SNP开发验证的研究方法和技术路线
snp基因芯片原理

snp基因芯片原理SNP基因芯片原理引言:随着基因组学和生物技术的快速发展,人类对于基因及其与疾病关联性的研究也越来越深入。
SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是一种常见的基因变异形式,它在人类遗传变异中占据重要地位。
为了研究SNP与疾病之间的关系,科学家们开发了SNP基因芯片,它是一种高通量、高灵敏度的分子生物学工具。
本文将详细介绍SNP基因芯片的原理以及应用。
一、SNP基因芯片的定义及分类SNP基因芯片是一种利用高通量平行杂交技术检测SNP位点的工具。
根据其设计原理和应用领域的不同,SNP基因芯片可以分为两类,即基于探针的SNP芯片和基于测序的SNP芯片。
1. 基于探针的SNP芯片基于探针的SNP芯片利用DNA探针与待测样品中的基因组DNA 序列特异性杂交的原理,通过检测杂交信号来识别不同基因型。
这种芯片设计简单、成本较低,适用于小规模的SNP检测。
2. 基于测序的SNP芯片基于测序的SNP芯片采用高通量测序技术,可以直接测定待测样品中的SNP位点。
这种芯片设计复杂、成本较高,但可以同时检测数百万个SNP位点,具有更高的灵敏度和准确性。
二、SNP基因芯片的工作原理SNP基因芯片的工作原理主要包括芯片设计、杂交反应、信号检测和数据分析四个步骤。
1. 芯片设计芯片设计是SNP基因芯片的关键步骤。
首先,需要确定待测SNP 位点的基因型信息,包括目标基因型和野生型等。
然后,根据基因型信息设计一组特异性的DNA探针,这些探针可以与待测样品中的目标SNP位点特异性杂交。
2. 杂交反应杂交反应是SNP基因芯片的核心步骤。
将待测样品中的DNA与芯片上的DNA探针进行杂交反应,使其结合形成DNA双链。
杂交反应的条件包括温度、时间和缓冲液成分等,需要根据具体实验要求进行优化。
3. 信号检测信号检测是SNP基因芯片的关键步骤。
通过荧光染料或放射性同位素等标记探针,使其与芯片上的杂交DNA结合,形成信号。
疾病相关基因SNP的分析与验证

疾病相关基因SNP的分析与验证随着技术的不断发展,生物信息学研究也日渐深入。
其中,SNP(单核苷酸多态性)成为研究生物学、药理学和医学中最重要的基因变异类型之一。
SNP分析已经成为了检测疾病和药物代谢的重要方法,而在研究人类遗传学和疾病相关基因中,SNP的应用更是不可或缺。
1. SNP的概念和分类SNP,即单个核苷酸的变异,也被称为基因突变或是基因多态性。
SNP是由单个碱基的变异所引起,通常在全基因组中有约1%的概率。
SNP被广泛应用于评估个体对疾病的易感性、药物代谢和肿瘤发生等领域。
SNP按照其在基因组中的位置分类,可分为外显子SNP、内含子SNP和调控SNP。
外显子SNP指的是存在于基因的外显子区域,可以直接影响蛋白质序列的结构和功能;内含子SNP存在于外显子和调节区域之间,通常对基因功能的影响较小;调控SNP存在于基因调节区域,可以影响基因的转录和表达,进而影响基因的功能。
2. SNP的分析SNP的分析通常包括三个步骤:SNP检测、基因型鉴定和统计分析。
其中SNP 检测是最为关键的一步,目前主要的检测技术有PCR-RFLP法、MassARRAY、SNP-PCR等。
在SNP检测的基础上,需要对检测结果进行基因型鉴定。
常见的基因型鉴定方法有PCR引物延伸分析、限制性片段长度多态性分析、基因芯片以及测序等。
最后,需要进行统计分析。
在统计分析中,最常用的是卡方检验和连锁不平衡分析。
卡方检验被广泛应用于检测基因型频率和疾病之间的关联性,而连锁不平衡分析则可以确定SNP之间的互连性。
3. SNP的验证SNP验证是保证SNP检测结果准确可靠的重要步骤。
SNP验证通常包括三个方面:测序验证、多样性验证和遗传流行病学验证。
测序验证是指通过测序对SNP检测结果进行验证。
这种验证方式直接检测SNP并确定其具体的位置和变异。
然而,测序验证的成本较高,时间较长,因此不适合高通量的SNP检测。
多样性验证是指将SNP检测结果与其他不同个体的SNP检测结果进行比较,以此确认SNP检测结果的可靠性。
SNP开发验证的研究方法和技术路线

SNP开发/验证的研究方法和技术路线1分子标记:分子标记,我想这部分是我们分子标记组最核心的任务。
现在,我们没有任何可用的标记检测我们的定位材料。
即使想要验证已经定位的QTLs,我们也需要相对应的区间内的分子标记,尤其是SNP 标记。
1.1 全基因组SNP—Affymetrix芯片:一套完整的全基因组的SNP芯片,相对于Douglas体系,其操作简单,高通量。
可以直接对定位群体进行初定位的扫描或是对育种材料的背景进行分析。
在国家玉米改良中心,有一套3k的Illumina芯片,就是用来对玉米材料进行高通量检测,基因型检测结果通常可以用来QTLs初定位,育种材料的群体划分与纯度鉴定以及低密度的关联分析等。
在此,我建议我们应该开发一套番茄基因型检测的芯片。
目前,只是查找到Illumina芯片有一套全基因SNP信息,包含7,720条探针。
而Affymetrix公司目前并没有相应的产品。
但是通过跟Affymetrix公司了解,可以利用Illumina芯片已有的结果进行开发。
番茄目前测序结果显示其全基因组大小为~760Mb,而玉米为~2,500Mb,但是他们包括的基因数目~30,000个,整体情况相近。
另外,番茄作为自交植物,其LD的衰减值应该更大,有效的历史重组会更少,遗传多样性低。
因此,综合考虑,我建议我们可以开发~3k芯片,应该可以满足大多数研究材料、育种材料的基因型检测需求。
虽然目前下一代测序技术蓬勃发展,但是对于用于基因型检测来讲,其数据分析与成本相对于芯片都要更复杂和更高。
总之,我们番茄处于刚刚发展阶段,我认为就基因型检测方面,芯片有其很高的应用价值。
即使像玉米,这样测序技术发展很多年的材料,芯片技术也在应用。
1.2全基因组SNP—Douglas:当用Affymetrix芯片检测鉴定完番茄基因型并完成基因型分析之后,1)对于优良的QTLs或是基因,页脚内容1我们可以直接选择覆盖整个区间的分子标记运行Douglas系统进行分子标记辅助育种,2)对于需要进一步验证的QTLs,我们也可以利用Douglas系统只检测材料覆盖定位区间的基因型,而不需要再一次利用Affymetrix芯片或是其他方法进行全基因检测(图1.1)。
SNP技术及发展和应用

• SNP技术概述 • SNP技术的分类 • SNP技术的发展趋势 • SNP技术在生物科学研究中的应用
• SNP技术在医学诊断中的应用 • SNP技术在农业科学研究中的应用
01
SNP技术概述
定义与特点
定义
SNP,即单核苷酸多态性,是指在基 因组水平上由单个核苷酸的变异所引 起的DNA序列的遗传变异。
特点
SNP具有普遍性、稳定性、遗传性等 特点,是遗传学和基因组学研究中的 重要遗传标记。
SNP技术的历史与发展
起源
20世纪70年代,科学家开始发现 SNP的存在。
发展历程
随着基因组学和生物信息学技术 的不断进步,SNP检测技术逐渐 发展成熟,并广泛应用于遗传学、 医学和生物信息学等领域。
未来展望
随着测序技术的不断革新,SNP 检测将更加快速、准确、自动化, 有望在更多领域发挥重要作用。
精准治疗
根据个体的基因变异情况,制定个性 化的治疗方案,提高治疗效果和减少 副作用。
04
SNP技术在生物科学研究中的 应用
遗传学研究
遗传疾病关联分析
通过SNP技术分析遗传疾病与基因变 异之间的关系,有助于深入了解疾病
的发病机制和遗传基础。
人类进化研究
利用SNP技术分析不同人群的基因变 异,揭示人类进化的历史和迁徙路线。
定义
单碱基替换型SNP(也称为二等位基因型SNP)是指 基因组中单个碱基的变异引起的基因型变化。
特点
这种类型的SNP通常只涉及一个碱基的替换,导致相 应氨基酸的改变,进而可能影响蛋白质的功能。
检测方法
通过直接测序或基于Taqman探针的SNP分型技术进 行检测。
插入或缺失型SNP
SNP检测方法汇总

TaqMan SNP基因分型技术方法:此技术是由美国Life technologies公司研发的SNP分型技术,其技术原理如下简介。
PCR 反应时,加入一对两端有不同荧光标记的特异探针来识别不同的等位基因(allele1和allele2),5’端为报告荧光基团(reporter),3’端为淬灭荧光基团(quencher)。
PCR过程中,两个探针能与正向引物和反向引物之间的互补序列特异退火结合。
当探针以完整形式存在时,由于能量共振转移,荧光基团只发出微弱荧光。
特异的探针与相应的等位基因杂合后,DNA聚合酶发挥5’到3’外切酶活性,把报告荧光基团切割下来,脱离了3’端淬灭荧光基团的淬灭作用(quench),从而发出荧光。
两个探针的5’端标有不同的荧光(FAM或VIC),3’端标有MGB 淬灭基团结合体。
根据检测到的不同荧光,可以判断相应的样本的SNP 等位基因型。
整个技术的示意图如下:应用领域:本方法适用于多个涉及到SNP分型的遗传研究领域。
尤其适合针对全基因组SNP 关联研究获得的初步阳性位点,以及全基因组测序得到的大量初筛突变位点进行进一步的大样品验证研究。
RFLP (多重荧光)SNP分型技术已知的很多SNP位点正好位于限制性内切酶的识别区域,针对这些SNP位点我们就可以使用此方法进行SNP分型。
尤其是对于大样品量的多个SNP分型来说,此方法的优势较为明显。
技术方法:RFLP技术于1980年由人类遗传学家Bostein提出。
它是第一代DNA分子标记技术。
Donis—Keller利用此技术于1987年构建成第一张人的遗传图谱。
DNA分子水平上的多态性检测技术是进行基因组研究的基础。
已被广泛用于基因组遗传图谱构建、基因定位以及生物进化和分类的研究。
RFLP是根据不同品种(个体)基因组的限制性内切酶的酶切位点碱基发生突变,或酶切位点之间发生了碱基的插入、缺失,导致酶切片段大小发生了变化,通过电泳将其区分。
SNP分析原理方法及其应用

SNPs检测方法
1.理想的检测SNPs的方法 的方法
——发现未知的SNPs,或 或检测已知的SNPs
(1) 灵敏度和准确度的要求 (2) 快速、简便、高通量 高通量、自动化 (3) 费用低廉
动脑筋的技术活,有意思 有意思,有趣,形式万变; 掌握基本原理,本质不离其中 本质不离其中
SNP分析原理方法及其应用 分析原理方法及其应用
卢大儒 复旦大学生命科学学院 遗传工程国家重点实验室
遗传分析内容
n
基因组: 染色体水平 DNA水平 基因突变 重复 重复, 缺失, 倒位,重排 甲基化 基因组不稳定 基因组不稳定 多态性: SNP STR, CNV
SNPs(single nucleotide polymorphisms) single nucleotide polymorphisms)
wt/w t wt/m
m/m
DNA序列上单个碱基对的置换、插入或缺失而引起可以遗传的基因结构的变化 插入或缺失而引起可以遗传的基因结构的变化
突变(Gene Mutation)
单核苷酸多态(SNP)
n<0.1% n<0.1% n生殖细胞或体细胞
n150% n生殖细胞
SNP及检测技术

SNP及检测技术1定义:单核苷酸多态性(single nucleotide polymorphism,SNP),主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。
它是人类可遗传的变异中最常见的一种。
占所有已知多态性的90%以上。
SNP在人类基因组中广泛存在,平均每500~1000个碱基对中就有1个,估计其总数可达300万个甚至更多。
SNP所表现的多态性只涉及到单个碱基的变异,这种变异可由单个碱基的转换(transition)或颠换(transversion)所引起,也可由碱基的插入或缺失所致。
但通常所说的SNP 并不包括后两种情况。
单核苷酸多态性(SNP)是指在基因组上单个核苷酸的变异,包括置换、颠换、缺失和插入。
所谓转换是指同型碱基之间的转换,如嘌呤与嘌呤( G2A) 、嘧啶与嘧啶( T2C) 间的替换;所谓颠换是指发生在嘌呤与嘧啶(A2T、A2C、C2G、G2T) 之间的替换。
从理论上来看每一个SNP 位点都可以有4 种不同的变异形式,但实际上发生的只有两种,即转换和颠换,二者之比为2:1。
SNP 在CG序列上出现最为频繁,而且多是C转换为T ,原因是CG中的C 常为甲基化的,自发地脱氨后即成为胸腺嘧啶。
一般而言,SNP 是指变异频率大于1 %的单核苷酸变异。
在人类基因组中大概每1000 个碱基就有一个SNP ,人类基因组上的SNP 总量大概是3 ×106个。
依据排列组合原理,SNP 一共可以有6种替换情况,即A/ G、A/ T、A/ C、C/ G、C/ T 和G/ T ,但事实上,转换的发生频率占多数,而且是C2T 转换为主,其原因是Cp G的C 是甲基化的,容易自发脱氨基形成胸腺嘧啶T , Cp G 也因此变为突变热点。
理论上讲,SNP既可能是二等位多态性,也可能是3个或4个等位多态性,但实际上,后两者非常少见,几乎可以忽略。
因此,通常所说的SNP都是二等位多态性的。
基因组survey的数据snp位点开发

基因组survey的数据snp位点开发
基因组survey的数据,包括SNP位点,是通过高通量测序技术来开发的。
这些技术可以同时检测整个基因组中的大量位点,包括SNP。
开发SNP位点数据需要对样本进行DNA测序,然后使用计算方法进行数据分析,以确定哪些位点是SNP。
这些数据可用于研究遗传学、疾病关联和基因组进化等领域。
具体来说,基因组survey的数据开发过程大致如下:
1.样本收集: 首先需要收集大量的DNA样本,这些样本可以来自
人类、动物或植物等生物。
2.DNA测序: 接下来对样本中的DNA进行测序。
目前,最常用的
测序技术是高通量测序技术,如NGS(next-generation
sequencing)。
这些技术可以同时检测整个基因组中的大量位
点。
3.数据分析: 测序得到的数据需要进行分析。
首先,使用软件将原
始数据进行质量控制和过滤,然后使用计算方法进行数据分析,确定哪些位点是SNP。
4.数据整理: 最后将分析得到的SNP数据整理成数据库或文件,供
研究人员使用。
这些数据可用于研究遗传学、疾病关联和基因组进化等领域。
需要注意的是,这是一个简化的过程描述,在实际操作中,还需要考虑很多其他因素,如数据格式、数据管理、数据可视化等。
SNP开发验证的研究方法和技术路线

SNP开发/验证的研究方法和技术路线1分子标记:分子标记,我想这部分是我们分子标记组最核心的任务。
现在,我们没有任何可用的标记检测我们的定位材料。
即使想要验证已经定位的QTLs,我们也需要相对应的区间内的分子标记,尤其是SNP标记。
1.1 全基因组SNP—Affymetrix芯片:一套完整的全基因组的SNP芯片,相对于Douglas体系,其操作简单,高通量。
可以直接对定位群体进行初定位的扫描或是对育种材料的背景进行分析。
在国家玉米改良中心,有一套3k 的Illumina芯片,就是用来对玉米材料进行高通量检测,基因型检测结果通常可以用来QTLs 初定位,育种材料的群体划分与纯度鉴定以及低密度的关联分析等。
在此,我建议我们应该开发一套番茄基因型检测的芯片。
目前,只是查找到Illumina芯片有一套全基因SNP信息,包含7,720条探针.而Affymetrix 公司目前并没有相应的产品.但是通过跟Affymetrix公司了解,可以利用Illumina芯片已有的结果进行开发。
番茄目前测序结果显示其全基因组大小为~760Mb,而玉米为~2,500Mb,但是他们包括的基因数目~30,000个,整体情况相近。
另外,番茄作为自交植物,其LD的衰减值应该更大,有效的历史重组会更少,遗传多样性低.因此,综合考虑,我建议我们可以开发~3k芯片,应该可以满足大多数研究材料、育种材料的基因型检测需求。
虽然目前下一代测序技术蓬勃发展,但是对于用于基因型检测来讲,其数据分析与成本相对于芯片都要更复杂和更高。
总之,我们番茄处于刚刚发展阶段,我认为就基因型检测方面,芯片有其很高的应用价值。
即使像玉米,这样测序技术发展很多年的材料,芯片技术也在应用。
1.2全基因组SNP—Douglas:当用Affymetrix芯片检测鉴定完番茄基因型并完成基因型分析之后,1)对于优良的QTLs 或是基因,我们可以直接选择覆盖整个区间的分子标记运行Douglas系统进行分子标记辅助育种,2)对于需要进一步验证的QTLs,我们也可以利用Douglas系统只检测材料覆盖定位区间的基因型,而不需要再一次利用Affymetrix芯片或是其他方法进行全基因检测(图1.1)。
snp 研究思路

snp 研究思路SNP(SingleNucleotidePolymorphism)是指单核苷酸多态性,是遗传学和分子生物学研究中的一个重要概念。
SNP 指的是基因组中某一个位点上存在的两种以上等位基因,每个等位基因只发生单个核苷酸的变异。
SNP 是遗传变异的一种形式,因此在人类疾病的研究中扮演着重要的角色。
SNP 研究的思路可以分为以下几个步骤:1. 确定研究对象和研究目的SNP 研究可以应用于不同的研究对象,包括人类、动物和植物等。
因此,在开始研究之前,需要明确研究对象和研究目的。
对于人类疾病的研究,研究目的可能是寻找和某种疾病相关的 SNP 位点,从而揭示疾病的发病机制。
2. 选择 SNP 位点在确定研究目的之后,需要从整个基因组中选择与研究对象相关的 SNP 位点。
选择 SNP 位点的方法有很多种,包括基于遗传关联的方法、基于功能预测的方法等。
在选择 SNP 位点时,需要考虑到 SNP 的遗传学特征、频率等因素。
3. 分析 SNP 的遗传学特征一旦选择了 SNP 位点,就需要对其进行遗传学特征的分析。
这些特征包括 SNP 的等位基因频率、遗传距离、连锁不平衡等。
这些信息有助于确定 SNP 在研究中的作用和意义。
4. 确定 SNP 的功能和作用SNP 的作用可以是直接影响某个基因的表达,也可以是通过调节基因之间的相互作用来影响生物学过程。
因此,确定 SNP 的功能和作用对于理解其在疾病发生机制中的作用至关重要。
5. 分析 SNP 与疾病的关联性最后一步是分析 SNP 与疾病的关联性。
这可以通过基于人群的关联研究或基于家系的遗传研究等方法来实现。
这些研究可以揭示SNP 对疾病的风险贡献、相互作用等信息。
综上所述,SNP 研究是一项复杂而重要的工作,需要综合运用遗传学、分子生物学和统计学等知识和技术。
通过对 SNP 的研究,可以深入理解疾病的发生机制,为疾病的预防、诊断和治疗提供重要的理论基础。
SNP分析原理方法及其应用

SNP分析原理方法及其应用SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是指在基因组中的一些位置上,不同个体之间存在的碱基差异,是常见的遗传变异形式之一、SNP分析是研究SNP在基因与表型之间关联性的方法,用于揭示SNP与遗传疾病、药物反应性等的关系。
本文将介绍SNP分析的原理、方法以及其应用。
一、SNP分析原理1.SNP检测技术:SNP检测技术包括基于DNA芯片的方法、测序技术、实时荧光PCR等。
其中,高通量测序技术是最常用的SNP检测方法,可以同时检测数千个SNP位点。
2.数据分析与统计学方法:通过SNP检测技术获得的数据可以分为基因型数据(AA、AB、BB等)和等位基因频率数据(A频率、B频率等)。
统计学方法常用的有卡方检验、线性回归、逻辑回归等,用于研究SNP与表型之间的关联性。
二、SNP分析方法1.关联分析:关联分析是研究SNP与表型之间关联性的基本方法。
常用的关联分析方法包括单基因型分析、单SNP分析、基因组关联分析(GWAS)等。
单基因型分析主要是比较单个SNP的基因型在表型不同组之间的差异;单SNP分析是研究单个SNP是否与表型相关;GWAS是通过分析数万个SNP与表型之间的关系来找到与表型相关的SNP。
2. 基因型预测:基因型预测是根据已有的SNP数据,通过统计模型来预测个体的基因型。
常用的基因型预测方法有HapMap、PLINK等。
3. 功能注释:功能注释是研究SNP位点的生物学功能,揭示SNP与基因功能、表达水平之间的关系。
常用的功能注释工具有Ensembl、RegulomeDB等。
三、SNP分析应用1.遗传疾病研究:SNP与遗传疾病之间存在着密切的关系。
通过SNP分析可以发现与遗传疾病相关的SNP位点,进一步揭示疾病发生的机制,为疾病的诊断、治疗提供依据。
2.药物反应性研究:个体对药物的反应性往往存在较大差异,这与个体的遗传背景密切相关。
snp鉴定流程

SNP(单核苷酸多态性)鉴定是研究基因变异和关联分析的重要方法。
SNP鉴定流程主要包括以下几个步骤:
1. 样本收集与DNA提取:从生物体(如血液、组织、细胞等)中提取DNA。
2. 基因组DNA定量:使用spectrophotometer(分光光度计)或其他相关设备,对提取的DNA进行定量,确保实验过程中的DNA浓度一致。
3. 基因组DNA酶切:根据实验需求,选择合适的酶切酶,对DNA进行酶切。
酶切后的DNA片段长度分布均匀,便于后续实验操作。
4. 连接酶切片段与荧光标记的适配子:将酶切后的DNA片段与荧光标记的适配子连接,形成复合物。
该步骤为后续杂交和检测打下基础。
5. 杂交与洗涤:将制备好的复合物在特定设备(如杂交箱)中进行杂交,然后洗涤去除未结合的荧光标记适配子。
6. 荧光检测与数据分析:将洗涤后的样本置于荧光检测设备中,检测荧光信号。
根据荧光信号的强弱,分析样本中的SNP位点。
7. 结果验证与分析:对检测结果进行验证,如PCR扩增、测序等。
进一步分析SNP位点的分布、频率等,探讨其与疾病、表型等因素之间的关系。
基因组学研究中SNP标记方法与数据分析

基因组学研究中SNP标记方法与数据分析SNP标记方法与数据分析在基因组学研究中起着重要的作用。
SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是基因组中最常见的变异形式,是导致个体间遗传差异的主要原因之一。
因此,对SNP标记方法和数据分析的研究对于揭示基因与表型之间的关联、为功能基因组学研究提供有效工具具有重要意义。
SNP标记方法主要分为两种:基于技术平台的方法和计算预测的方法。
技术平台包括传统的基因测序、SNP芯片和下一代测序。
传统的基因测序方法通过测序反应来确定SNP位点上的碱基,虽然准确性高,但费时费力。
SNP芯片是一种高通量的方法,可以同时检测多个SNP位点,准确性相对较低。
下一代测序则是目前最常用的方法,具有高通量、高分辨率、低成本的特点。
在SNP标记方法的选择上,需要根据研究对象、目标和预算来权衡不同方法的优缺点。
在SNP标记数据的分析中,主要涉及到数据的预处理、基因型分型和遗传关联分析。
首先,数据的预处理包括对原始数据进行质量控制、过滤掉低质量的SNP位点和个体,以及进行数据标准化和归一化。
这一步骤对后续的分析至关重要,能够减少误报率和漏报率,提高结果的可靠性。
其次,基因型分型是确定每个个体在每个SNP位点上的基因型。
由于SNP位点的碱基组合较多,需要运用一系列的算法和统计模型来进行基因型分型,其中包括Bayes算法、混合模型和机器学习方法等。
最后,遗传关联分析是研究SNP位点与表型之间关联的主要方法,可以通过构建模型、计算单个SNP的关联程度,或者进行基因组广义关联分析(GWAS),来揭示SNP位点与表型之间的关系。
在进行SNP标记方法和数据分析时,还需注意一些常见的挑战和问题。
首先,SNP标记的质量控制和过滤是一个关键的步骤,需要选择合适的阈值来确保数据的准确性。
同时,样本大小也是一个重要的考虑因素,在样本量较小时,可能会出现较大的偏差。
另外,SNP位点之间的连锁不平衡(Linkage Disequilibrium,LD)也需要在分析中进行考虑,以减少虚假关联的可能性。
基因多态性(SNP)meta分析stata流程及结果解释
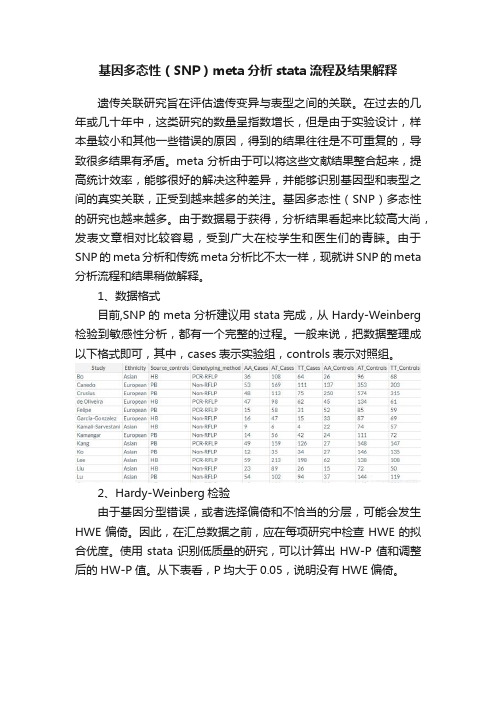
基因多态性(SNP)meta分析stata流程及结果解释遗传关联研究旨在评估遗传变异与表型之间的关联。
在过去的几年或几十年中,这类研究的数量呈指数增长,但是由于实验设计,样本量较小和其他一些错误的原因,得到的结果往往是不可重复的,导致很多结果有矛盾。
meta分析由于可以将这些文献结果整合起来,提高统计效率,能够很好的解决这种差异,并能够识别基因型和表型之间的真实关联,正受到越来越多的关注。
基因多态性(SNP)多态性的研究也越来越多。
由于数据易于获得,分析结果看起来比较高大尚,发表文章相对比较容易,受到广大在校学生和医生们的青睐。
由于SNP的meta分析和传统meta分析比不太一样,现就讲SNP的meta 分析流程和结果稍做解释。
1、数据格式目前,SNP的meta分析建议用stata完成,从Hardy-Weinberg 检验到敏感性分析,都有一个完整的过程。
一般来说,把数据整理成以下格式即可,其中,cases表示实验组,controls表示对照组。
2、Hardy-Weinberg检验由于基因分型错误,或者选择偏倚和不恰当的分层,可能会发生HWE偏倚。
因此,在汇总数据之前,应在每项研究中检查HWE的拟合优度。
使用stata识别低质量的研究,可以计算出HW-P值和调整后的HW-P值。
从下表看,P均大于0.05,说明没有HWE偏倚。
3、遗传模型给定两个等位基因(A,a),可能出现三种基因型(AA,Aa,aa)可以以不同方式产生不同的遗传模型。
基于生物学遗传模型进行不同模型的评估。
包括等位基因对比(A与a),隐性(AA与Aa + aa),显性(AA + Aa与aa)和超显性(Aa与AA + aa))遗传模型以及成对比较(AA与aa,AA与Aa和Aa与aa的比较)。
多次检验,使用Bonferroni方法调整P值。
4、异质性评估异质性的评估可以采用多种指标进行,一般来说有tau^2,Q值,I^2以及P值的计算,假如存在异质性,则可以使用亚组分析来解决。
分子生物学第五章 分子生物学的常规技术(四)SNP及其应用

原理:
当双链DNA在变性梯度凝胶中进行到与DNA变性 温度一致的凝胶位置时,DNA发生部分解链,电泳 迁移率下降,DNA链中有一个碱基改变时,会在不 同的时间发生解链,因影响电泳速度变化的程度 而被分离。
SNP的检测方法
Taqman法
以荧光共振能量传递(fluorescent resonance energy transfer,FRET)为基础的检测方法。
一个SNP表示在基因组某个位点上一个核苷酸的变 化, 作为一种碱基的替换,大多数为转换(C—T, G—A),也可能是颠换。 具有转换型变异的SNP约占SNP总量的2/3左右。 转换的发生率总是明显高于其它几种变异,而 且在CG序列上出现最为频繁,多是发生C—T的转换, 原因是CG中的C是甲基化的,它能自发的脱氨基而替 换为胸腺嘧啶。
同义cSNP: SNP所导致的编码序列改变并不影响其所翻 译的蛋白质的氨基酸序列,突变碱基与未突变碱 基的含义相同; 非同义cSNP: 指碱基序列的改变可使以其为蓝本翻译的蛋 白质序列发生改变,从而影响蛋白质的功能。这 种改变常常是导致生物性状改变的直接原因。 位于基因调控区的SNP则会影响基因表达量 的多少,因此,这两类SNP在功能和疾病发生发 展方面具有更重要的意义。
包括DNA聚合酶、硫酸化酶、萤光素酶和双磷
酸酶; 反应底物为adenosine 5‘phosphosulfate(APS) 和萤光素。 反应体系还包括待测序的DNA单链和测序引物。
在每一轮测序反应中,加入一种dNTP。 如该dNTP与模板配对,聚合酶就可以催化该dNTP掺入到 引物链中并释放焦磷酸基团(PPi)。 掺入的dNTP和释放的焦磷酸的物质的量相等,反应时 dATP由deoxyadenosine alfa-thio triphosphale(dATPaS) 替代. 因为DNA聚合酶对dATPaS的催化效率比对dATP的催化效 率高,且dATP是萤光素酶的底物,dATPctS不是。
SNP技术及发展和应用

基本步骤
1. 测序引物和 DNA 模板杂交( PCR 扩增的、单链的),与
酶和底物孵育。 2.四种dNTP(dATPS,dTTP,dCTP,dGTP)之一被加入反 应体系,如与模板配对,与引物的末端形成共价键, dNTP的焦磷酸基团(PPi)释放出来。 3.一系列的酶学反应,发出可见光信号。每个光信号的 峰高与反应中掺入的核苷酸数目成正比。 4. ATP和未掺入的dNTP由三磷酸腺苷双磷酸酶降解,淬 灭光信号,并再生反应体系。 5.然后加入下一种dNTP。
SNP在畜牧中的应用
分析病毒、细菌基因的多态性,有助于人们了 解病毒、细菌的感染发病机制与耐药性机制。 对于耐药基因的检测可从两个方面实现:一是 通过基因表达诺芯片检测药物诱导基因表达的 改变来分析其耐药性;二是寡核苷酸芯片检测 基因组序列的SNP位点来分析耐药性。目前已 经生产出乙肝耐药、结核杆菌耐药以及艾滋病 耐药的SNP检测基因芯片,用于指导临床对病 人实施个体化治疗。
SNP的概念
单核苷酸多态性(single nucleotide polymorphism,SNP),主要是指在基因组水平 上由单个核苷酸的变异所引起的DNA序列多态 性。它是人类可遗传的变异中最常见的一种。 占所有已知多态性的90%以上。SNP在人类基 因组中广泛存在,平均每500~1000个碱基对 中就有1个,估计其总数可达300万个甚至更 多。
SNP在牛QTL定位上的应用
出生体重、断奶前日增重和饲养期日增重是对肉牛生 产有重要影响的生长性状,这些基因QTL定位将提高 育种进程,在育种过程中应给予考虑。U等(2002a,b) 将出生重、断奶前日增重及饲养期日增重的QTL已经 精确定位于牛5号染色体上。Li等(2004)研究表明, my/5基因的SNP与生长性状之间显著相关。W.Ge 等(2000)根据牛生长激素受体基因cDNA序列和人类的 生长激素受体基因序列设计特异性引物,利用PCR— RFLP技术,检测到了牛生长激素受体基因上的4个 SNP,它们分别为T—C(位于76bp)、G—A(位于 22bp)、T—C(位于229bp)和A—G(位于257bp)。
干货分享SNP验证及使用_专题(上)

⼲货分享SNP验证及使⽤_专题(上)前⾔利⽤基因组测序(全基因组重测序、简化基因组测序)或基因芯⽚技术可获得⼤量的SNP标记,那么接下来⼤家都会考虑⼀个问题:我们该如何去验证和使⽤它们呢?针对不同实验⽬的,⼩编给出以下建议。
情境⼀由公司完成SNP分型并执⾏相关分析要求(如群体遗传分析)之后,如果我们后续的研究不再需要SNP标记进⾏辅助,仅单纯想验证⼀下SNP分型结果的准确性。
那么⼩编推荐使⽤Sanger 测序法、Taqman探针法、Sequenom、SNaPshot等技术,可⼀次对⼗⼏个或⼏⼗个SNP位点进⾏验证,并能对分型结果的可靠性作出较准确的评估。
情境⼆如果我们后续的研究中仍需要使⽤SNP标记进⾏辅助,如品种特异分⼦标记开发、图位克隆、分⼦标记辅助育种(MAS)等。
那么⼩编建议在验证SNP分型准确性的同时,可将其转化成以PCR为基础的分⼦标记,以⽅便各位在⾃⼰实验室开展后续⼯作。
此类型的⽅法主要有CAPS、dCAPS、AS-PCR三种,讲到这,本专题的3位主⾓终于登场。
鉴于前两位的技术原理类似,故将它们⼀起作为本专题的第⼀发与⼤家见⾯。
(满满的⼲货奉上 ~)CAPS标记1.全称CleavedAmplified Polymorphism Sequences(CAPS)——酶切扩增多态性序列2.原理简介若SNP刚好处于某⼀限制性内切酶的识别位点上,则可通过PCR技术与RFLP技术的结合,将SNP转化为CAPS标记。
基本步骤是在SNP位点的两侧设计引物,PCR特异性扩增,使⽤相应识别位点的限制性内切酶对PCR产物进⾏酶切后电泳检其多态性。
CAPS为共显性分⼦标记,可判断纯杂合。
(讲到这⾥⼤家可能还有较多疑惑,接着往下看)3.实例解析以拟南芥研究中经常会使⽤的两种⽣态型材料Col与Ler为例,经⾼通量测序发现两⽣态型材料在基因组某处存在纯合差异的SNP位点(灰⾊背景位置),观察发现在Col材料中该SNP位点刚好位于EcoRI的酶切识别位点上(红⾊字体,5’-G▼AATTC-3’),⽽Ler材料中该位点为G碱基,不存在EcoRI识别位点。
#分析流程#SNP芯片分析流程简析

#分析流程#SNP芯片分析流程简析许多的这些单核苷酸多态性可引起不同的遗传性状,即遗传的多态性(polymorphism),如ABO血型位点标记,白细胞HLA位点标记和个体药物代谢差异等。
了解这些DNA序列的差异和单核苷酸多态性以及这些差异所表现的意义将疾病的预测、诊断、预后和预防带来革命性的变化。
现已发现的单核苷酸多态性在人类基因组上就已经达到了三千万以上。
SNP分析无论是对于疾病的诊治、药物的开发还是物种群体的进化都具有十分重要的意义。
下面这张图展示了SNP芯片数据的分析主要过程。
1数据读取:通过扫描及读取芯片数据信息读取,获取杂交信号的相对强弱。
2数据标准化:数据标准化旨在除去数据中所包含的非生物学变化,这些变化可能来自实验的任何一步,包括芯片制作、RNA提纯、cDNA标记、DNA杂交、或者芯片扫描。
3SNP分析:通过信号强度,标准化的等位基因强度比,标记之前的距离和B等位基因的群体频率等参数推断SNP数目和特定染色体的区域,给出有统计意义的SNP列表。
4CNV/LOH分析:展示CNV在染色体上的分布情况及目标基因上所有CNV的分布及类型等位基因处于杂合时,会出现丢失或突变成另一个基因的趋势,称为杂合性缺失(LOH)。
杂合性缺失在肿瘤中是一种非常常见的DNA变异。
长期的细胞遗传学的研究证实,几乎所有的肿瘤细胞都存在染色体片断的非随机性丢失。
而基因拷贝数的变化,往往与神经功能、细胞生长的调节、新陈代谢以及某些疾病有关。
5COG/KOG功能注释及分类分析:对基因功能进行COG或KOG 分类,通过COG分类可以对变化基因所调节的功能有直观和感性的认识,从而了解待研究因子对于生物功能的影响,并对后续生物学实验的进行提供指导作用。
6GO Enrichment:对于得到的特定基因分类,采取DAVID、EasyGO等GO分析软件对所得基因进行功能富集分析,并得到可能的富集功能。
7KEGG Pathway分析:基于KEGG等数据库,采取超几何分布检验等统计手段,得到显著富集的生物信号通路或者代谢通路。
SNP检测方法综述

SNP检测方法综述SNP(Single Nucleotide Polymorphism)是人类基因组中最常见的遗传变异形式,它指的是在基因组中单一核苷酸的碱基发生变化所引起的遗传多态性。
SNP检测是一种用于研究个体间基因差异的重要技术,对于理解人类遗传多样性、疾病发生机制和药物反应等方面具有重要意义。
本文将综述常见的SNP检测方法,包括PCR-RFLP、TaqMan、MALDI-TOF、SNP芯片和基因测序方法。
PCR-RFLP(Polymerase Chain Reaction-Restriction Fragment Length Polymorphism)是最早也是最简单的SNP检测方法之一、它基于PCR技术扩增SNP位点的DNA片段,然后使用限制性内切酶切割扩增产物,并通过凝胶电泳的方法分离不同的限制性片段。
由于SNP会改变限制性内切酶切割位点,因此产生的限制性片段长度会有差别。
通过观察不同长度的片段,可以确定个体是否携带了该SNP。
TaqMan是一种基于荧光探针的SNP检测方法。
在TaqMan检测中,使用两个引物与单个碱基变异位点周围的DNA序列部分匹配。
其中一个引物带有FAM荧光标记,另一个带有VIC荧光标记。
当引物与模板DNA序列匹配时,TaqMan酶切的过程会释放出FAM标记的荧光信号。
然而,如果碱基变异导致引物无法与模板DNA匹配,则没有释放荧光信号。
通过荧光信号的检测,可以判断个体是否携带该SNP。
MALDI-TOF(Matrix-Assisted Laser Desorption/Ionization Time-of-Flight)是一种基质辅助激光解吸离子化飞行时间质谱技术,也可以用于SNP检测。
在SNP检测中,使用基于质谱分析的技术,先将PCR扩增的SNP位点DNA片段与一个特定的质量标准DNA片段混合。
通过质谱仪的离子化和飞行时间分析,可以确定SNP片段和质谱分析标准片段的质量和相对含量。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1分子标记:分子标记,我想这部分是我们分子标记组最核心的任务。
现在,我们没有任何可用的标记检测我们的定位材料。
即使想要验证已经定位的QTLs,我们也需要相对应的区间内的分子标记,尤其是SNP标记。
全基因组SNP—Affymetrix芯片:一套完整的全基因组的SNP芯片,相对于Douglas体系,其操作简单,高通量。
可以直接对定位群体进行初定位的扫描或是对育种材料的背景进行分析。
在国家玉米改良中心,有一套3k的Illumina芯片,就是用来对玉米材料进行高通量检测,基因型检测结果通常可以用来QTLs初定位,育种材料的群体划分与纯度鉴定以及低密度的关联分析等。
在此,我建议我们应该开发一套番茄基因型检测的芯片。
目前,只是查找到Illumina芯片有一套全基因SNP信息,包含7,720条探针。
而Affymetrix公司目前并没有相应的产品。
但是通过跟Affymetrix公司了解,可以利用Illumina芯片已有的结果进行开发。
番茄目前测序结果显示其全基因组大小为~760Mb,而玉米为~2,500Mb,但是他们包括的基因数目~30,000个,整体情况相近。
另外,番茄作为自交植物,其LD 的衰减值应该更大,有效的历史重组会更少,遗传多样性低。
因此,综合考虑,我建议我们可以开发~3k芯片,应该可以满足大多数研究材料、育种材料的基因型检测需求。
虽然目前下一代测序技术蓬勃发展,但是对于用于基因型检测来讲,其数据分析与成本相对于芯片都要更复杂和更高。
总之,我们番茄处于刚刚发展阶段,我认为就基因型检测方面,芯片有其很高的应用价值。
即使像玉米,这样测序技术发展很多年的材料,芯片技术也在应用。
全基因组SNP—Douglas:当用Affymetrix芯片检测鉴定完番茄基因型并完成基因型分析之后,1)对于优良的QTLs或是基因,我们可以直接选择覆盖整个区间的分子标记运行Douglas 系统进行分子标记辅助育种,2)对于需要进一步验证的QTLs,我们也而不需要再一次利用系统只检测材料覆盖定位区间的基因型,Douglas可以利用.)对于一些高信息量,。
3Affymetrix芯片或是其他方法进行全基因检测(图)一套可以用来构建番茄的指纹图谱。
因此,均匀分布在全基因的SNP分子标记,SNP标记是必不可少的。
完整能够与Affymetrix芯片相对应的SNP标记分布图图 QTL区间上的注:蓝色为深入片段,棕色为背景染色体。
SNP标记的开发Douglas系统下筛选具有一PCR反应体系,通过建立稳定、可靠的番茄DNA提取流程与优化致性、稳定性与多态性SNP分子标记引物,从而构建番茄全基因组SNP分子标记。
全基因组的SNP分子标记,可以用于番茄QTL定位群体的检测,分子标记辅助育种的选择以及全基因组选择的群体基因型的检测。
同时,从中挑选高质量,高信息量的分子标记用于构建一套完成的番茄指纹图谱,检测品种一致性。
供试材料:22份材料的DNA用作特异性引物筛选,2份水作为NTC(None Template Control),共计24份模板作为SNP引物的初期筛选。
以上实验在Q6仪器上进行。
对于筛选获得的一致性引物,在利用94株番茄自交系与2份水作为模板,进一步在Douglas仪器上验证。
DNA提取:1. 取~10mg 植物组织,加入研磨介质和400μl 裂解液,研磨5min,3000g离心5min。
2. 加入裂解液1/3 体积的提取液,旋涡振荡混匀,冰浴(或置于-20℃冰箱中)5min,于4℃,3200×g 离心10min,吸取上清300~400μl 加入到样品板中。
3. 按照下表在各96 孔板中加入试剂:样品及试剂体积成分板名板号板型300~400μl)1样品板(Microtiter deep wellLysis样品反应液无水乙醇96plate400μl800μlPW I(W1)洗涤板2Microtiter deep well30μl磁珠96plate800μlII(3W2)Microtiter deep well乙醇80%洗涤板96plate磁套50~100μl KingFisher 96 KF plate Elution洗脱板()44. 将各96 孔板按仪器提示顺序放入仪器中,运行程序。
5. 程序运行结束后,将DNA 溶液转移PCR 板中并测量浓度(>100ng/μl),进行后续实验或于-20℃保存待用。
分子标记命名:分子标记全部采用统一的命名规则,这样有利于结果的修改与维护。
例如:名称ID SNP位点正向引物反向引物模板序列SolBeckman-00001-V1Sol00001A/G引物合成:由上海生工公司负责引物合成。
SNP分子标记的开发:最近,Sim等人利用番茄的Illumina芯片检测410份自交系与16份番茄品种的基因型。
本研究在此基础上,利用Illumina开发的7,720条探针序列,选择其中丢失频率小于10%,等位基因频率大于10%的探针序列,获得~3,500条探针序列,作为SNP引物开发的模板序列(),每条序列的长度为~100bp,SNP 上下游各50bp。
初期,我们可以从~3500条探针中,选取~120探针序列作为模板发展引物,这120条序列要求分布在12条染色体上而且在每条染色体上尽量保证均匀分布。
如果实验成功,我们可以逐步地将剩余的探针序列发展成为SNP引物。
正向引物设计:由于我们需要使用KASP基因分型检测试剂盒,本试剂盒对正向引物已经确定为SNP位点上游~20bp(包括SNP位点),同时需要人工加上FAM或是HEX序列(图)。
因此,我们需要单独设计反向引物。
正向引物设计图反向引物设计:利用软件对模板序列进行引物选择,引物参数设定如下:;1 引物长度为18~25bp;2 GC含量为40~60%3 TM值58~62°C。
如果其上述模板序列不足以满足引物开发的需要:我们可以利用剩余~4,200条探针,继续开发新的引物。
另外,我们还可以利用PCR产物测序的方式获得gap 中序列,寻找新的SNP位点。
PCR反应:PCR反应体系:PCR反应体系需要保证均一化,这样可以满足将来在Douglas系统下进行SNP检测。
初期我们会在Q6仪器上进行预实验,摸索一致后,再在Douglas系统中进行。
Primer Mix反应体系:体积(μl)名称12FAM-Primer12HEX-Primer30Reverse-Primer46ddH2O100Total反应体系:PCR体积(μl)名称Reaction Mix Primer Mix ddH2O1DNA5Total反应程序: PCR保证均一化。
我们的PCR产物应该都为<100bp,因此我们可以采用统一设定,°C3采用两步法进行PCR反应,前期筛选引物我们设计个复性梯度:60°C、5755以及°C,以便找到最合适的复性温度。
的复性温度进行筛选:首先,利用60°C意义参数步骤反应前收集荧光 1:30min251°C 94°C 10min2预变性变性15s943°C复性,延伸1min60°C 453-4重复个循环40反应完收集荧光°C256 1:30min°C复性温度时扩增结果正常,则保留该引物在60引物在60°C如果SNP°C复性进行PCR反应。
如果得到引物扩增效果不好,则降低复性温度,利用57进行复性。
反应程序筛选:57 °C复性温度PCR 意义参数步骤反应前收集荧光°C251 1:30min预变性2°C 1940min394变性°C 15s复性,延伸°C 5741min40个循环5重复3-425°C反应完收集荧光 1:30min6°C复性温度时扩增结果正常,则保留该引物在57°C引物在如果SNP57°C55反应。
如果得到引物扩增效果不好,则降低复性温度,利用PCR复性进行引物剔除去复性温度时,反应结果不好,则该SNP进行复性。
最终,如果55°C 掉。
反应程序筛选:复性温度PCR55°C意义参数步骤25°C 1:30min1反应前收集荧光预变性294°C 10min变性315s94°C 复性,延伸1min554°C重复3-440个循环5反应完收集荧光25°C 1:30min6SNP分子标记准确性验证:引物,需要进一步在大群体中验证份模板SNPDNA扩增稳定的21对于已在,通过相同的份番茄自交系或品种的其稳定性、一致性、多态性。
选择84DNA SNP分子标记。
PCR方法,验证新开发的目标要求:SNP对特异的标记:在番茄的全基因上,我们获得番茄全基因组SNP1200能够已获得在覆盖番茄的全基因组。
/对染色体)DNA12份模板100引物(平均SNP分子标记,全部保留。
稳定扩增的分子标记辅助育种:2 ILs群体:渐渗系如果利用现在群体不再进一步构建亚群体,群体(ILsL pennellii。
但是群体)验证(图)QTLs~76个自交系,整个群体数目较小,不利于定位及M82对于这些材料的渗入片段都已经注视清楚,因此我们可以直接比较深入系与则可以定义为有深入片段引起的表型自交系的表型差异,如果存在显著性差异,在区间内。
QTL信息一致时,我们就认为此QTLs差异。
当鉴定结果与已定位的.同时我们要检索此QTL定位时的位置,以此为基础发展SNP标记,用于以后分子标记辅助育种。
对于每个QTL的区间,需要发展4~8个SNP标记覆盖其定位区间(图),其中,区间外两段各一个标记,内部2~6个SNP标记。
这样可以保证目标QTL被完整的应用。
SNP标记的开发,此时的SNP标记只需要在ILs群体的两个亲本中有多态性就可以应用。
引物设计同全基因组SNP引物设计相同。
性状验证:对于确定的QTLs,我们需要开发对应的SNP标记,利用这些标记重新验证定位结果:图深入系材料的基因型2.2IBLs群体:IBLs群体本身就是分离群体,因此可以直接用作定位(图)。
如果该群体基因型已经被鉴定,我们可以直接利用鉴定的基因型结合我们自己调查的表型,获得QTL初定位的结果。
同时,利用Affymetrix芯片进行全基因组的SNP检测,然后与表型数据相结合进行分析。
然后,需要查看定位的QTL区间内是否存在3 Douglas系统下SNP标记,如果满足分子标记辅助育种的应用,可以直接应用。
如果QTL区间内的没有或是过少的SNP标记,则需要重新开发SNP标记(标记开发同上)。
图 IBLs群体构建性状调查:从我的观点出发,对于分子育种比较有价值研究的QTL,应为育种家需求的性状所对应的QTL。