纳米材料粒度分析
纳米材料的粒度分析与形貌分析

随着粒度的减小,纳米材料的磁矫顽力通常会增加,这是因为小尺寸效应增强了磁畴壁的稳定性。此外,形貌对 纳米材料的磁各向异性也有重要影响,可以通过改变形貌来优化磁存储和磁传感器等应用中的性能。
THANKS FOR WATCHING
感谢您的观看
原料性质
原料纯度
原料中的杂质会影响纳米材料的生长过程,从而影响其粒度和形貌。高纯度的原料有利于获得粒度和形貌均匀的纳米 材料。
原料晶型
不同晶型的原料会生成不同结构和形貌的纳米材料。例如,采用硫化物原料制备的纳米材料多为立方状或球状,而采 用氧化物原料制备的纳米材料多为棒状或纤维状。
原料粒度
原料的粒度大小直接影响最终纳米材料的粒度和形貌。采用纳米级原料作为起始物,可以获得更小粒度 的纳米材料,同时也有利于保持形貌的规整性。
按维度分类
根据在纳米尺度上的维度数,纳米材 料可分为零维(0D)、一维(1D) 、二维(2D)等类型的纳米材料。
CHAPTER 02
粒度分析
粒度分析方法
激光散射法
利用激光照射纳米材料,通过 散射光的强度和分布测量粒径
。
小角X射线散射法
利用X射线照射纳米材料,通过 散射的X射线强度和角度测量粒 径。
纳米材料的粒度分析 与形貌分析
目 录
• 纳米材料的基本概念 • 粒度分析 • 形貌分析 • 纳米材料粒度与形貌的影响因素 • 粒度与形貌对纳米材料性能的影响
CHAPTER 01
纳米材料的基本概念
纳米材料定义
01
纳米材料是指在三维空间中至少 有一维处于纳米尺度范围(1100nm)或由它们作为基本单元 构成的材料。
量的重要手段。
粒度分析促进纳米科技发展
03
纳米材料粒度测试方法大全

纳米材料粒度测试方法大全目前,纳米材料已成为材料研发以及产业化最基本的构成部分,其中纳米材料的粒度则是其最重要的表征参数之一。
本文根据不同的测试原理阐述了8种纳米材料粒度测试方法,并分析了不同粒度测试方法的优缺点及适用范围。
1.电子显微镜法电子显微镜法是对纳米材料尺寸、形貌、表面结构和微区化学成分研究最常用的方法,一般包括扫描电子显微镜法(SEM)和透射电子显微镜法(TEM)。
对于很小的颗粒粒径,特别是仅由几个原子组成的团簇,采用扫描隧道电镜进行测量。
计算电镜所测量的粒度主要采用交叉法、最大交叉长度平均值法、粒径分布图法等。
优点:该方法是一种颗粒度观测的绝对方法,因而具有可靠性和直观性。
缺点:测量结果缺乏整体统计性;滴样前必须做超声波分散;对一些不耐强电子束轰击的纳米颗粒样品较难得到准确的结果。
2.激光粒度分析法激光粒度分析法是基于Fraunhofer衍射和Mie氏散射理论,根据激光照射到颗粒后,颗粒能使激光产生衍射或散射的现象来测试粒度分布的。
因此相应的激光粒度分析仪分为激光衍射式和激光动态散射式两类。
一般衍射式粒度仪适于对粒度在5μm以上的样品分析,而动态激光散射仪则对粒度在5μm以下的纳米、亚微米颗粒样品分析较为准确。
所以纳米粒子的测量一般采用动态激光散射仪。
优点:样品用量少、自动化程度高、重复性好, 可在线分析等。
缺点:不能分析高浓度的粒度及粒度分布,分析过程中需要稀释,从而带来一定误差。
3.动态光散射法动态光散射也称光子相关光谱,是通过测量样品散射光强度的起伏变化得出样品的平均粒径及粒径分布。
液体中纳米粒子以布朗运动为主,其运动速度取决于粒径、温度和黏度系数等因素。
在恒定温度和黏度条件下, 通过光子相关谱法测定颗粒的扩散系数就可获得颗粒的粒度分布,其适用于工业化产品粒径的检测,测量粒径范围为1nm~5μm的悬浮液。
优点:速度快,可获得精确的粒径分布。
缺点:结果受样品的粒度大小以及分布影响较大,只适用于测量粒度分布较窄的颗粒样品;测试中应不发生明显的团聚和快速沉降现象。
《纳米材料吸附热力学和动力学的粒度效应》范文

《纳米材料吸附热力学和动力学的粒度效应》篇一一、引言纳米材料因其独特的物理和化学性质,在众多领域中得到了广泛的应用。
其中,纳米材料的吸附性能是其重要的应用之一。
纳米材料的粒度对其吸附性能具有显著的影响,这既体现在热力学方面,也体现在动力学方面。
本文将就纳米材料吸附热力学和动力学的粒度效应进行详细的探讨。
二、纳米材料吸附热力学的粒度效应1. 吸附热与粒度的关系纳米材料的粒度对其吸附热具有显著影响。
一般来说,粒度越小,吸附热越大。
这是因为小粒度的纳米材料具有更高的比表面积,可以提供更多的吸附位点,使得吸附过程需要克服的能量障碍增加,从而提高了吸附热。
2. 吸附等温线的变化纳米材料的粒度还会影响其吸附等温线的形状。
随着粒度的减小,纳米材料的吸附等温线可能会从L型向S型转变。
这是因为小粒度的纳米材料具有更高的比表面积和更强的吸附能力,使得其在低浓度下的吸附量增大,进而改变等温线的形状。
三、纳米材料吸附动力学的粒度效应1. 吸附速率与粒度的关系纳米材料的粒度对其吸附速率具有显著影响。
一般来说,粒度越小,吸附速率越快。
这是因为小粒度的纳米材料具有更高的比表面积,可以提供更多的活性位点,使得吸附过程更容易进行。
此外,小粒度的纳米材料还具有更好的扩散性能,有利于吸附质在材料内部的扩散。
2. 粒度对吸附机理的影响纳米材料的粒度还会影响其吸附机理。
小粒度的纳米材料通常具有更高的表面能,使得其与吸附质之间的相互作用更强。
此外,小粒度的纳米材料还可能具有更多的缺陷和空位,这些缺陷和空位可以提供更多的吸附位点,从而影响吸附过程。
四、实验研究及结果分析为了验证上述理论,我们进行了一系列实验研究。
通过改变纳米材料的粒度,我们观察了其对吸附热和吸附速率的影响。
实验结果表明,随着粒度的减小,吸附热和吸附速率均有所增加。
此外,我们还发现小粒度的纳米材料在低浓度下的吸附量更大,这与上述理论相符。
五、结论本文探讨了纳米材料吸附热力学和动力学的粒度效应。
纳米材料的测试与表征-精选文档

光散射法粒度分析
• 测量范围广,现在最先进的激光光散射粒度 测试仪可以测量1nm~3000μm,基本满足 了超细粉体技术的要求 • 测定速度快,自动化程度高,操作简单,一般 只需1~1.5min • 测量准确,重现性好
• 可以获得粒度分布
激光相干光谱粒度分析法
• 通过光子相关光谱(PCS)法,可以测量粒子的 迁移速率。而液体中的纳米颗粒以布朗运动为主, 其运动速度取决于粒径,温度和粘度等因素。在 恒定的温度和粘度条件下,通过光子相关光谱 (PCS)法测定颗粒的迁移速率就可以获得相应 的颗粒粒度分布 • 光子相关光谱(PCS)技术能够测量粒度度为纳 米量级的悬浮物粒子,它在纳米材料,生物工程、 药物学以及微生物领域有广泛的应用前景
高分子纳米微球研究
沉降法粒度分析
沉降法的原理是基于颗粒在悬浮体系时,颗粒本 身重力(或所受离心力)、所受浮力和黏滞阻力三 者平衡,并且黏滞力服从斯托克斯定律 (F=6πrηv)来实施测定的,此时颗粒在悬浮体 系中以恒定速度沉降,且沉降速度与粒度大小的 平方成正比 重力沉降: 2~100μm的颗粒
• HRTEM是观察材料微观结构的方法。不仅 可以获得晶包排列的信息,还可以确定晶 胞中原子的位置。 • 200KV的TEM点分辨率为0.2nm,1000KV 的TEM点分辨率为0.1nm。 • 可以直接观察原子象
扫描探针显微镜(SPM)
• 扫描探针显微镜(Scanning Probe Microscope,SPM)是扫描隧道显微镜 (STM)及在扫描隧道显微镜的基础上发展 起来的各种新型探针显微镜(原子力显微 镜AFM,激光力显微镜LFM,磁力显微镜 MFM等等)的统称
• 对于不同原理的粒度分析仪器,所依据的测量原理不同, 其颗粒特性也不相同,只能进行等效对比,不能进行横向 直接对比。
纳米材料粒径评估方法

纳米材料粒径评估方法纳米材料是一种具有特殊结构和性质的材料,其粒径在纳米级别(1纳米=10^-9米)范围内。
粒径评估是对纳米材料进行表征和评价的重要方法之一,可以揭示纳米材料的尺寸分布、形貌特征以及与其他性质之间的关联。
本文将介绍几种常用的纳米材料粒径评估方法。
一、透射电子显微镜(Transmission Electron Microscope, TEM)透射电子显微镜是一种通过电子束穿透样品并形成显微图像的仪器。
利用TEM可以直接观察纳米材料的形貌和尺寸分布。
通过在TEM 中观察纳米材料的投影图像,可以测量出颗粒的直径,并进一步分析颗粒的尺寸分布情况。
二、扫描电子显微镜(Scanning Electron Microscope, SEM)扫描电子显微镜是一种利用电子束和样品表面相互作用产生信号来形成显微图像的仪器。
SEM可以对纳米材料进行表面形貌观察和尺寸评估。
通过SEM观察到的纳米材料表面形貌图像,可以通过测量颗粒的直径或者利用图像处理软件进行粒径分析。
三、动态光散射(Dynamic Light Scattering, DLS)动态光散射是一种利用激光束照射样品,测量散射光强度随时间的变化来评估颗粒粒径的方法。
纳米材料在DLS仪器中受到激光的照射后,颗粒会不断自发地进行热运动,散射出的光会随时间变化。
通过分析散射光强度的自相关函数,可以得到纳米颗粒的尺寸分布。
四、X射线衍射(X-ray Diffraction, XRD)X射线衍射是一种通过测量样品对入射X射线的衍射来确定晶体结构和晶格常数的方法。
对于纳米材料,XRD可以用来确定其结晶性质和晶体尺寸。
通过计算衍射峰的位置和强度,可以得到纳米材料的晶体尺寸。
五、原子力显微镜(Atomic Force Microscope, AFM)原子力显微镜是一种利用探针对样品表面进行扫描,并通过探针与样品之间的相互作用力来获得样品表面形貌和粒径信息的仪器。
纳米粒度_实验报告(3篇)

第1篇一、实验目的1. 了解纳米粒度仪的基本原理和操作方法。
2. 学习纳米粒度分析在材料科学、生物医学等领域的应用。
3. 通过实验,掌握纳米颗粒粒径和分布的测量方法。
二、实验原理纳米粒度仪是一种基于动态光散射(DLS)原理的仪器,通过测量颗粒在液体中布朗运动的速度,从而确定颗粒的大小和分布。
实验过程中,激光照射到悬浮颗粒上,颗粒对光产生散射,散射光经过光学系统被探测器接收,通过分析散射光的时间变化,可以得到颗粒的粒径和分布信息。
三、实验仪器与试剂1. 仪器:纳米粒度仪、激光光源、样品池、计算机等。
2. 试剂:纳米颗粒悬浮液、分散剂、滤纸等。
四、实验步骤1. 样品准备:将纳米颗粒悬浮液用滤纸过滤,去除杂质,确保样品的纯净度。
2. 仪器设置:打开纳米粒度仪,调整激光光源、样品池等参数,使仪器处于正常工作状态。
3. 样品测量:将处理好的纳米颗粒悬浮液注入样品池,设定测量时间,启动仪器进行测量。
4. 数据处理:将测量得到的数据导入计算机,利用纳米粒度仪自带软件进行数据处理,得到粒径和分布信息。
5. 结果分析:根据实验结果,分析纳米颗粒的粒径分布、平均粒径等参数,并与理论值进行对比。
五、实验结果与分析1. 纳米颗粒粒径分布:实验测得纳米颗粒的粒径分布如图1所示。
从图中可以看出,纳米颗粒的粒径主要集中在20-50nm范围内,符合实验预期。
图1 纳米颗粒粒径分布2. 纳米颗粒平均粒径:根据实验结果,纳米颗粒的平均粒径为30.5nm,与理论值相符。
3. 纳米颗粒分散性:实验测得纳米颗粒的分散性较好,说明样品在制备过程中未发生团聚现象。
六、实验讨论1. 实验过程中,纳米颗粒的粒径分布和平均粒径与理论值相符,说明实验方法可靠,仪器性能稳定。
2. 实验结果表明,纳米颗粒的分散性较好,有利于其在材料科学、生物医学等领域的应用。
3. 在实验过程中,应注意样品的制备和仪器操作,以保证实验结果的准确性。
七、结论本次实验成功测量了纳米颗粒的粒径和分布,验证了纳米粒度仪在材料科学、生物医学等领域的应用价值。
纳米材料的粒度分析与形貌分析(ppt 42页)

2.2 形貌分析的主要方法
• 扫描电子显微镜(SEM) • 透射电子显微镜(TEM) • 扫描隧道显微镜(STM) • 原子力显微镜(AFM)
3. 成分分析
3.1 成分分析方法与范围
类型(对象): 微量样品分析和痕量成分分析
取样量
待测成分的含量
(分析目的): 体相元素成分分析 表面成分分析 微区成分分析等方法
第六章 纳米材料的表征与检测技术
• 成分分析 • 形貌分析 • 粒度分析 • 结构分析 • 表面界面分析
1. 纳米材料的粒度分析
1.1 粒度分析的概念
①晶粒:是指单晶颗粒,即颗粒内为单相,无晶界。 ②一次颗粒:是指含有低气孔率的一种独立的粒子。
③团聚体:是由一次颗粒通过表面力或固体桥键作用而形成的更 大的颗粒。团聚体内含有相互连接的气孔网络。团聚体可分为硬 团聚体和软团聚体两种,团聚体的形成过程使体系能量下降。
1.2. 粒度分析的种类和适用范围
• 筛分法、显微镜法、沉降法 • 激光衍射法、激光散射法、光子相干光谱
法、电子显微镜图像分析法、基于布朗运 动的粒度测量法和质谱法
其中激光散射法和光子相干光谱法由于具有速度快、测量范 围广、数据可靠、重复性好、自动化程度高、便于在线测量 等测量而被广泛应用。
其测量颗粒最小粒径可以达到20nm和1nm。
• 体相元素组成分析方法: 原子吸收、原子发射、ICP质谱(破坏性) X射线荧光与衍射分析方法 (非破坏性)
3.2 X射线荧光光谱分析方法(XFS)
• 原理: X射线荧光的能量或波长是特征性的, 与元素有一一对应关系。
• 用途:定性和半定量
• 表面分析方法: X射线光电子能谱(XPS)分析方法 俄歇电子能谱(AES)分析方法 电子衍射分析方法 二次离子质谱(SIMS)分析方法等
纳米材料粒度测试方法大全

纳米材料粒度测试方法大全纳米材料粒度测试是纳米材料研究和应用中非常重要的一项工作,通过准确测量纳米材料的粒度可以了解其物理性质和化学性质,为纳米材料的合成、应用和性能优化提供数据支持。
下面将介绍几种常用的纳米材料粒度测试方法。
1.扫描电子显微镜(SEM):SEM是一种通过扫描纳米材料表面的高能电子束来观察和测量纳米材料粒度的方法。
该方法具有分辨率高、测量精度高、对纳米材料样品无需特殊处理等特点。
通过SEM观察到的纳米材料外观图像可以用于测量粒径、形貌和分布等参数。
2.透射电子显微镜(TEM):TEM是一种通过透射电子束观察纳米材料内部结构的方法,也可用于测量纳米材料的粒度。
TEM具有高分辨率,可以观察到纳米尺度的细节。
通过对TEM图像的分析,可以根据纳米材料的投影面积和长度等参数来计算纳米材料的粒径。
3.动态光散射(DLS):DLS是一种通过检测纳米材料颗粒在溶液中的布朗运动来测量纳米材料粒度的方法。
它利用激光束照射纳米颗粒溶液,测量散射光的强度和角度分布,从而得到纳米材料的尺寸分布。
DLS具有非接触式测量、快速、方便等特点,适用于纳米材料的溶液或悬浮液样品。
4.X射线衍射(XRD):XRD是一种通过测量材料晶体的衍射角度来确定晶体结构和晶粒尺寸的方法。
对于具有晶体结构的纳米材料,可以通过XRD图谱的峰宽来估算晶粒尺寸。
XRD具有无损测量、精度高等特点,适用于晶体结构明确的纳米材料。
5.傅里叶红外光谱(FTIR):FTIR是一种通过测量纳米材料在红外波段的吸收光谱来研究纳米材料结构和成分的方法。
纳米材料的粒度也可以通过红外吸收峰的强度和位置进行定性和定量分析。
FTIR具有所需样品量少、分辨率高等特点,适用于纳米材料的表面分析和组成分析。
6.水中悬浮液测定法:将纳米材料置于水中制备悬浮液,通过测量悬浮液的光学性质如透光率等,可以间接测得纳米材料的粒度。
该方法操作简单、快速,可用于大量样品的测量。
7.气相吸附法:纳米材料的比表面积可以通过气相吸附法来测量。
《纳米粒度分析》课件

优势
纳米粒度分析提供了快速、 准确的粒度数据。
局限性
纳米粒度分析受样品性质 和仪器限制。
Байду номын сангаас
发展趋势
纳米粒度分析将越来越广 泛应用于多个行业和领域。
参考文献
列出本课件中所引用的相关研究和文献。
讨论样品制备、仪器选择、数据分析和常见误差及排除方法。
1 样品制备
2 仪器选择
样品制备对纳米粒度分析结果有重要影响。
合适的仪器选择是确保准确分析的关键。
3 数据分析
4 常见误差及排除方法
正确的数据解析是得出可靠结果的前提。
介绍常见的误差来源和排除方法。
总结
概述纳米粒度分析的优势、局限性和未来发展趋势。
应用
纳米粒度分析广泛应用于材料科学、化学、 生物医学等领域。
纳米粒度分析方法
介绍纳米粒度分析的三种常见方法。
1
动态光散射(DLS)
通过测量光散射来分析纳米颗粒的尺寸和分布。
2
静态光散射(SLS)
使用静态光散射技术来获得纳米颗粒的尺寸数据。
3
激光粒度仪
利用激光光散射原理进行粒度分析的仪器。
DLS技术
DLS技术的原理、实验流程和数据分析。
原理
DLS利用光散射的强度和 频率变化来分析颗粒的尺 寸分布。
实验流程
包括样品制备、仪器设置 和数据采集。
数据分析
使用相关函数等方法解析 DLS测量数据。
SLS技术
SLS技术的原理、实验流程和数据分析。
原理
通过测量散射光强度的变化来分析纳米颗粒的尺寸。
实验流程
包括样品制备、仪器设置和数据采集。
数据分析
使用散射理论和数据拟合等方法进行数据分析和粒度计算。
纳米材料的测试与表征

高分子纳米微球研究
沉降法粒度分析
沉降法的原理是基于颗粒在悬浮体系时,颗粒本 身重力(或所受离心力)、所受浮力和黏滞阻力三 者平衡,并且黏滞力服从斯托克斯定律
(F=6πrηv)来实施测定的,此时颗粒在悬浮体
• STM通常被认为是测量表面原子结构的工具,具 有直接测量原子间距的分辨率。 STM还可以操纵 单个原子和分子
STM像
原子操纵
原子力显微镜AFM
• 原子力显微镜(AFM), 或者扫描力显微镜 (SFM)
• 跟所有的扫描探针显 微镜一样,AFM使用 一个极细的探针在样 品表面进行光栅扫描, 探针是位于一悬臂的 末端顶部,该悬臂可 对针尖和样品间的作 用力作出反应
原子吸收光谱法(AAS)
• 根据蒸气相中被测元素的基态原子对其原子共振 辐射的吸收强度来测定试样中被测元素的含量;
• 适合对纳米材料中痕量金属杂质离子进行定量测 定,检测限低 ,10-10-10-14 g/cm3
• 测量准确度很高 ,1%(3-5%) • 选择性好 ,不需要进行分离检测 • 分析元素范围广 ,70多种 • 不能同时进行多元素分析
• 其特点是样品使用量少,不仅可以获得样品的形 貌,颗粒大小,分布以还可以获得特定区域的元 素组成及物相结构信息
高分辨TEM
• HRTEM是观察材料微观结构的方法。不仅 可以获得晶包排列的信息,还可以确定晶 胞中原子的位置。
• 200KV的TEM点分辨率为0.2nm,1000KV 的TEM点分辨率为0.1nm。
电感耦合等离子体发射光谱法(ICP)
• ICP是利用电感耦合等离子体作为激发源,根据处于激发 态的待测元素原子回到基态时发射的特征谱线对待测元素 进行分析的方法
纳米粒子粒径评估方法

STM、SEM、SPM显微镜
形貌
X射线衍射法 IR、UV、拉曼光谱 法
核磁共振法
物相、构 造
纳米材料
化学成份
常规化学分析法(特征元素旳 解-滴定) 原子光谱分析法(吸收光谱、 射光谱) 质谱法 X射线特征分析法(X射线荧 光谱法、电子探针分析法) 光电子能谱法
性能
力学分析仪(显微硬度仪,力学万能试验 机等) 热综合分析仪,磁强分析仪,光谱仪等 多种性能测试仪
电镜照片 仪器照片
透射电镜旳构造
• 透射电镜旳外观照片。
• 一般透射电镜由电子光学 系统、电源系统、真空系 统、循环冷却系统和控制 系统构成,其中电子光学 系统是电镜旳主要构成部 分。
高辨别透射电子显微镜
• 透射电子显微镜发展旳另一种体现是辨别率旳不断提升。 目前200KV透射电子显微镜旳辨别率好于0.2nm,1000KV透 射电子显微镜旳辨别率到达0.1nm。
(a)恒高度模式;
(b)恒电流模式
S 为针尖与样品间距,I、Vb 为隧道电流和偏置电压,
Vz为控制针尖在 z 方向高度旳反馈电压。
6.2 原子力显微镜旳基本原理
• 原子力显微镜旳基本原理是:将一种对薄弱力极敏感 旳微悬臂一端固定,另一端有一微小旳针尖,针尖与样品 表面轻轻接触,因为针尖尖端原子与样品表面原子间存在 极薄弱旳排斥力,经过在扫描时控制这种力旳恒定,带有 针尖旳微悬臂将相应于针尖与样品表面原子间作用力旳等 位面而在垂直于样品旳表面方向起伏运动。
反馈控制是本系统旳关键工作 机制。
量子森林 • 该图是由托斯藤-兹欧姆巴在德国试验室中捕获旳图像,它展示了锗
硅量子点——仅高15纳米,直径为70纳米。
• 经过使用千万亿分之一秒旳激光脉冲撞击蓝宝石表面,蓝宝石被加热 了,表面留下了一道浅细旳陷坑之后,这块蓝宝石再次被撞击加热, 就产生了图中可见旳内部梯级构造
纳米材料的性能测试方法与分析技巧

纳米材料的性能测试方法与分析技巧在纳米科技领域中,纳米材料的性能测试是非常重要的。
随着纳米材料的广泛应用,准确评估其性能对于材料的研发和应用具有重要意义。
本文将介绍纳米材料性能测试的常用方法和分析技巧。
1. 粒径分析纳米材料的粒径是其最基本的性能参数之一。
常用的粒径分析方法包括动态光散射(DLS)、激光粒度分析仪(LPSA)和扫描电子显微镜(SEM)等。
其中,动态光散射是一种通过光粒度仪测量颗粒对粒径的分析方法。
激光粒度分析仪可以通过光学原理测量颗粒的大小分布。
扫描电子显微镜则通过高分辨率的图像展示颗粒的形态和大小。
这些方法可以帮助我们了解纳米材料的粒径分布情况,为性能的评估提供依据。
2. 表面形貌分析纳米材料的表面形貌对其性能具有重要影响。
扫描电子显微镜和透射电子显微镜(TEM)是常用的表面形貌分析方法。
扫描电子显微镜可以提供高分辨率的表面形貌图像,而透射电子显微镜则可以提供纳米级别的表面形貌信息。
通过这些方法可以观察到纳米材料的形状、表面结构和晶体结构等信息,为性能的评估提供基础数据。
3. 结构分析纳米材料的结构对其性能具有重要影响。
X射线衍射(XRD)和透射电子显微镜是常用的结构分析方法。
X射线衍射可以通过检测材料的晶体衍射峰来确定其晶体结构和晶格参数。
透射电子显微镜则可以通过对纳米材料的电子衍射图像进行分析,确定其晶体结构和晶格参数。
结构分析可以提供对纳米材料晶体结构的了解,为性能的评估提供依据。
4. 表面化学成分分析纳米材料的表面化学成分对其性能具有重要影响。
常用的表面化学成分分析方法包括能谱分析(EDS)和X射线光电子能谱(XPS)。
能谱分析可以通过分析材料发射的X射线能谱来确定其表面化学成分。
X射线光电子能谱则可以通过分析材料表面的光电子发射能谱来确定其表面化学成分。
这些方法可以帮助我们了解纳米材料的表面化学成分,为性能的评估提供依据。
5. 热性能分析纳米材料的热性能对其应用具有重要意义。
(完整版)纳米材料粒度分析

纳米材料粒度分析一、实验原理纳米颗粒材料(粒径<100nm )是纳米材料中最重要的一种,可广泛用于纳米复合材料制备中的填料、光催化颗粒、电池电极材料、功能性分散液等。
粒径(或粒度)是纳米颗粒材料的一个非常重要的指标。
测试颗粒粒径的方法有许多种,其中,电子显微镜法和激光光散射法均可用纳米材料粒度的测试,电子显微镜法表征纳米材料比较直观,可观察到纳米颗粒的形态,但需要通过统计计数(一般需统计1000个以上颗粒的粒径)方法来得到颗粒粒径,比较烦琐费时,尤其是在纳米颗粒的粒径分布较宽时,统计得到的粒径及粒径分布误差将增大。
激光光散射法得到的纳米颗粒粒径具有较好的统计意义,制样简单,测试速度快,但激光光散射法无法观察到颗粒形态,在测试非球形颗粒时测试误差也较大。
因此,上述两种纳米材料的测试方法各有优缺点。
本实验选用激光光散射法测试纳米材料的粒径及粒径分布。
所用仪器为Beckman-coulter N4 Plus 型激光粒度分析仪。
图1为N4 Plus 型激光粒度分析仪的测量单元组成图,主要由HeNe 激光光源、聚焦透镜、样品池、步进马达、光电倍增管(PMT)、脉冲放大器和鉴别器(PAD)、数字自相关器、6802微处理器和计算机组成。
图1 N4 Plus 型激光粒度测试仪的测量单元组成图N4 Plus 型激光粒度分析仪的测量原理主要基于颗粒的布朗(Brownian)运动和光子相关光谱(Photon Correlation Spectroscopy, PCS)现象。
在溶液中,粒子由热导致与溶剂分子发生随机碰撞所产生的运动称为布朗运动,由于布朗运动,粒子在溶液中可发生扩散移动。
在恒定温度及某一浓度下,粒子的平移扩散系数与颗粒的粒径成反比,即符合Stokes-Einstein 方程:d3Tk D B πη=(1)式中k B 为玻尔兹曼常数(1.38×10-16erg/︒K),T 为温度(︒K),η为分散介质(或稀释剂)粘度(poise),d 为颗粒粒径(cm)。
纳米材料的表征和分析方法分享

纳米材料的表征和分析方法分享纳米材料是指尺寸在纳米级别的材料,其具有独特的物理、化学以及生物学性质,广泛应用于能源、材料、生物医药等领域。
为了深入了解纳米材料的性质和优良特性,科学家们开发了多种表征和分析方法。
在本文中,我们将分享一些常用的纳米材料表征和分析方法。
一、纳米材料的表征方法1. 扫描电子显微镜(SEM):SEM可以获得材料表面形貌和微观结构的高分辨率图像。
通过SEM可以观察纳米颗粒的大小、形状以及表面形貌的变化,进而得出材料的结构特征和表面形貌。
2. 透射电子显微镜(TEM):TEM是一种高分辨率的表征技术,可用于观察纳米材料的晶体结构和颗粒形态。
通过TEM,可以实时观察纳米材料的形貌、尺寸和晶体结构,并进一步了解纳米材料的导电性、光学性质等。
3. 原子力显微镜(AFM):AFM可以直接观察纳米尺度下的表面形貌和表面力学性质。
通过扫描探针与样品表面的相互作用,AFM可以获得纳米尺度下的三维表面拓扑图像,同时还可以测量纳米材料的力学性能。
4. 粒度分析:粒度分析是用于确定纳米颗粒的尺寸分布和平均粒径的方法。
常见的粒度分析技术包括激光粒度仪、动态光散射仪等。
这些仪器可以通过散射光的特性来推断颗粒的大小,并计算出粒径分布图和平均粒径。
二、纳米材料的分析方法1. X射线衍射(XRD):XRD是一种常用的纳米材料分析方法,可以用于确定纳米材料的晶体结构、晶格参数和晶体缺陷。
通过分析材料对入射X射线的散射模式,可以得出材料的晶体结构和晶格常数,从而获得材料的结晶性质。
2. 红外光谱(IR):红外光谱是一种用于检测材料分子结构和化学键情况的分析方法。
通过测量材料在红外波段的吸收谱线,可以得知材料的化学成分、功能基团和化学键的状态,帮助研究人员了解纳米材料的化学性质和功能。
3. 核磁共振(NMR):核磁共振技术可以用于分析纳米材料的结构、组成和动力学性质。
通过测量材料中原子核的共振信号,NMR可以得到关于材料分子的信息,包括分子结构、化学位移等,从而为纳米材料的研究提供有价值的数据。
纳米粒度分析

纳米粒度分析纳米粒度分析是一种用于测量和分析纳米颗粒的技术。
纳米粒度是指颗粒的尺寸在1至100纳米之间。
纳米颗粒具有独特的物理和化学性质,因此对其进行准确的尺寸分析对于研究和应用纳米材料非常重要。
纳米粒度分析可以通过不同的方法进行,其中常用的包括光学显微镜、透射电子显微镜(TEM)、扫描电子显微镜(SEM)、动态光散射(DLS)和激光粒度分析仪(LPA)。
每种方法都有其特定的优点和限制,因此选择适当的方法取决于样品类型、尺寸范围和分析需求。
光学显微镜是一种便捷且经济的测量方法,可以直接观察和测量粒子在固定载玻片上的大小。
然而,由于光学显微镜的分辨率限制,只能测量大约200纳米以上的粒子。
透射电子显微镜(TEM)和扫描电子显微镜(SEM)是两种使用电子束的技术,可以提供更高的分辨率和更精确的粒子尺寸分析。
TEM通过通过样品的透射电子图像来进行分析,可以达到纳米尺度以下的分辨率。
SEM通过扫描电子束并检测从样品表面散射出的电子来获取图像和尺寸数据。
这两种方法可以对纳米颗粒进行直接的形貌和尺寸分析,但需要较复杂的样品制备和仪器操作。
动态光散射(DLS)是一种常用的液相纳米颗粒尺寸分析方法。
这种方法通过测量悬浊液中颗粒在热扰动下的光散射来确定粒子的尺寸分布。
DLS具有非接触测量、速度快和样品制备简单的优点,适用于纳米颗粒的溶液样品。
激光粒度分析仪(LPA)是一种利用粒子在激光束中散射光的方法进行尺寸分析的技术。
该仪器通过测量颗粒散射光的角度和强度来确定颗粒的尺寸分布。
LPA可以对固体和液体样品进行尺寸分析,且具有较高的分辨率和较广的尺寸范围。
除了上述方法,还有一些其他的纳米粒度分析技术,例如X射线衍射(XRD)、原子力显微镜(AFM)和场发射扫描电子显微镜(FESEM)。
这些方法在特定情况下也可以用于纳米颗粒的尺寸分析。
总而言之,纳米粒度分析是研究和应用纳米材料的重要手段。
选择合适的分析方法取决于样品类型、尺寸范围和分辨率要求等因素。
纳米材料粒度分析报告

纳米材料粒度分析一、实验原理纳米颗粒材料(粒径<100nm )是纳米材料中最重要的一种,可广泛用于纳米复合材料制备中的填料、光催化颗粒、电池电极材料、功能性分散液等。
粒径(或粒度)是纳米颗粒材料的一个非常重要的指标。
测试颗粒粒径的方法有许多种,其中,电子显微镜法和激光光散射法均可用纳米材料粒度的测试,电子显微镜法表征纳米材料比较直观,可观察到纳米颗粒的形态,但需要通过统计计数(一般需统计1000个以上颗粒的粒径)方法来得到颗粒粒径,比较烦琐费时,尤其是在纳米颗粒的粒径分布较宽时,统计得到的粒径及粒径分布误差将增大。
激光光散射法得到的纳米颗粒粒径具有较好的统计意义,制样简单,测试速度快,但激光光散射法无法观察到颗粒形态,在测试非球形颗粒时测试误差也较大。
因此,上述两种纳米材料的测试方法各有优缺点。
本实验选用激光光散射法测试纳米材料的粒径及粒径分布。
所用仪器为Beckman-coulter N4 Plus 型激光粒度分析仪。
图1为N4 Plus 型激光粒度分析仪的测量单元组成图,主要由HeNe 激光光源、聚焦透镜、样品池、步进马达、光电倍增管(PMT)、脉冲放大器和鉴别器(PAD)、数字自相关器、6802微处理器和计算机组成。
图1 N4 Plus 型激光粒度测试仪的测量单元组成图N4 Plus 型激光粒度分析仪的测量原理主要基于颗粒的布朗(Brownian)运动和光子相关光谱(Photon Correlation Spectroscopy, PCS)现象。
在溶液中,粒子由热导致与溶剂分子发生随机碰撞所产生的运动称为布朗运动,由于布朗运动,粒子在溶液中可发生扩散移动。
在恒定温度及某一浓度下,粒子的平移扩散系数与颗粒的粒径成反比,即符合Stokes-Einstein 方程:d 3T k D B πη=(1) 式中k B 为玻尔兹曼常数(1.38×10-16erg/︒K),T 为温度(︒K),η为分散介质(或稀释剂)粘度(poise),d 为颗粒粒径(cm)。
纳米材料粒径测试方法
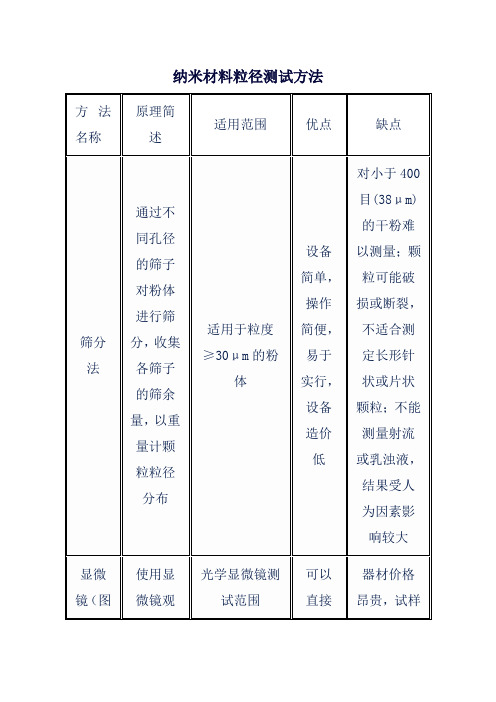
方法名称
原理简述
适用范围
优点
缺点
筛分法
通过不同孔径的筛子对粉体进行筛分,收集各筛子的筛余量,以重量计颗粒粒径分布
适用于粒度≥30μm的粉体
设备简单,操作简便,易于实行,设备造价低
对小于400目(38μm)的干粉难以测量;颗粒可能破损或断裂,不适合测定长形针状或片状颗粒;不能测量射流或乳浊液,结果受人为因素影响较大
显微镜(图像)法
使用显微镜观察颗粒形貌,并通过图像分析软件测量颗粒尺寸
光学显微镜测试范围0.8150μm,电学显微镜(SEM/TEM)检测范围0.001100μm
可以直接观察颗粒形貌,适合分布窄的样品,可获得球型度、长径比等Байду номын сангаас殊数据
器材价格昂贵,试样制备繁琐,操作复杂,速度慢,不宜分析粒度范围宽的样品,无法分析小于1微米的样品
沉降法
基于Stokes定律,通过颗粒在悬浮体系中的沉降速度计算粒径
重力沉降法适用于2100μm的颗粒,离心沉降法适用于0.0120μm的颗粒
分辨率高,对于粒径分布较宽的样本也能较好测试,准确性和重复性较好
测试时间较长,需精确控制温度,操作相对复杂;被测颗粒需满足球形单分散条件;不能处理不同密度的混合样品
测试范围为3~1000nm
操作简单,对于单分散悬浮液测试效果较好
对于多分散样本的测试结果较差,分辨率低,大颗粒光信号可能遮挡小颗粒信号,受样本本身性质影响
电阻法(库尔特计数法)
颗粒通过小孔时引起电阻变化,产生电压脉冲,脉冲幅值与颗粒粒径成正比
测量范围通常在0.5100μm,纳米库尔特测试范围可达502000nm
测量迅速,重复性好,分辨率高,几乎适用于所有类型颗粒测量
纳米材料的粒度分析

纳米材料的粒度分析1.1前言1.粒度分析的概念大部分固体材料均是由各种形状不同的颗粒构造而成,因此,细微颗粒材料的形状和大小对材料结构和性能具有重要的影响。
尤其对于纳米材料,其颗粒大小和形状对材料的性能起着决定性的作用。
因此,对纳米材料的颗粒大小、形状的表征和控制具有重要的意义。
一般固体材料颗粒大小可以用颗粒粒度概念来描述。
但由于颗粒形状的复杂性,一般很难直接用一个尺度来描述一个颗粒大小,因此,在粒度大小的描述过程中广泛采用等效粒度的概念。
对于不同原理的粒度分析仪器,所依据的测量原理不同,其颗粒特性也不相同,只能进行等效对比,不能进行横向直接对比。
如沉降式粒度仪是依据颗粒的沉降速度进行等效对比,所测的立径为等效沉速径,即用与被测颗粒具有相同沉降速度的同质球形颗粒的直径来代表实际颗粒的大小。
激光粒度仪则是利用颗粒对激光的衍射和散射特性作等效对比,所测出的等效粒径为等效散射粒径,即用与实际被测颗粒具有相同散射效果的球形颗粒的直径来代表这个颗粒的实际大小。
当被测颗粒为球形时,其等效粒径就是它的实际直径。
但由于粉体材料颗粒的形状不可能都是均匀球形的,有各种各样的结构,因此,在大多数情况下粒度分析仪所测的粒径是一种等效意义上的粒径,和实际的颗粒大小分布会有一定的差异,因此只具有相对比较的意义。
等效粒径(D)和颗粒体积(V)的关系可以用表达式D=1.24V1/3表示。
此外,各种不同粒度分析方法获得的粒径大小和分布数据也可能不能相互印证,不能进行绝对的横向比较。
由于粉体材料的颗粒大小分布较广,可以从纳米级到毫米级,因此在描述材料粒度大小时,可以把颗粒按大小分为纳米颗粒、超微颗粒、微粒、细粒、粗粒等种类。
依据这些颗粒的种类可以采用相应的粒度分析方法和仪器。
近年来,随着纳米科学和技术的迅速发展,纳米材料的颗粒分布以及颗粒大小已经成为纳米材料表征的重要指标之一,在普通的材料粒度分析中,其研究的颗粒大小一般在100nm~1um尺寸范围。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
纳米材料粒度分析一、实验原理纳米颗粒材料(粒径<100nm )是纳米材料中最重要的一种,可广泛用于纳米复合材料制备中的填料、光催化颗粒、电池电极材料、功能性分散液等。
粒径(或粒度)是纳米颗粒材料的一个非常重要的指标。
测试颗粒粒径的方法有许多种,其中,电子显微镜法和激光光散射法均可用纳米材料粒度的测试,电子显微镜法表征纳米材料比较直观,可观察到纳米颗粒的形态,但需要通过统计计数(一般需统计1000个以上颗粒的粒径)方法来得到颗粒粒径,比较烦琐费时,尤其是在纳米颗粒的粒径分布较宽时,统计得到的粒径及粒径分布误差将增大。
激光光散射法得到的纳米颗粒粒径具有较好的统计意义,制样简单,测试速度快,但激光光散射法无法观察到颗粒形态,在测试非球形颗粒时测试误差也较大。
因此,上述两种纳米材料的测试方法各有优缺点。
本实验选用激光光散射法测试纳米材料的粒径及粒径分布。
所用仪器为Beckman-coulter N4 Plus 型激光粒度分析仪。
图1为N4 Plus 型激光粒度分析仪的测量单元组成图,主要由HeNe 激光光源、聚焦透镜、样品池、步进马达、光电倍增管(PMT)、脉冲放大器和鉴别器(PAD)、数字自相关器、6802微处理器和计算机组成。
图1 N4 Plus 型激光粒度测试仪的测量单元组成图N4 Plus 型激光粒度分析仪的测量原理主要基于颗粒的布朗(Brownian)运动和光子相关光谱(Photon Correlation Spectroscopy, PCS)现象。
在溶液中,粒子由热导致与溶剂分子发生随机碰撞所产生的运动称为布朗运动,由于布朗运动,粒子在溶液中可发生扩散移动。
在恒定温度及某一浓度下,粒子的平移扩散系数与颗粒的粒径成反比,即符合Stokes-Einstein 方程:d3Tk D B πη=(1)式中k B 为玻尔兹曼常数(1.38×10-16erg/︒K),T 为温度(︒K),η为分散介质(或稀释剂)粘度(poise),d 为颗粒粒径(cm)。
当激光束照射到溶液中的悬浮颗粒上时,由于颗粒的随机布朗运动,颗粒产生的散射光强也将不断起伏波动,这种现象称作光子相光光谱现象,如图2所示。
布朗运动越强烈,散射光强随机涨落的速率也就越快,反之亦然。
利用光子相光光谱法测量的粒径是下限大约是3~5nm 。
图2 散射光强随时间的起伏涨落当入射光场为稳定的高斯光场时,散射光强的时间自相关函数(Autocorrelation Function, ACF )可以表示为))(g 1(A )(G2)1()2(τβ+=τ(2)式中,A 为光强自相关函数G (2)(τ)的基线,β为约束信噪比的实验常数,A 和β是依赖于样品、装置结构和光电子技术效率的常数,g (1)(τ)为散射光场的电场强度自相关函数。
通过数字相关仪测得的时间自相关函数G (2)(τ),即可得到被测颗粒的粒径信息。
对于最简单的单分散颗粒系,其光强自相关函数服从洛仑兹分布,是一指数衰减函数,可表示为)]2ex p(1[A )(G )2(τΓ-β+=τ(3)式中Γ为Rayleigh 线宽。
光强自相关函数G (2)(τ)如图3所示。
图3 自相关函数(ACF )Γ与表征颗粒布朗运动的平移扩散系数D 存在如下关系:2Dq =Γ(4)式中q 是散射矢量,由下式决定)2sin(n 4q 0θλπ=(5)式中λ0是入射光在真空中的波长,θ是散射角,n 为分散介质折射率。
根据Γ值,可从式(4)求得颗粒平移扩散系数D ,最后由式(1)求得被测颗粒试样的粒径。
需要注意的是,Stokes-Einstein 公式是在不存在其他作用里的条件下得到的。
为此,在应用PCS 法测量时溶液中的颗粒浓度应充分稀释,颗粒表面也不应有静电荷,以避免颗粒间的相互作用。
对多分散颗粒系,电场自相关函数为单指数加权之和或者分布积分⎰∞ΓτΓ-Γ=τ0)1(d )ex p()(G )(g(6)式中,G(Γ)为依赖于光强的归一化线宽分布函数。
由式(6)求得G(Γ)后,光强随颗粒粒径的分布函数G(D)可由Stokes-Einstein 关系式从G(Γ)中换算获得。
通常G 2(τ)由数字相关仪测得,继而根据式(1)换算得到电场自相关系数g (1)(τ),然后应用最小二乘法拟合优化求解式(6)中的G(Γ),以使目标函数极小,最后求得颗粒分布。
方程(6)称为第I 类Fredholm 积分方程,它的求解是一个病态问题,对同一个g (1)(τ)存在无限多个的符合G(Γ)的方程。
目前,学者们已经提出了多种不同的近似求解方法,如累积分析法、双指数法、直方图法、非负约束最小二乘法和CONTIN 法等。
N4 Plus 粒径分析仪数据处理方法[4]N4 Plus 粒径分析仪提供了两种粒径分析模式,即unimodal 和SDP(Size Distribution Processor)。
Unimodal 模式主要用于分析粒径分布较窄的颗粒,可得出强均粒径(mean intensity-weighted particle size)和标准偏差(standard deviation),其中标准偏差可在一定程度上反映粒径分布,但对于粒径分布较宽或存在多峰分布的颗粒误差较大。
SDP 模式分析可得到粒径及粒径分布,但这种方法与unimodal 相比,需要更精确的ACF 数据,因而需要较长的测试时间。
Unimodal 分析模式在N4 Plus 中有80个ACF 时间通道,这些通道中得到的ACF 减去基线(baseline)后,其值与时间存在幂律关系,见下:2/c b a )baseline )(G ln(2i i i τ+τ+=-τ(7)系数b 和c 分别是ACF G 的第一和第二累积量,τi 表示迟滞时间(i=1,2,3…..80)。
b 等于2Γ,b 的倒数与粒径平均值的倒数成比例关系,即:><=><≈d const d /11const b 1 (8) Tk 3.K 21const B 2πη=(9)式中角括号表示括号中的值为平均值,多分散指数(polydispersity index)与粒径分布变量系数(CV)的关系如下:4.I .P 211CV +⨯=(10)则标准偏差(standard deviation)可按下式计算:SD=d ×CV (11)SDP 分析模式Unimodal 分析模式对粒径分布较为复杂的颗粒精度不高,而SDP 分析可在无须任何假定条件下得到颗粒的粒径分布。
N4 Plus 不能对单独的颗粒进行记数,仪器必须在数学上分离由不同粒径产生的衰减时间。
这些衰减时间在不同时间的ACF 中是复合在一起的,数学分离比较困难。
在SDP 分析中的运算法则是一个称作CONTIN 的FORTRAN 程序,这个程序在分析PCS 数据中已得到大量应用。
SDP 分析结果得到的是一样品粒径分布的柱形图,可以用强均分布(intensity distribution)或重均分布(weight distribution)表示。
强均向重均转换需要用到精确的Mie 方程,需要输入颗粒的折光指数,如果颗粒折光指数未知,则只能近似转换。
强均粒径分布柱形图中的每个粒径下所显示的含量值与该粒径的颗粒光散射强度占整个光散射强度的百分数成正比。
重均粒径分布反映的是样品中不同粒径颗粒所占的相对重量分率,通常比强均还有用。
另外强均粒径与散射角度有关,而重均粒径与散射角度无关。
对于球形粒子,强均粒径转换成重均粒径需要用到颗粒和分散介质的折光指数及Mie 理论。
对于长径比小于3:1和粒径小于500nm 且长径比小于5:1的非球形粒子,Mie 理论仍可进行较好地近似转换。
对于长柱形或高度不对称型的长形颗粒,目前还没有好的方法来进行强均和重均之间的转换。
对于电解质或透明粒子,假定颗粒的折光指数为零,不需要输入折光指数。
如果折光指数未知,N4 Plus 仪器会依据Mie 理论提供一种近似的强均与重均粒径之间的转换,这种转换在很宽的折光指数围都具有较好的准确性。
在柱形粒径分布图中,每个峰的粒径是相应粒径围的颗粒粒径的平均值,即:∑∑=iiii aa d d (12)式中d 是峰的平均粒径,a i 是第i 级粒径柱的相对强度,d i 是相应i 级柱的粒径。
SD 定义为21i i2i i a )d d (a )d (SD ⎥⎥⎥⎦⎤⎢⎢⎢⎣⎡-=∑∑ (13)对于重均粒径分布图,与强均粒径分布计算类似。
除了每个峰的平均粒径、SD 和相对强度以外,还给出了整个颗粒样品的平均粒径和变量系数。
变量系数定义为:d)d (SD CV =(14)二、实验方法(1) 测试仪器及材料美国Beckman-coulter 公司生产的N4 Plus 粒径分析仪,见下图。
石英比色皿若干,无水乙醇和去离子水各500ml ,滴管3~4支,清洁纸若干,超声波清洗器一台。
图4 N4 Plus 粒径分析仪(2) 测试步骤① 制样:配制浓度为5%的气相白炭黑分散液,将其超声分散特定时间,制得预分散液,再将少量分散液放入比色皿中,用大量去离子水稀释,将比色皿放入样品池中,用软件检测其光学浓度,如浓度过高,继续稀释,直至在仪器的测试浓度围之(即5×104~1×106);② 启动:打开电脑及粒径分析仪的电源开关,平衡仪器10~20min ,启动粒径测试软件(PCS Soft),检查电脑与粒径分析仪之间是否已经连接;③ 参数设置:按SOM 快捷钮,输入测试温度、分散介质的粘度和折光指数,建立测试方法文件;④测试:在Run菜单中打开Set up run,设置数据输出文件名,操作者姓名,选取测试方法文件,按Start Run钮开始测试;⑤计算:分别用Unimodal distribution和SDP analysis or distribution分析模式对数据进行处理。
⑥记录:记录测试得到的不同粒径实验结果。
(3)清理工作将使用过的比色皿用无水乙醇清洗3次,再在清洁的无水乙醇中超声洗涤1分钟,将使用过的滴管也用无水乙醇洗涤干净,废液倒入废液瓶中,清理桌面,关闭粒径分析仪及计算机。
三、实验容测试气相白碳黑在水中的分散粒径,考察超声波(超声时间分别为5min和15min,分散液浓度5%)对粉体分散粒径的影响,每样测试2~3次,计算实验误差。
四、结果与讨论⒈四种粒径分析方式得到的测试结果:(1)Sample 1:浓度5%白炭黑,水介质,超声分散5min。
由表1.1可以看出,Unimodal模式用于分析气相白炭黑的粒径分布,可得出样品强均粒径为276.8±9.99,对应多分散指数PI=0.338±0.117,PI比较小,表示颗粒的粒径分布似乎较窄,但由于粒径存在多峰分布(表1.2可知)而且各峰的强度相当,单方测试误差其实是比较大的。