RepeatScou操作步骤(说明)
RepeatMasker网页版和命令行版使用说明(中文翻译版)

RepeatMasker网页版和命令行版使用说明(中文翻译版)引用自Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009 Mar;Chapter 4:Unit 4.10. doi: 10.1002/0471250953.bi0410s25.RepeatMasker是一款广泛应用于基因鉴定、分类和mask repetitive elements,包括低复杂度序列和散布重复序列。
RepeatMasker通过将数据库如:Repbase中已知的重复序列与输入的基因组序列比对来搜素重复序列。
在此我们描述两个基础协议,它对如何运用RepeatMasker去分析基因组序列的重复元件提供细节上的指导,而不论是通过网络界面还是通过Unix/Linux命令系统。
在RepeatMasker中的序列比较通常经过cross-match程序的序列比对来实现,对于较大序列这一过程需要大量处理时间。
交替协议描述的是通过应用诸如WU-BLAST这样的选择性比对程序来怎样减少处理时间。
而且RepeatMasker的优势、局限和已被发现的漏洞将在此进行讨论,最后提供理解其处理结果的指南。
在新的RepeatMasker程序包中添加了鉴定蛋白质序列的重复原件的程序。
要运行RepeatMasker,首先要选择重复库文件(repeat library files),这一文件包含重复元件共有序列。
目前,Repbase Update是最大的商业性(商购)重复库(free for academic use)并且包含了相当数量的包括人、啮齿动物、斑马鱼、果蝇以及拟南芥在内的生物体。
生物体的库文件中没有Repbase Update时,库文件会用RECON(Bao and Eddy, 2002; /recon.html)或RepeatScout (/repeatscout/; Price et al., 2005)从头产生。
GE公司CT操作台操作步骤

GE公司CT操作台操作步骤开机步骤:1先打开操作台下方的开关,待显示屏上出现“ OC initializing Please wait…….”在随后出现的“ Fastcal has not been performed within the last 24 hour”(在过去的24小时内尚未执行过快速空气校正)的提示框,点击OK,随后会在显示屏上出现“No scan have been take since 时间,Tube warmup should be run(至多少时间以来未进行过扫描,球管需要再次加热)”的提示框,再次点击OK;2、单击扫描监视器的右下角,单击“Daily Prep(日常准备)”键;在弹出的显示框中,单击“TUBE WARMUP(球管加热)”,单击“Accept & Run Tube Warmer-up(接受并执行球管加热),待键盘上的“Start scan”键亮起后,按下“Start scan”键。
待球管加热结束后,单击QUIT(退出)或进入第3步(快速空气校正)、单击扫描监视器的右下角,单击“Daily Prep(日常准备)”键,在弹出的显示框中,单击“F AST CAL(快速空气校正),键盘上的“Start scan”键亮起后,按下“Start scan”键。
待快速空气校正结束后,单击“QUIT(退出)”。
提示:开机后、球管未曝光连续2小时以上,需进行球管加热;为保证图像质量,建议每日进行一次快速空气校正。
关机步骤:按下右侧屏幕上方的红色“SHUTDOWN(关闭)”,在弹出的对话框中,选择“SHUTDOWN”,单击按下右侧屏幕上方的红色“SHUTDOWN(关闭)”OK;如果要重新启动,则在弹出的对话框中,选择默认的“RESTRART(重新启动)”。
在突然停电情况下,应确保控制台开关按钮处于OFF状态。
检查步骤:1、按下左侧屏幕左下方的“NEW PATIENT(新患者)”,在出现的信息栏中依次录入患者的信息,在录入PATIENT ID后,屏幕右侧人体解剖器被激活。
repeat submit

repeat submit
Repeat submit(重复提交)是指用户在同一时间内多次提交同一份表单或请求的行为。
常见的原因可能是由于网络不稳定或网站响应缓慢,导致用户点击了多次提交按钮;或者是用户自身的操作失误,不慎多次点击了提交按钮。
无论是哪种情况,这都会给网站带来负担和风险。
针对这种情况,网站开发者应采取相应措施,避免重复提交所带来的副作用。
一种常见的解决方案是引入防重复提交机制。
具体实现方式如下:
1. 在页面中加入 Token 字段。
每次提交时,服务器生成一个随机的Token 字符串,返回给前端页面。
前端将该值缓存到本地,作为提交表单时的 Token 值。
这样可以防止用户反复提交同一表单。
2. 前端限制按钮点击。
在第一次提交后,前端限制按钮变灰或隐藏,防止用户再次提交。
等到服务器返回结果后,再将按钮恢复正常。
3. 后端校验是否重复。
使用服务器处理框架的时候,可以在后端使用过滤器或拦截器等校验用户请求,判断是否重复提交。
如果是重复的请求,则返回错误码或跳转到错误页面。
4. 后端使用缓存技术。
在服务器端,可以使用缓存技术,将用户请求
的数据保存在缓存中。
如果重复提交,服务器可以从缓存中获取数据,返回给用户。
但是,需要注意缓存的过期时间,避免之后的请求对当
前操作造成影响。
总之,防止用户的重复提交对于网站的安全性和负荷能力至关重要。
只有采取合理的措施,才能有效地保护网站的稳定性和用户体验。
repeatmasker使用方法

repeatmasker使用方法RepeatMasker是一种常用的基因组序列分析工具,主要用于识别和屏蔽重复序列。
本文将介绍RepeatMasker的使用方法,帮助读者快速上手并了解该工具的基本原理和功能。
一、RepeatMasker简介RepeatMasker是一种基于序列比对的重复序列识别工具,可以识别和屏蔽基因组中的重复元件。
重复序列是指在基因组中存在多个拷贝的DNA片段,通常占据了基因组的大部分空间。
这些重复序列对于基因组结构和功能的研究具有重要意义,但在某些情况下也会对基因组注释和后续分析造成干扰。
RepeatMasker的作用就是将这些重复序列进行识别和屏蔽,以便更好地进行后续分析。
二、RepeatMasker的安装和运行1. 安装RepeatMasker:首先需要从RepeatMasker官方网站下载安装包,并按照官方提供的安装指南进行安装。
安装完成后,需要下载并安装相应的重复序列数据库,如RepBase等。
2. 准备输入序列:在运行RepeatMasker之前,需要准备好待分析的基因组序列文件(一般为FASTA格式),并确保序列文件中不包含非法字符或空行。
3. 运行RepeatMasker:打开终端或命令行窗口,输入以下命令运行RepeatMasker:repeatmasker -species [species] [input_file.fasta]其中,[species]为待分析基因组的物种信息,需要根据实际情况进行设置;[input_file.fasta]为待分析的基因组序列文件。
三、RepeatMasker结果解读RepeatMasker的运行结果主要包括以下几个文件:1. [input_file.fasta].masked:屏蔽后的输出序列文件,其中重复序列被替换为小写字母。
2. [input_file.fasta].out:注释文件,记录了每个重复序列的位置、类型、分数等信息。
Recovery 操作说明
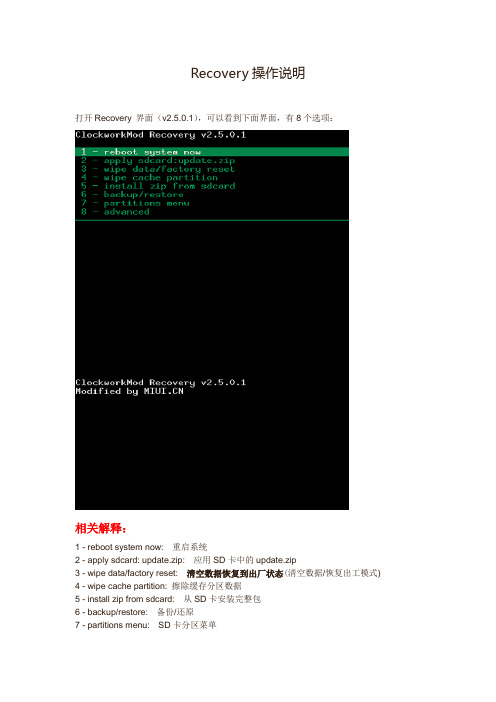
Recovery操作说明
打开Recovery 界面(v2.5.0.1),可以看到下面界面,有8个选项:
相关解释:
1 - reboot system now: 重启系统
2 - apply sdcard: update.zip: 应用SD卡中的update.zip
3 - wipe data/factory reset: 清空数据恢复到出厂状态(清空数据/恢复出工模式)
4 - wipe cache partition: 擦除缓存分区数据
5 - install zip from sdcard: 从SD卡安装完整包
6 - backup/restore: 备份/还原
7 - partitions menu: SD卡分区菜单
8 - advanced: 高级选项
几个补充:
1、reboot system now: 重启系统。
每次在recovery模式下完成任何操作都必须要使用到的!
2、apply sdcard: update.zip: 应用SD卡中的update.zip。
也就是大家刷机的时候常用到的一个选项!选择此项后大家就要等待一段时间!
3、wipe data/factory reset: 清空数据/设定工厂模式。
会把手机里的所有数据都清除!
4、backup rom to sdcard: 备份ROM到SD卡。
这个功能类似电脑上的GHOST,会把你手机上所有的数据都备份到SD卡上。
5、restore rom from sdcard: 从SD卡还原ROM。
会把原先备份好的ROM从SD卡上还原到手机上!。
语言复读机使用方法

语言复读机使用方法
使用语言复读机的方法很简单,只需要按照以下步骤操作:
1. 打开语言复读机软件或网站。
2. 在指定的文本框中输入要复读的内容。
3. 点击“复读”或类似的按钮,开始复读。
4. 语言复读机会逐句或逐词地将您输入的内容复读出来。
5. 您可以继续在文本框中输入新的内容,语言复读机会继续复读。
有些语言复读机会提供其他功能,如调整复读速度、选择不同的语音合成器或设置特定的重复次数。
您可以根据需要自行探索和使用这些功能。
另外,语言复读机通常可以通过复制和粘贴文本来复读长篇内容,这可以更方便地让您复读需要的文本。
注意:语言复读机使用的是计算机生成的语音合成技术,因此复读的语音可能不够自然或流畅。
不同的语言复读机可能有不同的质量和效果,请根据个人需求选择适合的语言复读机。
css中repeat的用法

css中repeat的用法一、什么是CSS的repeat属性repeat属性是CSS中常用的背景图像平铺方式之一。
它能够将一个背景图像在元素内部水平和垂直方向进行重复以填满整个区域。
使用repeat属性能够很方便地实现网页背景或元素纹理的效果。
二、repeat-x和repeat-y在CSS中,有两种常见的背景图像平铺方式,分别是repeat-x和repeat-y。
下面将详细介绍这两种方式。
1. repeat-x当使用repeat-x时,背景图像会在横向上进行重复,即使图像不足以填满整个区域也会进行重复。
这意味着横向无论多长,都会被完全覆盖。
示例代码:```cssdiv {background-image: url("image.jpg");background-repeat: repeat-x;}```通过上述代码,div元素将使用名为"image.jpg"的背景图像,并且该图像会在横向上进行重复以填充整个div区域。
2. repeat-y与repeat-x相似,当使用repeat-y时,背景图像会在纵向上进行重复。
即使图像高度不足以填满整个区域也会进行重复。
示例代码:```cssdiv {background-image: url("image.jpg");background-repeat: repeat-y;}```通过上述代码,div元素将使用名为"image.jpg"的背景图像,并且该图像会在纵向上进行重复以填充整个div区域。
三、repeat和no-repeat除了repeat-x和repeat-y之外,还有两个特殊的值:repeat和no-repeat。
1. repeat当使用repeat时,背景图像会同时在横向和纵向上进行重复。
这意味着无论图像多大,都会完全填满整个区域。
如果只想让背景图像单方向重复,请使用repeat-x或repeat-y。
logo语言repeat重复命令教案

2、小结学生学习情况,展示学生作品。
部分学生展示自己用LOGO命令画出图画。
4、要求学生上机操作,键入画正十边形的重复命令,观察画出的正十边形是否符合要求。
学生操作:用重复命令画正十边形,观察画出的正十边形是否符合要求。
3、用重复命令画圆
用重复命令画正三十六边形来模拟圆
1、用谈话法和讨论法,与学生讨论如何用重复命令画正三十六边形;
2、要求学生上机操作,画出正三十六边形;
3、T:通过观察你会发现,正三十六边形已经很像一个圆了,所以今后我们说画圆,就是让小海龟画一个正三十六边形来模拟一个圆;
拓展训练
1、与学生讨论教材“做一做”2中的四个图形,参考提示的内容,说一说它们是怎样画出来的;
2、要求学生上机操作,画出其中的一到两幅图画。
学生参加讨论;
学生上机操作,画出其中的一到两幅图画。
小结
1、小结本课内容:
(1)重复命令的使用格式;
(2)用重复命令画多种正多边的方法;
(3)用重复命令画圆的方法;
学生操作:键入画长方形的重复命令,观察画出的长方形是否正确。
2、用重复命令画正多边形
用重复命令画正多边形
1、示范讲解用重复命令画正三角形;
2、示范讲解用重复命令画正五边形;
3、示范讲解用重复命令画正六边形。
学生操作:用重复命令画正三角形;
学生操作:用重复命令画正三角形;
学生操作:用重复命令画正三角形。
总结用重复命令画正多边形的规律
1、通过以上用重复命令画正多边形的例子,总结出重复命令中每次转动角度和转动次数之间的关系:
转动角度X重复次数=360度
2、由这个规律,我们就能得到正多边形转角度数的计算方法:
RepeatMasker网页版和命令行版使用说明(中文翻译版)

RepeatMasker网页版和命令行版使用说明(中文翻译版)引用自Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009 Mar;Chapter 4:Unit 4.10. doi: 10.1002/0471250953.bi0410s25.RepeatMasker是一款广泛应用于基因鉴定、分类和mask repetitive elements,包括低复杂度序列和散布重复序列。
RepeatMasker通过将数据库如:Repbase中已知的重复序列与输入的基因组序列比对来搜素重复序列。
在此我们描述两个基础协议,它对如何运用RepeatMasker去分析基因组序列的重复元件提供细节上的指导,而不论是通过网络界面还是通过Unix/Linux命令系统。
在RepeatMasker中的序列比较通常经过cross-match程序的序列比对来实现,对于较大序列这一过程需要大量处理时间。
交替协议描述的是通过应用诸如WU-BLAST这样的选择性比对程序来怎样减少处理时间。
而且RepeatMasker的优势、局限和已被发现的漏洞将在此进行讨论,最后提供理解其处理结果的指南。
在新的RepeatMasker程序包中添加了鉴定蛋白质序列的重复原件的程序。
要运行RepeatMasker,首先要选择重复库文件(repeat library files),这一文件包含重复元件共有序列。
目前,Repbase Update是最大的商业性(商购)重复库(free for academic use)并且包含了相当数量的包括人、啮齿动物、斑马鱼、果蝇以及拟南芥在内的生物体。
生物体的库文件中没有Repbase Update时,库文件会用RECON(Bao and Eddy, 2002; )或RepeatScout(; Price et al., 2005)从头产生。
给Repeater控件里添加序号的5种方法

给Repeater控件里添加序号的5种方法.net是目前非常热门的一种程序编译语言,在.net培训中的众多知识点中,给Repeater控件里添加序号的5种方法是非常重要的一个。
下面就由达内的老师为大家介绍一下这方面的内容。
Repeater是我们经常用的一个显示数据集的数据控件,经常我们希望在数据前显示数据的序号,那么我们该怎么为Repeater控件添加序号呢?下面编辑为大家介绍几种常用的为Repeater控件添加序号的方法:方法一:利用Container.ItemIndex属性,代码如下:<Itemtemplate ><%# Container.ItemIndex + 1% ></Itemtemplate >方法二:利用Repeater的Items.Count属性,代码如下:<Itemtemplate ><%# this.Repeater.Items.Count + 1% ></Itemtemplate >方法三:利用JS在前台给一个Label标签赋值,代码如下:在.aspx中添加一个Label控件,用来显示序号。
<Label ID="label" runat="server" ></Label >JS代码:<body onload="show()" ><Script Language="javascript" >function show(){var bj = document.all.tags("Label经解释后生成的Html标签");for (i=0;i<obj.length;i++){document.all["Label经解释后生成的Html标签"][i].innerHTML=i+1;}}</script >该方法需注意的地方比较多,不推荐使用。
Repeater控件的用法

Repeater控件的用法Repeater 控件的用法Repeater 控件是一种常见的 Web 控件,它用于在页面上重复显示相同的 HTML 或自定义内容。
本文将介绍 Repeater 控件的用法,包括绑定数据源、设置模板、数据处理以及常用功能等。
一、绑定数据源Repeater 控件需要绑定数据源才能显示内容。
可以通过以下步骤进行数据源绑定:1. 在 页面上添加 Repeater 控件,在设计视图中或通过代码创建。
2. 在代码文件中定义一个数据源,可以是集合、数据集、数据库查询结果等。
3. 在 Page_Load 或其他事件中,将数据源绑定到 Repeater 控件。
示例代码如下:```csharpprotected void Page_Load(object sender, EventArgs e){if (!IsPostBack){List<string> data = GetDataFromDatabase(); // 从数据库获取数据 Repeater1.DataSource = data; // 将数据源绑定到 Repeater 控件 Repeater1.DataBind(); // 绑定数据}}```二、设置模板Repeater 控件使用模板来定义重复显示的 HTML 或自定义内容。
可以通过以下方式设置模板:1. 在 Repeater 控件内部,使用 <%# %> 语法绑定数据项的值。
2. 使用 <ItemTemplate> 标签来定义每个重复项的显示格式。
3. 在 <ItemTemplate> 中,可以使用任何有效的 HTML 标签和 控件。
示例代码如下:```html<asp:Repeater ID="Repeater1" runat="server"><ItemTemplate><div><h3><%# Eval("Title") %></h3><p><%# Eval("Content") %></p></div></ItemTemplate></asp:Repeater>```上述代码中,使用 Eval() 方法来绑定数据项的值,并在<ItemTemplate> 中定义了一个包含标题和内容的 div。
repeat 的用法

repeat 的用法一、什么是repeat的用法在编程领域,repeat是经常用到的一个功能。
它指的是重复执行某段代码或者某个任务。
无论是在编写脚本,还是在开发应用程序时,都会遇到需要重复执行特定代码块的情况。
使用repeat可以实现这一目的,并增加代码的灵活性与可视性。
下面将详细介绍repeat的用法和相关注意事项。
二、repeat循环结构Repeat循环结构通常由两部分组成:循环条件和循环体。
循环条件是一个逻辑表达式,决定了是否继续执行循环体;而循环体则包含了需要重复执行的代码逻辑。
1. Repeat-UntilRepeat-Until循环结构先执行循环体中的代码,然后再判断是否满足退出条件。
只有当退出条件为真时,才结束该次循环,否则继续执行下一次迭代。
示例:```repeat-- 循环体中要执行的代码print("Hello, World!")until condition```上述示例中,首先会输出"Hello, World!",然后再判断condition是否满足。
如果不满足,则会重新进入循环并再次输出"Hello, World!"直到满足退出条件为止。
2. Repeat-N-TimesRepeat-N-Times循环结构是指明确指定循环次数的重复执行方式。
这种方式下,循环体中的代码会被执行固定的次数。
示例:```repeat n times-- 循环体中要执行的代码print("Hello, World!")end repeat```上述示例中,`n`代表了循环的次数。
每次循环都会输出"Hello, World!",直到达到指定的循环次数。
三、Repeat用法的注意事项尽管repeat是一个非常强大和灵活的工具,但在使用它时需要注意以下几点:1. 设置退出条件确保为每个repeat循环设置了合适的退出条件,否则可能导致无限循环或者出现其他不可预料情况。
repeat的用法及搭配 (2)

repeat的用法及搭配一、repeat的基本用法repeat是一个常见的英语动词,其基本含义是“重复”或“再做一次”。
在日常生活中,我们经常使用repeat来表达某个动作、事件或任务的重复发生。
例如,当我们想要重复播放一首歌曲时,我们可以说:“Can you repeat that song?”二、repeat表示行为和动作的重复1. repeat后接名词:当repeat后接名词时,表示重复进行某项工作或行为。
例如,“He repeated the experiment several times to ensure accuracy.”(他进行了几次实验以确保准确性。
)2. repeat后接动词不定式:当repeat后接动词不定式时,表示重复做某事。
例如,“She repeated to herself that everything would be fine.”(她反复对自己说一切都会好起来的。
)3. repeat oneself:这是一个习语用法,表示某人反复讲同样的话或陈述同样的观点。
例如,“The teacher got annoyed because the studen t kept repeating himself during class.”(学生在上课期间反复讲同样的话使老师感到恼火。
)三、repeat与其他动词搭配1. repeat a phrase/sentence/word:表示重复某个短语/句子/单词。
例如,“Could you please repeat that sentence? I didn't catch it."(你能再说一遍那句话吗?我没有听清楚。
)2. repeat an action/task:表示重复某个动作/任务。
例如,“The coach asked the players to repeat the exercise three times.”(教练要求球员们将这个练习重复三次。
让繁琐变方便——重复命令REPEAT(一)

正五边形
5
360
360/5
用重复命令画正多边形的方法:
REPEAT 边数[FD 步长 LT(RT) 360/边数]
图形1:
REPEAT 18[FD 10 RT 360/36] 图形2:
1、REPEAT 36[FD 10 RT 360/36] 2、REPEAT 36[FD 10 LT 360/36]
REPEAT 3[FD 100 RT 360/3]
REPEAT 4[FD 100 RT 360/4]
72° 108°
72°
100步
REPEAT 5[FD 100 RT 360/5] REPEAT 5[FD 100 RT 72]
几何图形
正三角形 正四边形
边数
3 4
总共旋转 度数 360 360
每次旋转 旋转公式 角度 120 90 72 360/3 360/4
一复习1隐藏小海龟命令2显示小海龟命令3前进命令4后退命令5左转命令6右转命令二新课导入如果一个图形是用几个命令重复几次几十次上百次甚至上千次画成的要一遍遍地输入多麻烦啊
让繁琐变方便 ——重复命令REPEAT(一)
第一课时
授课人:周杰
小海龟常用命令
隐藏小海龟命令(
HT ) 显示小海龟命令( ST ) 前进命令( FD ) 后退命令( BK ) 左转命令( LT ) 右转命令( RT )
用重复命令(REPEAT)画出的图,总是每次 走的步数相同,转的角度相同。只要设置好下面三 个数,就可以正确使用重复命令:
重复的次数; 每次走的步数;
每次转动的角度。
100步
1、FD 2、FD 3、FD 4、FD
《REPEAT命令教案》小学信息技术人教 课标版 三年级起点六年级上册教案

一、REPEAT命令教案教学目标知识和技能:1.掌握重复命令repeat的书写格式。
2.弄懂重复命令repeat的执行过程。
3.掌握画正多边形的方法。
过程和方法:1.让学生通过参与本课的学习活动,初步学会小海龟画正多边形、画拱桥的方法。
2.让学生通过“画小桥”的活动,进一步理解用程序解决问题的基本思路。
情感态度和价值观:1.引导学生学好本领、用好本领、服务社会。
2.通过开展小组学习活动,使学生们从中体会合作交流好处,培养他们竞争与协作相融的学习氛围。
学情分析教学对象是小学五年级的学生,他们活泼好动、好奇心强,具有一定的自学能力。
经过前面几节课的学习,学生对PC Logo已经有一定的认识,并学会了Logo语言中前进、后退、左转、右转等15个基本的命令,能用命令来画一些简单的图形。
本节课只要能让学生亲身体验整个活动的过程,必能激发起他们强烈的求知欲,从而达到教学的目的。
重点难点教学重点:懂得用重复命令编写程序画出拱桥。
解决措施:采取以教师为主导,以学生操作为根本,以小组互助为主线的教学策略。
教学难点:运用所学命令将台阶和桥拱连接起来。
解决措施:先让学生回顾已学知识,利用教师给同学们准备好的学习卡,尝试做一做。
当学生出现困惑时,发挥小组的互助力量帮忙解决疑难,教师从中适当指导。
教学过程4.1 教学活动【导入】讲故事引出学习任务1.故事引入:上两节课我们帮助了小海龟画了它梦想家园,有屋子、绿树、河流。
今天,动物们准备来小海龟家里作客,可是前面有条小河,过不去,同学们,你们觉得该怎么做呢?对,那今天我们就来给小海龟建一座小桥,让大伙能开开心心过河。
2.板书课题。
3.出示任务:这节课我们要把小桥建出来,就要完成两大任务:(1)建台阶(2)建桥拱【讲授】小海龟画台阶1.课件出示桥的台阶部分(一个台阶)。
2.完成一个台阶的命令是怎么样的?请你把命令写在课本P99页的“细心想”的横线上。
出示命令:fd20rt90fd20lt903.图中有多少个台阶?(5个)总结:那就是要重复5次就能把五个台阶画出来了。
repeater控件用法(二)

repeater控件用法(二)repeater控件用法1. 什么是repeater控件?Repeater控件是一种用于在 Web表单中绑定数据的控件。
它允许我们以自定义的方式显示重复的数据源,构建灵活的用户界面。
2. repeater控件的基本结构Repeater控件由三个主要元素组成: - <HeaderTemplate>:定义重复区域的头部模板; - <ItemTemplate>:定义重复区域的项模板,用于显示每个数据项; - <FooterTemplate>:定义重复区域的尾部模板。
3. 使用repeater控件绑定数据使用repeater控件绑定数据的步骤如下: 1. 将数据源绑定到repeater控件; 2. 在ItemTemplate中定义每个数据项的显示方式;3. 在Page_Load事件中设置repeater控件的数据源并调用DataBind 方法。
4. repeater控件的常用属性•DataSource:设置或获取repeater的数据源;•HeaderTemplate:设置或获取repeater的头部模板;•ItemTemplate:设置或获取repeater的项模板;•FooterTemplate:设置或获取repeater的尾部模板;•SeparatorTemplate:设置或获取repeater的分隔符模板。
5. 使用repeater控件展示列表Repeater控件最常用的场景是展示列表。
我们可以将数据源中的每个项通过ItemTemplate进行展示,例如:<asp:Repeater ID="rptList" runat="server"><ItemTemplate><div><h3><%# Eval("Title") %></h3><p><%# Eval("Content") %></p></div></ItemTemplate></asp:Repeater>在上述示例中,我们使用ItemTemplate定义了一个适用于每个数据项的HTML结构,通过<%# Eval("FieldName") %>语法将数据源中的字段绑定到模板中。
六年级下册信息技术教案3重复命令川教版

六年级下册信息技术教案3重复命令川教版1教学目的1.知识与技艺:能用repeat命令画正方形、长方形、正多边形和圆;2.进程与方法:经过解说、探求、演示、协作练习、实际操作等方式,让先生学会小组协作学习,能具有发明性的学习和思索3.情感态度与价值观:在独立思索的基础上,同窗之间相互协作,养成积极进取的学习习气。
培育先生丰厚的想象力与发明力,促进先生自主、协作、探求才干的提高。
2学情剖析先生在前面的学习进程中已对LOGO言语有一定的了解,能停止编写复杂的命令。
小学六年级的先生具有一定的探求才干,能独立思索,但是思索还不是特别片面,还需求适当的加以提示。
六年级的先生对LOGO的学习积极很高,但是要和数学结合起来,关于数学基础不是特别好的,有一定难度。
3重点难点repeat重复命令的运用格式和功用;角度的计算 n>的学习积极很高,但是要和数学结合起来,关于数学基础不是特别好的,有一定难度。
4教学进程4.1 第一学时4.1.1教学活动活动1【导入】温习引入温习引入(8分钟)行进命令( )前进命令( )左转命令( )右转命令( )复位命令( )清屏命令( )抬笔命令( )落笔命令( )你会画正方形吗?请启动LOGO软件,画一个边长为80步的正方形。
(2分钟内完成)入手操作、演示。
让成功的先生将自己的命令写在黑板上。
2.先生动脑思索。
(3分钟)效果一:FD 80是正方形的什么?效果二:RT 90呢?效果三:细心观察屏幕上的命令,你发现它们有什么规律呢?相反的命令重复了4次。
要重复四次,那我们有没有什么方法让这个重复更精简点,只写一次。
请大家看到教材17页第三课«重复命令»活动2【讲授】新课学习操作义务一:请大家阅读教材17-18页内容,请大家自己用repeat重复命令再画一个边长为100步的正方形,要求:只能有一串命令哟!(5分钟)剖析画正方形重复执行的内容是什么?FD 80 RT 90重复命令的格式正方形 repeat 4 [FD 80 RT 90]重复命令的格式:REPEAT N[重复执行的命令]操作义务二:用重复命令画一个长100宽80的长方形,看看哪个组最快的全部完成呢?(4分钟)操作义务三:(5分钟)接上去请一二小组用重复命令画一个边长为80的正三角形三四小组用重复命令画一个边长为80的正五边形五六小组用重复命令画一个边长为80的正六边形七八小组用重复命令画一个边长为80的正八边形并思索,画正多边形有什么规律?小组内商量讨论,构成分歧意见向全班汇报。
六年级信息技术下册第3课重复命令课件2川教版

用repeat命令画下面图形
画多角星的通式:
REPEAT 角数[FD 线段长度 RT 180-180/角数] 转角度分为奇数与偶数: 奇数角: 180-180/角数 偶数角: 180-360/角数
动脑筋,画一画
⑴
⑵
⑹
⑶
⑺
⑻
⑽
三、画出奇妙的图案
在重复命令中,还可以再使用重复命令,这叫做重复命令 的嵌套。Logo语言允许重复命令嵌套多层,即一级一级地嵌套 下去,但最经常使用的是二级嵌套与三级嵌套。为绘制各种复 杂的有规律的组合图形提供非常简单方法,这就是嵌套的魅力。
【知识点】:重复命令嵌套 [命令格式]:重复命令1[重复命令2[重复命令3[…]…]…] [执行方式]:先执行最里层,后逐级向外执行。
以正方形作为基本图形为例,分析下列四种组合图形的规律性。
⑴
1
⑵
2
2
1
⑶
2
1
⑷
▲
动脑筋,画一画
⑴
⑵
⑶
⑷
⑸
⑹
⑺
ห้องสมุดไป่ตู้
神奇的重复命令
六年级下册 第四课
L O G O语言程序设计
复习
• 重复命令的格式 • Repeat _重__复__次_数___[ 重复的命令 ]
任务一
画出一个长50步,宽100步的长方形。
重复的命令: fd 50 rt 90 fd 100 rt 90
50
Repeat 2[fd 501r0t090 fd 100 rt 90]
Ericsson Vehicular Repeater 操作指南说明书

Operator’s ManualVRCV ehicular RepeatereNOTICE!This manual covers Ericsson and General Electric products manufactured and sold by Ericsson Inc.NOTE!Repairs to this equipment should be made only by an author-ized service technician or facility designated by the supplier. Any repairs, alterations or substitution of recommended parts made by the user to this equipment not approved by theThis manual is published by Ericsson Inc., without any warranty. Improvements and changes to this manual necessitated by typographical errors, inaccuracies of current information, or improvements to programs and/or equipment, may be made by Ericsson Inc., at any time and without notice. Such changes will be incorporated into new editions of this manual. No part of this manual may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying and recording, for any purpose, without the express written permission of Ericsson Inc. Copyright © January 1997, Ericsson Inc.2SAFETY INFORMATIONThe operator of any mobile radio should be aware of certain hazards common to the operation of vehicular radio transmis-sions.A list of the possible hazards are:1. Explosive AtmospheresJust as it is dangerous to fuel a vehicle with the motor running, be sure to turn the radio off while fueling the vehicle. Do Not carry containers of fuel in the trunk of the vehicle when the radio is mounted in the trunk.2. Interference To V ehicular Electronic SystemsElectronic fuel injection systems, electronic anti-skid breaking sys-tems, electronic cruise control systems, etc., are typical of the types of electronic devices that may malfunction due to the lack of protec-tion from radio frequency energy present when transmitting. If the vehicle contains such equipment, consult the dealer for the make of vehicle and enlist his aid in determining if such elec-tronic circuits perform normally when the radio is trans-mitting.3. Dynamite Blasting CapsDynamite blasting caps may be caused to explode by operating a radio within 500 feet of the blasting caps.Always obey the "Turn Off Two Way Radio" signs posted where dynamite is being used. When transporting blasting caps in your vehicle:a. Carry the blasting caps in a closed metal box with a softlining.b.Leave the radio OFF whenever the blasting caps arebeing put into or removed from the vehicle.4.Radio Frequency EnergyTo prevent burns or related physical injury from radio frequency energy, do not operate the transmitter when anyone outside of the vehicle is within two feet of the antenna.3OPERATING RULES AND REGULATIONSTwo way FM radio systems must be operated in accordance with the rules and regulations of the Federal Communications Commission (FCC). As an operator of two way radio equip-ment, the user must be thoroughly familiar with the rules that apply to the intended type of radio operation. Following these rules will help to eliminate confusion, assure the most efficient use of existing radio channels, and result in a smoothly func-tioning radio network.When using the radio, remember these rules:1.It is a violation of FCC rules to interrupt any distress oremergency mes-sage. In conventional mode the radio operates in much the same way as a telephone "party line"therefore always listen to make sure that the line is clear--that no one else is on the air--before sending any messages. If someone is sending an emergency message--such as reporting a fire or asking for help in an accident--KEEP OFF THE AIR!2. Use of profane or obscene language is prohibited byFederal Law.3. It is against the law to send false call letters or a falsedistress or emergency message.4. The FCC requires that conversations be brief and con-fined to business. To save time, use coded messages whenever possible.5. Using the radio to send personal messages (except in anemergency) is a violation of FCC rules. Only those messages essential for the business operation may be sent.46. It is against the Federal law to repeat or otherwise makeknown anything overheard on the radio. Conversations between others sharing a commu-nications channel must be regarded as confidential.7. The FCC also requires that the caller be identified atcertain specific times by means of call letters. Refer to the rules that apply to the particu-lar type of operation for the proper procedure.8. No changes or adjustment shall be made to the equipmentexcept by an authorized or certified electronic technician.5INTRODUCTIONThe V ehicular Repeater Conventional (VRC) is designed to provide extended portable communication coverage by re-peating conventional transmissions in both directions through an existing high power mobile radio.VRC FRONT PANELThe VRC has 8 LEDs to indicate the current status of the repeater:CPU Flashes at approximately 1 Hz rate to indicate proper operation of the on-board microprocessor.Pri Indicates the VRC is at priority status and will repeat all transmissions, portable-to-base and base-to-port-able. In multi-vehicle applications, only one VRC/mo-bile combination should be at priority status. RepeaterCor Indicates the VRC is receiving carrier from a portable (Tone LED should also be on) or another VRC (CorLED only).Tone Indicates the VRC is decoding the sub-audible tone from the portable. This LED should only be on whenthe repeater Cor LED is also on. If the VRC is thepriority unit, the Mob ile Tx LED should also be on. TX Indicates when the VRC is transmitting to the portable. 6Mob ileCor Indicates that a transmission is being received by the mobile. If the Pri LED is on, the Rptr Tx LED shouldalso be on. If the VRC is not at priority status, the RptrCor LED should be on indicating that another repeateris handling the transmission.Tx Indicates when the mobile is being keyed by the VRC. Opt The option LED is used to indicate program mode (on steady) when the programming cable is inserted, or toindicate a problem with the radio portion of the repeater(rapid flashing). If the Opt LED flashes at approxi-mately 10 Hz rate, contact your service center.FUNCTIONAL DESCRIPTIONGenerally, vehicular repeaters are used as mobile extenders in cross-band operation; i.e., the link is UHF and the mobile is low band or VHF. In-band operation is possible, but care must be taken to prevent interference between the mobile’s higher power transmitter and the repeater receiver. Proper frequency selection and antenna placement are important even in cross-band operation, but especially for in-band use. The use of low power pre-selector cavities may be placed in line with the repeater antenna cable since it is simplex and low power.The VRC operates on UHF simplex frequencies. The port-ables must transmit with a CTCSS tone, but should be carrier squelch receive, or use a decode tone different from the trans-mit tone. Part of the multi-vehicle format dictates that all of the VRCs must be able to monitor all link traffic on site and be able to determine if a portable is transmitting, or if other repeaters are transmitting.7When the user leaves the vehicle, the VRC must be acti-vated via the mobile radio front panel or a separate switch. When the mobile radio is receiving carrier and proper CTCSS tone, the VRC will begin transmitting on the portable’s receive frequency. The user is able to hear and respond to all radio messages, including other portables at the site. The VRC can be programmed to give the portables priority in a conversation by periodically sampling for portable activity (carrier and proper CTCSS tone) during base-to-portable transmissions. During sampling, if the VRC detects a portable transmission, it will cease transmission, key the mobile radio and repeat portable-to-base. This allows the portable to respond during repeater hang time or during full duplex interconnect calls. Priority sampling can be enabled/disabled through PC pro-gramming and interval can be programmed between 0.25 seconds and 2.5 seconds in 0.25 second increments.The VRC has a fixed 3-minute time out timer for base-to-portable transmissions. If the mobile carrier operated relay (COR) is active for more than 3 minutes (and the VRC is the priority unit), the VRC will send a double blip and cease transmission until the mobile COR is inactive. The 3 minute time-out is in effect regardless of whether the VRC is pro-grammed for priority sampling or not.MULTI-VEHICLE OPERATIONWhen the VRC is first activated, it will transmit a short “lock tone” that alerts the user that the system is functioning. It will then assume the priority status and be ready to repeat any base-to-portable or portable-to-base transmissions. If an-other unit arrives on scene and is activated, it too will transmit a “lock tone”. When the first VRC detects the “lock tone” from the second unit, it will increment a “priority counter” and will 8no longer repeat any transmissions. The recently arrived unit will be the priority repeater, and the first unit will be one count away from priority. This process will continue for each unit that arrives at the scene, creating a priority hierarchy for up to 256 vehicles, each with a unique count and only one unit at priority status. The VRC will not transmit it’s “lock tone’ if the radio channel is busy when first enabled. It will wait in non-priority status until all transmissions cease, then send it’s “lock tone” and become the priority unit.Even though the other VRCs are not a priority status, they will continue to monitor the channel for activity. If the priority unit leaves the scene or becomes disabled, the other units will detect the condition to repeat and determine that there is no priority unit repeating the transmission. They will then begin decrementing their priority counters until one of them reaches the priority status and begins repeating the transmission. Since the VRCs are all at different counts, only one will reach priority status and begin transmitting. The other units will sense the new priority repeater and cease counting down, preserving the priority hierarchy.If another unit were to arrive from a different scene and it is still the active priority, there will be two active repeaters on the air when a condition to repeat exists. When one of the VRCs unkeys to check for portable activity, it will detect the presence of the other active VRC and increment it’s priority counter and cease transmission. This is the self-clearing mode to prevent radio interference.If the portable user is out of the vehicle and the mobile radio is keyed by the local microphone inside the vehicle, the VRC will detect the local PTT and repeat the transmission to the other portables so that both sides of the conversation will be9heard by everyone on the link. The local microphone repeat function can be enabled/disabled via PC programming.The VRC also has a local receive audio speaker jack that enables a person inside the vehicle to monitor portable-to-base transmissions that are being repeated through the mobile.If portable users wish to communicate portable-to-portable without accessing the mobile VRC repeater, the may transmit on the same frequency without CTCSS tone (or a different CTCSS tone); the VRC only responds to carrier and proper CTCSS tone from the portables.10LIMITED W ARRANTY AND REPAIR INFORMATIONAn explanation of the Limited Warranty’s benefits and exclusions follows.What does your warranty cover?•Any defect in material or workmanship.For how long after the original purchase of the equipment?•One (1) year.What will we do?•Repair your Equipment or provide you with a new or, at our option, a reconditioned unit in the event repairs cannot be made.•The exchange unit (repaired or replacement) is warranted for the remainder of your product’s original one (1) year warranty period.How do you make a warranty claim?•Contact Pyramid Communications [(714) 901-5462] for return authorization prior to returning any defective merchandise.•Properly pack your unit. Include any cables and other parts and accessories which were originally provided with the product. We recommend using the original carton and packing materials.•Include in the package your name and address, a description of the defect and a copy of the sales receipt or other evidence of date of original purchase.•Ship the unit standard UPS or equivalent to:Pyramid Communications5142 Bolsa Avenue #103Huntington Beach, CA 92649•Pay any charges billed to you for service not covered by the warranty. Returned units that are out of warranty shall be deemed as authorization for repair and sender shall be responsible for all reasonable repair costs.•The repaired (or new or reconditioned) unit will be shipped to you prepaid freight.What does your warranty not cover?•Customer instruction. Y our Operator’s Manual provides information regarding operating instruc-tions and user controls. For additional information, ask your dealer.•Any labor charges incurred in removal/installation of defective or repaired units.•Installation and set-up service adjustments.•Damage from misuse or neglect.•Products which have been modified or incorporated into other products.•Products purchased or serviced outside the USA.•Changes that provide improvements or enhance performance.•Any unit which is not new when sold to the first end user or unit whose serial number has been altered or removed.•Damage or loss occurring during shipment (claims must be presented to the carrier) or shipping charges to return defective units for repair.11Ericsson Inc.Private Radio SystemsMountain View RoadLynchburg, Virginia 24502AE/LZT 123 3245 /1 R1A 1-800-592-7711 (Outside USA, 804-592-7711)Printed in U.S.A.。
repeat的用法和固定搭配

repeat的用法和固定搭配一、repeat的基本用法在英语中,repeat是一个常见的动词,它表示重复或再次做某事。
当我们想要表达某个动作发生多次时,可以使用repeat来描述。
下面将详细介绍repeat的基本用法以及一些固定搭配。
1. repeat用作及物动词在这种情况下,repeat后面通常跟着一个名词、代词或动词不定式,用于说明需要重复的内容。
例如:- He repeated the instructions to the students.(他对学生们再次重复了指示。
)- Could you repeat that, please?(你能再说一遍吗?)2. repeat用作不及物动词此时,repeat后面通常跟随介词或副词短语,并且不需要宾语。
它意味着进行某个动作的重复。
例如:- The song repeated over and over again.(那首歌一遍又一遍地重复播放。
)- The alarm keeps repeating every five minutes.(警报每五分钟就会响一次。
)二、固定搭配1. repeat oneself这个搭配中,oneself是反身代词形式,意为“自己”。
当一个人再次说同样的话或表达相同的观点时使用这个固定短语。
例如:- He tends to repeat himself when he's tired.(他累了的时候,容易重复说同样的话。
)2. repeat after someone这个搭配常用于教育或学习过程中,表示一个人跟随另一个人的发言或声音来进行模仿。
例如:- Repeat after me: "I can do it!"(跟着我说:“我能做到!”)3. repeat history这个搭配常用于指某个事件或行为在不同时间或地点重复发生。
它暗示了对历史循环性的警醒。
例如:- If we don't learn from our mistakes, we are doomed to repeat history.(如果我们不能从错误中吸取教训,我们注定会重演历史。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
The purpose of the RepeatScout software is to identify repeat familysequences from genomes where hand-curated repeat databases (a laRepBase update) are not available. In fact, the output of this programcan be used as input to RepeatMasker as a way of automatically maskingnewly-sequenced genomes.Included in this package is a more or less arbitrary 3 Mb of the human X chromosome for your testing and debugging purposes.Wget下载,tar -xzvf解压,直接进入make一下即可,安装后文件如下Running RepeatScout proceeds in four phases.First, build_lmer_table creates a file that tabulates the frequency of all l-mers in these quence to be analyzed.第一步:用build_lmer_table命令把整个基因组生成一个频率表格,把所有有过重复的kmer都找出来,EG:build_lmer_table -sequence genome.fa -freq genome.fqSecond, RepeatScout takes this table and the sequence and produces a fasta file that contains all the repetitive elements that it could find.第二步:用RepeatScout 这个命令根据生成的频率表格和基因组序列产生一个包含有所有的能找到的重复元件的文件。
EG:RepeatScout -sequence genome.fa -output repeats.fa -freq genome.fqThird, the "filter-stage-1.prl" scriptis run on the output of RepeatScout to remove low-complexity and tandem elements;RepeatMasker is run on the sequence of interest using this filtered RepeatScout library.The program "filter-stage-2.prl"then filters out any repeat element that does not appear a certain number of times (by default, 10).第三步:用filter-stage-1.prl这个脚本过滤掉低复杂度和串联重复元件。
EG:filter-stage-1.prl repeats.fa &> repeats.fa.filter_1Finally, the locations of the repeats found by RepeatMasker are used, in conjuction with GFF files that describe segmental duplications or exons or other such "uninteresting" regions to remove sequences from the library that are likely to not be mobile elements; the program "compare-out-to-gff.prl" does exactly this.第四步Repeat ScoutRepeatScout is run in several steps using several different programs. This tutorial assumes you are using a fasta file named genome.fa you could, however, use any fasta file simple replacing genome.fa with the name of the fasta file being used. Also, running any of the listed commands without any additional input (or running them with the -h option) will display the help message including a description of the program, the usage, and all options.1) Count every 12 base pair sequence in the genome, and when it is done, look at the result file:$build_lmer_table -sequence genome.fa -freq genome.freq $less genome.freq2) Extend the 12 base pair sequences to form initial consensus repeats. This will take a minute.$RepeatScout -sequence genome.fa -freq genome.freq -output genome.firstrepeats.lib3) Filter out simple and tandem repeats from the library$cat genome.firstrepeats.lib | filter-stage-1.prl > genome.filtered.lib4) Count the number of times each repeat in the filtered library appears. The output file genome.fa.out will contain a column with the count of each repeat type.$RepeatMasker -pa [number of processors] -libgenome.filtered.lib -nolow genome.fa*The -pa option tells RepeatMasker how many processors to use. If you are only using one processor, do not use the -pa option. If you were running RepeatMasker on 3 processors, you would use -pa 3.**The -nolow option stops RepeatMasker from masking low complexity repeats. Since we are only concerned with the number of times the generated repeats appear, masking low complexity repeats simply adds more time.5) Filter out any repeats that appear less than 10 times. Note, because our sample data is so small, not much will be repeated 10 times, so check the results when the first command finishes.$cat genome.filtered.lib | filter-stage-2.prl --cat genome.fa.out > genome.secondfilter.lib$Less genome.secondfilter.lib6) Mask the genome file with the new repeat library.$RepeatMasker -pa [number of processors] -libgenome.secondfilter.lib genome.fa7) To view the length and frequency of repeats, open genome.fa.tbl. Total length, %GC, and percent bases masked (i.e. percent in repeats) are shown. Also shown are number and total length occupied by different kinds of repeats.8) Because the repeat library made from our five scaffolds was so small, it is possible that the repeat library from step 5 and 7 is blank. Run RepeatMasker again using repeats found in all species so far. These are already in the virtual machine.$RepeatMasker -pa [number of processors] -species all genome.fa。