n2吸附脱附
n2吸附脱附等温线

n2吸附脱附等温线N2吸附脱附等温线是用于研究材料表面性质的一种重要手段。
本文将按照以下步骤,阐述N2吸附脱附等温线所包含的内容及其应用。
1. 什么是N2吸附脱附等温线?N2吸附脱附等温线是利用氮气对材料表面进行吸附和脱附实验,得到的吸附量与氮气压力的关系曲线。
这条曲线多数呈N形,其中有两个重要的节点:吸附量最大值和相应的氮气压力。
2. N2吸附脱附等温线的意义与应用N2吸附脱附等温线可以说明材料的孔结构、比表面积、孔径大小及分布等信息。
吸附量最大值可表示材料的比表面积,节点处的氮气压力则可表示孔径大小。
比表面积是影响材料催化、吸附等性质的重要因素之一。
通过测量比表面积,可以了解材料的催化活性、吸附能力、分离成分等性能特征。
孔径大小及分布则可影响材料的分离功能、传质性质、稳定性等。
3. 实验步骤进行N2吸附脱附实验的过程分为以下几个步骤:(1)样品处理:样品必须在真空环境中处理,将样品置于高温高真空条件下去除其水、气等杂质。
(2)吸附方法:以具有已知饱和压力和温度的氮气气体为吸附剂,将氮气不断注入试样,直至达到吸附平衡。
(3)脱附方法:在采用凝析法进行N2脱附时,降低温度或升高压力即可导致N2从孔中排放出来,直至脱附完毕。
(4)数据处理:利用吸附、脱附实验得到的数据(吸附量、氮气压力等),绘制出N2吸附脱附等温线。
4. 注意事项进行N2吸附脱附实验时需要注意以下几点:(1)确保样品表面处理干净,避免产生不必要的误差。
(2)严格控制温度、压力等实验条件,确保实验的准确性。
(3)根据所需测试范围,选择合适的测试量程和测试区间,避免测试数据超过量程或过分集中在某一范围内。
总之,N2吸附脱附等温线是解析材料性质的重要方法,通过实验的手段可以准确地了解材料的孔结构、比表面积、孔径大小及分布等信息,以此为依据,进行更深入的材料表面性质研究与应用。
N2吸脱附曲线说明

关于氮气等温吸脱附计算比概况积、孔径分布的若干说明之迟辟智美创作我们拿到的数据,只有吸脱附曲线是真实的,比概况积、孔径分布、孔容之类的都是带有主观人为色彩的数据.经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=0.05~0.35之间的一小段用传说中的BET公式处置了一下,获得单层吸附量数据Vm,然后据此算出比概况积,如此而已.◆六类吸附等温线类型几乎每本类似参考书城市提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将年夜量等温线归为五类,阶梯状的第六类为Sing增加.每一种类型城市有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为高压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段.那么吸附曲线在:高压端偏Y轴则说明资料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;高压端偏X轴说明与资料作用力弱(ІІІ型,Ⅴ型).中压端多为氮气在资料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子聚积发生的孔,有序或梯度的介孔范围内孔道.BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子聚积水平,如І型中如最后上扬,则粒子未必均匀.平常获得的总孔容通常是取相对压力为0.99左右时氮气吸附量的冷凝值.◆几个常数※※※ STP每mL氮气例:BET方法获得的比概况积则是S/(平方米每克)=4.354*Vm,其中Vm由BET方法处置可知Vm=1/(斜率+截距)◆以SBA-15分子筛的吸附等温线为例加以说明此等温线属IUPAC 分类中的IV型,H1滞后环.从图中可看出,在高压段吸附量平缓增加,此时N2 分子以单层到多层吸附在介孔的内概况,对有序介孔资料用BET方法计算比概况积时取相对压力p/p0 = 0.10~0.29比力适合.在p/p0 =0.5~0.8左右吸附量有一突增.该段的位置反映了样品孔径的年夜小,其变动宽窄可作为衡量中孔均一性的根据.在更高p/p0时有时会有第三段上升,可以反映出样品中年夜孔或粒子聚积孔情况.由N2-吸脱附等温线可以测定其比概况积、孔容和孔径分布.对其比概况积的分析一般采纳BET (Brunauer-Emmett-Teller)方法.孔径分布通常采纳BJH (Barrett-Joiner- Halenda)模型.◆Kelvin方程Kelvin方程是BJH模型的基础,由Kelvin方程得出的直径加上液膜厚度就是孔道直径.弯曲液面曲率半径R‘=2γVm/,若要算弯曲液面发生的孔径R,则有R’Cosθ=R,由于分歧资料的接触角θ分歧,下图给出的不考虑接触角情况弯曲液面曲率半径R‘和相对压力p/po对应图:◆滞后环※滞后环的发生原因这是由于毛细管凝聚作用使N2 分子在低于常压下冷凝填充了介孔孔道,由于开始发生毛细凝结时是在孔壁上的环状吸附膜液面上进行,而脱附是从孔口的球形弯月液面开始,从而吸脱附等温线不相重合,往往形成一个滞后环.还有另外一种说法是吸附时液氮进入孔道与资料之间接触角是前进角,脱附时是后退角,这两个角度分歧招致使用Kelvin方程时呈现不同.固然有可能是二者的共同作用,个人倾向于认同前者,至少直觉上(玄乎?)前者说得通些.※滞后环的种类滞后环的特征对应于特定的孔结构信息,分析这个比力考验对Kelvin方程的理解.H1是均匀孔模型,可视为直筒孔便于理解.但有些同学在解谱时会说由H1型滞后环可知SBA-15具有有序六方介孔结构,这是毛病的说法.H1型滞后环可以看出有序介孔,可是否是六方、四方、三角就不知道了,六方是小角XRD看出来的工具,这是明显的张冠李戴;H2比力难解释,一般认为是多孔吸附质或均匀粒子聚积孔造成的,多认为是“ink bottle”,等小孔径瓶颈中的液氮脱附后,束缚于瓶中的液氮气体会骤然逸出;H3与H4相比高压端吸附量年夜,认为是片状粒子聚积形成的狭缝孔;H4也是狭缝孔,区别于粒子堆集,是一些类似由层状结构发生的孔.※中压部份有较年夜吸附量但不发生滞后环的情况在相对压力为0.2-0.3左右时,根据Kelvin方程可知孔半径是很小,有效孔半径只有几个吸附质分子年夜小,不会呈现毛细管凝聚现象,吸脱附等温线重合,MCM-41孔径为2、3个nm时有序介孔吸脱附其实不呈现滞后环.◆介孔分析通常采纳的都是BJH模型(Barrett-Joiner- Halenda),是Kelvin方程在圆筒模型中的应用,适用于介孔范围,所得结果比实际偏小.针对MCM-41、SBA-15孔结构分析的具更高精度的KJS(Kruk-Jaroniec-Sayari)及其修正方法,KJS出来时用高度有序的MCM41为资料进行孔分析,结合XRD结果,得出了比BJH有更高精度的KJS方程,适用孔径分析范围在2-6.5nm之间.后来又做了推广,使之有较年夜的适用范围,可用于SBA-15孔结构(4.6-30nm)的表征.◆关于t-Plot和αs方法是对整条吸附或脱附曲线的处置方法,t-Plot可理解为thickness图形法,以氮气吸附量对单分子层吸附量作图,凝聚时形成的吸附膜平均厚度是平均吸附层数乘以单分子层厚度(0.354nm),比概况◆微孔分析含微孔资料的微孔分析对真空度,控制系统,温度传感器有分歧的要求,测试时间也比力长,时间可能是普通样品的十倍甚至二十倍.由于微孔尺寸和探针分子年夜小相差有限,部份微孔探针分子尚不能进入,解析方法要根据分歧的样品来定,需要时可借鉴相关文献方法来参考,再则自己做一批样品采纳的是一种分析方法,结果的趋势多半是正确的.现在用一种模型来分析所有范围的孔径分布还是有些困难,非线性密度泛涵理论(NLDFT)听说是可以,但论文中采纳的较少.★送样提醒★明确测试目的:比概况积和孔结构对活性中心分布,反应物产物停留发生影响,因焙烧温度等处置招致比概况分歧等情况,不要没事随便做个比概况的孔分布看看.基本上以做实验时所用的状态为准,烘干送样,要说明样品种类,年夜约比概况积,一般要提供够100平方米概况积的样品(如活性炭比概况一般是700左右,则提供0.2g足够).如果平时所用催化剂是成型后再做反应,则成型后送样,这样结果比力客观.分子筛样品一般不用成型,特别是介孔分子筛成型时则不能用较年夜压力,否则辛辛苦苦做出来可能毁于一压.◆论坛相关帖子,谢谢各位!。
n2 物理吸附-脱附表征

n2 物理吸附-脱附表征
物理吸附-脱附表征是指利用吸附-脱附技术来研究材料的表面
性质和孔隙结构。
这种技术主要用于研究吸附剂、催化剂和多孔材
料等。
在物理吸附-脱附表征中,常用的技术包括氮气吸附法(BET 法)、氩气吸附法、比表面积测定法、孔体积测定法等。
首先,物理吸附-脱附表征可以通过氮气吸附法来评估材料的比
表面积。
氮气吸附法是利用氮气在不同相对压力下吸附到材料表面
的原理来测定材料的比表面积。
通过绘制吸附等温线和脱附等温线,可以计算出材料的比表面积,进而了解材料的表面活性和孔隙结构。
其次,物理吸附-脱附表征也可以通过氩气吸附法来评估材料的
孔体积。
氩气吸附法利用氩气分子在不同相对压力下进入材料孔隙
的原理,来测定材料的孔体积分布。
通过分析吸附等温线和脱附等
温线的形状,可以得到材料的孔体积分布信息,从而了解材料的孔
隙结构特征。
另外,物理吸附-脱附表征还可以结合比表面积测定法和孔体积
测定法来全面评估材料的吸附性能。
比表面积和孔体积是影响材料
吸附性能的重要因素,通过综合分析两者的数据,可以更全面地了
解材料的吸附-脱附特性,为材料的应用提供重要参考。
总的来说,物理吸附-脱附表征是一种重要的材料表征技术,通过测定材料的比表面积和孔体积等参数,可以全面了解材料的表面性质和孔隙结构特征,为材料的研究和应用提供重要的参考依据。
n2吸附–脱附曲线

n2吸附–脱附曲线n2吸附–脱附曲线是研究材料吸附性能的重要手段之一。
它通过实验测定材料在不同温度下对氮气的吸附量,从而得到吸附–脱附曲线。
这条曲线可以反映材料的孔隙结构、比表面积以及吸附能力等信息,对于研究材料的吸附特性和应用具有重要意义。
n2吸附–脱附曲线实验通常采用比表面积测定仪进行,其原理是利用氮气在温度和压力控制下与材料表面发生吸附和脱附的过程。
在实验过程中,首先将待测样品放入比表面积测定仪中,然后通过控制温度和压力来实现氮气的吸附和脱附。
在吸附过程中,氮气会进入材料的孔隙结构中,使得样品的质量增加;而在脱附过程中,氮气会从孔隙结构中释放出来,导致样品质量减少。
通过测量样品质量的变化,可以得到不同温度下的吸附–脱附曲线。
n2吸附–脱附曲线的形状可以分为四个区域:I区、II区、III区和IV区。
在I区,随着相对压力的增加,吸附量迅速增加,这是因为氮气分子首先进入孔隙结构较大的部分;在II区,吸附量的增加逐渐减缓,这是因为较小孔隙的填充速度较慢;在III区,相对压力接近于1时,吸附量基本稳定,这是因为所有孔隙都已经填充满;在IV区,随着相对压力的减小,脱附过程开始,吸附量逐渐减少。
n2吸附–脱附曲线中的一些重要参数包括比表面积、孔径分布和孔容等。
比表面积是指单位质量或单位体积材料表面积的大小,它可以通过BET方法计算得到。
孔径分布是指不同孔径大小的孔隙在材料中所占的比例,它可以通过BJH法计算得到。
孔容是指单位质量或单位体积材料中孔隙所占的体积比例,它可以通过样品的准确质量和密度计算得到。
n2吸附–脱附曲线在材料科学和化学工程领域有着广泛的应用。
首先,它可以用于研究材料的孔隙结构和比表面积等性质,从而评价材料的储气、储能和分离等应用潜力。
其次,它还可以用于研究材料的吸附性能,例如用于催化剂和吸附剂等方面的研究。
此外,n2吸附–脱附曲线还可以用于评估材料的质量和稳定性,例如用于药物载体和生物材料等方面的研究。
n2吸附脱附等温线

n2吸附脱附等温线N2吸附脱附等温线是一种非常重要的表征物质吸附性能的方法,它可以揭示吸附剂和被吸附物质之间的相互作用和特性,为材料科学和化学领域的研究提供有力的支持。
下面我们来详细了解一下N2吸附脱附等温线的含义和应用。
一、N2吸附脱附等温线的基本概念N2吸附脱附等温线是指在一定温度下,将氮气吸附在样品表面并测量其吸附量和压力的变化,绘制出的吸附等温线图,也称为氮气吸附等温线。
其中,吸附等温线的坐标轴分别表示吸附剂(N2)的相对压力和吸附剂占据吸附剂和被吸附物质之间孔隙体积的百分比,即孔隙率。
通过N2吸附脱附等温线,我们可以获得吸附材料的许多物理性质,如孔径分布、比表面积、介孔体积、孔隙体积分布等等。
此外,它还可以用于评估吸附材料的性能,如吸附机理、吸附剂敏感性、吸附剂稳定性等等。
二、N2吸附脱附等温线的类型N2吸附脱附等温线通常可以分为五种类型,分别是:1.类型I等温线:由于大孔单一,吸附剂分子直接填充孔隙,通常见于膨胀型和树脂类物质。
2.类型II等温线:在低相对压力下出现很平缓的区域,表示大孔口发生准孔道狭窄,通常见于有孔型结构物质。
3.类型III等温线:表明孔径分布主要集中在介孔范围内,峰值很明显,通常见于沸石类、硅铝酸盐和金属有机骨架类材料等。
4.类型IV等温线:表示孔径分布主要在微孔范围内,吸附值随着相对压力增加快速上升,并达到饱和。
该类型等温线通常见于炭类和氧化钨等材料。
5.类型V等温线:由于孔径很小,需要强烈的吸附力才能使吸附剂进入其孔内。
该类型等温线通常见于碳分子筛和介孔硅材料等。
三、N2吸附脱附等温线的应用N2吸附脱附等温线在科学研究和工业应用中具有广泛的应用。
以下为具体应用:1. 材料表征:N2吸附脱附等温线可以揭示材料的孔径分布、比表面积、孔隙体积分布等,为材料表征提供了重要的信息。
2. 催化剂研究:N2吸附脱附等温线可以对催化剂进行活性研究,评估催化剂的比表面积和孔隙结构等性质,进而优化催化剂性能。
N2-吸附脱附

氮气等温吸脱附计算比★★注意★★我们拿到的数据,只有吸脱附曲线是真实的,比表面积、孔径分布、孔容之类的都是带有主观人为色彩的数据。
经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=0.05~0.35之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
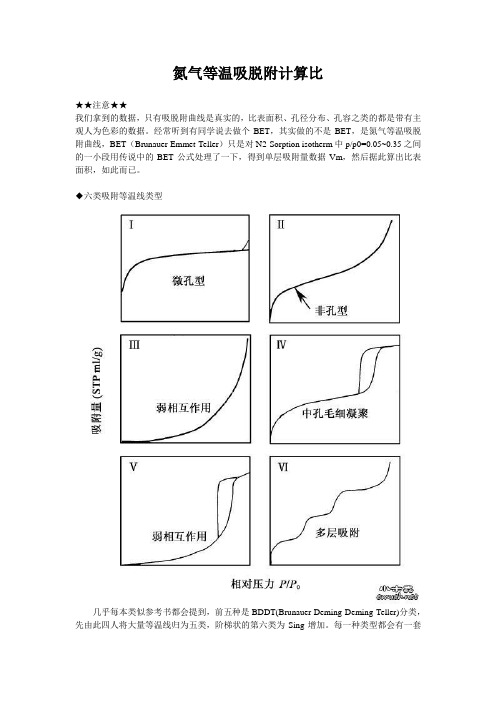
◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing增加。
每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段。
那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)。
中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生的孔,有序或梯度的介孔范围内孔道。
BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到的总孔容通常是取相对压力为0.99左右时氮气吸附量的冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积0.162平方纳米,形成单分子层铺展时认为单分子层厚度为0.354nm※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为0.001547mL例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*0.001547=400/654=约0.61mL※STP每mL氮气分子铺成单分子层占用面积4.354平方米例:BET方法得到的比表面积则是S/(平方米每克)=4.354*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)。
N2 吸附脱附
氮气等温吸脱附计算比★★注意★★我们拿到的数据,只有吸脱附曲线是真实的,比表面积、孔径分布、孔容之类的都是带有主观人为色彩的数据。
经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=0.05~0.35之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing增加。
每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段。
那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)。
中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生的孔,有序或梯度的介孔范围内孔道。
BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到的总孔容通常是取相对压力为0.99左右时氮气吸附量的冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积0.162平方纳米,形成单分子层铺展时认为单分子层厚度为0.354nm※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为0.001547mL例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*0.001547=400/654=约0.61mL※STP每mL氮气分子铺成单分子层占用面积4.354平方米例:BET方法得到的比表面积则是S/(平方米每克)=4.354*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)◆以SBA-15分子筛的吸附等温线为例加以说明此等温线属IUPAC 分类中的IV型,H1滞后环。
n2吸附脱附曲线

n2吸附脱附曲线N2吸附脱附曲线是一种重要的实验手段,用于研究材料的吸附性能以及孔结构特征。
在该实验中,通常采用BET理论(Brunauer-Emmett-Teller)来确定材料的比表面积。
下面,我将用简体中文详细介绍N2吸附脱附曲线的原理、实验步骤和数据分析方法。
一、N2吸附脱附曲线原理N2吸附脱附曲线是通过测量吸附剂材料在不同相对压力下的吸附量,从而分析材料的比表面积和孔隙特性。
该实验基于吸附剂材料与气体分子之间的相互作用力,当气体分子与吸附剂表面发生相互作用时,会引起吸附。
BET理论认为,当相对压力P/P0较低时,吸附剂与气体分子的相互作用主要是吸附在表面上的单层分子之间的相互作用。
吸附量与相对压力之间的关系可以用BET方程来描述:X/M = X/M0 * (1 - C * P/P0) / (1 - P/P0)其中,X/M是吸附量与样品质量之比,X/M0是吸附平台,C是BET 常数,P是吸附剂的相对压力,P0是饱和蒸汽压力。
二、N2吸附脱附曲线实验步骤1.准备样品:将需要测试的吸附剂样品(如催化剂、吸附剂等)进行预处理,去除表面杂质和水分。
2.测定吸附剂样品质量:将待测样品的质量进行称量,并记录下来。
3.真空处理:将待测样品置于高真空条件下,去除吸附剂内部和表面的杂质和水分。
4.温度调控:根据吸附剂样品的性质,设置合适的实验温度。
5.吸附实验:将N2气体通过烧杯等装置导入到吸附剂样品中,控制吸附剂样品的相对压力。
分别测定吸附时的相对压力和吸附量。
6.脱附实验:通过减少N2气体的流量或者停止导入气体,使得气体分子从吸附剂样品的表面脱附。
同样,测定脱附时的相对压力和吸附量。
7.数据处理:根据实验所得的数据,计算出吸附剂样品的比表面积和孔隙容积,并绘制N2吸附脱附曲线。
三、N2吸附脱附曲线数据分析方法1. BET比表面积计算:根据BET方程,绘制吸附量与相对压力之间的曲线。
当绘制的吸附量与相对压力之间的线性范围较大时,可使用线性拟合方法计算BET常数和BET比表面积。
N2吸附脱附测试技术及分析

H1型迟滞回线
❖ 均匀大小且形状规则 的孔
❖ 吸附时吸附质一层一 层的吸附在孔的表面 (孔径变小)
❖ 脱附时为弯月面
圆筒状孔道毛细凝聚(a)吸附(b)脱附示意 图
H2型迟滞回线
❖ 吸附分支由于发生毛细凝 聚现象儿逐渐上升;吸附 时凝聚在空口的液体为孔 体的吸附和凝聚提供蒸汽 。
❖ 脱附分支在较低的相对压 力下突然下降;脱附时, 空口的液体阻挡孔体蒸发 处的气体,必须等到压力 小到一定程度。
开始凝聚 开始蒸发
H3和H4型迟滞回线
❖ 形状和尺寸均匀的孔呈现H4迟 滞环
❖ H4也是狭缝孔,区别于粒子堆 集,是一些类似由层状结构产 生的孔。
❖ 开始凝聚时,由于气液界面是 大平面,只有当压力接近饱和 蒸汽压时才发生毛细凝聚(吸 附等温线类似Ⅱ型)。蒸发时 ,气液界面是圆柱状,只有当 相对压力满足时,蒸发才能开 始。
2.吸附理论
II型等温线:S 型等温线
❖ 大孔材料 ❖ 相对压力较低时,主
要是单分子层吸附,B 点对应于单分子层的 饱和吸附量 ❖ 饱和蒸汽压时,吸附 层无限大
2.吸附理论
III型等温线: 在整个压力范围内凸向下,曲线没有拐点B
❖ 在憎液性表面发生多分子 层,或固体和吸附质的吸 附相互作用小于吸附质之 间的相互作用时
❖ 低比压区与单层吸附有关,由于单层吸 附的可逆性,所以在低比压区不存在迟 滞现象
❖ 吸附时由孔壁的多分子层吸附和在空 中凝聚两种因素产生,而脱附仅由毛 细管凝聚所引起。
❖ 吸附时首先发生多分子层吸附,只有当 孔壁上的吸附层达到足够厚度时才能发 生凝聚现象。
❖ 在与吸附相同的比压下脱附时仅发生在 毛细管中的液面上的蒸汽,却不能使相 同比压下吸附的分子脱附。要使其脱附 ,就需要更小的比压,故出现脱附的滞 后现象。
n2 吸脱附dft孔径分布计算

n2 吸脱附dft孔径分布计算下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!n^2 吸脱附 DFT 孔径分布计算简介在材料科学和化学领域,通过分子动力学模拟和密度泛函理论(DFT)计算,了解吸附和脱附过程对于材料的重要性日益凸显。
N2吸脱附曲线说明

N2吸脱附曲线说明计算氮等温吸附和解吸的比表面积和孔径分布的几点注意事项我们获得的数据只是真实的吸收-解吸曲线。
比表面积、孔径分布、孔容等都是主观和人为的数据。
经常听到一些学生说要做一个BET,但他们实际做的不是BET,而是氮气等温吸附-解吸曲线。
BET(Brunauer-Emmet-Teller)只处理N2-N2-吸附等温线中p/p0=0.05~0.35之间的一小段,用著名的BET公式获得单层吸附数据Vm,然后根据它计算比表面积,如此而已。
◆六种吸附等温线几乎每一本类似的参考书都会提到前五类是BDDT(布鲁纳-戴明-戴明-泰勒)。
首先,他们四个人把大量的等温线分成五类,而第六类台阶状的是星升。
每种类型都有一组语句。
事实上,可以理解,相对压力是X轴,氮吸附量是Y轴。
X轴相对压力大致分为三个部分:低压(0.0-0.1)、中压(0.3-0.8)和高压(0.90-1.0)。
那么吸附曲线为:低压端偏离y轴表明材料对氮气有很强的作用力。
类型??类型,类型iv),许多微孔由于微孔隙中的强吸附势,显示在吸附曲线的起点?类型;材料作用力(?)对低压端偏离x轴的解释较弱??类型五)。
中压端主要是氮气在材料孔隙中的冷凝和积聚。
中孔分析来自这些数据,包括样品颗粒堆积产生的孔和有序或梯度中孔内的孔。
BJH方法基于本节获得的孔径数据。
高压段可以大致看出颗粒堆积的程度,如?如果模型最终上升,粒子可能不均匀。
通常,当相对压力约为0.99时,获得的总孔体积通常是氮吸附的冷凝值。
◆几个常数※液氮温度为77K时,液氮六方密堆积态氮分子的横截面积为0.162平方纳米,形成单层铺展时,单层厚度为0.354纳米※在标准温度和压力下冷凝1毫升氮气后(假设冷凝密度不变),体积为0.001547毫升例如,当吸附曲线p/p0在下面的吸附图中最大时,如果氮气的吸附容量为约400毫升,可以看出总孔体积= 400 * 0.001547 = 400/654 =约0.61毫升STP占地4.354平方米/毫升的氮分子铺砌成单层。
N2 吸附脱附复习课程

N2吸附脱附氮气等温吸脱附计算比★★注意★★我们拿到的数据,只有吸脱附曲线是真实的,比表面积、孔径分布、孔容之类的都是带有主观人为色彩的数据。
经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=0.05~0.35之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing增加。
每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段。
那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)。
中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生的孔,有序或梯度的介孔范围内孔道。
BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到的总孔容通常是取相对压力为0.99左右时氮气吸附量的冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积0.162平方纳米,形成单分子层铺展时认为单分子层厚度为0.354nm※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为0.001547mL 例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*0.001547=400/654=约0.61mL※ STP每mL氮气分子铺成单分子层占用面积4.354平方米例:BET方法得到的比表面积则是S/(平方米每克)=4.354*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)◆以SBA-15分子筛的吸附等温线为例加以说明此等温线属IUPAC 分类中的IV型,H1滞后环。
氮气吸附脱附原理

氮气吸附脱附原理氮气吸附脱附是一种常见的气体分离技术,广泛应用于工业生产和实验室研究中。
它基于氮气在吸附剂上的吸附和脱附过程,通过控制气体在吸附剂上的停留时间来实现对气体的分离。
本文将介绍氮气吸附脱附的原理及其在实际应用中的重要性。
氮气吸附脱附的原理主要涉及到吸附剂和氮气分子之间的相互作用。
吸附剂通常是一种多孔材料,比如活性炭、硅胶等,它具有大量的微孔和介孔结构,能够提供足够的表面积来吸附氮气分子。
当氮气通过吸附剂时,氮气分子会与吸附剂表面发生吸附作用,被固定在吸附剂表面上。
这一过程是一个物理吸附过程,主要依靠分子间的范德华力来实现。
随着氮气分子的不断吸附,吸附剂表面上逐渐形成了一层致密的氮气分子膜。
随着氮气分子在吸附剂上的积累,吸附剂的吸附能力会逐渐达到饱和状态。
这时需要进行脱附操作,将吸附剂上的氮气分子释放出来。
脱附过程通常通过降低压力或者升高温度来实现。
当压力降低或者温度升高时,吸附剂表面上的氮气分子会逐渐失去吸附能力,从而被释放出来。
这一过程是一个可逆过程,释放出的氮气分子可以再次被吸附剂吸附。
氮气吸附脱附的原理可以用于气体分离和纯化。
在实际应用中,通过控制吸附剂的类型、结构和操作条件,可以实现对不同气体的分离。
比如,可以利用氮气吸附脱附技术来分离空气中的氮气和氧气,从而获得高纯度的氮气。
此外,氮气吸附脱附还可以应用于石油化工、医药制药、食品加工等领域,用于分离和纯化各种气体。
总之,氮气吸附脱附是一种重要的气体分离技术,它基于气体分子在吸附剂上的吸附和脱附过程,通过控制操作条件来实现对气体的分离和纯化。
在工业生产和实验室研究中具有广泛的应用前景,对于提高气体纯度、降低生产成本具有重要意义。
希望本文对氮气吸附脱附原理有所帮助,谢谢阅读。
n2 物理吸附-脱附表征

n2 物理吸附-脱附表征
物理吸附-脱附表征是一种对吸附剂表面上的物理吸附和脱附过程进行表征的方法。
物理吸附是指气体或液体分子在吸附剂表面上通过范德华力或静电作用吸附,并形成吸附层的过程。
脱附则是指吸附剂表面上的吸附分子从吸附位点离开的过程。
常用的物理吸附-脱附表征方法包括吸附等温线、脱附等温线、BJH 孔径分布等。
吸附等温线是通过测量在不同温度和压力下吸附剂吸附气体或液体的吸附量来获得的。
吸附等温线可以反映出吸附剂对不同气体或液体的吸附性能,包括吸附量随压力的变化趋势、吸附平衡常数等。
脱附等温线则是在吸附等温线基础上,通过降低压力或增加温度来脱附吸附剂上的分子,测量吸附剂脱附的量。
脱附等温线可以反映出吸附剂对吸附分子的脱附性能,包括脱附量随压力的变化趋势、脱附平衡常数等。
BJH孔径分布是一种通过脱附等温线数据计算得出的吸附剂孔径分布的方法。
根据吸附分子在孔道内的脱附速率与孔道半径的关系,可以通过拟合脱附等温线数据得到吸附剂孔径的分布情况。
这可以提供吸附剂孔径大小和分布的信息,对于研究吸附剂的孔道结构和性能具有重要意义。
物理吸附-脱附表征方法可以用于研究吸附剂的吸附性能、孔道结构
以及与吸附过程相关的物理性质。
这对于吸附剂的设计和应用具有指导意义,例如在催化剂、气体分离、废水处理等领域中的应用。
N2吸脱附曲线说明

关于氮气等温吸脱附计算比表面积、孔径分布的若干说明观人为色彩的数据。
经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=0.05~0.35之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing增加。
每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段。
那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)。
中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生的孔,有序或梯度的介孔范围内孔道。
BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到的总孔容通常是取相对压力为0.99左右时氮气吸附量的冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积0.162平方纳米,形成单分子层铺展时认为单分子层厚度为0.354nm※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为0.001547mL例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*0.001547=400/654=约0.61mL例:BET方法得到的比表面积则是S/(平方米每克)=4.354*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)此等温线属IUPAC 分类中的IV型,H1滞后环。
N2吸脱附曲线说明

关于氮气等温吸脱附计算比概况积、孔径分布的若干说明之杨若古兰创作我们拿到的数据,只要吸脱附曲线是真实的,比概况积、孔径分布、孔容之类的都是带有客观人为色彩的数据.经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=0.05~0.35之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比概况积,如此而已.◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing添加.每一品种型都会有一套说法,其实可以这么理解,以绝对压力为X轴,氮气吸附量为Y轴,再将X轴绝对压力粗略地分为低压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段.那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强感化力(І型,ІІ型,Ⅳ型),较多微孔存在时因为微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料感化力弱(ІІІ型,Ⅴ型). 中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包含样品粒子堆积发生的孔,有序或梯度的介孔范围内孔道.BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最初上扬,则粒子未必均匀.平常得到的总孔容通常是取绝对压力为0.99摆布时氮气吸附量的冷凝值.◆几个常数※※※ STP每mL氮气例:BET方法得到的比概况积则是S/(平方米每克)=4.354*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)◆以SBA-15分子筛的吸附等温线为例加以说明此等温线属IUPAC 分类中的IV型,H1滞后环.从图中可看出,在低压段吸附量平缓添加,此时N2 分子以单层到多层吸附在介孔的内概况,对有序介孔材料用BET方法计算比概况积时取绝对压力p/p0 = 0.10~0.29比较适合.在p/p0 =0.5~0.8摆布吸附量有一突增.该段的地位反映了样品孔径的大小,其变更宽窄可作为衡量中孔均一性的根据.在更高p/p0时有时会有第三段上升,可以反映出样品中大孔或粒子堆积孔情况.由N2-吸脱附等温线可以测定其比概况积、孔容和孔径分布.对其比概况积的分析普通采取BET (Brunauer-Emmett-Teller)方法.孔径分布通常采取BJH (Barrett-Joiner- Halenda)模型.◆Kelvin方程Kelvin方程是BJH模型的基础,由Kelvin方程得出的直径加上液膜厚度就是孔道直径.曲折液面曲率半径R‘=2γVm/,若要算曲折液面发生的孔径R,则有R’Cosθ=R,因为分歧材料的接触角θ分歧,下图给出的不考虑接触角情况曲折液面曲率半径R‘和绝对压力p/po对应图:◆滞后环※滞后环的发生缘由这是因为毛细管凝聚感化使N2 分子在低于常压下冷凝填充了介孔孔道,因为开始发生毛细凝结时是在孔壁上的环状吸附膜液面上进行,而脱附是从孔口的球形弯月液面开始,从而吸脱附等温线不相重合,常常构成一个滞后环.还有另外一种说法是吸附时液氮进入孔道与材料之间接触角是前进角,脱附时是后退角,这两个角度分歧导导致用Kelvin方程时出现差别.当然有可能是二者的共同感化,个人倾向于认同前者,至多直觉上(玄乎?)前者说得通些.※滞后环的品种滞后环的特征对应于特定的孔结构信息,分析这个比较考验对Kelvin方程的理解.H1是均匀孔模型,可视为直筒孔便于理解.但有些同学在解谱时会说由H1型滞后环可知SBA-15具有有序六方介孔结构,这是错误的说法.H1型滞后环可以看出有序介孔,但是否是六方、四方、三角就不晓得了,六方是小角XRD看出来的东西,这是明显的张冠李戴;H2比较难解释,普通认为是多孔吸附质或均匀粒子堆积孔形成的,多认为是“ink bottle”,等小孔径瓶颈中的液氮脱附后,束缚于瓶中的液氮气体会突然逸出;H3与H4比拟高压端吸附量大,认为是片状粒子堆积构成的狭缝孔;H4也是狭缝孔,区别于粒子堆集,是一些类似由层状结构发生的孔.※中压部分有较大吸附量但不发生滞后环的情况在绝对压力为0.2-0.3摆布时,根据Kelvin方程可知孔半径是很小,无效孔半径只要几个吸附质分子大小,不会出现毛细管凝聚景象,吸脱附等温线重合,MCM-41孔径为2、3个nm时有序介孔吸脱附其实不出现滞后环.◆介孔分析通常采取的都是BJH模型(Barrett-Joiner- Halenda),是Kelvin方程在圆筒模型中的利用,适用于介孔范围,所得结果比实际偏小. 针对MCM-41、SBA-15孔结构分析的具更高精度的KJS (Kruk-Jaroniec-Sayari)及其批改方法,KJS出来时用高度有序的MCM41为材料进行孔分析,结合XRD结果,得出了比BJH有更高精度的KJS方程,适用孔径分析范围在2-6.5nm之间.后来又做了推广,使之有较大的适用范围,可用于SBA-15孔结构(4.6-30nm)的表征.◆关于t-Plot和αs方法是对整条吸附或脱附曲线的处理方法,t-Plot可理解为thickness图形法,以氮气吸附量对单分子层吸附量作图,凝聚时构成的吸附膜平均厚度是平均吸附层数乘以单分子层厚度(0.354nm),比概况◆微孔分析含微孔材料的微孔分析对真空度,控制零碎,温度传感器有分歧的请求,测试时间也比较长,时间可能是普通样品的十倍甚至二十倍.因为微孔尺寸和探针分子大小相差无限,部分微孔探针分子尚不克不及进入,解析方法要根据分歧的样品来定,须要时可借鉴相干文献方法来参考,再则本人做一批样品采取的是一种分析方法,结果的趋势多半是准确的.此刻用一种模型来分析所有范围的孔径分布还是有些困难,非线性密度泛涵理论(NLDFT)听说是可以,但论文中采取的较少.★送样提醒★明确测试目的:比概况积和孔结构对活性中间分布,反应物产品停留发生影响,因焙烧温度等处理导致比概况分歧等情况,不要没事随便做个比概况的孔分布看看. 基本上以做实验时所用的形态为准,烘干送样,要说明样品品种,大约比概况积,普通要提供够100平方米概况积的样品(如活性炭比概况普通是700摆布,则提供0.2g足够).如果平时所用催化剂是成型后再做反应,则成型后送样,如许结果比较客观.分子筛样品普通不必成型,特别是介孔分子筛成型时则不克不及用较大压力,否则辛辛劳苦做出来可能毁于一压.◆论坛相干帖子,感谢各位!。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
氮气等温吸脱附计算比★★注意★★我们拿到的数据,只有吸脱附曲线是真实的,比表面积、孔径分布、孔容之类的都是带有主观人为色彩的数据。
经常听到有同学说去做个BET,其实做的不是BET,是氮气等温吸脱附曲线,BET(Brunauer-Emmet-Teller)只是对N2-Sorption isotherm中p/p0=~之间的一小段用传说中的BET公式处理了一下,得到单层吸附量数据Vm,然后据此算出比表面积,如此而已。
◆六类吸附等温线类型几乎每本类似参考书都会提到,前五种是BDDT(Brunauer-Deming-Deming-Teller)分类,先由此四人将大量等温线归为五类,阶梯状的第六类为Sing增加。
每一种类型都会有一套说法,其实可以这么理解,以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压()、中压、高压()三段。
那么吸附曲线在:低压端偏Y轴则说明材料与氮有较强作用力(І型,ІІ型,Ⅳ型),较多微孔存在时由于微孔内强吸附势,吸附曲线起始时呈І型;低压端偏X轴说明与材料作用力弱(ІІІ型,Ⅴ型)。
中压端多为氮气在材料孔道内的冷凝积聚,介孔分析就来源于这段数据,包括样品粒子堆积产生的孔,有序或梯度的介孔范围内孔道。
BJH方法就是基于这一段得出的孔径数据;高压段可粗略地看出粒子堆积程度,如І型中如最后上扬,则粒子未必均匀。
平常得到的总孔容通常是取相对压力为左右时氮气吸附量的冷凝值。
◆几个常数※液氮温度77K时液氮六方密堆积氮分子横截面积平方纳米,形成单分子层铺展时认为单分子层厚度为※标况(STP)下1mL氮气凝聚后(假定凝聚密度不变)体积为例:如下面吸脱附图中吸附曲线p/p0最大时氮气吸附量约为400 mL,则可知总孔容=400*=400/654=约※ STP每mL氮气分子铺成单分子层占用面积平方米例:BET方法得到的比表面积则是S/(平方米每克)=*Vm,其中Vm由BET方法处理可知Vm=1/(斜率+截距)◆以SBA-15分子筛的吸附等温线为例加以说明此等温线属IUPAC 分类中的IV型,H1滞后环。
从图中可看出,在低压段吸附量平缓增加,此时N2 分子以单层到多层吸附在介孔的内表面,对有序介孔材料用BET方法计算比表面积时取相对压力p/p0 = ~比较适合。
在p/p0 =~左右吸附量有一突增。
该段的位置反映了样品孔径的大小,其变化宽窄可作为衡量中孔均一性的根据。
在更高p/p0时有时会有第三段上升,可以反映出样品中大孔或粒子堆积孔情况。
由N2-吸脱附等温线可以测定其比表面积、孔容和孔径分布。
对其比表面积的分析一般采用BET(Brunauer-Emmett-Teller)方法。
孔径分布通常采用BJH(Barrett-Joiner- Halenda)模型。
◆Kelvin方程Kelvin方程是BJH模型的基础,由Kelvin方程得出的直径加上液膜厚度就是孔道直径。
弯曲液面曲率半径R‘=2γVm/[RT*ln(p0/p)],若要算弯曲液面产生的孔径R,则有R’Cos θ=R,由于不同材料的接触角θ不同,下图给出的不考虑接触角情况弯曲液面曲率半径R ‘和相对压力p/po对应图:◆滞后环※滞后环的产生原因这是由于毛细管凝聚作用使N2 分子在低于常压下冷凝填充了介孔孔道,由于开始发生毛细凝结时是在孔壁上的环状吸附膜液面上进行,而脱附是从孔口的球形弯月液面开始,从而吸脱附等温线不相重合,往往形成一个滞后环。
还有另外一种说法是吸附时液氮进入孔道与材料之间接触角是前进角,脱附时是后退角,这两个角度不同导致使用Kelvin方程时出现差异。
当然有可能是二者的共同作用,个人倾向于认同前者,至少直觉上(玄乎)前者说得通些。
※滞后环的种类滞后环的特征对应于特定的孔结构信息,分析这个比较考验对Kelvin方程的理解。
H1是均匀孔模型,可视为直筒孔便于理解。
但有些同学在解谱时会说由H1型滞后环可知SBA-15具有有序六方介孔结构,这是错误的说法。
H1型滞后环可以看出有序介孔,但是否是六方、四方、三角就不知道了,六方是小角XRD看出来的东西,这是明显的张冠李戴;H2比较难解释,一般认为是多孔吸附质或均匀粒子堆积孔造成的,多认为是“ink bottle”,等小孔径瓶颈中的液氮脱附后,束缚于瓶中的液氮气体会骤然逸出;H3与H4相比高压端吸附量大,认为是片状粒子堆积形成的狭缝孔;H4也是狭缝孔,区别于粒子堆集,是一些类似由层状结构产生的孔。
※中压部分有较大吸附量但不产生滞后环的情况在相对压力为左右时,根据Kelvin方程可知孔半径是很小,有效孔半径只有几个吸附质分子大小,不会出现毛细管凝聚现象,吸脱附等温线重合,MCM-41孔径为2、3个nm时有序介孔吸脱附并不出现滞后环。
◆介孔分析通常采用的都是BJH模型(Barrett-Joiner- Halenda),是Kelvin方程在圆筒模型中的应用,适用于介孔范围,所得结果比实际偏小。
针对MCM-41、SBA-15孔结构分析的具更高精度的KJS(Kruk-Jaroniec-Sayari)及其修正方法,KJS出来时用高度有序的MCM41为材料进行孔分析,结合XRD结果,得出了比BJH 有更高精度的KJS方程,适用孔径分析范围在之间。
后来又做了推广,使之有较大的适用范围,可用于SBA-15孔结构的表征。
◆关于t-Plot和αs方法是对整条吸附或脱附曲线的处理方法,t-Plot可理解为thickness图形法,以氮气吸附量对单分子层吸附量作图,凝聚时形成的吸附膜平均厚度是平均吸附层数乘以单分子层厚度,比表面积=*单分子层吸附量*阿伏加德罗常数。
样品为无孔材料时,t-Plot是一条过原点直线,当试样中含有微孔,介孔,大孔时,直线就会变成几段折线,需要分别分析。
αs方法中的下标是standard的意思,Sing提出用相对压力为时的吸附量代替单分子层吸附量,再去作图,用这种方法先要指定一个标准,或是在仪器上做一个标样,处理方法和图形解释两种方法是类似的。
两则之间可以相互转化,t=αs◆微孔分析含微孔材料的微孔分析对真空度,控制系统,温度传感器有不同的要求,测试时间也比较长,时间可能是普通样品的十倍甚至二十倍。
由于微孔尺寸和探针分子大小相差有限,部分微孔探针分子尚不能进入,解析方法要根据不同的样品来定,需要时可借鉴相关文献方法来参考,再则自己做一批样品采用的是一种分析方法,结果的趋势多半是正确的。
现在用一种模型来分析所有范围的孔径分布还是有些困难,非线性密度泛涵理论(NLDFT)听说是可以,但论文中采用的较少。
★送样提醒★明确测试目的:比表面积和孔结构对活性中心分布,反应物产物停留产生影响,因焙烧温度等处理导致比表面不同等情况,不要没事随便做个比表面的孔分布看看。
基本上以做实验时所用的状态为准,烘干送样,要说明样品种类,大约比表面积,一般要提供够100平方米表面积的样品(如活性炭比表面一般是700左右,则提供足够)。
如果平时所用催化剂是成型后再做反应,则成型后送样,这样结果比较客观。
分子筛样品一般不用成型,特别是介孔分子筛成型时则不能用较大压力,否则辛辛苦苦做出来可能毁于一压。
列一些参考资料书目,有兴趣可找一下看看:1.刘维桥孙桂大《固体催化剂实用研究方法》中国石化出版社2.刘振宇等,多孔炭的纳米结构及其解析,化学进展,2001,13(1)3.近藤精一,石川达雄,安部郁夫著. 李国希译.《吸附科学》北京化学工业出版社,20054.陈诵英孙予罕丁云杰周仁贤罗孟飞《吸附与催化》河南科学技术出版社,20015. Gregg,., Sing,《吸附、比表面与孔隙率》化学工业出版社,1989W. J., Crittenden B.. Adsorption Technology and Design, Elsevier Science& Technology 1998. M. Principles of Adsorption and Adsorption Processes, John Wiley & Sons (1984) M., Adsorption Engineering, Elsevier (1990)论文, S., Emmett P. H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc., 1938, 60:309-319E. P.; Joyner L. G.; Halenda P. P.. The determination of pore volume and area distributions in porous substances: I. computations from nitrogen isotherms . J. . Soc., 1951, 73:373-380K, Everett D, Haul R, Moscou L, Pierotti R, Rouquerol J, Siemieniewska T. Pure Appl. Chem, 1985, 57: 603-619M., Jaroniec M., Sayari A.. Application of large pore MCM-41 molecularsieves to improve pore size analysis using nitrogen adsorption measurements. Langmuir, 1997, 13:6267-6273M., Solovyov L. A.. Improvement of the Kruk-Jaroniec-Sayari Method forpore size analysis of ordered silicas with cylindrical mesopores. Langmuir, 2006, 22:6757-6760A. Adv. Colloid Interface Sci., 2001, 93: 135-224◆论坛相关帖子,谢谢各位!【原创】我所理解的BJHbbs/tid=2609274【求助】请教吸附滞后环的问题bbs/tid=2529404bbs/tid=2529404【求助】催化剂的比表面积以及孔体积与其粒径存在必然关系吗bbs/tid=2469677【求助】N2脱吸附曲线滞后环不闭合的原因bbs/tid=1361358请教:关于孔分布吸附-脱附的滞后环bbs/tid=1317581【求助】N2吸脱附曲线中如何分别计算微孔面积和介孔面积呢bbs/tid=2068501【求助】孔径分布曲线怎么得到bbs/tid=1584583&fpage=0&view=&highlight=&page=1【资源】催化剂比表面和孔结构的相关分析bbs/tid=2619868【资源】关于催化剂比表面测试的方法简介bbs/tid=2621460。