参与人员参考和撰写。
新药临床试验流程SOP

新药临床试验流程SOP1. 前期准备1.1 药物研发阶段- 药物发现:进行药物的初步筛选和评估,包括药理学、毒理学等研究。
- 前期开发:完成药物的化学合成或生物技术制备,并进行初步的安全性和有效性评估。
1.2 临床试验申请- 临床试验申请(CTA):向国家药品监督管理局提交新药临床试验的申请。
- 伦理审查:提交给伦理委员会审查,确保试验符合伦理要求。
2. 临床试验分期新药临床试验通常分为I、II、III、IV期,每一期都有其特定的目的和要求。
2.1 I期临床试验- 目的:评估药物的安全性、耐受性和药代动力学特性。
- 受试者人数:通常20-30人。
2.2 II期临床试验- 目的:评估药物的初步疗效和安全性。
- 受试者人数:通常数十到数百人。
2.3 III期临床试验- 目的:确认药物的疗效和安全性,为药物上市提供充分的证据。
- 受试者人数:通常数百到数千人。
2.4 IV期临床试验- 目的:上市后监测药物的长期疗效和安全性。
- 受试者人数:广泛的人群。
3. 试验执行3.1 试验设计- 随机对照试验(RCT):比较试验药物与对照药物或安慰剂的疗效。
- 队列研究:观察试验药物的疗效和安全性。
3.2 数据收集- 日记卡:记录受试者的症状、体征和药物不良反应。
- 实验室检测:血液、尿液等生物样本的分析。
3.3 数据管理- 电子数据捕捉系统(EDC):实时收集和分析数据。
- 中央实验室:确保实验室检测的一致性和质量。
4. 结果分析4.1 数据分析计划- 统计分析:预先设定数据分析的方法和指标。
- 医学分析:评估药物的疗效和安全性。
4.2 结果报告- 临床试验报告:详细记录试验的设计、执行和结果。
- 总结报告:向药品监督管理局提交药物临床试验的总结报告。
5. 后续流程5.1 药物上市申请- 新药上市申请(NDA):提交药物的完整临床试验数据,申请药物上市。
5.2 药品审批- 审批决定:国家药品监督管理局审查NDA,做出是否批准药物上市的决定。
制定医疗器械临床试验标准操作规程的标准操作规程

制定医疗器械临床试验标准操作规程的标准操作规程1目的:统一标准,明确职责,保障医疗器械临床试验的运行条件,提高临床试验的运行质量,确保临床试验数据完整、准确、真实、可靠。
2范围:适用于本机构医疗器械临床试验相关标准操作规程的制定。
3规程:3.1医疗器械临床试验通用标准操作规程(StandardOperatingProcedure,SOP)由机构办公室负责起草制定;专业特色Sc)P由专业负责人或其委派研究者进行起草制定,并交机构办公室审定,由机构负责人批准后颁发,并注明制定日期、审核日期、批准日期、颁发日期及生效日期。
3.2颁发日期与生效日期之间应有适当的过渡期,以便相关人员学习掌握。
3.3Sc)P应按照统一格式制定,字体大小及序号的使用严格按照《临床试验文件编制规范》的规定进行。
3.4医疗器械临床试验通用SOP的文件编号为GCP-QX-SOP-文件序号-版本号。
例如GCP-QX-SOP-OoI-1O表示第一个SOP文件的第一版。
专业特色Sc)P的文件编号为GCP-QX-ZY-SOP-文件序号-版本号。
3.5SOP应具有可操作性,有详细的操作步骤以便遵从。
3.6临床试验前对所有参加试验的人员进行相关的SOP的培训I,并在试验开始阶段认真监查SOP的执行,在执行中应对SOP的适用性和有效性进行系统的检查,对确认不适用的SOP进行修改或补充。
3.7临床试验机构办公室对SOP进行归档保存,并定期进行审查,填写审查记录。
3.8发现Sc)P不适用或可操作性差时,应对SoP进行修订,并填写修订记录,注明修订内容、修订人和修订日期等重要信息。
3.9新的SC)P颁发生效后,旧的SOP同时废除,并统一由机构办公室回收,保留一份其余销毁。
临床试验标准操作规程

临床试验标准操作规程GCP作为一种质量管理规范,必须保证药物临床试验数据和结果的质量,即科学性、可靠性、准确性和完整性。
为达到保证临床试验质量的目标,就必须制订进行临床试验的标准操作规程(standard operation procedure, SOP), 规范临床试验的整个过程、各个环节、每个步骤和各项操作,以保证临床试验各项行为的规范性,保证临床试验数据与结果的可追溯性。
根据GCP的要求,每一临床试验的申办公司(或CRO)及承担临床研究的机构均应制订并实施一整套标准操作规程。
我国《药物临床试验机构资格认定办法》也把标准操作规程的制订和实施作为申请单位的重要条件。
标准操作规程详细地规定了申办者或研究机构是如何严格按照GCP原则来进行其临床试验的。
SOP—经制订就具有内部法规性质,有关人员必须知晓并严格遵守。
本章重点探讨药物临床试验SOP制订的意义、范围、程序和应遵循的原则。
1 、SOP的概念标准操作规程是为了有效地实施和完成临床试验而针对每一工作环节、阶段或具体操作制订的标准和详细的书面规程。
2、制订SOP的意义制订SOP最根本的目的就是保证临床试验按照GCP规范实施,有助于严格控制临床试验中存在的或出现的各种影响试验结果的主、客观因素,尽可能地降低误差或偏差,确保得到真实可靠的研究资料,提高临床试验各项结果的评价质量。
按照SOP进行标准化操作,既有利于专家判断现行方法是否可靠,也有利于实验室自身査找分析误差的原因,以保证研究过程中数据的准确性。
制订SOP的重要意义体现在:2.1用SOP统一新药临床试验的标准,使不同研究部门或实验室的研究方法、同种实验操作和管理制度规范化,使研究人员、试验人员、管理人员的工作有据可依,以规范操作者的行为。
尽量减少操作方法上的差异性或随意性造成的误差,提高不同单位、部门或人员以及不同时期研究工作间的可比性。
2.2用SOP明确规定各个不同部门及各类不同人员的职责,建立“左右成纲不乱,上下成线不断”的完善的组织形式,使其各尽其责,互相衔接,默契配合,循规蹈矩,防止差错,确保临床研究工作的有序开展,提高新药研究资料的可信性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附件A06标准操作规程(SOP目录SOP (Standard Operating Procedure ):为有效地实施和完成某一临床试验中每项工作所拟定的标准和详细的书面规程。
药物临床试验机构标准操作规程制订、修订及编码的操作规程1、药物临床试验机构办公室指定人员起草或修订机构总的SOP2、起草人或修订人按照GCP的要求,根据医院的实际情况起草或修订SOP3、药物临床试验机构办公室主任审核。
4、药物临床试验机构对SOP实行统一编码。
5、编码格式为:“JGSOIP XX”,“JG'代表“机构” ;“XXX”为顺序号。
例如:“标准操作规程的制订,修订及编码操作规程”的编码为:JGSOPO016、SOP经专家小组讨论通过,由药物临床试验机构主任审核批准后生效。
7、药物临床试验机构办公室对通过的SOP归档保存。
8 新的SOP S过后,旧的SOP同时废除,并统一由办公室回收。
10、药物临床试验机构办公室组织相关人员学习SOP由药物临床试验机构办公室监督SOP的实施。
各专业标准操作规程的制订、修订及编码的操作规程1、药物临床试验机构专业科室指定人员起草或修订本专业的SOP2、起草人或修订人按照GCP的要求,根据本专业的实际情况起草或修订SOP3、对SOP实行统一编码。
4、编码格式为:“YYSO P XX”,“YY”为本专业前两个字的汉语拼音的第一个字母(大写);“XXX”为本专业SOP的顺序号。
例女口:心血管专业的第一个SOP的编号为:“ XXSOPO01。
5、SOP经本专业专家小组讨论通过,专业负责人审核,上报药物临床试验机构主任批准后生效6、归档保存(一份存本专业办公室,一份存医院机构办公室)。
7、新的SOP S过后,旧的SOP同时废除。
8 本专业组织相关人员学习SOP并组织实施。
1、申办者制定初步试验方案。
2、药物临床试验机构办公室接到申办者提供的初步试验方案及相关临床前资料,通过审核后,由药物临床试验机构主任指派的试验负责人确定研究小组成员。
3、试验负责人指定研究小组成员或本人与申办者共同起草试验方案。
4、试验方案应包括以下内容:(1)试验题目;(2)试验目的,试验背景,临床前研究中有临床意义的发现和与该试验有关的临床试验结果、已知对人体的可能危险与受益,及试验药物存在人种差异的可能;(3)申办者的名称和地址,进行试验的场所,研究者的姓名、资格和地址;(4)试验设计的类型,随机化分组方法及设盲的水平;(5)受试者的入选标准,排除标准和剔除标准,选择受试者的步骤,受试者分配的方法;(6)根据统计学原理计算要达到试验预期目的所需的病例数;(7)试验用药品的剂型、剂量、给药途径、给药方法、给药次数、疗程和有关合并用药的规定,以及对包装和标签的说明;(8)拟进行临床和实验室检查的项目、测定的次数和药代动力学分析(9)试验用药品的登记与使用记录、递送、分发方式及储藏条件;(10)临床观察、随访和保证受试者依从性的措施;(11)中止临床试验的标准,结束临床试验的规定;(12)疗效评定标准,包括评定参数的方法、观察时间、记录与分析;(13)受试者的编码、随机数字表及病例报告表的保存手续;(14)不良事件的记录要求和严重不良事件的报告方法、处理措施、随访的方式、时间和转归;(15)试验用药品编码的建立和保存,揭盲方法和紧急情况下破盲的规定;(16)统计分析计划,统计分析数据集的定义和选择;(17)数据管理和数据可溯源性的规定;(18)临床试验的质量控制与质量保证;(19)试验相关的伦理学;(20)临床试验预期的进度和完成日期;(21)试验结束后的随访和医疗措施;(22)各方承担的职责及其他有关规定;5、试验负责人审核试验方案。
6、试验负责人将审核的试验方案上报药物临床试验机构办公室。
7、药物临床试验机构办公室填写“药物临床试验方案讨论请示件” 上报药物临床试验机构主任。
&药物临床试验机构决定参加试验方案讨论人员(试验负责人、各协作单位项目负责人及统计人员必须参加),机构主任在“药物临床试验方案讨论请示件”签字。
9、药物临床试验机构办公室根据药物临床试验机构主任的决定,组织专家人员和申办者参加试验方案的讨论,药物临床试验机构办公室做好签到记录、会议记录并做好会议纪要。
10、各方在试验方案讨论意见表上签字。
11、根据讨论意见,研究小组对试验方案作必要的修改。
12、药物临床试验机构办公室将签字后的试验方案上报伦理委员会审批后实施。
1、知情同意书随试验方案制定,报药物临床试验机构办公室。
2、知情同意书随试验方案经各方专家讨论通过。
3、药物临床试验机构办公室将知情同意书随试验方案经伦理委员会批准。
4、由试验小组成员与受试者签署知情同意书,签署知情同意书时必须向受试者说明有关临床试验的详细情况:(1)受试者参加试验应是自愿的,而且有权在试验的任何阶段随时退出试验而不会遭到歧视或报复,其医疗待遇与权益不会受到影响。
(2)受试者参加试验及在试验中的个人资料均属保密。
必要时,药品监督管理部门、伦理委员会或申办者,按规定可以查阅参加试验的受试者资料。
(3)试验目的、试验过程与期限、检查操作、受试者预期可能的受益和风险,告知受试者可能被分配到试验的不同组别。
(4)必须给受试者充分的时间以便考虑是否愿意参加试验,对无能力表达同意的受试者,应向其法定代理人提供上述介绍与说明。
知情同意过程应采用受试者或法定代理人能理解的语言和文字,试验期间,受试者可随时了解与其有关的信息资料。
(5)如发生与试验相关的损害时,受试者可以获得治疗和相应的补偿。
5、经充分和详细解释试验的情况后获得知情同意书。
⑴ 由受试者或其法定代理人在知情同意书上签字并注明日期,执行知情同意过程的研究者也需在知情同意书上签署姓名和日期。
⑵ 对无行为能力的受试者,如果伦理委员会原则上同意、研究者认为受试者参加试验符合其本身利益时,则这些病人也可以进入试验,同时应经其法定监护人同意并签名及注明日期。
⑶ 儿童作为受试者,必须征得其法定监护人的知情同意并签署知情同意书,当儿童能做出同意参加研究的决定时,还必须征得其本人同意。
⑷在紧急情况下,无法取得本人及其合法代表人的知情同意书,如缺乏已被证实有效的治疗方法,而试验药物有望挽救生命,恢复健康,或减轻病痛,可考虑作为受试者,但需要在试验方案和有关文件中清楚说明接受这些受试者的方法,并事先取得伦理委员会同意。
⑸如发现涉及试验药物的重要新资料则必须将知情同意书作书面修改送伦理委员会批准后,再次取得受试者同意。
原则:做什么,写什么,怎么做,怎么写要求:真实、及时、准确、清晰,规范。
规程:1、原始资料包括病历报告表(另行规定)、各种会议记录、程序记录、资料存档记录、实验记录以及其它相关的原始资料等。
2、准备各种记录本,包括:机构办公室会议记录本,质量检查记录本,资料归档保存登记本,资料借阅登记本,资料收发登记本,药品收发登记本,不良事件报告登记本,业务培训登记本。
3、制定各种登记本的记录要求,在每本登记本前注明。
4、准备实验记录本,实验记录具有页码。
5、设计各种记录表格:药物临床试验方案讨论请示件,试验方案讨论意见表,试验启动前质量检查表,试验过程中质量检查表,试验结束后质量检查表。
6、及时、规范记录各种记录和表格,记录时必须注明日期、记录人、发生的事等。
7、原始资料存档保存。
要求:真实、及时、准确、规范规程:1、试验数据及时、准确填写在规定的记录本或预先设计的表格中。
2、试验数据记录字迹清晰,填写国家规定的计量单位。
3、本人或他人复核一次记录。
4、如发现记录错误,不得涂改,应在原记录上划一斜杠,保证能看清原记录,然后记录修改后的数据,并签名。
5、临床试验记录应标有正常值,并附有临床判断。
(正常、异常无临床意义、异常有临床意义、未查)。
6、记录人签名,填写日期。
要求:1、填写病例报告表应使用的规定墨水颜色(蓝黑色或黑色)。
2、填写病例报告表要及时。
3、字迹清楚和易于辨认。
4、填写病例报告表要求填写的所有信息,如有特殊情况无法填写要求内容,应填写原因,如晨间血压未查,那么血压这一格不应空着,应写明“未作”,必要时,应写明未作的理由。
5、所有的注释应填在病历报告表特定的注释区或注释页,对未执行方案要求的访问和检查也应在病例报告表中注释。
6、指定专人负责更正和修改病例报告表。
7、病例报告表不能涂改,只能用附加说明的方式,所有的更正和修改均要有更正者的签名和注明日期。
8只有在没有更准确的方法收集到临床试验的资料时,才会从受试者日记中获得信息。
如果病例报告表使用受试者日记的信息,研究人员应和受试者一起对这些数据进行复核,更正和改动患者日记中的信息应有患者的签名和日期。
9、一旦申办者收回病例报告表后,申办者和研究人员都不得单独更改病例报告表,除非经特定的程序。
10、非正常的改动(例如原始文件未出现支持这一改动的记录)应对更改作出解释11、所有的病例报告表更正必须有原始记录信息证明时才是正当的。
标准操作规程:1、研究小组的医生负责病例报告表的书写。
2、仔细阅读临床试验方案,了解试验内容、目的。
3、受试者入选时,填写入选标准和排除标准,确定病人是否可以入选。
4、确定入选者,签定知情同意书,填写试验中心号,随机顺序号,患者姓名拼音字母,医生签名,填写日期。
5、按病例报告表设计要求,填写基线检查内容:病史、病情记录,体检,血尿常规,肝肾功能等。
6、按病例报告表设计要求,填写治疗期的内容。
7、按病例报告表设计要求,填写随访期的内容。
8如有填写错误需更改,按更正程序和要求更改。
9、结束时交主管医师审核,主管医师签字。
不良事件:不良事件是病人或临床试验的受试者接受一种药品后出现的不良医学事件,但不一定与治疗有因果关系。
严重不良事件:是试验过程中发生需住院治疗、延长住院时间、伤残、影响工作能力、危及生命或死亡、导致先天畸形等事件。
处理及报告程序1、申办者提供该药物的临床前安全性研究资料及其它与安全性有关的资料,并列入研究者手册。
2、在设计方案中对不良事件应作出明确的定义,并说明不良事件严重程度的判断标准,判断不良事件与试验药物关系的分类标准(如肯定有关、可能有关、可能无关、无关和无法判定)。
方案中要求研究者必须如实填写不良事件记录表,记录不良事件的发生时间、严重程度、持续时间、采取的措施和转归。
3、试验开始前,研究小组成员必须熟悉该防范和处理医疗中受试者及突发事件预案的内容。
4、遇有严重不良事件,临床医师必须在第一时间(2小时内)向项目负责人和药物临床试验机构办公室报告,药物临床试验机构办公室应在24小时内向省食品药品监督管理部门、伦理委员会、申办单位报告。
在原始资料中应记录何时、以何种方式(如电话、传真或书面)、向谁报告了严重不良事件。