肢带型肌营养不良症1例报告及浅析
易误诊为炎性肌病的肢带型肌营养不良2B型一例报告

伸侧可见明显色素沉着ꎮ 既往史、 个人史、 家族史
无特殊ꎮ 入 院 后 完 善 相 关 检 查: ALT 87 5 U / Lꎬ
AST 95 U / Lꎬ CK 8 416 U / Lꎬ 肌 酸 激 酶 同 工 酶
(CK ̄MB) 137 U / Lꎬ 乳酸脱氢酶 ( LDH) 419 U / Lꎬ
好转ꎮ 一周后再发上述症状ꎬ 遂于当地医院就诊ꎬ
完善常规检查时偶然发现肌酸激酶 (CK) 8 167 U / Lꎬ
因当时患者无胸闷、 胸痛ꎬ 心电图正常ꎬ 无肌痛、
肌无力ꎬ 无皮疹、 技工手等特殊症状ꎬ 故未予特殊
处理ꎮ 1 周后复查谷丙转氨酶 ( ALT) 175 4 U / Lꎬ
谷草转氨酶 (AST) 271 2 U / Lꎬ CK 达 11 348 U / Lꎬ
点
[1]
ꎮ LGMD2B 多在 19 ~ 27 岁起病ꎬ 早期可以表
查 CK 波动在 8 000 ~ 11 000 U / L 之间ꎮ 为求进一步
门诊就诊ꎬ 门诊拟考虑 “ 肌酸激酶升高查因: 炎
性肌病? 其他?” 收住院ꎮ 患者近一年来偶感写作
业久时双上臂酸痛不适ꎮ 偶感进食哽噎感ꎬ 无肌无
现为无症状高肌酸激酶血症ꎬ 中后期绝大部分患者
遂于 2018 年 12 月就诊于邵阳市中心医院风湿免疫
科门诊ꎬ 考 虑: “ 肌 酸 激 酶 升 高 查 因: 多 发 性 肌
炎? 其他?” 完善抗核抗体 ( ANA) 谱示阴性ꎮ 肌
炎抗体谱 ( 肌炎特异性 + 肌炎相关性自身抗体) 示
阴性ꎮ 红细胞沉降率 ( ESR) 、 C 反应蛋白 ( CRP)
c ̄ANCA ( 胞浆型) 、 肌炎抗体谱 ( 肌炎特异性 + 相
CK异常增高肌营养不良症1例分析

It a dN vmbr 01V 1 2N .9 n L bMe,oe e 1,o. , o1 J 2 3
・
个 案 与短篇 ・
乙型 肝 炎病 毒 与肝癌 的相 关性
尚 守 亮 ຫໍສະໝຸດ ( 江苏省 滨海县 中医院检 验 科
文献 标 识 码 : C 肝 癌 是 我 国 常见 的 癌 症 , 普 查 资 料 肝 癌 年死 亡率 仅 次 于 据 胃癌 和 肺 癌居 第 3位 。而 每 年 肝 癌 新 发 病 中我 国 占全 球 4 , 5 成 为世 界上 肝 癌 发 病 最 集 中 的 国 家 l 。 为 探 讨 乙 型 肝 炎 病 毒 1 】 ( V) HB 与肝 癌 的 相关 关 系 , 肝 癌 患者 进 行 研究 , 道 如 下 。 对 报
( MD) 称 严 重 性 假 肥 大 型 肌 营 养 不 良症 , 乎 仅 见 于男 性 D 也 几
国际检 验 医学杂志 21 年 1 月 第 3 卷 第 1 01 1 2 9期
It a dNoe br 01V 13, o1 n L bMe, vm e 21, o 2N .9 J .
险 与 大 三 阳患 者 相 似 , 至 高 于 大 三 阳 ; V D 甚 HB NA 拷 贝 数 小 于 1。 o y mI 占 4 . % , 明 依 然 可 能 有 较 高 的 肝 癌 风 险 0 cp / , 55 说 发 生 ; 性 活动 性 乙型 肝 炎 患 者 占 9 . , 明慢 性 活 动 性 乙 慢 50 说 型肝炎患者发生肝癌的风险性高。
儿 童 。母 亲若 为基 因携 带 者 ,O 男 性 子 代 发 病 通 常 始 于 2 5 ~ 8岁 , 期 感 觉 走 路 笨 拙 , 跌 倒 , 立 时 脊 髓 前 凸 , 部 挺 出 , 初 易 站 腹 步 行缓 慢 摇 摆 , 特 殊 的 “ 步 ” 态 。 ②B ce 型 肌 营 养 不 良 呈 鸭 步 ek t
肢带型肌营养不良症个案护理

提高患者的自我管理能力 加强患者的心理支持和社会支持 提高患者的家庭护理能力
评估患者病情:了解患者的病情、症状、 治疗情况等
实施护理措施:按照护理计划,对患者 进行护理,包括药物治疗、康复训练、 心理支持等
制定护理目标:根据患者的病情和需求, 制定具体的护理目标
单击此处_
汇报人:刀客特万
01 患 者 病 情 评 估
02 护 理 计 划 的 制 定
03 护 理 措 施 的 实 施
04 护 理 效 果 的 评 估
05 护 理 总 结 与 建 议
患者病情评估
观察患者的肌肉力量和耐力 评估患者的日常生活活动能力 评估患者的心理状态和情绪
评估患者的营养状况和饮食情况 评估患者的呼吸功能和肺功能 评估患者的心脏功能和血压情况
康复训练频率:根据患者的 恢复情况调整训练频率
康复训练注意事项:注意患 者的身体状况,避免过度训
练导致肌肉损伤
营养均衡:保 证蛋白质、脂 肪、碳水化合 物等营养素的
均衡摄入
食物选择:选 择易消化、高 营养的食物, 如鸡蛋、牛奶、
瘦肉等
饮食规律:定 时定量,避免
暴饮暴食
饮食卫生:注 意饮食卫生, 避免食物中毒
观察患者日常生活能力变化 评估患者肌肉力量和耐力 观察患者呼吸功能变化 评估患者心理状态和情绪变化
评估指标:包括生活质量、功能状态、心理状态等 评估方法:采用问卷调查、访谈、观察等方式 评估周期:定期进行,如每月、每季度等 评估结果:根据评估指标和评估方法,对护理效果进行量化和定性分析
定期评估患者的病 情和功能状态
评估护理效果:定期评估护理效果,根 据评估结果调整护理计划
肢带型肌营养不良症

康复治疗
物理治疗:包括运动疗法、按摩疗法等,帮助 患者恢复肌肉力量和功能
言语治疗:针对言语障碍患者,帮助恢复言语 功能
职业治疗:针对日常生活活动能力受损的患者, 提供辅助器具和训练,帮助恢复生活自理能力
心理治疗:针对心理障碍患者,提供心理支持 和辅导,帮助患者适应疾病和康复过程
2
护理措施
点击此处添加正文,文字是您思想的提炼,为了演示发布的良好效果, 请言简意赅的阐述您的观点。
定期监测:定期监测体重、身高、
血常规等指标,了添加正文,文字是您思想的提炼,为了演示发布的良好效果, 请言简意赅的阐述您的观点。
预后情况
01
预后较差,多数患者在20-30岁之间失去行走能力
02
早期诊断和治疗可以延缓病情发展,提高生活质量
03
定期体检和康复治疗可以改善预后
肢带型肌营养 不良症
演讲人
目录
01. 治疗方法 02. 护理措施 03. 预后及预防
1
治疗方法
点击此处添加正文,文字是您思想的提炼,为了演示发布的良好效果, 请言简意赅的阐述您的观点。
药物治疗
皮质类固醇:如泼尼松,可减轻症状,但长 期使用有副作用
免疫抑制剂:如环磷酰胺,可抑制免疫系统, 减轻症状
理压力
01
02
03
04
定期进行身体 检查,监测病
情变化
2020
定期进行康复 治疗,改善生
活质量
2022
谢谢
抗肌萎缩蛋白药物:如依达拉奉,可改善肌 肉功能,但价格昂贵
基因疗法:如AAV基因疗法,可改善肌肉功 能,但尚在研究阶段
物理治疗
运动疗法:通过锻 炼增强肌肉力量和
耐力
肢带型肌营养不良症1例

[ 7 ] 牟章兵 , 吴宁玲, 莫静. 降眼压药 物预防 L A S I K术后 屈光 回退
[ J ] . 眼科 新 进 展 , 2 0 1 2, 6 ( 3 2 ): 5y a K, A i z a w a D, I g a r a s h i A, e t a 1 . E f e c t s f o a n t i g l a u c o I n a d r u g s
的研究 [ J ] . 眼科新进展 , 2 0 0 3, 2 3 ( 3) : 1 9 1—1 9 3 .
[ 3 ] Hu n a g X, He X, T n a X. R e s e a r c h o f c o r n e a l e c t a s i a f o l l o w i n g l a s e r i n s i t u k e r a t o m i l e u s i s i n r a b b i t s l J I . E h e S c i e n l e , 2 0 0 2 , 2: 1 1 9 .
后角膜后表 面前 凸变 化 的研 究 [ J ] . 宁 夏 医学 杂 志 , 2 0 1 1 , 6
[ 收稿 日期] 2 0 1 2—1 2— 2 0
[ 责任编辑 ] 李 洁
文章 编号 : 1 0 0 1 — 5 9 4 9 ( 2 0 1 3 ) 0 6— 0 5 2 6— 0 1
可逐渐缓解 , 无感觉异常 , 无尿 、 便 障碍 , 无发热 , 无肌肉疼痛及 吞咽困难。发病以来 体重 无明显 变化 。既往史 : 平 素身体健
和脂肪及结缔组织 增生 , 肌 肉无 异常代 谢产 物堆 积。 目前 ,
・
病例报告 ・
肢 带 型肌 营 养不 良症 1例
duchenne型肌营养不良1例诊断体会
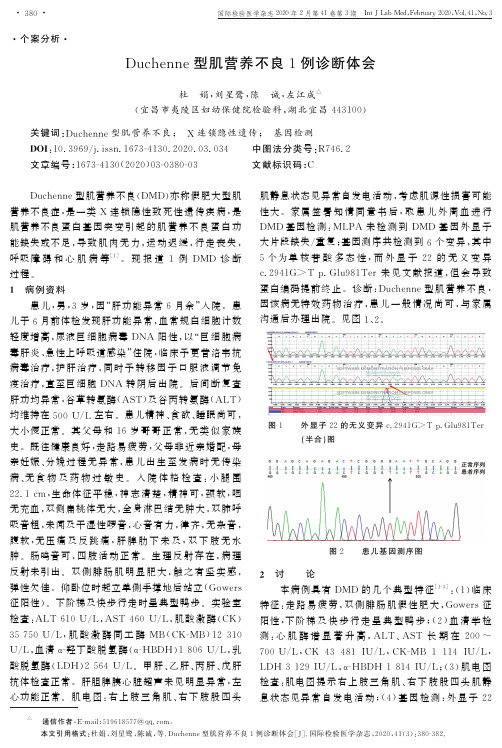
ә通信作者,E -m a i l :519618577@q q.c o m ㊂ 本文引用格式:杜娟,刘星鹭,陈诚,等.D u c h e n n e 型肌营养不良1例诊断体会[J ].国际检验医学杂志,2020,41(3):380-382.㊃个案分析㊃D u c h e n n e 型肌营养不良1例诊断体会杜 娟,刘星鹭,陈 诚,左江成ә(宜昌市夷陵区妇幼保健院检验科,湖北宜昌443100) 关键词:D u c h e n n e 型肌营养不良; X 连锁隐性遗传; 基因检测D O I :10.3969/j.i s s n .1673-4130.2020.03.034中图法分类号:R 746.2文章编号:1673-4130(2020)03-0380-03文献标识码:CD u c h e n n e 型肌营养不良(D M D )亦称假肥大型肌营养不良症,是一类X 连锁隐性致死性遗传疾病,是肌营养不良蛋白基因突变引起的肌营养不良蛋白功能缺失或不足,导致肌肉无力,运动迟缓,行走丧失,呼吸障碍和心肌病等[1]㊂现报道1例D M D 诊断过程㊂1 病例资料患儿,男,3岁,因 肝功能异常6月余 入院㊂患儿于6月前体检发现肝功能异常,血常规白细胞计数轻度增高,尿液巨细胞病毒D N A 阳性,以 巨细胞病毒肝炎㊁急性上呼吸道感染 住院,临床予更昔洛韦抗病毒治疗,护肝治疗,同时予转移因子口服液调节免疫治疗,直至巨细胞D N A 转阴后出院㊂后间断复查肝功均异常,谷草转氨酶(A S T )及谷丙转氨酶(A L T )均维持在500U /L 左右㊂患儿精神㊁食欲㊁睡眠尚可,大小便正常㊂其父母和16岁哥哥正常,无类似家族史㊂既往健康良好,走路易疲劳,父母非近亲婚配,母亲妊娠㊁分娩过程无异常,患儿出生至发病时无传染病㊁无食物及药物过敏史㊂入院体格检查:小腿围22.1c m ,生命体征平稳,神志清楚,精神可,颈软,咽无充血,双侧扁桃体无大,全身淋巴结无肿大,双肺呼吸音粗,未闻及干湿性啰音,心音有力,律齐,无杂音,腹软,无压痛及反跳痛,肝脾肋下未及,双下肢无水肿㊂肠鸣音可,四肢活动正常㊂生理反射存在,病理反射未引出㊂双侧腓肠肌明显肥大,触之有坚实感,弹性欠佳㊂仰卧位时起立单侧手撑地后站立(G o w e r s 征阳性)㊂下阶梯及快步行走时呈典型鸭步㊂实验室检查:A L T 610U /L ,A S T 460U /L ,肌酸激酶(C K )35750U /L ,肌酸激酶同工酶M B (C K -M B )12310U /L ,血清α-羟丁酸脱氢酶(α-H B D H )1806U /L ,乳酸脱氢酶(L D H )2564U /L ㊂甲肝㊁乙肝㊁丙肝㊁戊肝抗体检查正常㊂肝胆脾胰心脏超声未见明显异常,左心功能正常㊂肌电图:右上肢三角肌㊁右下肢股四头肌静息状态见异常自发电活动,考虑肌源性损害可能性大㊂家属签署知情同意书后,取患儿外周血进行D M D 基因检测:M L P A 未检测到D M D 基因外显子大片段缺失/重复;基因测序共检测到6个变异,其中5个为单核苷酸多态性,而外显子22的无义变异c .2941G>T p .G l u 981T e r 未见文献报道,但会导致蛋白编码提前终止㊂诊断:D u c h e n n e 型肌营养不良,因该病无特效药物治疗,患儿一般情况尚可,与家属沟通后办理出院㊂见图1㊁2㊂图1 外显子22的无义变异c .2941G>T p .G l u 981T e r(半合)图图2 患儿基因测序图2 讨 论本病例具有D M D 的几个典型特征[1-3]:(1)临床特征:走路易疲劳,双侧腓肠肌假性肥大,G o w e r s 征阳性,下阶梯及快步行走呈典型鸭步;(2)血清学检测:心肌酶谱显著升高,A L T ㊁A S T 长期在200~700U /L ,C K 43481I U /L ,C K -M B 1114I U /L ,L D H 3129I U /L ,α-H B D H 1814I U /L ;(3)肌电图检查:肌电图提示右上肢三角肌㊁右下肢股四头肌静息状态见异常自发电活动;(4)基因检测:外显子22无义变异c .2941G>T p .G l u 981T e r,可导致蛋白质编码提前终止㊂基于以上几点,该患儿可明确诊断为D M D ㊂但该患儿确诊耗时近6个月时间,主要原因可能有以下几方面原因:(1)家庭重视不够:患儿平常身体无明显不适,但走路易疲劳,仰卧需单手撑地才能站立等症状早有出现,但患儿年龄尚小,被认为发育迟,并未考虑存在疾病而被忽视未就医;(2)临床医生经验不足:该患儿因体检发现肝功能异常入院,一段时间仅关注肝脏功能及相关检查较为局限,加之巨细胞病毒阳性,而忽略其他如心肌酶谱㊁体格检查等;(3)患儿运动发育基本正常,会独立行走;(4)无特殊家族史,其同母哥哥体健㊂这些原因均导致了该患儿疾病的延迟确诊㊂D M D 遗传方式为X 连锁隐性遗传,发病率在各个国家㊁地区和人种间无明显差异,每3600~6000例出生男婴中有1例发病㊂中国的发病率约为1/3853,估算全国患者约70000人[4]㊂该病常于2~5岁起病,常于20岁左右死于心力衰竭或呼吸功能不全[5]㊂该病在诊疗过程中重点要与其他类型肌营养不良㊁脊肌萎缩症㊁炎性肌病和代谢性肌病进行鉴别诊断,但通过临床表现㊁血肌酶㊁肌肉活检和基因检测能够进行鉴别[4]㊂D M D 基因主要有3种突变类型[6-8]:(1)大片段缺失型:最为常见,突变发生频率约占所有突变的60%;(2)大片段重复型:较少见,约占所有突变的10%;(3)微小突变:包括单个或数个核苷酸置换㊁缺失或插入等,约占所有突变的30%㊂本病例应用M L P A 技术检测大片段缺失和大片段重复,结合二代测序技术对D M D 基因外显子及周围内含子进行微小突变分析㊂能够明确病因,但目前尚无有效的治疗方法㊂对其鉴别诊断,对患者的康复治疗有一定的指导作用,同时,能够提供一定的遗传咨询与指导㊂通过对本例D M D 的确诊,提示临床在诊疗过程中,对不明原因的肝功能异常,且经抗病毒㊁护肝等治疗无效者,要加强体格检查及其他辅助检查,如血清肌酶㊁肌电图及肌活检,必要时行基因检测,提高罕见病的诊疗水平㊂参考文献[1]B I R N K R A N T D J ,B U S H B Y K ,B A N N C M ,e t a l .D i a g-n o s i s a n d m a n a g e m e n t o f d u c h e n n e m u s c u l a r d y s t r o p h y,p a r t 1:d i a gn o s i s ,a n d n e u r o m u s c u l a r ,r e h a b i l i t a t i o n ,e n d o -c r i n e ,a n d g a s t r o i n t e s t i n a l a n d n u t r i t i o n a l m a n a ge m e n t [J ].L a n c e t N e u r o l ,2018,17(3):251-267.[2]B A X T E R P .D i a g n o s i s a n d m a n a ge m e n t of d u c h e n n e m u s c u l a r d y s t r o p h y [J ].D e v M e d C h i l d N e u r o l ,2010,52(4):313.[3]张成.‘中国假肥大型肌营养不良症诊治指南“解读[J ].中国现代神经疾病杂志,2018,18(07):475-479.[4]中华人民共和国国家卫生健康委员会.罕见病诊疗指南(2019年版)[M ].北京:人民卫生出版社,2019:627-631.[5]董奇超,陈慧敏,金欣.D u c h e n n e 型肌营养不良基因治疗研究进展[J ].中国当代儿科杂志,2018,20(8):691-697.[6]A L MOMA N I R ,S T O E P N ,B A K K E R E ,e t a l .R a pi d a n d c o s t e f f e c t i v e d e t e c t i o n o f s m a l l m u t a t i o n s i n t h e D M Dg e n e b y h i g h r e s o l u t i o n m e l t i n g c u r v e a n a l y s i s [J ].N e u r o -m u s c u l D i s o r d ,2009,19(6):383-390.[7]N I C O B ,MA R Z U L L O A ,C O R S I P ,e t a l .A p o s s i b l er o l e o f t r y p t a s e i n a n g i o ge n e s i s i n t h e b r a i n of m d x m o u s e ,a m o d e l o f D u c h e n n e m u s c u l a r d y s t r o p h y [J ].N e u r o s c i e n c e ,2004,123(3):585-588.[8]Z HO U L ,L U H .T a r g e t i n g Fi b r o s i s i n D u c h e n n e M u s -c u l a r D y s t r o p h y [J ].J N e u r o p a t h o l E x p Ne u r o l ,2010,69(8):771-776.(收稿日期:2019-07-16 修回日期:2019-10-19)本文引用格式:丁宁,王玉月.质谱快速鉴定两例A I D S 合并真菌性血流感染病例分析[J ].国际检验医学杂志,2020,41(3):380-382.㊃个案分析㊃质谱快速鉴定两例A I D S 合并真菌性血流感染病例分析丁 宁1,王玉月2(1.常州市妇幼保健院检验科,江苏常州213000;2.常州市第一人民医院检验科,江苏常州213003) 关键词:质谱鉴定; A I D S ; 真菌性血流感染D O I :10.3969/j.i s s n .1673-4130.2020.03.035中图法分类号:R 512.91;R 519文章编号:1673-4130(2020)03-0382-03文献标识码:C获得性免疫缺陷综合征(A I D S )是由人免疫缺陷病毒(H I V )引起的严重危害人类健康的传染病,以损。
先天性肌营养不良1A型1例临床与基因分析

Congenital muscular dystrophy type 1A: a report of one case with literature review JIANG Shiyuan,XIANG Na (Department of Paediatrics,Shanxian Hygeia Hospital,Shanxian 274300,Shandong, China)
1 临床资料
患儿,男,5 岁 2 个月,足月剖宫产,母孕期及出生 时未见异常,出生后喂养困难,2 岁可独坐,早期出现 关节萎缩,至今不能独走。就诊时体格检查 :神清,眼 距宽,鼻梁低,上下牙齿不合 ;双侧呼吸运动对称,未 闻及啰音 ;心律齐,心音有力,心脏各听诊区均未闻 及杂音 ;腹软,肝脾肋下未触及 ;四肢肌力及肌张力 均降低,膝腱反射未引出,病理反射(-)。实验室检 查 :肌酸激酶(creatine kinase,CK)491 U/L(参考值 25 ~ 195 U/L),肌酸激酶同工酶(creatinine kinase MB isoenzyme,CK-MB)41 . 8 U/L(参考值 0 ~ 25 U/L);肌 电图检测发现肌源性损害可能 ;头颅 MRI 提示大脑白 质异常信号,主要累及侧脑室前后角。根AMA2 基因变异导致先天性肌营养不良的临床、实验室检查及遗传学特点。方法 回顾分 析 1 例先天性肌营养不良 1 A 型患儿的临床资料,并复习相关文献。结果 患儿,男,5 岁 2 个月,临床表现为运动发育落后, 2 岁时可独坐,不能独走 ;肌力及肌张力低下,早期出现关节挛缩。生化检测发现肌酸激酶(CK)升高(491 U/L),其同工 酶 CK-MB 升高(41 . 8 U/L);肌电图提示肌源性损害可能 ;头颅 MRI 提示大脑白质异常信号。基因检测发现 LAMA2 存在 复杂杂合突变,c. 2045 - 2046 delAG 杂合缺失,来自母亲,为已报道的致病变异 ;exon 5 存在杂合缺失,来自父亲,为未报道 的新变异,软件功能预测提示为致病性变异。结论 LAMA2 基因变异导致先天性肌营养不良,患儿以运动发育落后起病, CK 升高,高通量基因检测有助于明确诊断。
名老中医陈卫川治疗肢带型肌营养不良症经验举隅

名老中医陈卫川治疗肢带型肌营养不良症经验举隅1. 引言1.1 疾病背景肢带型肌营养不良症,是一种罕见的遗传性疾病,主要表现为肌肉无力、脂肪代谢异常等症状。
这种疾病常常在婴幼儿期就会出现,严重影响患者的生活质量。
肢带型肌营养不良症是一个非常棘手的疾病,目前西医治疗方法并不十分有效,而且长期使用药物会有一定的副作用。
患者常常因为治疗困难而感到沮丧和无助。
在面对这种困扰患者已久的疾病时,中医提供了一种全新的治疗思路和方法。
中医治疗肢带型肌营养不良症侧重于调理患者的脏腑功能,通过中药调理和针灸理疗来改善症状。
相比于西医的药物治疗,中医的治疗方法更加温和,也更加注重患者的整体身体状况。
接下来我们将介绍中医在治疗肢带型肌营养不良症方面的特点和具体方法。
【字数:241】1.2 治疗困难肢带型肌营养不良症是一种罕见的遗传性疾病,常见于婴幼儿。
由于病因复杂,症状多样,临床表现不一,使得该病的诊断和治疗都存在一定的困难。
病情的轻重不一及病人的个体差异也给治疗带来挑战。
肢带型肌营养不良症的患者往往需要长期治疗和康复,治疗过程中需要考虑到患者的年龄、身体状况、并发症等多方面因素,这也增加了治疗的复杂性。
当前西医在治疗肢带型肌营养不良症的效果不稳定,常常只能通过对症治疗来缓解症状,而无法根治疾病。
这些治疗困难使得患者和家属感到沮丧和无助,也给医生带来了较大的挑战。
寻找更有效的治疗方法和改善患者生活质量的途径是当前亟需解决的问题。
2. 正文2.1 中医治疗特点中医治疗肢带型肌营养不良症的特点包括个性化治疗、综合调理、长期疗效和少副作用等方面。
中医治疗肢带型肌营养不良症注重个性化治疗。
中医通过辨证施治,根据患者的体质、病情、年龄等因素进行个性化的治疗方案制定,针对不同的患者采取针对性的治疗措施,有效解决了患者的症状和体质问题。
中医治疗肢带型肌营养不良症强调综合调理。
中医认为疾病的发生是由于人体内部的阴阳失衡导致的,因此治疗不仅仅局限于治疗症状,更重要的是调理人体内部的阴阳平衡,提高身体的免疫力和自愈能力,从根本上解决疾病问题。
常染色体隐性遗传肢带型肌营养不良症分子遗传学研究进展解读

常染色体隐性遗传肢带型肌营养不良症分子遗传学研究进展进行性肌营养不良症(progressive muscular dystrophy)是一组原发于肌肉组织的遗传病,特点为进行性加重的肌肉萎缩、无力。
最常见的类型是X连锁的Duchenne/Becker肌营养不良症(Duchenne muscular dystrophy, DMD/BMD),另一类较常见的类型为常染色体遗传的肢带型肌营养不良症,临床表现复杂,男女均可受累。
我们所在研究组的临床资料中,约有25%~45%的常染色体遗传肌营养不良症有待深入研究。
1常见的进行性肌营养不良症分类及分子基础50~60年代时,基于不断有女性进行性肌营养不良患者的报道,其临床表现与DMD极相似,故称这类患者为类杜氏肌营养不良(Duchenne-like muscular dystr-ophy, DLMD),除了在少数女患者中发现X染色体异常或X染色体与常染色体异位外,多数患者很难用通常的X连锁遗传规律解释。
因而在分析了大量有女性DLMD患者的家系资料基础上,有人提出这类疾病可能存在常染色体隐性遗传。
Ben Othmane等1992年首次将在突尼斯人中确定的此类疾病称为儿童期重型常染色体隐性遗传肌营养不良症(severe childhood autosomal recessive muscular dystrophy, SCARMD),之后不断在北非、中东地区、欧洲、日本、南美和北美人群中有此类疾病的报道。
在近年的相关文献多数已用SCARMD取代了DLMD[1-5]。
常染色体遗传的肢带型肌营养不良症(limb-girdle muscular dystrophy LGMD)是进行性肌营养不良症中除DMD/BMD外最常见的类型。
该类型特点是髋肩带肌无力萎缩,进行性发展至远端肌肉,男女均可受累,有明显的异质性,不仅表现在遗传方式有显性和隐性之差别,而且其临床表现也有很大不同,轻型可以在儿童期也可在20岁以后发病,肌萎缩无力进展速度可能非常缓慢,60岁以后仍可行走不影响寿命,与BMD相似。
肌营养不良症疾病研究报告

肌营养不良症疾病研究报告疾病别名:肌营养不良症所属部位:全身就诊科室:神经内科,脑外科病症体征:不能露齿及突唇,肌肉肥大,肌肉萎缩,无力,眼睑下垂,易跌倒疾病介绍:什么是肌营养不良症?肌营养不良症按照字面意思是指消瘦或者肌肉萎缩,肌营养不良症是一组以进行性加重的肌无力和支配运动的肌肉变性为特征的遗传性疾病群,肌营养不良症有很多种类型,有些是在出生时就可以观察到的先天性肌营养不良症,而其他的则是在青春期才观察到的,不考虑发病的确切时间,有些肌营养不良症会导致运动受损甚至瘫痪症状体征:肌营养不良症有什么症状?(一)假肥大型呈性环连隐性遗传,男性罹病,女性携带。
通常在幼儿期起病。
表现为能走路的年龄推迟,行走缓慢、易跌,跌倒后不易爬起。
多数有小腿肌的肥大,病初肥大肌肌力相对稍强。
臀中肌受累而致骨盆左右上下摇动;跟腱挛缩而足跟不能着地;腰大肌受累而腹部前凸,脑后仰。
呈鸭型步态。
从蹲位只有靠两手撑着自己身体而逐步站直大腿,然后逐步挺起身子。
继骨盆带肌肉受累之后,逐步出现肩胛带肌肉萎缩、无力,两臂举高不能。
菱形肌、前锯肌、肩胛肌、岗上、岗下肌萎缩而使肩胛游离、肩胛骨呈翼状耸起,称翼状肩。
病程逐步发展,某些儿童可能由于本身生长发育的影响,出现病程的相对稳定或好转。
多数病孩在10岁时已丧失行走能力,依靠轮椅或坐卧不起,出现脊柱和肢体畸形。
晚期,四肢挛缩,活动完全不能。
常因伴发肺部感染、褥疮等于20岁之前丧生。
智商常有不同程度减退。
半数以上可伴心脏损害,心电图异常。
早期呈现心肌肥大,除心悸外一般无症状。
(二)面一肩一肱型呈常染色体显性遗传。
男女均可罹病。
病情严重程度不一,轻者可无任何主诉,在偶然机会或医师进行家谱分析时发现。
幼年或青春期隐匿起病,常在发病后数年才被引起注意。
面肌受累较早,表现为睡眠时闭眼不紧、吹气无力、苦笑脸容。
逐步出现颈肌、肩胛带肌、肱肌的萎缩、无力。
肩胛带和肱部肌肉萎缩,两侧肩峰隆突明显。
整个肩胛部酷似衣架。
肢带型肌营养不良症1例报告及分析

肢带型肌营养不良症1例报告及分析
刘铭柏;方坚;孙许宝;叶江琳
【期刊名称】《基层医学论坛》
【年(卷),期】2009(013)023
【摘要】@@ 1 病例资料rn患者,男,1971年出生,因"四肢上段进行性肌萎缩18年"人院.患者15岁时不能抬米袋上楼,未引起注意,于J991年起发现右肘无力,渐觉四肢上段肌肉萎缩无力,并渐加重,1999年行肌电图提示肌源性损害,运动单位时限缩短,电压偏低.股四头肌病理活检示:轻度炎症性改变,肌源性萎缩.
【总页数】1页(P756)
【作者】刘铭柏;方坚;孙许宝;叶江琳
【作者单位】广州中医药大学附属第三医院芳村分院;广州中医药大学附属第三医院芳村分院;广州中医药大学附属第三医院芳村分院;广州中医药大学附属第三医院芳村分院
【正文语种】中文
【中图分类】R5
【相关文献】
1.肢带型肌营养不良症一家系五代26例报告 [J], 江晓云
2.肢带型肌营养不良症2A型临床前期两例临床表型及基因突变分析 [J], 李欢;朱瑜龄;利婧;王倞;何若洁;林金福;张成
3.肢带型肌营养不良症2D型一家系临床表型及基因突变分析 [J], 欧俐羽;孙毅明;利婧;王倞;李欢;曾缨;梁颖茵;张成
4.肢带型肌营养不良症一家系10例报告 [J], 黄树其;黄流清;赵忠新
5.肢带型肌营养不良症2B型与免疫介导的坏死性肌病临床及影像学差异分析 [J], 赵亚雯;王艳莉;王朝霞;张巍;袁云
因版权原因,仅展示原文概要,查看原文内容请购买。
肢带型肌营养不良2A型1例临床、病理及基因分析(附家系基因)

• 168 •J Apoplexy and Nervous Diseases,February 2017 ,Vol 34,No. 2文章编号:1003-2754(2017)02-0168~02 中图分类号:R746.2肢带型肌营养不良2A型1例临床、病理及基因分析(附家系基因)黄志强、尤红2,葛力2肢带型肌营养不良症(limb-girdle muscular dystrophy,LG-RID)是一类临床表现为进行性骨盆带和肩胛带肌无力的遗传性疾病。
目前国内对该病2A型的临床、病理和基因研究较少,我们对确诊为该型患者的临床、病理、基因测序进行分析。
1临床资料患者,男,14岁,学生,因“进行性双下肢乏力伴步态异常2 y余”人院。
患者2 y前无明确诱因出现双下肢乏力,以右 下肢为著,行走时右脚尖先着地,左脚尖着地正常,无足内翻,无明显跛行,当时家属未予重视。
后双下肢无力进行性加重,出现行走时双脚尖着地,上楼及蹲位站立困难,运动后无疲乏、无肌肉酸痛、无“晨轻暮重”、无肌肉肥大和萎缩症状,遂来我院就诊。
人院体检:生命体征平稳,发育正常,智力正常,营养中等,鸭步行走,双足下垂,右手背可见一直径约5 c m类圆形黑色素沉积,心肺腹查体未见明显异常,双踝关节畸形,呈弓形足。
专科查体:高级智能中枢未见异常,颅神经检查提示Webei■试验偏左,双侧R innie试验均气导 > 骨导,全身肌肉无明显萎缩,双上肢肌张力和肌力正常,双下肢肌张力增高,双下肢近端肌力4级,远端肌力5级,双上肢和左下肢浅、深感觉正常,右下肢浅、深感觉减退,浅反射正常,双上肢腱反射正常,双下肢腱反射消失,病理征和脑膜刺激征阴性,自主神 经功能正常。
辅助检查:血常规、凝血、大小便常规、甲状腺功能、肾脏功能、血脂、血糖均未见明显异常。
生化:谷丙转氨酶(A LT) :104 U/L,谷草转氨酶(A ST) :75 U/L,碱性磷酸酶(A LP) :314 U/L,肌酸激酶(C K) :4728.5 U/L,乳酸脱氢酶(LD H) :515 U/L,CK同工酶M B: 112. 6 U/L。
肢带型肌肉营养不良如何解决

如对您有帮助,可购买打赏,谢谢
生活常识分享肢带型肌肉营养不良如何解决
导语:肢带型肌肉营养不良的情况主要原因就是由于我们对于自己的饮食,没有引起高度重视,这种疾病发生在小孩子的身上比较高,小孩子养成了挑食的
肢带型肌肉营养不良的情况主要原因就是由于我们对于自己的饮食,没有引起高度重视,这种疾病发生在小孩子的身上比较高,小孩子养成了挑食的习惯,就不能全面的摄入一些营养,加大了自身出现肢带型肌肉营养不良的几率,普及一下肢带型肌肉营养不良的治疗方法吧。
饮食宜清淡、营养丰富,忌食或少食油腻厚味过热、伤津耗液及损伤脾胃之品,可多食鱼类、蛋类、鸡肉、瘦猪肉等。
基础方法
包括系列中药制剂为主的药物治疗,中药汤剂,大灸和按摩等方法是疾病各时期都要应用的方法,已开发出十多种临床制剂可适用于各种不同证型的病人;
康复训练方法
包括肌营养不良操和肌腱康复训练法。
肌腱康复训练法已形成了针对疾病各个时期的不同方法,包括肌腱曲伸训练、站立肌肉训练、移步训练等;
物理疗法
主要有导平仪、中药治疗仪和自制夹板,自制的简易关节夹板,用来固定膝关节和踝关节辅助康复训练;
以上为我们介绍了肢带型肌肉营养不良的最好的治疗方法,日常一定要注意多吃一些肉蛋奶,等富含高蛋白的食物,可以有效的帮助我们补充身体所需要的营养,对于促进我们骨骼的发育非常有帮助,可以提高我们骨骼的韧性与强度。
肢带型肌营养不良与致病机制

分类
按照遗传方式可以分为常染色体显性遗传性肢带型肌营 养不良症(LGMD1 型)和常染色体隐性遗传性肢带型肌营养 不良症(LGMD2 型):
LGMD
LGMD1 LGMD2
LGMD1A-1H
10%
LGMD2A-2Z
90%
检测方法
诊断和鉴别
肢带型肌营养不良症主要出现近端肩胛带肌和骨盆带肌受累倾向,亦可累及 远端肌肉。除骨骼肌受累外,还可以合并全身多系统表现,主要包括:
LGMD2P 型: 目前仅见1例个案报道,3岁发病,伴小头畸形、脊柱前凸、踝关节挛缩和显著智
力发育迟滞。
LGMD2型临床特征
LGMD2Q 型: 通常于2~3岁发病,约20岁丧失行走能力,心功能正常。
LGMD2R 型: 青少年期发病,伴关节挛缩、翼状肩胛、脊柱侧弯,而心肌病少见。
LGMD2S 型: 通常于学龄早期发病,伴肌肉疼痛和肌肉痉挛,部分伴骨骼异常,包括髋关节发育不
良和脊柱侧弯,亦可伴白内障、集合性斜视,部分患者可出现智力发育迟滞以及舞蹈样动 作、手足徐动、震颤和肌张力障碍性姿势,亦可出现癫痫发作,而心肺功能正常。
LGMD2T 型: 通常于出生后至成年期发病,部分伴阵发性横纹肌溶解症、轻度智力发育迟滞、癫痫发
作、小头畸形、白内障、眼震等,心肺功能正常。
LGMD2U 型: 通常于儿童早期发病,多伴广泛性肌肉肥大,部分伴舌体肥大,并出现翼状肩胛、脊柱
LGMD2型临床特征
LGMD2H 型 : 通常于70岁左右发病,部分伴周围神经病变,电生理学表现为神经传导速度(NCV)
减慢。血清肌酸激酶水平正常或仅轻度升高。
LGMD2I 型: 发病年龄9~23岁,下肢以屈髋肌群和大腿内收肌群受累显著,上肢以肩内收肌群
先天性肌营养不良1A型1例临床与基因分析

先天性肌营养不良1A型1例临床与基因分析江士远;向娜【摘要】目的报道1例LAMA2基因变异导致先天性肌营养不良的临床、实验室检查及遗传学特点.方法回顾分析1例先天性肌营养不良1A型患儿的临床资料,并复习相关文献.结果患儿,男,5岁2个月,临床表现为运动发育落后,2岁时可独坐,不能独走;肌力及肌张力低下,早期出现关节挛缩.生化检测发现肌酸激酶(CK)升高(491 U/L),其同工酶CK-MB升高(41.8 U/L);肌电图提示肌源性损害可能;头颅MRI提示大脑白质异常信号.基因检测发现LAMA2存在复杂杂合突变,c.2045-2046delAG 杂合缺失,来自母亲,为已报道的致病变异;exon5存在杂合缺失,来自父亲,为未报道的新变异,软件功能预测提示为致病性变异.结论 LAMA2基因变异导致先天性肌营养不良,患儿以运动发育落后起病,CK升高,高通量基因检测有助于明确诊断.%Objective To investigate the clinical features and genetic tests of a case with congenital muscular dystrophy type 1A (MDC1A). Methods Clinical data of proband were collected, and genetic change were tested using next generation sequencing, and literatures pertinent to the epidemiology, mechanisms, especially genetic testing of lisencephaly were reviewed. Results A 5 year and 2 month old boy present with normal intelligence and delayed motor development, he can be sit alone but not walk at two years old. Physical examination showed normal mental reaction, muscular dystrophy, hypotonia, and joint contracture at early age. From biochemical tests, we found creatine kinase (CK) and CK-MB were increased (491U/L, 41.8U/L). EMG test suggested possible muscle-derived damage. Brain MRI showed white matter abnormality. And a heterozygousmutation (c.2045-2046delAG) inherited from his mother in LAMA2 gene, and another novel heterozygous mutation (del Exon5) inherited from his father were identified by genetic test. Conclusions LAMA2 gene deficiency can lead to MDC1A, and gene testing can help diagnosis.【期刊名称】《临床儿科杂志》【年(卷),期】2017(035)005【总页数】3页(P369-371)【关键词】先天性肌营养不良;临床特点;分子诊断【作者】江士远;向娜【作者单位】单县海吉亚医院儿科山东单县 274300;单县海吉亚医院儿科山东单县 274300【正文语种】中文先天性肌营养不良(congenital muscular dystrophy,CMD)是一类出生后早期起病、主要影响骨骼肌功能的疾病,不同的肌营养不良虽有相似的临床表型,但遗传差异很大[1]。
名老中医陈卫川治疗肢带型肌营养不良症经验举隅

名老中医陈卫川治疗肢带型肌营养不良症经验举隅
甘佳乐 贾爱民 陈卫川
宁夏回族自治区中医医院暨中医研究院脑病科,宁夏 银川 750021
2019年 1月 23日 初 诊:患 者 双 下 肢 酸 软 无 力,近端明显,上楼及蹲位站立困难,行走不稳, 腰部酸 困 无 力,纳 可,睡 眠 可,二 便 调。舌 红, 苔黄 腻,脉 沉 细 数。脉 症 合 参,证 属 肝 肾 不 足, 兼有湿热,治以健脾化湿、滋补肝肾为法。处方: 薏苡仁 30g,苍术 15g,怀牛膝 12g,黄柏 12g, 泽泻 12g,天麻 12g,砂仁 10g,木香 12g,木瓜 15g,鸡血藤 30g,狗脊 20g,丝瓜络 15g,路路 通 12g,石菖蒲 12g,五加皮 12g,赤芍 12g,甘 草 12g。7剂,日 1剂,凉水煎煮 400mL,分早晚 2次饭后 30min温服。并嘱咐患者每日适度锻炼。
肢 带 型 肌 营 养 不 良 症 (Imb-girdlemuscular dystrophy,LGMD) 是一种常染色体显性或隐性遗 传的肌肉变性疾病,属中医 “痿证” 范畴。多在 10~20岁 起 病, 临 床 表 现 为 缓 慢 进 行 性 加 重 的 骨 盆带肌肉无 力 和 萎 缩, 腰 椎 前 凸, 后 逐 渐 累 及 肩 胛带肌肉,平均起病 20年后逐渐丧失劳动能力。 目前尚无特异性治疗方法。陈卫川主任系全国名 老中医,全国第二批名老中医药专家学术经验传 承指导老师,从医 60余载,在治疗神经内科疑难 杂症方面屡 起 沉 疴。 笔 者 有 幸 师 从 陈 老, 将 其 辨 治肢带 型 肌 营 养 不 良 症 的 学 术 思 想 及 临 床 经 验, 总结如下。
肢带型肌营养不良2d型1例临床与基因型分析

doi:10.3969/j.issn.1000-3606.2020.02.009肢带型肌营养不良2D型1例临床与基因型分析徐 敏1 郭 虎1 高修成2 卢孝鹏1南京医科大学附属儿童医院 1.神经内科,2.影像科(江苏南京 210008)摘要: 目的 提高对肢带型肌营养不良2D型临床及基因型特征的认识。
方法 回顾分析1例肢带型肌营养不良2D型患儿的临床资料,并复习相关文献。
结果 男性患儿,5岁7个月起病。
有进行性肌无力、腓肠肌肥大、Gower征阳性。
磷酸肌酸激酶20 602 U/L;肌电图示肌源性损害;肌肉磁共振示双下肢皮下脂肪层及肌肉内见条片状压脂序列高信号,肌肉层著。
MLPA检测DMD基因阴性;全外显子测序示SGCA基因第3外显子c.234-235AC>GA纯合突变,该位点未见报道,美国医学遗传学与基因组学学会(ACMG)评级为致病性变异,其父母均为杂合携带。
患儿最终诊断为肢带型肌营养不良2D型。
结论 SGCA c.234-235AC>GA变异可能为肢带型肌营养不良2D型新的致病性突变。
肌酶明显升高时,除假肥大型肌营养不良症外,还应注意肢带型肌营养不良2D型。
关键词: 肢带型肌营养不良2D型; SGCA基因; 磷酸肌酸激酶Clinical and genotypic analysis of a patient with limb-girdle muscular dystrophy type 2D XU Min1, GUO Hu1, GAO Xiucheng2, LU Xiaopeng1 (1.Department of Neurology, 2.Department of Medical Imaging, Children’s Hospital of Nanjing Medical University, Nanjing 210008, Jiangsu, China)Abstract: Objective To improve the understanding of the clinical and genotypic characteristics of limb-girdle muscular dystrophy type 2D (LGMD2D). Methods The clinical manifestations, laboratory tests, electromyography, muscle MRI and genetic test results of a patient with LGMD2D were retrospectively analyzed, and the related literature were reviewed. Results The patient was a 5 years and 7 months old male, who presented with progressive muscle weakness, gastrocnemius hypertrophy, Gower sign positive, phosphocreatine kinase 20602 U/L, electromyography showed myogenic damage, muscle MRI showed a high signal of double lower limb subcutaneous fat layer and the muscles on the fat suppressed sequence, and the muscles were more obvious. The MLPA result of Duchenne muscular dystrophy (DMD) gene test was negative, and the whole exom sequencing showed a homozygous c.234-235AC>GA mutation in exon 3 of SGCA gene which has not been reported. According to ACMG rating, this variant was pathogenic, and his parents were all heterozygous carriers. The patient was diagnosed as LGMD2D.Conclusion SGCA c.234-235AC>GA variation may be a novel pathogenic mutation in LGMD2D. For children with clinically suspected muscular dystrophy and significantly increased muscle enzyme, in addition to DMD, we should also pay attention to the LGMD2D.Key words: limb-band muscular dystrophy 2D; SGCA gene; phosphocreatine kinase肢带型肌营养不良症2D型(limb-girdle mus-cular dystrophy type 2D,LGMD2D)是一种罕见的常染色体隐性遗传性肌病,由α-sarcoglycan基因(SGCA)突变引起,于1994年Roberds等[1]首次报道。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
肢带型肌营养不良症1例报告及浅析
发表时间:2019-11-05T09:55:58.847Z 来源:《医师在线》2019年8月15期作者:谈伟1 朱亚楠1 吴静1 陈孚伟1 戴军1 张瑞刘朗[导读] 分析肢带型肌营养不良症治疗方式。
谈伟1 朱亚楠1 吴静1 陈孚伟1 戴军1 张瑞刘朗1 赵艳2 赵红东3
(1泗阳康达医院;江苏宿迁223800;2南京大学附属鼓楼医院;3南京医科大学附属南京医院;江苏南京210000)【摘要】目的:分析肢带型肌营养不良症治疗方式。
方法:针对一例肢带型肌营养不良症患者展开分析,确定病症特点,根据患者的实际情况,制定相应的治疗方案。
结果:经过针对性治疗之后,患者在一年之内病情没有加重,在一段时期之内得到了有效控制。
结论:通过对肢带型肌营养不良症患者展开分析,深入了解肢带型肌营养不良症的临床表现、治疗方式以及今后发展趋势,为相关研究人员提供
研究条件,提升我国肢带型肌营养不良症的治疗水平。
【关键词】肢带型肌营养不良症;治疗方法;临床表现
肢带型肌营养不良症简称LGMD,该疾病主要发生在患者的髋部和肩部肌肉,发病原因较为复杂,发病时间的范围较广,包括儿童期、青春期、成年期甚至是老年期,男女的患病概率大致相同。
如果肢带型肌营养不良症发病时期为儿童期,则病症的进展速度会更快,严重的会导致儿童残疾,如果发病是为青春期或者是成年期,则病情发展速度相对缓慢,能够为医生提供一定的治疗时间。
1.资料与方法
1.1一般资料
本次研究的肢带型肌营养不良症患者为女性,22岁,四肢在6年前出现近端无力等现象,近阶段病情加重,无法正常的抬头、蹲起和上楼。
由于在发病6年中,病情没有对正常生活产生较大影响,因此没有采取相应的控制措施。
在2年之内病情逐渐加重,四肢肌肉逐渐萎缩,尤其是在近端部位。
没有出现排尿、排便障碍,发病之后没有出现发热、皮肤斑疹以及大幅度体重变化的情况。
父母是近亲结婚,患者没有外伤手术史、输血史以及肝炎等病症,未婚,月经周期正常,没有发生痛经。
患者的母亲和外祖母走路轻微摇晃,属于肌无力症状,但是进程较为缓慢。
1.2方法
对患者DNA进行性肌营养不良目的基因捕获二代测序,生物信息学分析致病基因,Sanger法对患者验证。
目前采用的治疗方法主要包括以下几种,第一,临床康复训练,尽可能长时间的保持肌肉活性,并将其与其他治疗方法相结合,其中包括物理治疗法、职业治疗法、呼吸训练疗法以及吞咽功能训练等,根据患者的实际情况,制定针对性的治疗方案[1]。
第二,力量训练。
力量训练能够帮助患者锻炼肌肉,延缓其发生萎缩的速度,并且该种治疗方式没有副作用。
但是在训练中需要注意,由于患者的肌肉发生萎缩,采用高强度的训练方式可能会出现肌肉损伤甚至是肌无力等情况。
所以需要根据患者的肌肉萎缩程度,适当进行力量训练,合理确定训练时间以及训练强度。
提升肌肉使用效率的同时,降低肌肉中存在的疲劳感[2]。
1.3观察指标
本次研究的观察指标主要为患者在完成治疗一年之后,各项功能水平的对比情况。
2.结果
2.1患者基因检测结果(见表1)
通过表1分析能够看出,该患者的神经肌肉病相关367个基因未检测到明确致病基因,但根据患者临床表现,符合肌营养不良表现,仍继续康复训练。
2.2患者功能恢复情况(见表2)
3.讨论
目前可以将肢带型肌营养不良症分为十二种类型,导致该病症出的主要原因为基因缺陷。
基因位于染色体中,能够帮助人们体内产生各种蛋白质以及编码。
同样,基因可以表根据肌肉的不同功能,合成相应的蛋白质。
一旦基因出现缺陷,则合成的蛋白质也会发生变异,导致人体中的整个肌细胞无法正常工作。
因此,肢带型肌营养不良症患者通常会出现肌肉无力等情况,随着病症的加重,患者的肌无力症状也会逐渐加重[3]。
该疾病属于致命疾病,除了行动独立性会受到阻碍之外,患者的心脏和呼吸功能也会受到影响,例如心律不齐以及心肌病等。
一旦心肌以及呼吸肌出现萎缩,则会严重威胁患者的生命安全,因此在实际治疗中需要注意这一问题,制定针对性的解决方案,尽量延长患者的生命。
目前已经基本掌握了该疾病的发展模式,医生在治疗中可以通过疾病进展,对患者状态展开监督管理,调整治疗方案,延缓病症的发展速度[4]。
针对该患者的基本情况,医生根据检查报告分析确定,该患者四肢近端肌无力现象登记为3+-4-级,远端肌无力现象4+级,多处肌肉发生萎缩,四肢的反应速度减慢。
左股外侧的肌肉中,肌束衣和肌内衣结缔组织出现增生情况,肌纤维的大小不一,发生不同程度的肌肉萎缩以及肌肉增生。
根据患者家族病史,确定患者患有肢带型肌营养不良症,通过对患者的DNA进行分析发现,患者符合常染色体遗传的现象,因此该疾病的主要病因为基因突变[5]。
通过以上分析能够看出,在经过控制治疗之后,患者的病情得到控制,并没有发生明显的加重情况,发展速度较为缓慢。
在整个治疗中主要采用肌肉锻炼以及药物治疗相结合的方式,对病情展开了有效控制。
该疾病目前在我国并没有出现有效的治疗方式,通常将支持治疗作为主要方法,治疗重点在控制病情发展中。
也就是说当今针对该疾病并没有有效的根治方法,一旦患病,平均在20年左右,患者会逐渐失去劳动能力。
参考文献:
[1]李欢,朱瑜龄,利婧,王倞,何若洁,林金福,张成.肢带型肌营养不良症2A型临床前期两例临床表型及基因突变分析[J].中国现代神经疾病杂志,2018,18(07):506-513.
[2]欧俐羽,孙毅明,利婧,王倞,李欢,曾缨,梁颖茵,张成.肢带型肌营养不良症2D型一家系临床表型及基因突变分析[J].中国现代神经疾病杂志,2017,17(08):609-615.
[3]杨钊,董继宏.常染色体显性遗传性肢带型肌营养不良症研究进展[J].中国临床神经科学,2017,24(01):84-88.
[4]罗苏珊. 肢带型肌营养不良症2A型的诊断方法及临床特点研究[D].复旦大学,2017.
[5]王丹,周敬华,曹学兵.常染色体隐性遗传肢带型肌营养不良症致病基因研究进展[J].国际神经病学神经外科学杂志,2017(03):277-280.。