FDA新药申报
美国FDA新药注册申报NDA流程简介

美国FDA新药注册申报NDA流程简介
美国FDA(Food and Drug Administration)是负责监管药品的安全性和有效性的机构。
NDA(New Drug Application)是制药公司向FDA提交的新药注册申报文件,它是提交给FDA以申请新药上市的必要步骤。
NDA的流程可以大致分为以下几个步骤:
1.前期研究:制药公司在研发新药之前通常会进行多个阶段的临床试验,以评估新药的安全性和有效性。
这些临床试验的结果将用于NDA中。
2.提交申请:在完成所有必要的研究和试验后,制药公司将准备并提交NDA申请给FDA。
这个申请包括新药的所有相关信息,如药物成分、制造过程、稳定性、临床试验结果、药品标签等。
3.文件审查:FDA会对提交的NDA进行审查,确保申请文件的完整性和合规性。
如果发现不完整或有疑问的地方,FDA可能会请求补充信息或进一步解释。
4.审查评估:FDA的药品审评员会对申请进行详细评估,根据临床试验数据、药物的安全性和有效性等因素,来判断是否批准新药上市。
审评员可能会与制药公司进行沟通,提出问题或寻求进一步资料。
5.决策:基于审评员的评估和建议,FDA会做出决策,即批准或拒绝新药的上市申请。
如果批准,FDA会制定药品标签和使用说明等要求。
需要注意的是,整个NDA流程可能需要一段时间,通常需要
几年的时间来完成。
此外,FDA还有一些快速审评程序,如加速审评、优先审评等,可以加快某些重要新药的审批速度。
【医疗药品管理】FDA新药申请指南
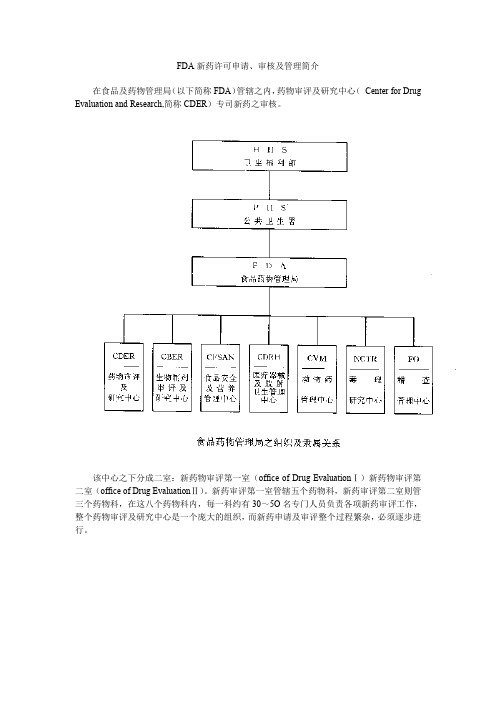
FDA新药许可申请、审核及管理简介在食品及药物管理局(以下简称FDA)管辖之内,药物审评及研究中心(Center for Drug Evaluation and Research,简称CDER)专司新药之审核。
该中心之下分成二室:新药物审评第一室(office of Drug EvaluationⅠ)新药物审评第二室(office of Drug EvaluationⅡ)。
新药审评第一室管辖五个药物科,新药审评第二室则管三个药物科,在这八个药物科内,每一科约有30~5O名专门人员负责各项新药审评工作,整个药物审评及研究中心是一个庞大的组织,而新药申请及审评整个过程繁杂,必须逐步进行。
药物审评及研究中心(Center for Drug Evalution and Research,CDER)之组织说明如下:1.秘书室(Executive Secretariant Staff)2.总务室(Office of Management)(l)药物资记中心(Drug Information Resource)(2)医学图书馆(Medical Library)(3)总务及王计(Management and Budget)(4)资讯系统设计(Information System Design)3.专业进修室(Professional Development)4.顾问团(Advisors and Consultants Staff)5·前导性新药审评(Pilot Drug Evaluation)6.非处方药审评室(office of OTC Drug Evaluation)(1)单篇非处方药审评室(MonograPh Review Staff)(2)非处方药政策性科(OTC Drug Policy Staff)(3)医学审评科(Medical Review Staff)7.非专利处方药室(office of Generic Drugs)(1)化学第一科(ChemistryⅠ)(2)化学第二科(ChemistryⅡ)(3)生体相等性科(Bioequivalence)8.研究发展室(Office of Research Resources)(1)研究及试验科(Research and Testing)(2)药品分析科(Drug Analysis)(3)生体药学科(Biopharmaceutics)(4)临床药理科(Clinical pharmacology)9.新药标准室(Office of Drug Standards)(l)新药市场广告及信息科(Drug Marketing,Advertising and Communications)10.新药合法性室(Office of Compliance)(1)新药信息科(Drug Labeling Compliance)(2)新药品质审核科(Drug Quality Evaluation)(3)新药产品及制造品质科(Mahufacturing and Product Quality)(4)科学性侦查科(Scientific Investigations)(5)新药管理科(Regulatory Affairs)11.新药流行学及统计生物学室(Office of Epidemiology and Biostatistics)(1)流行学及监视科(Epidemiology and Surveillances)(2)生物统计科(Biometrics)12.新药审评第一室(Office of Drug EvaluationⅠ,ODEI)(1)心脏药物科(Division of Cardio-Renal Drug Products)(2)神经药物科(Division of Neuropharmacological Drug Products)(3)肿瘤肺药物科(Division of Oncology and Pulmonary Drug Products)(4)影像、手术及牙药物科(Division of Medical Imaging Surgical and Dental Drug Products)(5)胃肠及凝血药物科(Division of Gastrointestinal and Coagulation Drug Products)13.新药审评第二室(Office of Drug Evaluation Ⅱ, ODE Ⅱ)(1)新陈代谢及内分泌药物科(Division of Metabolism and Endocrine Drug ProductS)(2)抗传染药物科(Division of Anti-Infective Drug Products )(3)抗病毒药物科(Division of Anti—Viral Drug ProductS)二、新药申请(一)药物的定义依据联邦食品药物及化妆品法第二章第201节,药物的定义如下:l.美国药典,同种治疗法药典,或者国家处方集(National Formulary)中所列的物质。
FDA新药审批流程简述

FDA新药审批流程简述FDA(美国食品药品监督管理局)负责监管并审批新药的上市。
下面将对FDA新药审批流程进行简述。
1.阶段Ⅰ临床试验:新药首先在健康志愿者身上进行,评估药物的安全性和耐受性,并确定药物的剂量范围。
2.阶段Ⅱ临床试验:新药在患者身上进行,评估药物的疗效和副作用。
试验时间较长,研究人员需要收集更多的数据,以确定新药的安全性和效力。
3.阶段Ⅲ临床试验:新药在大规模患者群体身上进行,以证明其疗效和安全性是否持久有效,并与现有的治疗方法进行比较。
4.新药申请:药企将试验结果提交给FDA,并申请新药上市批准。
包括药物的数据和试验结果,使用方法等。
FDA会评估申请材料。
5.NDA审批:NDA(新药申请)包括对药物研究的细节、试验结果等的描述。
FDA对NDA进行评估,确定药物是否符合上市标准。
此过程可能需要数月或数年。
6.审查:FDA将药物进行详细审查,并与药企进行沟通,以充分了解药物的性质和潜在的风险。
7.审查会议:FDA可能会召开药物审查会议,邀请专家、学者和公众就药物的疗效和风险发表意见。
8.确认上市:如果FDA认为新药符合上市标准,将发出批准通知,允许药企将新药投入市场销售。
以上是FDA新药审批流程的简要概述。
在流程中,FDA扮演着保障公众健康和安全的角色,确保新上市的药物是安全有效的。
FDA New Drug Approval Process OverviewThe FDA (U.S. Food and Drug Administration) is responsible for regulating and approving new drugs for market. The following is an overview of the FDA's new drug approval process.1. Phase I Clinical Trials: The new drug is first tested on healthy volunteers to assess its safety and tolerability and determine the dosage range.5. NDA Approval: The NDA (New Drug Application) includes a detailed description of the drug's research, trial results, etc. The FDA evaluates the NDA to determine if the drug meets the approval standards. This process can take months or years.7. Review Meetings: The FDA may hold drug review meetings where experts, scholars, and the public are invited to provide opinions on the drug's efficacy and risks.9. Post-Market Surveillance: The FDA continues to monitor the drug's safety and effectiveness, collects feedback from the market, and takes necessary actions, including further research and updating warning labels.The above is a brief overview of the FDA's new drug approval process. Throughout the process, the FDA plays a crucial role insafeguarding public health and safety, ensuring that newly marketed drugs are safe and effective.。
FDA新药审批流程简述

FDA新药审批流程简述FDA(美国食品药品监督管理局)是负责监督和管理美国食品和药品安全的联邦机构。
它负责确保市场上的药品是安全有效的,并且符合严格的审批标准。
新药审批是FDA的主要职责之一,它是一个复杂且漫长的过程,通常包括以下几个步骤:1.提交新药申请(NDA)首先,药物制造商需要通过NDA向FDA提交有关新药的完整资料。
这些资料通常包括药物的化学成分、制造工艺、药理学研究、临床试验数据以及用于治疗的适应症等信息。
此外,还需要提供药物的质量、安全和有效性的证据。
2.评估申请文件一旦FDA收到NDA,会对申请文件进行评估。
这个过程通常包括对文件的完整性和合规性的审查,例如核实所有必需的资料是否齐全。
如果缺少必要的信息,FDA可能会要求制造商提供补充材料。
3.优先审批对于一些药物,FDA可能会给予优先审批待遇。
例如,对于治疗一些严重疾病的新药,FDA可能会加快审批速度,以符合患者的迫切需求。
4.临床试验阶段一旦FDA确认NDA文件完整无误,药物制造商可以开展临床试验。
临床试验是评估药物疗效和安全性的关键步骤,通常分为三个阶段(I、II、III)。
这些试验需要遵守严格的方案和伦理规定,以确保患者的安全和药物的有效性。
5.申请审核委员会的审查在临床试验结束后,药物制造商将向FDA提交试验结果,并要求FDA审核审查委员会对其进行审查。
审查委员会是由FDA专家组成的独立机构,他们会仔细评估试验结果以及相关数据和文献,并发表意见。
这些意见对于FDA的最终决策具有重要影响。
6.申请批准在经过临床试验和审查后,FDA将根据收集到的数据和顾问委员会的意见,决定是否批准新药上市。
如果FDA认为药物的风险和益处之间的平衡是积极的,它将批准新药,并颁发批准证书。
7.监督上市后安全性一旦新药获得批准,FDA对其进行监督,以确保其安全性和真实性。
制造商需要持续向FDA提供有关药物的安全性和有效性的信息。
此外,FDA还通过药物安全盛会进行监测,并与制造商和医疗专业人员合作,收集和分析有关药物的副作用和其他安全问题的信息。
fda_new_drug_application(新药申请指南)
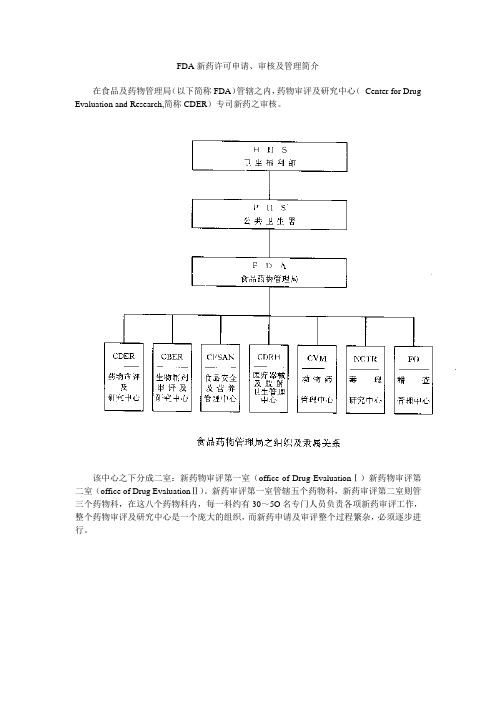
FDA新药许可申请、审核及管理简介在食品及药物管理局(以下简称FDA)管辖之内,药物审评及研究中心(Center for Drug Evaluation and Research,简称CDER)专司新药之审核。
该中心之下分成二室:新药物审评第一室(office of Drug EvaluationⅠ)新药物审评第二室(office of Drug EvaluationⅡ)。
新药审评第一室管辖五个药物科,新药审评第二室则管三个药物科,在这八个药物科内,每一科约有30~5O名专门人员负责各项新药审评工作,整个药物审评及研究中心是一个庞大的组织,而新药申请及审评整个过程繁杂,必须逐步进行。
药物审评及研究中心(Center for Drug Evalution and Research,CDER)之组织说明如下:1.秘书室(Executive Secretariant Staff)2.总务室(Office of Management)(l)药物资记中心(Drug Information Resource)(2)医学图书馆(Medical Library)(3)总务及王计(Management and Budget)(4)资讯系统设计(Information System Design)3.专业进修室(Professional Development)4.顾问团(Advisors and Consultants Staff)5·前导性新药审评(Pilot Drug Evaluation)6.非处方药审评室(office of OTC Drug Evaluation)(1)单篇非处方药审评室(MonograPh Review Staff)(2)非处方药政策性科(OTC Drug Policy Staff)(3)医学审评科(Medical Review Staff)7.非专利处方药室(office of Generic Drugs)(1)化学第一科(ChemistryⅠ)(2)化学第二科(ChemistryⅡ)(3)生体相等性科(Bioequivalence)8.研究发展室(Office of Research Resources)(1)研究及试验科(Research and Testing)(2)药品分析科(Drug Analysis)(3)生体药学科(Biopharmaceutics)(4)临床药理科(Clinical pharmacology)9.新药标准室(Office of Drug Standards)(l)新药市场广告及信息科(Drug Marketing,Advertising and Communications)10.新药合法性室(Office of Compliance)(1)新药信息科(Drug Labeling Compliance)(2)新药品质审核科(Drug Quality Evaluation)(3)新药产品及制造品质科(Mahufacturing and Product Quality)(4)科学性侦查科(Scientific Investigations)(5)新药管理科(Regulatory Affairs)11.新药流行学及统计生物学室(Office of Epidemiology and Biostatistics)(1)流行学及监视科(Epidemiology and Surveillances)(2)生物统计科(Biometrics)12.新药审评第一室(Office of Drug EvaluationⅠ,ODEI)(1)心脏药物科(Division of Cardio-Renal Drug Products)(2)神经药物科(Division of Neuropharmacological Drug Products)(3)肿瘤肺药物科(Division of Oncology and Pulmonary Drug Products)(4)影像、手术及牙药物科(Division of Medical Imaging Surgical and Dental Drug Products)(5)胃肠及凝血药物科(Division of Gastrointestinal and Coagulation Drug Products)13.新药审评第二室(Office of Drug Evaluation Ⅱ, ODE Ⅱ)(1)新陈代谢及内分泌药物科(Division of Metabolism and Endocrine Drug ProductS)(2)抗传染药物科(Division of Anti-Infective Drug Products )(3)抗病毒药物科(Division of Anti—Viral Drug ProductS)二、新药申请(一)药物的定义依据联邦食品药物及化妆品法第二章第201节,药物的定义如下:l.美国药典,同种治疗法药典,或者国家处方集(National Formulary)中所列的物质。
FDA NDA新药Ⅰ期临床试验申报资料

新药Ⅰ期临床试验申报资料的内容及格式要求1995年11月美国FDA发布2009年6月药审中心组织翻译诺华制药有限公司翻译北核协会审核药审中心最终核准目录I. 引言 (1)II. 资料内容和格式 (1)A. 封面 (1)B. 目录 (1)C. 介绍性声明与整体研究计划 (1)D. 研究者手册 (2)E. 研究方案 (2)F. 化学、生产和控制信息 (2)G. 药理毒理信息 (5)H. 研究药物既往在人体中使用的经验 (6)I. 其他相关说明 (6)新药Ⅰ期临床试验申报资料的内容及格式要求I. 引言美国FDA于1995年11月颁布了《GUIDANCE FOR INDUSTRY:CONTNET AND FORMAT OF INVESTIGATIONAL NEW DRUG APPLICATIONS(IND)FOR PHASE 1 STUDIES OF DRUGS,INCLUDING WELL-CHARACTERIZED,THERAPEUTIC,BIOTECHNOLOGY-DERIVED PRODUCTS》,为进一步促进我国新药研发和审评能力的提高,本指导原则系在参考FDA上述指导原则的基础上,结合我国《药品注册管理办法》的相关要求,经翻译转化,以指导、规范我国新药I期临床试验申报资料的内容及格式要求。
本指导原则阐述了将研究药物(包括已进行结构确证的治疗性生物工程类产品)申请用于人体研究时,所需要提供的资料内容和格式要求1。
按照《药品注册管理办法》,属注册分类1和2的药品,应当进行临床试验。
在满足我国现行法规对申报临床试验资料要求的基础上,按照药物研究阶段的不同,将申报资料的要求加以分类,将有助于加速新药批准进入临床试验的时间。
对于拟进行I期临床试验的注册申请,所提交的申报资料应能够满足安全性评价的需要,其中最重要的两部分内容包括:1)毒理学研究汇总报告,基于已完成的动物研究结果,为人体研究提供初步的支持依据;2)适合于I期临床试验研究用样品的制备资料。
一文读懂FDA新药注册流程

一文读懂FDA新药注册流程FDA(美国食品药品监督管理局)是一个负责监管和审批新药上市的机构。
其新药注册流程遵循一系列严格的标准和程序,确保新药的安全性和有效性。
以下是一份详细的描述FDA新药注册流程的1200字以上文章。
FDA新药注册流程是一个复杂而严格的过程,旨在保护公众的健康和安全。
该流程通常分为三个主要阶段:实验室研究、临床试验和申请审批。
首先,在实验室研究阶段,新药的发现和开发开始于基础科学研究。
科学家们进行一系列的实验,以验证一些化合物是否具有潜在的治疗效果。
这个阶段的目标是鉴定和筛选出可能的药物候选。
一旦确定了潜在的候选者,科学家会继续开展研究,并了解药物的药动学、药理学和安全性。
接下来,进入临床试验阶段。
在这个阶段,新药候选者将在人体中进行测试。
临床试验是一系列严格控制的研究,其目的是评估药物的安全性和有效性。
临床试验分为三个阶段,每个阶段都有不同的目标和参与人数。
第一阶段的临床试验是对一小群健康志愿者进行的,目的是评估药物的安全性和耐受性,并确定适当的剂量。
这个阶段通常只涉及数十名志愿者,并在医院或临床研究中心进行。
第二阶段的临床试验是在相对较大的患者群体中进行的,通常包括数百名患者。
这个阶段的主要目标是确定药物的疗效,并进一步评估安全性。
经过第二阶段的临床试验,确定了相对安全和有效的药物剂量和用法。
第三阶段的临床试验是最大规模的研究,通常包括数千名患者。
在这个阶段,药物的疗效和安全性将在各种人群中进行全面评估。
这些试验旨在确认药物是否比现有疗法更安全或更有效。
此阶段可能需要数年时间,并需收集大量的数据和信息。
一旦临床试验完成,并证明了药物的安全性和有效性,便可以进入申请审批阶段。
在这一阶段,该药物的制造商将向FDA提交一份申请,申请批准该药物上市。
申请中需要包括所有先前实验室和临床试验的数据和报告。
申请审批是一个详细和复杂的过程。
FDA的专业人员将仔细研究申请中的数据,确保其准确性和可靠性。
FDA新药申请程序

FDA新药申请程序FDA(美国食品药品监管局)是负责监管美国食品和药品的联邦机构,其主要职责是确保公众使用安全、高效的药品和食品。
当制药公司开发一种新药时,他们必须按照FDA的一系列规定和程序来提交新药申请。
下面将详细介绍FDA新药申请程序。
新药开发的第一阶段是前临床研究,该阶段的目标是确定是否有足够的证据表明药物是安全的,并且有望对特定疾病或疾病群体产生治疗效果。
前临床研究主要包括体外实验和动物实验。
在这个阶段,研究人员必须向FDA提交一份“药物开发准备报告”(Investigational New Drug Application,简称IND),包括药物的化学成分、药理学相关数据以及试验设计。
FDA对IND进行评估,如果认为药物的风险低于潜在获益,那么该公司将被允许进行下一阶段的研究。
下一个阶段是临床试验,主要分为三个阶段:I期、II期和III期。
I期试验通常在健康志愿者身上进行,目的是评估药物的安全性和耐受性,以及确定适当的剂量范围。
II期试验是在患者中进行,旨在确定药物的治疗效果和剂量。
III期试验是在更大的病人群体中进行,以进一步确认药物的效果和安全性,并与标准治疗进行对比。
在三个阶段的试验中,制药公司负责收集试验数据,并根据FDA的规定提交适当的数据报告。
FDA对NDA进行评估的时间通常为10个月,在此期间,FDA的专家组织会就各个方面对申请进行审查,包括药物的质量、安全性和有效性。
如果发现问题或需要补充资料,FDA可能会要求制药公司提供额外的信息。
一旦NDA获得FDA的批准,该药物就可以在美国市场上销售和使用。
此外,FDA还设有一项称为“加速批准”(Fast Track)的计划,该计划旨在加快新药上市的进程,特别是对于那些用于治疗重大或无法治愈疾病的药物。
加速批准要求制药公司在研发和临床试验过程中紧密合作,并提供进行中的试验数据。
加速批准使得制药公司可以更早地让新药上市,以满足患者的需求。
FDA新药申请程序

FDA新药申请程序FDA(美国食品药品监管局)是负责确保美国公众获得安全和有效药物的监管机构。
为了确保药物的安全性和疗效,FDA对新药申请进行了严格的审查和批准程序。
新药开发是一个复杂和昂贵的过程,通常需要多次临床试验和大量数据收集。
下面将详细介绍FDA新药申请的程序。
1.基础研究和药物发现阶段:在此阶段,科学家通过实验室研究、细胞培养和动物实验等方法鉴定出一种具有潜在治疗作用的新药物。
2.预临床研究(药物毒理学和药代动力学研究):在此阶段,新药物的体内代谢、毒理学和药代动力学特性进行评估。
该阶段的研究数据将用于设计后续临床试验。
3.临床试验:临床试验是评估新药疗效和安全性的关键步骤。
该阶段分为三个阶段:I期、II期和III期。
I期试验旨在评估药物在人体内的代谢和安全性。
II期试验考察药物对特定疾病的疗效。
III期试验是最后一个阶段,目的是确定药物的疗效、剂量和安全性。
5.NDA审查:一旦NDA被提交给FDA,它将分配给一个专门的评审团队进行审查。
该团队会仔细审查文档中的所有数据和信息,并进行评估。
他们将对药物的质量、安全性和疗效进行评估,并提出问题和需要进一步证明的地方。
审查的时限通常为10个月,但在一些情况下可能会延长。
6.审查结果:FDA会向药企发出一封通知信,信中解释审查团队的评估和意见。
如果药物符合FDA的安全性和有效性要求,FDA将批准NDA,并核发许可证。
如果存在问题或缺失,FDA可能要求补充信息或重新评估。
7.药物上市:一旦FDA批准了新药的NDA,该药物即可在美国市场上市销售。
药企将开始生产、推广和销售药物,并严格遵守FDA的监管要求。
8.后续监管:药物上市后,FDA将继续监督其安全性和有效性。
FDA在市场上设立了不同的监测系统,包括药理学、药效学和不良事件报告。
如果出现安全问题或重大风险,FDA有权采取必要的措施,包括药品召回、警告公告和流行病学调查等。
总之,FDA的新药申请程序旨在确保药物的安全性和疗效。
美国FDA简略新药申请(ANDA)概览

美国FDA简略新药申请(ANDA)概览摘要:本文通过对美国简略新药申请流程,原料药、制剂技术审评要求及部分注册资料关键控制点进行介绍以构建出美国FDA简略新药申请(ANDA)的概览。
关键词 FDA ANDA 简略新药申请1.背景介绍美国食品药品监督管理局(U.S. Food & Drug Administration)简称FDA,是药品监管相当严苛的市场,只有经过FDA批准的药品,才可以在美国上市销售。
简略新药申请(Abbreviated New Drug Application)简称ANDA,与新药申请(New Drug Application,简称NDA)相比新药申请(New Drug Application,简称NDA)不需要开展非临床试验研究及临床试验研究。
美国负责ANDA审评的机构为FDA下属的仿制药办公室(Office of Generic Drugs, OGD)。
新化合物(New Chemical Entity,简称NCE)专营期从NDA获批开始计时,用于阻止ANDA和505(b)(2)含有此化合物产品进入市场。
5年NCE专营期内FDA不接受ANDA递交,或者4年后,ANDA可以通过专利挑战来递交申请。
按照《药品价格竞争和专利期恢复法》法案,对挑战专利的、首个仿制药申请可能获得180天专营期。
180天的计算开始时间是申请人开始商业上市的日期,或法院判决专利无效、不侵权的日期,两者中较早的时间。
通用技术文件(Common Technical Document,简称 CTD),FDA要求ANDA以CTD格式提交注册资料。
CTD是人用药品注册技术要求国际协调会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,简称ICH)为协调ICH地区注册申报资料的格式而制定的,其实施的目的是规范注册申请,统一技术格式要求,减少不必要浪费。
美国新药申请审评程序

美国新药申请审评程序美国新药申请审评程序在美国, 一个普通的新化合物从最初的发现到申请上市, 大约需要经过15 年的时间, 其中FDA用于审评的时间大约为6~12 个月。
FDA 将最终决定该新药能否进入市场, 因此除了实施有效的新药开发计划外, 熟悉FDA 的新药审评程序对于申请人来说也是非常重要的一个环节。
美国创新药物的申请包括两个步骤:①新药研究申请(investigational new drug application , IND) , 相当于我国的临床研究申请。
②新药申请( new drugapplication , NDA) , 相当于我国的新药上市申请,在三期临床结束后提出。
本文介绍的是FDA 对新药申请(即NDA) 的审评程序。
1、预备会议(pre - NDA meeting)在三期临床结束后, 申请人就可以进行新药申请, 在申请人提交NDA 之前, FDA 新药审评部门通常会和申请人举行一次会议。
在会议上, 申请人提交临床试验的概述报告, 让FDA 及评阅人员了解所申请新药的NDA 格式和内容。
2、新药申请的审评程序(New Drug Application review process)2.1 、申请书的受理(filing of NDA)在收到新药申请书以后的60 天内, FDA 首先会对新药申请书的完整性作出评价。
如果FDA 认为申请书的内容完整可以受理作进一步的评价, 则书面通知申请人。
如果FDA 发现申请书中存在下列任何原因, 将拒绝受理此申请书, 并书面告知相关理由:①没有完成FDA 申请表(356h 表) ;②申请书不符合规定的格式;③申请书不完整, 所要求的一项或多项内容缺失;④缺少一份完整的环境评估报告, 或申请人请求FDA 放弃对此部分内容的要求,但没有能够提交充分的信息来说服FDA ;⑤对于非英语的申请书, 没有准确而完整的英文译本;⑥没有指出是否在符合GLP 的条件下开展每个非临床试验;⑦没有指出每个临床试验是否符合伦理委员会规定(institutional review board regulations) 和知情同意书规定(informed consent regulations) ;⑧FDA 已经批准了该新药的申请书或简化申请书;⑨对于应该用简化申请书的形式来递交的新药, 如果申请人想以NDA 的形式来递交, 那么可以在30 天内书面要求和FDA 就此问题举行一次讨论会。
fda 新药审评流程

fda 新药审评流程
FDA(美国食品药品监督管理局)对于新药的审评流程如下:
1. 提交新药申请(NDA)或生物类似药申请(BLA):药企
将新药的相关数据和研究结果提交给FDA,并请求批准该药
物上市。
2. 预评审:FDA的药物评估员会对申请进行初步评估,确定
是否符合进一步的审评要求。
3. 审评启动:一旦FDA确认了申请的完整性和合规性,审评
启动。
FDA会组建一个药物评估团队进行审评。
4. 审评过程:药物评估团队会对申请提交的各个方面进行细致审查,包括临床试验数据、药理学和毒理学数据、生产过程以及药物的风险与效益。
5. 交流和补充材料:在审评过程中,FDA可能会与药企进行
多次沟通,以解答问题或要求提供额外的信息和数据。
6. 专家会议:FDA可能会召集一个独立的专家委员会或咨询
委员会,就药物的安全性和有效性等问题进行讨论和评估。
7. 审评报告:药物评估团队最终将审评结果整理成一份审评报告,提出批准与否的建议。
8. 审核和批准:FDA的高层管理人员会评估审评报告,最终
决定是否批准该药物上市。
如果批准,就会颁发批准文件,并确定适用的标签和使用指导。
9. 监管管理:一旦批准上市,FDA会进行监管管理,包括制定监管要求、监测药物的安全性和有效性、对药物进行质量控制和市场监管等。
需要注意的是,整个审评流程的时间长度是不确定的,可能会因为各种因素而有所变化。
FDA新药审核流程

FDA新药审核流程FDA(美国食品药品监督管理局)是美国联邦政府的一个机构,负责审批并监督所有食品、药品、医疗器械、化妆品和其他消费品的安全性和有效性。
新药审核是FDA的一个重要职责,涉及多个环节和步骤。
下面将详细介绍FDA新药审核的流程。
1. 前期准备:在提交新药申请(New Drug Application,NDA)之前,药厂需要进行大量的研发工作,包括药物的预临床实验、临床试验、效力和安全性评估等。
这个过程通常持续数年。
2.提交NDA:一旦药厂认为新药具备了提交FDA审批的条件,他们将向FDA提交NDA。
NDA需要包含大量的数据和信息,包括药物的化学成分、制备方法、质量控制等。
3.优先审查:FDA对一些新药进行优先审查,这些药物通常是用于治疗罕见病或临床需求紧迫的药物。
优先审查可以缩短FDA的审批时间。
4.首次评审:FDA将对NDA进行首次评审,评估药物的有效性和安全性。
评审小组通常由FDA的药物评审员和其他专家组成。
5.补充材料:在首次评审之后,FDA可能会要求药厂提供额外的数据和信息,以填补信息不完整或不足的方面。
6.药物评估:FDA的药物评审员将针对NDA中的各个方面进行全面评估,包括活性成分、药物代谢、药动学、药效学、不良反应等。
评审员还会研究临床试验数据,以评估药物的效果和安全性。
8.二次评审:在考虑了所有相关数据和信息之后,FDA可能会对NDA进行二次评审,并向药厂提供评审意见和建议。
此时,FDA可能会提出一些需要满足的条件,才能最终批准该药物上市。
9.批准与上市:如果通过了二次评审,FDA将批准新药上市,并授予该药物一个专利保护期。
这意味着在一定的时间内,其他药厂不能生产和销售相同的药物。
10.监测与后市场研究:一旦药物上市,FDA将继续监测该药物的有效性和安全性。
药厂也需要承担监测和报告药物的不良反应的责任。
此外,FDA可能要求药厂进行后市场研究,以更好地了解药物的效果和安全性。
FDA新药申请程序

FDA新药申请程序美国食品药品监督管理局(FDA)是负责监管和批准新药上市的机构。
在美国,所有新药都必须经过FDA的审查和批准程序,以确保其安全性和有效性。
以下是FDA新药申请程序的详细步骤:第一步:发现和基础研究新药的开发通常从科学家对疾病的基础研究开始。
在这个阶段,科学家会尝试理解疾病的发生机制,并尝试找出治疗该疾病的潜在药物。
第二步:临床前研究在将药物应用于人体之前,需要进行大量的临床前研究。
这些研究包括体外和动物试验,以评估药物的安全性和有效性。
如果临床前研究表明药物有治疗潜力,并且不会对人体造成严重的安全风险,那么可以开始提交FDA新药申请。
第三步:IND(Investigational New Drug)申请IND申请是一份详细的文件,其中包含了新药的临床前研究数据、药物特性、疾病的治疗需求以及计划进行的临床试验的详细计划。
一旦IND申请被FDA接受,开发者可以开始进行临床试验。
第四步:临床试验临床试验是确定新药是否安全、有效以及对疾病产生治疗效果的关键步骤。
它分为三个不同的阶段:I期、II期和III期。
在这些试验中,药物将被应用于患者身上,并且监测其效果和副作用。
临床试验的设计必须符合FDA的规定,并且需要得到患者的知情同意。
第五步:申请审查在临床试验结束后,开发者将提交新药申请(NDA)给FDA。
NDA是一份详细的文件,其中包含了从临床试验中获得的所有数据,包括药物的安全性和有效性数据。
这些数据需要经过严密的分析和解释,以证明药物的治疗效果。
第六步:审查过程一旦NDA被提交,FDA将进行审查。
审查过程包括多个阶段,包括初步审查、详细审查和面板审查。
FDA将评估所有提供的数据,并就药物的安全性、有效性和适应症提出问题。
在这个过程中,开发者和FDA之间可能会进行多次的沟通和交流,以澄清问题和提供进一步的信息。
第七步:批准和上市经过审查过程,如果FDA认为新药是安全有效的,并且对疾病产生治疗效果,那么他们将批准其上市。
fda ind申报流程

fda ind申报流程英文回答:FDA IND (Investigational New Drug) application process can be quite complex and involves several steps. As an individual who has gone through this process, I can provide you with a detailed explanation.1. Pre-IND Meeting: Before submitting the IND application, it is advisable to have a pre-IND meeting with the FDA. This meeting allows you to discuss your drug development plan, study design, and any concerns or questions you may have. It helps in clarifying regulatory requirements and expectations.2. Preparing the IND Application: The IND applicationis a comprehensive document that provides detailed information about the drug, its manufacturing process, preclinical and clinical data, and proposed study protocols. It also includes information about the investigators, studysites, and patient population. It is essential to ensure that the application is complete and meets the FDA's requirements.3. Submission of the IND Application: Once the IND application is prepared, it needs to be submitted to the FDA. The application is typically submitted electronically through the FDA's Electronic Submissions Gateway (ESG). It is crucial to follow the FDA's guidelines for submission and provide all the necessary documents and forms.4. FDA Review: After the IND application is submitted, the FDA reviews the application to assess the safety and efficacy of the investigational drug. The FDA has specific timelines for reviewing the IND application, and they may request additional information or clarification during the review process.5. IND Safety Reporting: Once the IND is in effect, the sponsor is responsible for monitoring and reporting any adverse events or safety concerns related to the investigational drug. This includes submitting expeditedsafety reports for serious and unexpected adverse events.6. Clinical Trials: If the IND application is approved by the FDA, the sponsor can proceed with conductingclinical trials. These trials are conducted in phases, starting with Phase 1 studies to assess safety and dosage, followed by Phase 2 and Phase 3 studies to evaluateefficacy and safety in larger patient populations.7. NDA (New Drug Application) Submission: If theresults of the clinical trials are positive, the sponsorcan submit a New Drug Application (NDA) to the FDA for approval to market the drug. The NDA includes comprehensive data on the drug's safety, efficacy, and manufacturing process.8. FDA Review of NDA: The FDA reviews the NDA to determine whether the drug should be approved for marketing. The review process includes evaluating the data fromclinical trials, assessing the drug's benefits and risks, and ensuring that the manufacturing process meets quality standards.9. FDA Approval: If the FDA determines that the drug is safe and effective for its intended use, they grantapproval for marketing the drug. This allows the sponsor to commercialize the drug and make it available to patients.中文回答:FDA IND(Investigational New Drug)申请流程相当复杂,涉及多个步骤。
美国fda申报流程和时限

美国fda申报流程和时限下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!美国FDA申报流程和时限在美国,食品和药物管理局(FDA)是负责监管和批准药品、医疗器械以及食品安全的重要机构。
fda申报资料的模板和例子 -回复

fda申报资料的模板和例子-回复如何撰写FDA申报资料的模板和提供实例。
第一步:选择申报资料的类型在开始之前,首先我们需要确定申报资料的具体类型。
FDA申报资料通常涵盖新药申请(NDA)、通用名药物申请(ANDA)、生物类似药物申请(BLA)等多个种类。
根据所需申报资料的类型,我们可以制定相应的模板。
第二步:制定申报资料的大纲在编写申报资料之前,制定一个清晰的大纲非常重要。
大纲可以帮助我们更好地组织和安排申报资料的内容,并确保所需信息得以全部覆盖。
下面是一个示例大纲,供参考:I. 申报资料概述A. 资料简介B. 资料目的C. 申报类型II. 药物简介A. 药物名称和化学结构B. 药理作用和机制C. 适应症和临床需求III. 非临床研究A. 药品制备及质量验证B. 药品稳定性研究C. 药理学和药代动力学研究IV. 临床试验A. 试验设计和方法B. 试验对象招募C. 试验安全性和有效性评估D. 试验结果和统计分析V. 药物质量控制A. 质量控制方法和标准B. 药品生产过程和设备C. 药品质量监控和验证VI. 安全性评价A. 不良事件监测与报告B. 严重不良事件的评估和处理C. 药物安全风险评估VII. 申报资料总结A. 结论和建议B. 对未来研究的展望第三步:填写申报资料的详细内容根据制定的大纲,我们可以开始填写申报资料的详细内容。
以下是一些可能的例子,用于说明如何填写各个部分的内容:申报资料概述:在这一部分,我们可以向FDA介绍我们的资料内容、目的以及所属的申报类型。
可以简单介绍申报资料的意义和用途。
药物简介:这里需要提供药物的名称、化学结构以及药理作用和机制。
也可以解释药物的适应症和临床需求,并阐明为什么该药物对患者有益。
非临床研究:该部分应描述药物的制备过程、质量验证、稳定性研究以及药理学和药代动力学研究结果。
这些研究数据将有助于证明药物的质量和安全性。
临床试验:在这一部分,应详细描述临床试验的设计和方法、试验对象的招募过程、试验的安全性和有效性评估以及试验结果和统计分析。
fda ind申报流程

fda ind申报流程
1. 准备阶段
- 确定产品性质和适用法规
- 选择合适的IND类型(如治疗用IND、研究用IND等)
- 组建研发团队,包括临床医生、科学家、监管专家等
- 收集并评估现有的非临床和制造数据
2. 提交IND申请
- 填写1571表格(Cover Sheet)
- 编写调查用新药简介(Investigational New Drug Application) - 准备相关支持性文件,如非临床研究报告、制造信息等
- 提交电子版或纸质版IND申请材料
3. FDA审查阶段
- FDA在30天内对IND申请材料进行初审
- 如有缺陷,会向申请人发出临床安持通知
- 如无重大缺陷,IND申请将获得默认通过
4. 临床试验阶段
- 按照IND申请中规定的方案开展临床试验
- 向FDA及时报告严重不良反应等重大安全信息
- 根据需要对试验方案进行修订,并获得FDA批准
5. 持续更新IND
- 定期向FDA报告临床试验进度
- 及时更新IND相关信息,如制造变更、非临床新数据等
- 在适当阶段,根据数据申请开展后续的临床试验阶段
IND申报是新药研发进入临床阶段的关键步骤,需要做大量的准备工作,并与FDA保持紧密沟通,确保临床试验按计划顺利推进。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
File NDA at FDA
File IND at FDA
Review process/ approval
Additional postmarketing testing required by FDA
Success Rate
1 approved
BY BERTRAM A. SPILKER, M.D., PH.D., SENIOR VICE PRESIDENT FOR SCIENTIFIC
THE DRUG DEVELOPMP R O VA L P R O C E S S
R
E G U L A T O R Y
A
F F A I R S
, P
H
RMA
T
he U.S. system of new drug approvals is perhaps the most rigorous in the world. On average, it costs a company $802 million to get one new medicine from the laboratory to U.S. patients, according to a November 2001 report by the Tufts Center for the Study of Drug Development. It takes 10 to 15 years on average for an experimental drug to travel from lab to U.S. patients, according to the Tufts Center for the Study of Drug Development, based on drugs approved from 1994 through 1998. Only five in 5,000 compounds that enter preclinical testing make it to human testing. And only one of those five is approved for sale. Once a new compound has been identified in the laboratory, medicines are developed as follows: Preclinical Testing. A pharmaceutical company conducts laboratory and animal studies to show biological activity of the compound against the targeted disease, and the compound is evaluated for safety. Investigational New Drug Application (IND). After completing preclinical testing, a company files an IND with the U.S. Food and Drug Administration (FDA) to begin to test the drug in people. The IND becomes effective if FDA does not disapprove it within 30 days. The IND shows results of previous experiments; how, where and by whom the new studies will be conducted; the chemical structure of the compound; how it is thought to work in the body; any toxic effects found in the animal studies; and how the compound is manufactured. All clinical trials must be reviewed and approved by the Institutional Review Board (IRB) where the trials will be conducted. Progress reports on clinical trials must be submitted at least annually to FDA and the IRB.
Clinical Trials Discovery/ Preclinical Testing Years Test Population Purpose 6.5 Laboratory and animal studies Assess safety, biological activity and formulations 5,000 compounds evaluated Phase I 1.5 20 to 100 healthy volunteers Determine safety and dosage Phase II 2 100 to 500 patient volunteers Evaluate effectiveness, look for side effects 5 enter trials Phase III 3.5 1,000 to 5,000 patient volunteers Confirm effectiveness, monitor adverse reactions from long-term use FDA 1.5 15 Total Phase IV
E W
Clinical Trials, Phase I. These tests involve about 20 to 100 normal, healthy volunteers. The tests study a drug’s safety profile, including the safe dosage range. The studies also determine how a drug is absorbed, distributed, metabolized, and excreted as well as the duration of its action. Clinical Trials, Phase II. In this phase, controlled trials of approximately 100 to 500 volunteer patients (people with the disease) assess a drug’s effectiveness. Clinical Trials, Phase III. This phase usually involves 1,000 to 5,000 patients in clinics and hospitals. Physicians monitor patients closely to confirm efficacy and identify adverse events. New Drug Application (NDA). Following the completion of all three phases of clinical trials, a company analyzes all of the data and files an NDA with FDA if the data successfully demonstrate both safety and effectiveness. The NDA contains all of the scientific information that the company has gathered. NDAs typically run 100,000 pages or more. By law, FDA is allowed six months to review an NDA. The average NDA review time for new molecular entities approved in 2001 was 16.4 months. Approval. Once FDA approves an NDA, the new medicine becomes available for physicians to prescribe. A company must continue to submit periodic reports to FDA, including any cases of adverse reactions and appropriate quality-control records. For some medicines, FDA requires additional trials (Phase IV) to evaluate long-term effects. Discovering and developing safe and effective new medicines is a long, difficult, and expensive process. The research-based pharmaceutical industry invested more than $30 billion in research and development in 2001. 23
N
M
E D I C I N E S
I N
D
E V E L O P M E N T
F O R
2002
T H E D R U G D I S C O V E RY, D E V E L O P M E N T
AND
APPROVAL PROCESS
It takes 10-15 years on average for an experimental drug to travel from the lab to U.S. patients. Only five in 5,000 compounds that enter preclinical testing make it to human testing. One of these five tested in people is approved.