蛋白纯化(his标签)说明书
his蛋白纯化操作步骤全

His蛋白的纯化一目的蛋白样液的收集1:-20度冰箱中取出pet30a-p26甘油菌2:菌种复苏,将5ul P26菌种加入到5ml K+ LB培养基中(1:1000加入卡纳抗生素的LB培养基),以未加菌的K+ LB培养基作空白对照,37度摇床培养12-16h。
3:扩大培养,将5ml过夜培养的P26菌液按照1:100的比例接种到500ml K+ LB培养基中,摇床培养至OD值为0.6(2h左右)4:诱导表达,扩培后的菌种中加入1mg/ml iptg,使iptg最终浓度1ug/ml(1mmol/l ) 37度摇床培养4h,置-70度保存或立即进行下步操作5:收集包涵体,离心,5000r 15min,弃去上清液(可置于-20度保存)6:溶解包涵体,用PBS重悬细胞沉淀(约每毫升沉淀加5mlPBS),放在冰上用超声波细胞粉碎机破菌(有效时间30-40min,超5s,停10s,及时换冰防止温度过高使蛋白质变性)7:蛋白提取液的收集,离心,5000r 15min取上清(可4度保存),菌体保存,测菌体中目的蛋白含量大小使用8:介质处理,取出底帽滴出多余液体,柱子顶部朝上直立固定好,蒸馏水洗涤一次,再用2倍树脂体积的1*binding buffer平衡柱子,最后用1*binding buffer配置成50%的树脂。
9:介质与核酸结合,从柱子上部加入蛋白提取液,4度旋转混匀1h,收集流出液(若需要流出液再重新过柱一次,以最大限度提高结合力)10:取10ml 1*binding buffer洗树脂,洗2次,流出液标记洗涤顺序保存11:样品的收集,取10个1.5mlEP管分别标记1,2,3……10,每次取1ml 1*elution buffer加入树脂内滴入标记1-10的EP管内每管6滴(第8管的目的蛋白含量已经很低,10管即可,若需要可适当增加)12:树脂的保存,若样品体积大于柱子体积,树脂10倍体积的PBS重新平衡后可马上使用,若不使用可用蒸馏水洗涤树脂,加入20%乙醇10ml -20度保存(一种蛋白树脂可重复使用,注意不要超过树脂的结合能力)二检测收集的样品中目的蛋白量的大小13:取13个1.5mlEP管,分别标记1#,2#,3#……10#,A,B,C从1号EP 管内取30ul样液于1#号EP管内,依次移取至10号于10#号,取30ul 10步中第一次收集的流出液于标记 A 的EP管中,取30ul第二次收集的流出液于标记 B 的EP管中,将7步中保留的菌体用少量pbs稀释并取30ul于标记C的EP管内。
6xHis Tag 融合蛋白纯化说明书

Code No. Product No. supplied 17-5247-01 HisTrap HP 1 ml 5 × 1 ml 17-5247-05 HisTrap HP 1 ml 100 × 1 ml* 17-5248-01 HisTrap HP 5 ml 1 × 5 ml 17-5248-02 HisTrap HP 5 ml 5 × 5 ml 17-5248-05 HisTrap HP 5 ml 100 × 5 ml* * Pack size available by special order.Connector kitConnectors supplied Usage No. supplied 1/16” male/luer female Connection of syringe to top ofHiTrap column 1 Tubing connector Connection of tubing (e.g. Peristalticflangeless/M6 female Pump P1) to bottom of HiTrap column* 1 Tubing connector Connection of tubing (e.g. Peristalticflangeless/M6 male Pump P1) to top of HiTrap column** 1 Union 1/16” female/ Connection to original FPLC SystemM6 male through bottom of HiTrap column 1 Union M6 female/ Connection to original FPLC System1/16” male through top of HiTrap column 1 Stop plug female, 1/16” Sealing bottom of HiTrap column 2, 5 or 7 * Union 1/16” female/M6 male is also needed.** Union M6 female/1/16” male is also needed.Table of contents1. Description 32. General Considerations 63. Operation 74. Optimization 115. Stripping and recharging 126. Cleaning-in-place 137. Scaling-up 138. Storage 149. Troubleshooting 1410. Further Information 1711. Ordering Information 18p. 21. DescriptionMedium propertiesHisTrap HP 1 ml and 5 ml columns are prepacked with Ni Sepharose High Performance, which consists of 34 µm highly cross-linked agarose beads with an immobilized chelating group. The medium has then been charged with Ni2+-ions.Several amino acids, for example histidine, form complexes with many metal ions. Ni Sepharose High Performance selectively binds proteins if suitable complex-forming amino acid residues are exposed on the proteinsurface. Additional histidines, such as in the case of (histidine)6 -tag,increase affinity for Ni2+ and generally make the histidine-tagged proteinthe strongest binder among other proteins in e.g. an E. coli extract. Column propertiesHisTrap HP columns are made of biocompatible polypropylene that doesnot interact with biomolecules. Columns are delivered with a stopper on the inlet and a snap-off end on the outlet. The columns have porous top and bottom frits that allow high flow rates. They cannot be opened or refilled. Columns can be operated with either a syringe and the supplied luer adapter, a peristaltic pump, or a chromatography system such asÄKTAdesign or FPLC System.Note: To prevent leakage, ensure that the adapter is tight.p. 3Table 1. HisTrap HP characteristics.Matrix Highly cross-linked spherical agarose, 6% Average particle size 34 µmMetal ion capacity ~15 µmol Ni2+/ml mediumDynamic bindingcapacity* At least 40 mg (histidine)6-tagged protein/ml mediumColumn volumes 1 ml or 5 mlColumn dimensions i.d. × H: 0.7 × 2.5 cm (1 ml)1.6 ×2.5 cm (5 ml)Recommended flow rate 1 and 5 ml/min for 1 and 5 ml column respectively Max. flow rates 4 and 20 ml/min for 1 and 5 ml column respectively Max back pressure††0.3 MPa, 3 barCompatibility during use Stable in all commonly used buffers, reducing agents,denaturants, and detergents. See Table 2.Chemical stability§§§0.01 M HCl, 0.1 M NaOH. Tested for 1 week at 40 ºC.1 M NaOH, 70% acetic acid. Tested for 12 hours.2% SDS. Tested for 1 hour.30% 2-propanol. Tested for 30 min.Avoid in buffers Chelating agents, e.g. EDTA, EGTA, citrate (see Table 2) pH stability§§§short term (< 2 hours): 2–14long term (< 1 week): 3–12Storage 20% ethanolStorage temperature +4 to +30 ºC* Dynamic binding capacity conditions:Sample: 1 mg/ml (histidine)6-tagged pure protein (Mr28 000 or 43 000) inbinding buffer (QB, 10% determination) or (histidine)6-tagged proteinbound from E. coli extractColumn volume: 0.25 ml or 1 mlFlow rate: 0.25 ml/min or 1 ml/minBinding buffer: 20 mM sodium phosphate, 0.5 M NaCl, 5 mM imidazole, pH 7.4Elution buffer: 20 mM sodium phosphate, 0.5 M NaCl, 0.5 M imidazole, pH 7.4 Note: Dynamic binding capacity is protein-dependent.††H2O at room temperature§§§Ni2+-stripped mediumThe Ni2+-charged medium is compatible with all commonly used aqueous buffers, reducing agents, denaturants such as 6 M Gua-HCl and 8 M urea, and a range of other additives (see Table 2).p. 4Table 2. Ni Sepharose High Performance is compatible with the following compounds, at least at the concentrations given.Reducing agents* 5 mM DTE5 mM DTT20 mM ß-mercaptoethanol5 mM TCEP10 mM reduced glutathioneDenaturing agents 8 M urea††6 M guanidine hydrochloride††Detergents 2% Triton X-100 (nonionic)2% Tween 20 (nonionic)2% NP-40 (nonionic)2% cholate (anionic)1% CHAPS (zwitterionic)Other additives 500 mM imidazole20% ethanol50% glycerol100 mM Na2SO41.5 M NaCl1 mM EDTA§§§60 mM citrate§§§Buffer substances 50 mM sodium phosphate, pH 7.4100 mM Tris-HCl, pH 7.4100 mM Tris-acetate, pH 7.4100 mM HEPES, pH 7.4100 mM MOPS, pH 7.4100 mM sodium acetate, pH 4††* See Notes and blank run, p. 10–11.††Tested for 1 week at +40 °C.§§§The strong chelator EDTA has been used successfully in some cases, at 1 mM.Generally, chelating agents should be used with caution (and only in the sample,not the buffers). Any metal-ion stripping may be counteracted by addition of a smallexcess of MgCl2 before centrifugation/filtration of the sample. Note that stripping effectsmay vary with applied sample volume.p. 52. General considerationsHisTrap HP is supplied precharged with Ni2+ ions. In general, Ni2+ is thepreferred metal ion for purification of recombinant histidine-taggedproteins. Note, however, that in some cases it may be wise to test othermetal ions, e.g. Zn2+ and Co2+, as the strength of binding depends onthe nature of the histidine-tagged protein as well as the metal ion (seeOptimization).We recommend binding at neutral to slightly alkaline pH (pH 7–8) in thepresence of 0.5–1.0 M NaCl. Sodium phosphate buffers are often used.Tris-HCl can generally be used, but should be avoided in cases where the metal-protein affinity is very weak, since it may reduce binding strength.Avoid chelating agents such as EDTA or citrate in buffers, see Table 2.Including salt e.g. 0.5–1.0 M NaCl in the buffers and samples, eliminatesion-exchange effects but can also have a marginal effect on the retention of proteins.Imidazole at low concentrations is commonly used in the binding and the wash buffers to minimize binding of host cell proteins. For the same reason, it is important to also include imidazole in the sample (generally, at the same concentration as in the wash buffer). At somewhat higher concentrations, imidazole may also decrease the binding of histidine-tagged proteins. The imidazole concentration must therefore be optimized to ensure the best balance of high purity (low binding of host cell proteins) and high yield(binding of histidine-tagged target protein). This optimal concentration is different for different histidine-tagged proteins, and is usually slightly higher for Ni Sepharose High Performance than for similar IMAC media on themarket (see Data File 18-1174-40). Use highly pure imidazole; such imidazole gives essentially no absorbance at 280 nm.As alternatives to imidazole elution, histidine-tagged proteins can be eluted from the medium by several other methods or combinations of methods – a lowering of pH within the range of 2.5–7.5 can be used, for example. At pH values below 4, metal ions will be stripped off the medium.Note:If the proteins are sensitive to low pH, we recommend collection of the eluted fractions in tubes containing 1 M Tris-HCl, pH 9.0(60–200 µl/ml fraction) to restore the pH to neutral.p. 6Chelating agents such as EGTA or EDTA will also strip metal ions from the medium and thereby cause protein elution, but the target protein pool will then contain Ni2+ ions. In this case, Ni2+ ions can be removed by desalting ona HiTrap™ Desalting, a PD-10 Desalting Column, or HiPrep™ 26/10 Desalting, (see Table 4).Leakage of Ni2+ from Ni Sepharose High Performance is very low underall normal conditions, lower than for other IMAC media tested. For applications where extremely low leakage during purification is critical, leakage can be even further reduced by performing a blank run (see page 11).Likewise, a blank run should also be performed before applying buffers/ samples containing reducing agents (see page 11).Whatever conditions are chosen, HisTrap HP columns can be operated witha syringe, peristaltic pump, or chromatography system.Note:If Peristaltic Pump P-1 is used, the maximum flow rate that can be run on a HisTrap HP 1 ml column is 3 ml/min.3. OperationBuffer preparationWater and chemicals used for buffer preparation should be of high purity. Filter buffers through a 0.22 µm or a 0.45 µm filter before use.Use high purity imidazole as this will give very low or no absorbance at 280 nm.If the recombinant histidine-tagged protein is expressed as inclusion bodies, include 6 M Gua-HCl or 8 M urea in all buffers and sample. On-column refolding of the denatured protein may be possible.p. 7Recommended conditionsBinding buffer: 20 mM sodium phosphate, 0.5 M NaCl, 20–40 mMimidazole, pH 7.4 (The optimal imidazoleconcentration is protein-dependent; 20–40 mM issuitable for many proteins.)Elution buffer: 20 mM sodium phosphate, 0.5 M NaCl, 500 mMimidazole, pH 7.4 (The imidazole concentrationrequired for elution is protein-dependent). Sample preparationFor optimal growth, induction, and cell lysis conditions for your recombinant histidine-tagged clones, please refer to established protocols.Adjust the sample to the composition and pH of the binding bufferby: Adding buffer, NaCl, imidazole, and additives from concentratedstock solutions; by diluting the sample with binding buffer; or by bufferexchange, (see Table 4). Do not use strong bases or acids for pH-adjustment (precipitation risk). Filter the sample through a 0.22 µm or a 0.45 µm filter and/or centrifuge it immediately before applying it to the column.To prevent the binding of host cell proteins with exposed histidine, it isessential to include imidazole at a low concentration in the sample andbinding buffer (see Optimization).p. 8p. 9T a b l e 4. P r e p a c k e d c o l u m n s f o r d e s a l t i n g a n d b u f f e r e x c h a n g e .C o d e N o C o l u m nL o a d i n g v o l u m e E l u t i o n v o l u m e C o m m e n t sA p p l i c a t i o n17-1408-01 H i T r a p 0.1–1.5 m l 1.3–4.0 m l P r e p a c k e d w i t h F o r d e s a l t i n g a n d b u f f e r D e s a l t i n g S e p h a d e x ™ G -25 e x c h a n g e o f p r o t e i n S u p e r f i n e . R e q u i r e s e x t r a c t s (M r >5000). a s y r i n g e o r p u m p t o r u n . 17-5087-01 H i P r e p U p t o 15–20 m l P r e p a c k e d w i t h F o r d e s a l t i n g a n d 26/10 15 m l S e p h a d e x G -25 b u f f e r e x c h a n g e o f D e s a l t i n g F i n e . R e q u i r e s a p r o t e i n e x t r a c t s p u m p t o r u n . (M r >5000).17-0851-01 P D -10 2.5 m l 3.5 m l P r e p a c k e d w i t h F o r d e s a l t i n g a n d D e s a l t i n g S e p h a d e x G -25. b u f f e r e x c h a n g e R e q u i r e s o n l y o f p r o t e i n e x t r a c t s g r a v i t y t o r u n . (M r >5000).17-0855-01 N I C K ™ 0.1 m l 0.4 m l P r e p a c k e d w i t h F o r s e p a r a t i o n o f S e p h a d e x G -25. p r o t e i n s (M r >5000) a n d n i c k t r a n s l a t e d D N A f r o m r a d i o l a b e l l e d n u c l e o t i d e s n o t s h o r t e r t h a n 120 m e r s , a n d s i m i l a r s e p a r a t i o n s .17-0853-01 N A P ™-5 0.5 m l 1.0 m l P r e p a c k e d w i t h F o r p u r i f i c a t i o n o f 17-0854-01 N A P -10 1.0 m l 1.5 m l S e p h a d e x G -25 p r o t e i n s (M r >5000),17-0852-01 N A P -25 2.5 m l 3.5 m l D N A g r a d e . D N A a n d o l i g o - R e q u i r e s o n l y n u c l e o t i d e s g r e a t e rg r a v i t y t o r u n . t h a n 10 b a s e s i n l e n g t h .Purification1. Fill the syringe or pump tubing with distilled water. Remove the stopperand connect the column to the syringe (use the adapter provided),laboratory pump or chromatography system tubing “drop-to-drop” toavoid introducing air into the system.2. Remove the snap-off end at the column outlet.3. Wash the column with 3–5 column volumes of distilled water.4. Equilibrate the column with at least 5 column volumes of binding buffer.Recommended flow rates are 1 ml/min or 5 ml/min for the 1 and 5 mlcolumns respectively.In some cases a blank run is recommended before final equilibration/sample application (see page 11).5. Apply the pretreated sample using a syringe or a pump.6. Wash with binding buffer until the absorbance reaches a steadybaseline (generally, at least 10–15 column volumes).Note:Purification results are improved by using imidazole in sample and binding buffer (see Optimization).7. Elute with elution buffer using a one-step or linear gradient.Five column volumes are usually sufficient if the protein of interest iseluted by a one-step gradient. A shallow gradient, e.g. a linear gradient over 20 column volumes or more, may separate proteins with similarbinding strengths.Note:If imidazole needs to be removed from the protein, use HiTrap Desalting, a PD-10 Desalting Column, or HiPrep 26/10 Desaltingdepending on the sample volume (see Table 4).Note:Ni Sepharose High Performance is compatible with reducing agents.However, removal of any weakly bound Ni2+ ions by performing ablank run without reducing agents (as described on page 11) beforeapplying buffer/sample including reducing agents is recommended.Do not leave HisTrap HP columns with buffers including reducingagents when not in use.p. 10Note:Leakage of Ni2+ from Ni Sepharose High Performance is low under all normal conditions. The leakage is lower than for other IMAC mediatested (see Data File Ni Sepharose High Performance, 18-1174-40).For very critical applications, leakage during purification can be even further diminished by performing a blank run (as described below)before loading sample.Blank run:Use binding buffer and elution buffer without reducing agents.1. Wash the column with 5 column volumes of distilled water (to removethe 20% ethanol).2. Wash with 5 column volumes of elution buffer.3. Equilibrate with 10 column volumes of binding buffer.4. OptimizationImidazole at low concentrations is commonly used in the binding and the wash buffers to minimize binding of host cell proteins. For the same reason, it is important to also include imidazole in the sample (generally, at the same concentration as in the wash buffer). At somewhat higher concentrations, imidazole may also decrease the binding of histidine-tagged proteins. The imidazole concentration must therefore be optimized to ensure the best balance of high purity (low binding of host cell proteins), and high yield (binding of histidine-tagged target protein). This optimal concentration is different for different histidine-tagged proteins, and is usually slightly higher for Ni Sepharose High Performance than for similar IMAC media on the market (see Data File Ni Sepharose High Performance, 18-1174-40). Finding the optimal imidazole concentration for a specific histidine-tagged protein is a trial-and-error effort, but 20–40 mM in the binding and wash buffers is a good starting point for many proteins. Use a high purity imidazole, such imidazole gives essentially no absorbance at 280 nm.Ni2+ is usually the first choice metal ion for purifying most (histidine)6-taggedrecombinant proteins from nontagged host cell proteins, and also the ion most generally used. Nevertheless, it is not always possible to predict whichp. 11metal ion will be best for a given protein. The strength of binding between a protein and a metal ion is affected by several factors, including the length, position, and exposure of the affinity tag on the protein, the type of ion used, and the pH of buffers, so some proteins may be easier to purify with ions other than Ni2+.A quick and efficient way to test this possibility and optimize separation conditions is to use HiTrap IMAC HP 1 ml columns, which are packed with IMAC Sepharose High Performance (not charged with metal ions). Each column can be charged with different metal ions, e.g. Cu2+, Co2+, Zn2+, Ca2+, or Fe2+. Instructions are included with each column.A study to compare the purification of six (histidine)6-tagged recombinantproteins, including three variants of maltose-binding protein, with different metal ions has indicated that Ni2+ generally gives best selectivity between (histidine)-tagged and nontagged host-cell proteins (see Application Note 18-1145-18).5. Stripping and rechargingNote:The column does not have to be stripped and recharged between each purification if the same protein is going to be purified; it issufficient to strip and recharge it after 5–7 purifications, dependingon the cell extract, extract volume, target protein, etc. Recommended stripping buffer: 20 mM sodium phosphate, 0.5 M NaCl,50 mM EDTA, pH 7.4Strip the column by washing with at least 5–10 column volumes of stripping buffer. Wash with at least 5–10 column volumes of binding buffer and 5–10 column volumes of distilled water before recharging the column. Recharge the water-washed column by loading 0.5 ml or 2.5 ml of 0.1 MNiSO4 in distilled water on HisTrap HP 1 ml and5 ml column, respectively.Salts of other metals, chlorides, or sulfates, may also be used (seeOptimization). Wash with 5 column volumes distilled water, and 5 column volumes binding buffer (to adjust pH) before storage in 20% ethanol.p. 126. Cleaning-in-placeWhen an increase in back-pressure is seen, the column can be cleaned. Before cleaning, strip off Ni2+ ions using the recommended procedure described above.After cleaning, store in 20% ethanol (wash with 5 column volumes) or recharge with Ni2+ prior to storage in ethanol.The Ni2+-stripped column can be cleaned by the following methods;• Remove ionically bound proteins by washing the column with several column volumes of 1.5 M NaCl; then wash the column with approx. 10column volumes of distilled water.• Remove precipitated proteins, hydrophobically bound proteins, and lipoproteins by washing the column with 1 M NaOH, contact time usually 1–2 hours (12 hours or more for endotoxin removal). Then wash thecolumn with approx. 10 column volumes of binding buffer, followed by5–10 column volumes of distilled water.• Remove hydrophobically bound proteins, lipoproteins, and lipids by washing the column with 5–10 column volumes 30% isopropanol forabout 15–20 minutes. Then wash the column with approx. 10 columnvolumes of distilled water.Alternatively, wash the column with 2 column volumes of detergent in a basic or acidic solution. Use, for example, 0.1–0.5% nonionic detergentin 0.1 M acetic acid, contact time 1–2 hours. After treatment, alwaysremove residual detergent by washing with at least 5 column volumes of 70% ethanol. Then wash the column with approx. 10 column volumes of distilled water.7. Scaling-upTwo or three HisTrap HP 1 ml or 5 ml columns can be connected in series for quick scale-up (note that back-pressure will increase).Ni Sepharose High Performance, the medium prepacked in HisTrap HP columns, is supplied preswollen in 25 and 100 ml lab packs (see ordering information). An alternative scale-up strategy is thus to pack the medium in empty columns – Tricorn™ and XK columns are suitable for this purpose.p. 138. StorageStore HisTrap HP columns in 20% ethanol at +4 to +30 °C.9. TroubleshootingThe following tips may be of assistance. If you have any further questions about your HisTrap HP column, please visit/hitrap, contact our technical support, or your local representative.Note:When using high concentrations of urea or Gua-HCl, protein unfolding generally takes place. Refolding on-column (or afterelution) is protein-dependent.Tips:To minimize dilution of the sample, solid urea or Gua-HCl can be added.Tips:Samples containing urea can be analyzed directly by SDS-PAGE whereas samples containing Gua-HCl must be buffer-exchanged toa buffer with urea before SDS-PAGE.Column has clogged:• Cell debris in the sample may clog the column. Clean the column according to the section Cleaning-in-place.• It is important to centrifuge and/or filter the sample through a 0.22 µm or a 0.45 µm filter, see Sample preparation.Sample is too viscous:• If the lysate is very viscous due to a high concentration of host nucleic acid, continue sonication until the viscosity is reduced, and/or addDNase I to 5 µg/ml, Mg2+ to 1 mM, and incubate on ice for 10–15 min.Alternatively, draw the lysate through a syringe needle several times. p. 14Protein is difficult to dissolve or precipitates during purification:• The following additives may be used: 2% Triton™ X-100, 2% Tween™20, 2% NP-40, 2% cholate, 1% CHAPS, 1.5 M NaCl, 50% glycerol, 20 mMß-mercaptoethanol, 1–3 mM DTT or DTE (up to 5 mM is possible butdepends on the sample and the sample volume), 5 mM TCEP, 10 mMreduced glutathione, 8 M urea, or 6 M Gua-HCl. Mix gently for 30 min toaid solubilization of the tagged protein (inclusion bodies may requirelonger mixing). Note that Triton X-100 and NP-40 (but not Tween) have a high absorbance at 280 nm. Furthermore, detergents cannot be easilyremoved by buffer exchange.No histidine-tagged protein in the purified fractions:• Elution conditions are too mild (histidine-tagged protein still bound): Elute with an increasing imidazole gradient or decreasing pH todetermine the optimal elution conditions.• Protein has precipitated in the column: For the next experiment, decrease amount of sample, or decrease protein concentration byeluting with linear imidazole gradient instead of imidazole steps. Trydetergents or changed NaCl concentration, or elute under denaturing(unfolding) conditions (add 4–8 M urea or 4–6 M Gua-HCl).• Nonspecific hydrophobic or other interaction: Add a nonionic detergent to the elution buffer (e.g. 0.2% Triton X-100) or increase theNaCl concentration.• Concentration of imidazole in the sample and/or binding buffer is too high: The protein is found in the flow-through material. Decrease theimidazole concentration.• Histidine-tag may be insufficiently exposed: The protein is found in the flowthrough material; perform purification of unfolded protein in urea or Gua-HCl as for inclusion bodies.• Buffer/sample composition is incorrect: The protein is found in the flowthrough material. Check pH and composition of sample and binding buffer. Ensure that chelating or strong reducing agents are not presentin the sample at too high concentration, and that the concentration ofimidazole is not too high.p. 15SDS-PAGE of samples collected during the preparation of the bacterial lysate may indicate that most of histidine-tagged protein is located in the centrifugation pellet. Possible causes and solutions are:• Sonication may be insufficient: Cell disruption may be checked by microscopic examination or monitored by measuring the release ofnucleic acids at A260. Addition of lysozyme (up to 0.1 volume of a 10 mg/mllysozyme solution in 25 mM Tris-HCl, pH 8.0) prior to sonication mayimprove results. Avoid frothing and overheating as this may denaturethe target protein. Over-sonication can also lead to copurification of host proteins with the target protein.• The protein may be insoluble (inclusion bodies): The protein can usually be solubilized (and unfolded) from inclusion bodies usingcommon denaturants such as 4–6 M Gua-HCl, 4–8 M urea, or strongdetergents.Prepare buffers containing 20 mM sodium phosphate, 8 M urea,or 6 M Gua-HCl, and suitable imidazole concentrations, pH 7.4–7.6.Buffers with urea should also include 500 mM NaCl. Use these buffersfor sample preparation, as binding buffer and as elution buffer. Forsample preparation and binding buffer, use 10–20 mM imidazole or theconcentration selected during optimization trials (including urea or Gua-HCl).The protein is collected but is not pure (multiple bands on SDS polyacrylamide gel):• Partial degradation of tagged protein by proteases: Add protease inhibitors (use EDTA with caution, see Table 2).• Contaminants have high affinity for nickel ions: Elute with a stepwise or linear imidazole gradient to determine optimal imidazoleconcentrations to use for binding and for wash; add imidazole to thesample in the same concentration as the binding buffer. Wash beforeelution with binding buffer containing as high concentration of imidazole as possible, without causing elution of the tagged protein. A shallowimidazole gradient (20 column volumes or more), may separate proteins with similar binding strengths. If optimized conditions do not removecontaminants, further purification by ion exchange chromotographyp. 16(HiTrap Q HP or HiTrap SP HP) and/or gel filtration (Superdex™ Peptide,Superdex 75 or Superdex 200) may be necessary.• Contaminants are associated with tagged proteins: Add detergent and/or reducing agents before sonicating cells. Increase detergent levels(e.g. up to 2% Triton X-100 or 2% Tween 20), or add glycerol (up to 50%)to the wash buffer to disrupt nonspecific interactions.Histidine-tagged protein is eluted during sample loading/wash:• Buffer/sample composition is incorrect: Check pH and composition of sample and binding buffer. Ensure that chelating or strong reducingagents are not present in the sample at a too high concentration, andthat the concentration of imidazole is not too high.• Histidine-tag is partially obstructed: Purify under denaturing conditions (use 4–8 M urea or 4–6 M Gua-HCl).• Column capacity is exceeded: Join two or three HisTrap HP 1 ml columns together or change to a HisTrap HP 5 ml column.10. Further informationVisit /hitrap for further information. Several handbooks also contain useful information, see ordering information.p. 1711. Ordering InformationProduct No. supplied Code No.HisTrap HP 5 × 1 ml 17-5247-01 HisTrap HP 100 × 1 ml * 17-5247-05 HisTrap HP 1 × 5 ml 17-5248-01 HisTrap HP 5 × 5 ml 17-5248-02 HisTrap HP 100 × 5 ml * 17-5248-05 Related products No. supplied Code No.Ni Sepharose High Performance 25 ml 17-5268-01 Ni Sepharose High Performance 100 ml 17-5268-02 HiTrap Desalting 5 × 5 ml 17-1408-01 HiTrap Desalting 100 × 5 ml * 11-0003-29 PD-10 Desalting Column 30 17-0851-01 HiPrep 26/10 Desalting 1 × 53 ml 17-5087-01 HiPrep 26/10 Desalting 4 × 53 ml 17-5087-02 HisTrap FF 5 × 1 ml 17-5319-01 HisTrap FF 100 × 1 ml * 17-5319-02 HisTrap FF 5 × 5 ml 17-5255-01 HisTrap FF 100 × 5 ml * 17-5255-02 HisTrap FF crude 5 × 1 ml 11-0004-58 HisTrap FF crude 100 × 1 ml * 11-0004-59 HisTrap FF crude 5 × 5 ml 17-5286-01 HisTrap FF crude 100 × 5 ml * 17-5286-02 HisTrap FF crude Kit 1 kit 28-4014-77 HisPrep™ FF 16/10 1 × 20 ml 17-5256-01 * Pack size available by special order.p. 18。
6X组氨酸标记(His-tag)蛋白纯化试剂盒产品说明书(中文

1. 本产品为 5 次操作 2. 操作时,须戴手套 3. 整个操作在 4℃温度下进行 4. 如果用户需要获得变性标记蛋白,建议使用变性液(Reagent D),整个操作在室温下进行 5. 用户扩增的细菌可溶性总蛋白样品须完全可溶状态,否则影响蛋白的结合(例如包涵体);建议使用
不溶性融合蛋白溶解试剂盒( 30016 )帮在纯化前,须进行目标蛋白鉴定试验,例如 IPTG 测试;建议使用
2
白浓度 14.本公司提供系列融合蛋白处理试剂产品
质量标准
1. 本产品经鉴定性能长期稳定 2. 本产品经鉴定结合效果满意
HIS 标记蛋白扩增试剂盒( HL30018.1 ) 7. 用户扩增的细菌可溶性总蛋白样品避免含有二硫苏糖醇(DTT)、巯基乙醇(BME)、EDTA、EGTA 等
化学成分 8. 亲和液(Reagent B)建议保存在-20℃冰箱里,避免反复冻融,建议分装 9. 亲和液(Reagent B)每次使用 1 毫升,可结合 5 至 10 毫克目标蛋白,回收率高达 95%。
产品内容
平衡液(Reagent A) 亲和液(Reagent B) 洗脱液(Reagent C) 产品说明书
XX 毫升 XX 毫升 XX 毫升 1份
保存方式
保存在-20℃冰箱里;有效保证 6 月
用户自备
变性液(Reagent D):用于变性融合蛋白纯化 透析液(Reagent E):用于蛋白纯化后透析 细菌可溶性总蛋白样品:用于纯化的待测样品 1.5 毫升离心管:用于反应操作的容器 50 毫升锥形离心管:用于胶体溶液配制的容器 平式摇荡仪:用于混匀反应物 4℃台式离心机:用于沉淀反应物 4℃微型台式离心机:用于沉淀反应物
6 X 组氨酸标记(His-tag)蛋白纯化试剂盒产品说明书(中文版)
GE组氨酸标签蛋白纯化手册
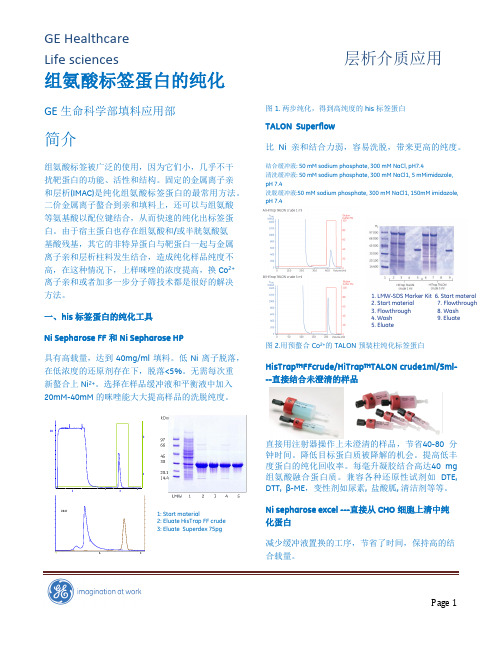
%0.120.0组氨酸标签蛋白的纯化GE 生命科学部填料应用部简介组氨酸标签被广泛的使用,因为它们小,几乎不干扰靶蛋白的功能、活性和结构。
固定的金属离子亲和层析(IMAC)是纯化组氨酸标签蛋白的最常用方法。
二价金属离子螯合到亲和填料上,还可以与组氨酸等氨基酸以配位键结合,从而快速的纯化出标签蛋白。
由于宿主蛋白也存在组氨酸和/或半胱氨酸氨基酸残基,其它的非特异蛋白与靶蛋白一起与金属离子亲和层析柱料发生结合,造成纯化样品纯度不高,在这种情况下,上样咪唑的浓度提高,换Co 2+离子亲和或者加多一步分子筛技术都是很好的解决方法。
一、his 标签蛋白的纯化工具Ni Sepharose FF 和Ni Sepharose HP具有高载量,达到40mg/ml 填料。
低Ni 离子脱落,在低浓度的还原剂存在下,脱落<5%。
无需每次重新螯合上Ni 2+。
选择在样品缓冲液和平衡液中加入20mM-40mM 的咪唑能大大提高样品的洗脱纯度。
1: Start material2: Eluate HisTrap FF crude 3: Eluate Superdex 75pg图1. 两步纯化,得到高纯度的his 标签蛋白TALON Superflow比Ni 亲和结合力弱,容易洗脱,带来更高的纯度。
结合缓冲液: 50 mM sodium phosphate, 300 mM NaCl, pH7.4清洗缓冲液: 50 mM sodium phosphate, 300 mM NaCl1, 5 mMimidazole, pH 7.4洗脱缓冲液:50 mM sodium phosphate, 300 mM NaCl1, 150mM imidazole, pH 7.4图2.用预螯合Co 2+的TALON 预装柱纯化标签蛋白HisTrap™FFcrude/HiTrap™TALON crude1ml/5ml---直接结合未澄清的样品分钟时间。
HisTag_融合蛋白纯化(默克新版)

默克生命科学服务热线:400 820 8872 bioteam@
高纯度包涵体的制备 以下操作可用于任何 BugBuster®系列产品抽提的包涵体纯化。 1. 如可溶蛋白抽提步骤1-4进行操作。 2. 将步骤“4”所得到的沉淀重悬于BugBuster(货号70584),BugBuster的量与当初重悬细胞糊的体积相同。
默克生命科学服务热线:400 820 8872 bioteam@
一、亲和纯化样品的前处理
1. 菌液体积-起始目的蛋白量
纯化条件的优化需考虑多个因素,包括 His·Tag 融合蛋白表达水平和上样量。若目的蛋白未能高效表达,需要采 用一个较高的浓缩系数(concentration factor)进行菌的裂解,即在较大培养体系中,按一定比例加入一定体积 的裂解/结合缓冲液。浓缩系数定义为菌液体积与裂解/结合缓冲液体积之比。不同目的蛋白表达水平推荐使用的 裂解/结合缓冲液体积、对应的浓缩系数大小,请参考表 1。 例如,某蛋白表达水平约为 0.1mg/ml,需要在变性条件下进行小批提纯,则 100ml 的培养物离心获得的菌体, 按浓缩 100 倍比例重悬于含变性剂的 1ml 裂解/结合缓冲液中。 在非变性条件下进行纯化时,要准确预计裂解液中可溶蛋白的含量比较困难,一般建议采用 50-100 倍浓缩。
产品使用说明书
His·Tag 融合蛋白纯化操作手册
采用 pET 系统进行原核蛋白表达,蛋白的表达量达到 20mg/100ml 培养物并不是困难的事。 在大肠杆菌中表达的目的蛋白,其可溶性(可溶蛋白或包涵体)、细胞定位(细胞质、细胞周 质、培养基上清),都会对后续的纯化策略造成影响。我们建议研究者在蛋白表达后,首先进 行目的蛋白的细胞定位(请参考 pET 系统操作手册);在进行大量纯化之前,小量纯化蛋 白,摸索确定适合于具体蛋白的纯化条件,也是值得推荐的好方法。外源蛋白在大肠杆菌中表 达,可能以可溶形式存在,也可能以包涵体形式存在。尤其在高水平表达的条件下,更容易形 成包涵体。包涵体的形成与外源蛋白本身性质、载体、宿主菌、以及表达水平都有关系,可以 通过选择不同表达载体和 E.coli 宿主菌组合,摸索生长条件和适宜诱导条件,达到优化蛋白表 达的目的。His·Tag®融合蛋白,可以在天然条件或变性条件下用 NTA His·Bind 树脂或 IDA His·Bind 树脂进行纯化。
His-Tag可溶性蛋白纯化方法

可溶性蛋白VP60-N的纯化方法一、Ni2+琼脂糖吸附树脂处理:1.打开柱帽使酒精溶液完全流出;2.加入8ml灭菌水重悬树脂(缓慢颠倒纯化柱),4℃平放5分钟;3.沉淀树脂(垂直静置5~10min),流出洗液,重复洗3次;4.加6ml binding buffer 重悬树脂;5.沉淀树脂(同3),流出洗液;6.重复步骤4、5共3次。
二、细菌细胞裂解:在菌泥中加入30ml 1×纯化buffer和500ul溶菌酶(10mg/ml)反复冻融4次(每次冻融各30min);离心(11000rpm,30min)收集上清液到新管子准备纯化并取10ul(或更多)-20℃保存(for SDS-PAGE analysis)。
(在细胞裂解时可以进行步骤一,同时处理透析袋:将透析袋用50%酒精进行煮沸处理10min,之后用水将其洗净,最后用超纯水将透析袋里面冲洗5次,外面冲洗3次,放入超纯水中备用。
透析袋用完后用自来水冲洗干净,在用超纯水如上冲洗后放入50%酒精中室温保存。
)三、蛋白纯化:(整个过程都在4℃进行)1.在纯化柱中加入10ml上述获得的细胞裂解液,重悬树脂(同上),4℃平放2h,揭开上下两个帽子,收集流出物,待柱内液体流尽后再加入10ml裂解液(处理同前,直至加完裂解液),将收集的流出液再过同一根柱子三遍,并取流出物10ul(或更多)-20℃保存(for SDS-PAGE analysis);2.加入8ml wash buffer 重悬树脂,静置平放在4℃5min,打开帽子使流尽洗液,同时收集10ul(或更多)流出物(for SDS-PAGE analysis);3.重复步骤2 3~4次;4.在纯化柱内加入5ml Elution buffer,打开帽子,收集流出物并取10ul(或更多)-20℃保存(for SDS-PAGE analysis);5.将步骤4收集的流出物装入透析袋在4℃PBS(pH7.4)溶液中透析过夜,收集透析后溶液(-20℃保存)同时取10ul(或更多)-20℃保存(for SDS-PAGE analysis);6.在进行步骤5时进行纯化柱保存处理:向柱内加入0.5M NaOH 8ml重悬树脂,静置平放在4℃30min后使流出,用灭菌水重悬树脂3次(4℃),加入25%酒精水溶液,4℃保存。
His标签融合蛋白纯化步骤

His标签融合蛋白纯化步骤(ni-nta琼脂糖凝胶亲和层析纯化法)1.缓冲液配制◆lysisbuffer1(undernativecondition)0.5mmtris.hcl0.5mnacl5%(w/v)glycerol10mmimidazol100mg(1mg/ml)lysozyme(purificationof6xhis-taggedproteinsfrome.coli)1%nonidetp40(np40=igepalca-630)0.25%tween20(ortriton100)0.02%nan3(optional)200µg(2µg/ml)rnasea(optional)1mg(10µg/ml)dnase1(optional)50mmnaf(optional)1mmn a3vo4(optional)+ddh2oto100ml,adjustphto8.0usingnaoh此lysisbuffer适用于于从e.coli、哺乳动物细胞和昆虫细胞中提纯拎his标签的蛋白质。
仅在用作水解e.coli细菌时,重新加入溶菌酶。
na3vo4是磷酸酶抑制剂,保护磷酸化的蛋白不被磷酸酶还原。
naf是酯酶抑制剂,保护脂蛋白不被酯酶降解。
特别注意:当采用镍琼脂糖凝胶提纯拎his标签的蛋白时,缓冲液中无法提edta,因edta能够并使镍从螯合物上瓦解,从而并使拆分介质丧失效果。
◆lysisbuffer2(underdenaturecondition)50mmtris-cl8murea(or6mgu-hcl)10mmimidazole0.05%tween20adjustphto8.0usingnaoh◆dialysis/tevcleavagebuffer50mmtris-cl,ph8.0100mmnacl(dependsonsolubilityofprotein)2.5%glycerol0.5mmedta0.5mmdttortcep◆缓冲液a:50mmtris.hcl0.5mnacl5%glycerol0.05%tween20用高浓度hcl调节ph值至8.0。
Ni柱纯化His-tag蛋白质

Ni柱纯化His-tag蛋白质一非变性条件下抽提His-Tag蛋白质下列方法适用于从细菌中抽提通常含有连续6个组氨酸尾(His Tag Protein)的具有天然结构的可溶性重组蛋白质(Native Protein)。
其他来源的蛋白质样品,如果目标蛋白质具有His-Tag结构,提供的层析方法可供参考。
1.样品准备1) 准备细胞,接种,诱导表达。
对于实验室用的pET载体表达系列,在细胞OD600=0.5-0.8时,用1mM IPTG诱导1-3小时,可以获得理想的表达效率。
收集细胞,置于-70度或立即进行步骤2操作。
2) 加入1/20细胞生长体积的NTA-0 Buffer(20mM Tris-HCl pH7.9,0.5M NaCl,10% Glycerol)和PMSF。
PMSF用无水乙醇配制成200mM储存液,-20度或4度保存;PMSF使用的工作浓度位1mM;注意:PMSF见水分解,需要在使用前加入。
该步骤冰上操作。
3)将细胞悬浮起来,加入溶菌酶(工作浓度位0.2-0.4mg/ml,如果表达的宿主细胞内含pLysS或pLysE,可以不加溶菌酶),混匀,冰上放置30分钟,超声或匀浆破碎细胞。
该步骤冰上操作。
4)加入10% Triton X-100, 使Triton的终浓度为0.05%,充分混匀,冰上放置15分钟,间或混匀);冰上超声破碎细胞,同时降低粘稠度。
5)加入1M MgCl2,使MgCl2的终浓度为1mM,混匀。
加入DNase, 使DNase 的终浓度为10mg/ml,混匀,室温放置10分钟。
6)5000转/分(20,000xg以上),4度离心15分钟以上。
取上清,置于冰上备用或-20度保存。
2.层析1) 将NTA树脂装入合适的层析柱,层析用10倍NTA体积的NTA-0 Buffer洗。
2)将样品(步骤2(6))加到NTA层析柱中,流速控制在15ml/小时左右,收集穿透部分,用于SDS/PAGE分析蛋白质的结合情况。
His标签蛋白纯化亲和层析介质-纳微

His标签蛋白纯化亲和层析介质使用说明书Uni®IDA-80NiUni®NTA-80NiUni®IDA-80LUni®NTA-80L全国咨询热线:400-828-1622苏州纳微科技有限公司中文网站:English Website: /E-mail:info@公司总部地址:苏州工业园区百川街2号苏州中国通用缩略术语:280 nm:在指定波长的紫外吸收M:物质的量的浓度单位,mol/l的简写mM: 物质的量的浓度单位,mmol/l的简写BV:柱体积,bed volume的简写Mr:相对分子量MPa:兆帕斯卡Bar:压强单位,工程上“公斤力”的单位psi:压强单位,磅每平方英寸压强单位之间换算:1 MPa = 10 bar ≈ 145 psi索引目录1. 标签蛋白纯化亲和层析产品简介 (4)2. 层析纯化操作步骤 (4)2.1 层析柱装填 (4)2.2 柱效评价 (5)2.3 清洗 (5)2.4 平衡 (5)2.5 金属螯合 (5)2.6上样 (6)2.7洗脱 (6)2.8 再生 (6)2.9 在位清洗 (6)3. 储存 (7)4. 故障排除 (7)5. 订购信息 (8)1.标签蛋白纯化亲和层析产品简介重组蛋白的富集和纯化一般采用已知大小的标签,通过重组表达之后,利用标签和填料的特异性相互作用,达到分离纯化效果。
固定金属离子或金属螯合亲和层析(MAC)利用蛋白表面的某些氨基酸(如组氨酸、色氨酸、半胱氨酸等)和固定在层析介质上的过渡金属离子(Ni2+,Cu2+,Zn2+,Co2+,Fe3+等)以配位键结合,从而实现对不同标签蛋白质的分离。
影响蛋白质与金属离子鳌合作用大小的因素,主要是蛋白质表面可结合的氨基酸(种类、数目和分布)、金属离子的种类和密度、层析条件(pH、盐的种类和浓度、添加剂等)。
固定金属离子亲和层析介质具有吸附容量大、选择性好、分辨率高、易于再生、成本较低等优点,广泛用于生物制药和生物制品的下游蛋白质、核酸及多肽的分离纯化,尤其是组氨酸标记(His-Tag)蛋白质的分离纯化。
His·Tag_融合蛋白纯化操作手册

50ml培养液采用2.5ml抽提试剂。如果是小规模培养,则采用约1/5培养体积的抽提试剂重悬沉淀(例如, 1.5ml培养液采用300µl抽提试剂)。如果抽提试剂过量也没有什么副作用。 选做:加蛋白酶抑制剂。BugBuster Master Mix与蛋白酶抑制剂兼容。如果目的蛋白后续要用凝血酶(货号 69671),Xa因子(货号69036)或重组肠激酶(货号69066)处理,就应该避免使用丝氨酸蛋白酶抑制剂。尽 管纯化过程可能去除活性抑制剂,建议在酶切前最好做透析或凝胶过滤。 3. 室温下将重悬的细胞液在摇板或低速搅拌器上孵育10-20分钟。 注意:孵育后获得的抽提物不是粘稠的。 4. 4℃下16,000g离心20分钟以去除不溶的细胞碎片。如果需要,沉淀可以留作“包涵体纯化”(见以下说明) 的材料。 5. 将上清转入另一个新试管。这样抽提得到的可溶蛋白溶液可以直接上样于Novagen的纯化树脂(以及其它很 多类似纯化系统)。蛋白溶液在冰上可以短时存放(2-3小时),也可在-20℃长时间存放直至下步分析。蛋 白抽提液应该根据目的蛋白的活性要求的温度存放,有些蛋白经冻融会失活。
25µg
50×
0.1mg/ L
0.8%
100ml
10g
100×
天然条件
>1mg/ L
>1%
50ml
>50g
50×
<1mg/ L
<1%
100ml
<100g
100×
与其它亲和纯化介质一样,His·Bind 树脂在接近其结合载量时使用,可以获得最好蛋白分离效果。所以在蛋白纯化前估计细菌 抽提物中目的蛋白的含量,有利于确定上样量、选择合适体积的亲和树脂或预装柱。SDS-PAGE,Western blot,S·TagTM Rapid Assay,FRETWorksTM S·Tag Assay 等方法都可用于确定抽提物中目的蛋白的含量。
his标签蛋白纯化说明

His标签重组蛋白纯化常规方法参考目的蛋白预测等电点选择PBS或Tris-HCl溶液做为超声破碎缓冲体系,以下操作步骤以PBS缓冲体系为例:1.离心收菌1.1离心机开机预热5min,设置参数:4号转子、6500rpm、30℃、2min;1.2取诱导培养后新鲜菌液45ml左右倒入50ml离心管中,配平,离心,弃上清;1.3重复步骤1.2,至菌液内菌体全部收集于离心管中,-20℃保存。
1.4保存时间不宜超过10天。
2.超声破碎2.1取冻存于50ml离心管中的菌液沉淀加入25ml 1×PBS 移液枪吹吸沉淀至悬浮;2.2冰浴,超声破碎,参数:6号超声探头,超声3s,间歇3s,功率55%,时间15min,报警温度60℃;2.3超声破碎结束后观察菌液超声前后变化,离心收集上清,离心参数:4号转子、10 000rpm、4℃、10min;2.4转移上清液至50ml离心管,取上清样品28ul,加入7ul 5×SDS-PAGE loading buffer,吹吸混匀,此样品做为超声破碎后上清样品;2.5在离心沉淀中加入25ml PBS,移液枪吹吸混匀,取沉淀样品28ul,加入7ul 5×SDS-PAGE loading buffer,吹吸混匀,此样品做为超声破碎后沉淀样品。
3.亲和层析3.1处理Ni柱固定Ni柱及废液收集器皿,流出柱内20%乙醇,加入PBS 1CV清洗Ni柱。
3.2上柱取超声破碎后上清溶液上柱,控制流速在150cm/h,可适当采取注射器针、移液器枪头控制流速,接取流穿液样品28ul,加入7ul 5×SDS-PAGE loading buffer,吹吸混匀,此样品做为流穿样品。
3.3洗杂取PBS配制25mM咪唑溶液1-1.5CV清洗Ni柱,接取洗杂液样品28ul,加入7ul 5×SDS-PAGE loading buffer,吹吸混匀,此样品做为洗杂样品。
6X组氨酸标记(His-tag)蛋白纯化试剂盒产品说明书(中文
产品内容
平衡液(Reagent A) 亲和液(Reagent B) 洗脱液(Reagent C) 产品说明书
XX 毫升 XX 毫升 XX 毫升 1份
保存方式
保存在-20℃冰箱里;有效保证 6 月
用户自备
变性液(Reagent D):用于变性融合蛋白纯化 透析液(Reagent E):用于蛋白纯化后透析 细菌可溶性总蛋白样品:用于纯化的待测样品 1.5 毫升离心管:用于反应操作的容器 50 毫升锥形离心管:用于胶体溶液配制的容器 平式摇荡仪:用于混匀反应物 4℃台式离心机:用于沉淀反应物 4℃微型台式离心机:用于沉淀反应物
1
实验步骤
实验开始前,将试剂从-20℃冰箱里取出,在冰槽里融化。然后进行下列操作。
1. 从-70℃冰箱里取出用户自备的通过细菌扩增分离的细菌可溶性总蛋白样品 2. 置于室温下 15 分钟,直至融化(使蛋白重新折叠) 3. 移出 5 毫升样品(等于 1 升菌液萃取物)到 50 毫升锥形离心管 4. 加入 XX 毫升 平衡液(Reagent A),用手混匀 5. 加入 XX 毫升 亲和液(Reagent B),用手混匀 6. 平放在平式摇荡仪上,在 4℃温度下孵育 XX 分钟,速度为 XX RPM 7. 放进 4℃台式离心机离心 XX 分钟,速度为 XX g 8. 小心抽去上清液 9. 加入 XX 毫升 平衡液(Reagent A),用手混匀 10.放进 4℃台式离心机离心 XX 分钟,速度为 XX g 11.重复实验步骤 9 至 10 四次 12.加入 XX 毫升 洗脱液(Reagent C),用手混匀 13.平放在平式摇荡仪上,在 4℃温度下孵育 XX 分钟,速度为 XX RPM 14.转移到 1.5 毫升离心管 15.放进 4℃微型台式离心机离心 XX 分钟,速度为 XX g(或 XX RPM,例如 eppendorf 5415R) 16.小心移取上清液到预冷的新的 1.5 毫升离心管,置于冰槽里 17.或保存纯化的蛋白在-70℃冰箱里 18.或移取 5 微升进行 SDS-PAGE 电泳鉴定以及 BRADFORD 蛋白定量 19.(选择步骤)透析处理
His标签融合蛋白

默克生命科学服务热线:400 820 8872 bioteam@
高纯度包涵体的制备 以下操作可用于任何 BugBuster®系列产品抽提的包涵体纯化。 1. 如可溶蛋白抽提步骤1-4进行操作。 2. 将步骤“4”所得到的沉淀重悬于BugBuster(货号70584),BugBuster的量与当初重悬细胞糊的体积相同。
重复两次。再次重悬,于4℃,16,000g离心15分钟并去除上清。 7. 重悬最终的沉淀(即纯化的包涵体)于选定的缓冲液,最好是能与后续纯化方法兼容的缓冲液。包涵体可以
重悬于Novagen的蛋白重折叠试剂盒(货号70123)中的1×溶解缓冲液(参考操作手册TB234)或其它变性 剂。不溶的His•Tag®融合蛋白可以用含有变性剂的1×结合缓冲液重悬用于His•Bind®纯化(IDA或NTA)。
产品使用说明书
His·Tag 融合蛋白纯化操作手册
*建议同时参考本说明书英文原文(说明书编号TB273)。
采用 pET 系统进行原核蛋白表达,蛋白的表达量达到 20mg/100ml 培养物并不是困难的事。 在大肠杆菌中表达的目的蛋白,其可溶性(可溶蛋白或包涵体)、细胞定位(细胞质、细胞周 质、培养基上清),都会对后续的纯化策略造成影响。我们建议研究者在蛋白表达后,首先进 行目的蛋白的细胞定位(请参考 pET 系统操作手册);在进行大量纯化之前,小量纯化蛋 白,摸索确定适合于具体蛋白的纯化条件,也是值得推荐的好方法。外源蛋白在大肠杆菌中表 达,可能以可溶形式存在,也可能以包涵体形式存在。尤其在高水平表达的条件下,更容易形 成包涵体。包涵体的形成与外源蛋白本身性质、载体、宿主菌、以及表达水平都有关系,可以 通过选择不同表达载体和 E.coli 宿主菌组合,摸索生长条件和适宜诱导条件,达到优化蛋白表 达的目的。His·Tag®融合蛋白,可以在天然条件或变性条件下用 NTA His·Bind 树脂或 IDA His·Bind 树脂进行纯化。
histag蛋白纯化步骤

histag蛋白纯化步骤
His-tag 蛋白纯化是一种常用的蛋白质纯化方法,其基本步骤如下:
1. 表达和收集:通过基因工程技术在目标蛋白的 N 端或 C 端添加 His-tag 序列,并在适合的宿主细胞中表达目标蛋白。
收集表达后的细胞或细胞培养上清液。
2. 破碎细胞:使用适当的方法破碎表达细胞,以释放出目标蛋白。
3. 离心和过滤:对破碎后的细胞裂解液进行离心,去除细胞碎片和不溶性物质。
然后,通过过滤去除残留的固体杂质。
4. 结合:将离心和过滤后的上清液与含有镍离子的亲和层析树脂混合,使 His-tag 与树脂上的镍离子结合。
5. 洗涤:用适当的缓冲液洗涤树脂,以去除未结合的杂质。
6. 洗脱:使用含有咪唑或其他竞争配体的缓冲液洗脱结合在树脂上的目标蛋白。
7. 浓缩和透析:对洗脱下来的目标蛋白进行浓缩和透析,以去除残留的咪唑和其他杂质。
8. 纯度鉴定:通过 SDS-PAGE、Western blotting 等方法鉴定纯化后的目标蛋白的纯度。
9. 保存:根据需要,将纯化后的目标蛋白保存于适当的条件下,如缓冲液、冷冻保存等。
His-Tag蛋白纯化步骤

变性条件下从大肠杆菌中纯化多聚组氨酸标签蛋白(主要以包涵体的形式表达)的样品制备1、用1X PBS重悬细胞沉淀(约每毫升沉淀加5ml 1 MBS),并按上述方法进行超声破菌。
2、12000 rpm离心10 min收集包涵体。
若有必要,用1 >PBS洗包涵体几次。
3、用Binding/Wash Buffer (约每毫升沉淀加5ml 1 XPBS)溶解包涵体,并在室温下孵育30〜60分钟。
若使沉淀充分溶解,有必要进行机械或超声均质。
4、12000rpm离心30min,取上清至一干净管中。
His 标签蛋白的重力纯化流程1ml柱子的总体积为10ml,只需加入介质。
如果样品体积大于柱子体积,可重复利用,注意不要超过树脂的结合能力。
1、平衡柱子的工作温度。
应在室温或4C下进行纯化。
2、取出底帽,倒出多余的液体,直立固定好柱子,让柱子顶部朝上。
3、用2 倍树脂体积的Binding/Wash Buffer 平衡柱子,以0.5〜1 ml/min 的流速过柱。
4、从柱子上部加入经Binding/Wash Buffer 处理的大肠杆菌裂解物或蛋白提取物,收集流出液。
若需要,让流出液重新过柱一次,以最大限度地提高结合力。
5、用两倍树脂体积的Binding/Wash Buffer洗涤树脂并收集流出液。
重复该步骤,用一新的收集管收集流出液。
直到流出液的吸光度在280 nm基线处。
6、用两倍树脂体积的Elution Buffer 将His 标签蛋白从树脂上洗脱下来。
重复此步骤两次,并单独收集每次洗脱出来的液体。
7、用Modified Coomassie Bradford Assay Kit (No SK3041)。
洗脱的蛋白可直接进行SDS-PAGE 分析。
注意:洗脱获得的蛋白可用凝胶过滤(如No BSP090 gravity Desalting Column)或透析去除咪唑以便后续应用。
SDS-PAGE分析前,含6M盐酸胍的样品必须用含8 M 尿素的缓冲液透析。
his蛋白纯化操作步骤全
His蛋白的纯化一目的蛋白样液的收集1:-20度冰箱中取出pet30a-p26甘油菌2:菌种复苏,将5ul P26菌种加入到5ml K+ LB培养基中(1:1000加入卡纳抗生素的LB培养基),以未加菌的K+ LB培养基作空白对照,37度摇床培养12-16h。
3:扩大培养,将5ml过夜培养的P26菌液按照1:100的比例接种到500ml K+ LB培养基中,摇床培养至OD值为0.6(2h左右)4:诱导表达,扩培后的菌种中加入1mg/ml iptg,使iptg最终浓度1ug/ml(1mmol/l ) 37度摇床培养4h,置-70度保存或立即进行下步操作5:收集包涵体,离心,5000r 15min,弃去上清液(可置于-20度保存)6:溶解包涵体,用PBS重悬细胞沉淀(约每毫升沉淀加5mlPBS),放在冰上用超声波细胞粉碎机破菌(有效时间30-40min,超5s,停10s,及时换冰防止温度过高使蛋白质变性)7:蛋白提取液的收集,离心,5000r 15min取上清(可4度保存),菌体保存,测菌体中目的蛋白含量大小使用8:介质处理,取出底帽滴出多余液体,柱子顶部朝上直立固定好,蒸馏水洗涤一次,再用2倍树脂体积的1*binding buffer平衡柱子,最后用1*binding buffer配置成50%的树脂。
9:介质与核酸结合,从柱子上部加入蛋白提取液,4度旋转混匀1h,收集流出液(若需要流出液再重新过柱一次,以最大限度提高结合力)10:取10ml 1*binding buffer洗树脂,洗2次,流出液标记洗涤顺序保存11:样品的收集,取10个1.5mlEP管分别标记1,2,3……10,每次取1ml 1*elution buffer加入树脂内滴入标记1-10的EP管内每管6滴(第8管的目的蛋白含量已经很低,10管即可,若需要可适当增加)12:树脂的保存,若样品体积大于柱子体积,树脂10倍体积的PBS重新平衡后可马上使用,若不使用可用蒸馏水洗涤树脂,加入20%乙醇10ml -20度保存(一种蛋白树脂可重复使用,注意不要超过树脂的结合能力)二检测收集的样品中目的蛋白量的大小13:取13个1.5mlEP管,分别标记1#,2#,3#……10#,A,B,C从1号EP 管内取30ul样液于1#号EP管内,依次移取至10号于10#号,取30ul 10步中第一次收集的流出液于标记 A 的EP管中,取30ul第二次收集的流出液于标记 B 的EP管中,将7步中保留的菌体用少量pbs稀释并取30ul于标记C的EP管内。
His-tag 蛋白纯化磁珠-说明书

Service@
400-600-3979
BeaverBeads TM 金属离子螯合磁珠纯化组氨酸标签蛋白操作流程
细胞(菌体) 粗蛋白样品 重悬,裂解 + 加入Buffer A 3.目标蛋白结合 重悬后 翻转混合 30min 磁性分离
1.样品处理
2.磁珠预处理
4.磁珠洗涤
收集上清
目标蛋白量 2mg 样品体积 ~10 mL 5%磁珠悬液用量 1~2mL
产品特性
1. 基本信息
产品名称 货号 磁珠粒径范围 螯合金属离子 金属离子密度 蛋白结合量 注1 工作温度 储液浓度 保存液 保存温度 BeaverBeads Nickel 镍离子螯合磁珠 71205,71225,71275 Ni 2+ ~40 mg/mL(100%磁珠)
User Manual
BeaverBeads™ His-tag Protein Purification
海狸金属离子螯合磁珠是专为高效、快速纯化组氨酸标签蛋白而设计的一种新型功能化材料,可通过 磁性分离方式直接从生物粗样品中一步纯化出高纯度的目标蛋白,极大地简化纯化工艺和提高纯化效 率,适合科研和工业领域便捷地进行组氨酸标签蛋白的纯化工作。 与传统的柱层析纯化方式相比,采用海狸磁珠纯化组氨酸标签蛋白,无需对粗蛋白样品进行多次长时 间的高速离心,也不需要过滤操作;样品与磁珠的特异性结合、洗涤和目标蛋白洗脱也变得非常简 单、快速、易操作,无需控制流速,更不需要昂贵的层析设备。对于熟练的操作者来说,在一小时内 就能获得高纯度的目标蛋白,且能轻松实现多样品平行处理,实现高通量蛋白纯化。 本产品系列包括镍离子(Nickel)、钴离子(Cobalt)和铜离子(Copper)共三种金属离子的螯合磁 珠,它们在目标蛋白载量和非特异性吸附等性能上有所区别,用户可根据目标蛋白与不同种类金属离 子的结合性能,以及对目标蛋白的产量和纯度的要求,选择不同种类的金属离子螯合磁珠。具体产品 信息和规格参考本说明书的《相关产品信息表》。
cOmplete His-Tag Purification Resin中文说明书

cOmplete His-Tag Purification Resin 是一种即用型的镍离子螯合纯化树脂,其固相支持介 质为琼脂糖,适用于小规模至大规模的 His 标签融合蛋白纯化。能从粗制裂解液中一步 法纯化获得高纯度的蛋白。 cOmplete His-Tag Purification Resin 使用了一种新的化学螯合物和技术,使 Ni2+在树脂上 的结合更加牢固。与最常用的螯合物氮川乙酸(NTA)和亚氨二乙酸(IDA)不同,cOmplete His-Tag Purification Resin 使用的螯合物能保护 Ni2+免受巯醇还原,使 Ni2+的脱落可能性降 至最低(图 1)。因此,应用 His-Tag Purification Resin 进行蛋白纯化后的缓冲液和蛋白洗 脱液中几乎没有 Ni2+存在,Ni2+存在于蛋白溶液中将催化氧化反应,导致蛋白破坏。在 纯化反应后,无需进行 Ni2+去除。 与其他金属螯合蛋白纯化树脂不同的是,His-Tag Purification Resin 与含有 EDTA(金属螯 合蛋白酶抑制剂)、DTT(还原剂)的溶液完全兼容(图 1)。这意味着您可根据您的蛋 白特性,灵活地选择缓冲液条件和参数。
即使在严苛的缓冲条件下,5 次蛋白纯化操作后,cOmplete His-Tag Purification Resin 依 然可保持其蛋白结合载量,且无需进行镍离子的重新螯合。 cOmplete His-Tag Purification Resin 可以与罗氏蛋白酶抑制剂混合片剂 cOmplete ULTRA Table(t 含 EDTA,抑制金属蛋白酶活性),以及罗氏磷酸酶抑制剂混合片剂 PhosSTOP Tablet 一起使用,在纯化过程中,全面保护蛋白免受蛋白酶水解和磷酸化作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Instruction ManualProBond TM Purification SystemFor purification of polyhistidine-containing recombinant proteinsCatalog nos. K850-01, K851-01, K852-01, K853-01, K854-01,R801-01, R801-15Version K2 September200425-0006iiTable of ContentsKit Contents and Storage (iv)Accessory Products (vi)Introduction (1)Overview (1)Methods (2)Preparing Cell Lysates (2)Purification Procedure—Native Conditions (7)Purification Procedure—Denaturing Conditions (11)Purification Procedure—Hybrid Conditions (13)Troubleshooting (15)Appendix (17)Additional Protocols (17)Recipes (18)Frequently Asked Questions (21)References (22)Technical Service (23)iiiKit Contents and StorageTypes of Products This manual is supplied with the following products:Product CatalogNo.ProBond™ Purification System K850-01ProBond™ Purification System with Antibodywith Anti-Xpress™ Antibody K851-01with Anti-myc-HRP Antibody K852-01with Anti-His(C-term)-HRP Antibody K853-01with Anti-V5-HRP Antibody K854-01ProBond™ Nickel-Chelating Resin (50 ml) R801-01ProBond™ Nickel Chelating Resin (150 ml) R801-15ProBond™Purification System Components The ProBond™ Purification System includes enough resin, reagents, and columns for six purifications. The components are listed below. See next page for resin specifications.Component Composition Quantity ProBond™ Resin 50% slurry in 20% ethanol 12 ml5X NativePurification Buffer250 mM NaH2PO4, pH 8.02.5 M NaCl1 × 125 ml bottleGuanidinium LysisBuffer6 M Guanidine HCl20 mM sodium phosphate, pH 7.8500 mM NaCl1 × 60 ml bottleDenaturingBinding Buffer8 M Urea20 mM sodium phosphate, pH 7.8500 mM NaCl2 × 125 ml bottlesDenaturing WashBuffer8 M Urea20 mM sodium phosphate, pH 6.0500 mM NaCl2 × 125 ml bottlesDenaturing ElutionBuffer8 M Urea20 mM NaH2PO4, pH 4.0500 mM NaCl1 × 60 ml bottle3 M Imidazole,20 mM sodium phosphate, pH 6.0500 mM NaCl1 × 8 ml bottlePurificationColumns10 ml columns 6Continued on next pageivKit Contents and Storage, ContinuedProBond™Purification System with Antibody The ProBond™ Purification System with Antibody includes resin, reagents, and columns as described for the ProBond™ Purification System (previous page) and 50 µl of the appropriate purified mouse monoclonal antibody. Sufficient reagents are included to perform six purifications and 25 Western blots with the antibody.For more details on the antibody specificity, subclass, and protocols for using the antibody, refer to the antibody manual supplied with the system.Storage Store ProBond™ resin at +4°C. Store buffer and columns at room temperature.Store the antibody at 4°C. Avoid repeated freezing and thawing of theantibody as it may result in loss of activity.The product is guaranteed for 6 months when stored properly.All native purification buffers are prepared from the 5X Native PurificationBuffer and the 3 M Imidazole, as described on page 7.The Denaturing Wash Buffer pH 5.3 is prepared from the Denaturing WashBuffer (pH 6.0), as described on page 11.Resin and ColumnSpecificationsProBond™ resin is precharged with Ni2+ ions and appears blue in color. It isprovided as a 50% slurry in 20% ethanol.ProBond™ resin and purification columns have the following specifications:• Binding capacity of ProBond™ resin: 1–5 mg of protein per ml of resin• Average bead size: 45–165 microns• Pore size of purification columns: 30–35 microns• Recommended flow rate: 0.5 ml/min• Maximum flow rate: 2 ml/min• Maximum linear flow rate: 700 cm/h• Column material: Polypropylene• pH stability (long term): pH 3–13• pH stability (short term): pH 2–14ProductQualificationThe ProBond™ Purification System is qualified by purifying 2 mg of myoglobinprotein on a column and performing a Bradford assay. Protein recovery mustbe 75% or higher.vAccessory ProductsAdditionalProductsThe following products are also available for order from Invitrogen:Product QuantityCatalogNo.ProBond™ Nickel-Chelating Resin 50 ml150 mlR801-01R801-15Polypropylene columns(empty)50 R640-50Ni-NTA Agarose 10 ml25 ml R901-01 R901-15Ni-NTA Purification System 6 purifications K950-01 Ni-NTA Purification Systemwith Antibodywith Anti-Xpress™ Antibody with Anti-myc-HRP Antibody with Anti-His(C-term)-HRP Antibodywith Anti-V5-HRP Antibody 1 kit1 kit1 kit1 kitK951-01K952-01K953-01K954-01Anti-myc Antibody 50 µl R950-25 Anti-V5 Antibody 50 µl R960-25 Anti-Xpress™ Antibody 50 µl R910-25 Anti-His(C-term) Antibody 50 µl R930-25 InVision™ His-tag In-gel Stain 500 ml LC6030 InVision™ His-tag In-gelStaining Kit1 kit LC6033Pre-Cast Gels and Pre-made Buffers A large variety of pre-cast gels for SDS-PAGE and pre-made buffers for your convenience are available from Invitrogen. For details, visit our web site at or contact Technical Service (page 23).viIntroductionOverviewIntroduction The ProBond™ Purification System is designed for purification of 6xHis-tagged recombinant proteins expressed in bacteria, insect, and mammalian cells. Thesystem is designed around the high affinity and selectivity of ProBond™Nickel-Chelating Resin for recombinant fusion proteins containing six tandemhistidine residues.The ProBond™ Purification System is a complete system that includespurification buffers and resin for purifying proteins under native, denaturing,or hybrid conditions. The resulting proteins are ready for use in many targetapplications.This manual is designed to provide generic protocols that can be adapted foryour particular proteins. The optimal purification parameters will vary witheach protein being purified.ProBond™ Nickel-Chelating Resin ProBond™ Nickel-Chelating Resin is used for purification of recombinant proteins expressed in bacteria, insect, and mammalian cells from any 6xHis-tagged vector. ProBond™ Nickel-Chelating Resin exhibits high affinity and selectivity for 6xHis-tagged recombinant fusion proteins.Proteins can be purified under native, denaturing, or hybrid conditions using the ProBond™ Nickel-Chelating Resin. Proteins bound to the resin are eluted with low pH buffer or by competition with imidazole or histidine. The resulting proteins are ready for use in target applications.Binding Characteristics ProBond™ Nickel-Chelating Resin uses the chelating ligand iminodiacetic acid (IDA) in a highly cross-linked agarose matrix. IDA binds Ni2+ ions by three coordination sites.The protocols provided in this manual are generic, and may not result in 100%pure protein. These protocols should be optimized based on the bindingcharacteristics of your particular proteins.Native VersusDenaturingConditionsThe decision to purify your 6xHis-tagged fusion proteins under native ordenaturing conditions depends on the solubility of the protein and the need toretain biological activity for downstream applications.• Use native conditions if your protein is soluble (in the supernatant afterlysis) and you want to preserve protein activity.• Use denaturing conditions if the protein is insoluble (in the pellet afterlysis) or if your downstream application does not depend on proteinactivity.• Use hybrid protocol if your protein is insoluble but you want to preserveprotein activity. Using this protocol, you prepare the lysate and columnsunder denaturing conditions and then use native buffers during the washand elution steps to refold the protein. Note that this protocol may notrestore activity for all proteins. See page 14.1MethodsPreparing Cell LysatesIntroduction Instructions for preparing lysates from bacteria, insect, and mammalian cellsusing native or denaturing conditions are described below.Materials Needed You will need the following items:• Native Binding Buffer (recipe is on page 8) for preparing lysates undernative conditions• Sonicator• 10 µg/ml RNase and 5 µg/ml DNase I (optional)• Guanidinium Lysis Buffer (supplied with the system) for preparing lysatesunder denaturing conditions• 18-gauge needle• Centrifuge• Sterile, distilled water• SDS-PAGE sample buffer• Lysozyme for preparing bacterial cell lysates• Bestatin or Leupeptin, for preparing mammalian cell lysatesProcessing Higher Amount of Starting Material Instructions for preparing lysates from specific amount of starting material (bacteria, insect, and mammalian cells) and purification with 2 ml resin under native or denaturing conditions are described in this manual.If you wish to purify your protein of interest from higher amounts of starting material, you may need to optimize the lysis protocol and purification conditions (amount of resin used for binding). The optimization depends on the expected yield of your protein and amount of resin to use for purification. Perform a pilot experiment to optimize the purification conditions and then based on the pilot experiment results, scale-up accordingly.Continued on next page2Preparing Bacterial Cell Lysate—Native Conditions Follow the procedure below to prepare bacterial cell lysate under native conditions. Scale up or down as necessary.1. Harvest cells from a 50 ml culture by centrifugation (e.g., 5000 rpm for5 minutes in a Sorvall SS-34 rotor). Resuspend the cells in 8 ml NativeBinding Buffer (recipe on page 8).2. Add 8 mg lysozyme and incubate on ice for 30 minutes.3. Using a sonicator equipped with a microtip, sonicate the solution on iceusing six 10-second bursts at high intensity with a 10-second coolingperiod between each burst.Alternatively, sonicate the solution on ice using two or three 10-secondbursts at medium intensity, then flash freeze the lysate in liquid nitrogen or a methanol dry ice slurry. Quickly thaw the lysate at 37°C andperform two more rapid sonicate-freeze-thaw cycles.4. Optional: If the lysate is very viscous, add RNase A (10 µg/ml) andDNase I (5 µg/ml) and incubate on ice for 10–15 minutes. Alternatively,draw the lysate through a 18-gauge syringe needle several times.5. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.Note: Some 6xHis-tagged protein may remain insoluble in the pellet, and can be recovered by preparing a denatured lysate (page 4) followed bythe denaturing purification protocol (page 12). To recover this insolubleprotein while preserving its biological activity, you can prepare thedenatured lysate and then follow the hybrid protocol on page 14. Notethat the hybrid protocol may not restore activity in all cases, and should be tested with your particular protein.6. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or freeze at -20°C. When ready to use, proceed to theprotocol on page 7.Continued on next page3Preparing Bacterial Cell Lysate—Denaturing Conditions Follow the procedure below to prepare bacterial cell lysate under denaturing conditions:1. Equilibrate the Guanidinium Lysis Buffer, pH 7.8 (supplied with thesystem or see page 19 for recipe) to 37°C.2. Harvest cells from a 50 ml culture by centrifugation (e.g., 5000 rpm for5 minutes in a Sorvall SS-34 rotor).3. Resuspend the cell pellet in 8 ml Guanidinium Lysis Buffer from Step 1.4. Slowly rock the cells for 5–10 minutes at room temperature to ensurethorough cell lysis.5. Sonicate the cell lysate on ice with three 5-second pulses at high intensity.6. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris.Transfer the supernatant to a fresh tube.7. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or at -20°C. When ready to use, proceed to the denaturingprotocol on page 11 or hybrid protocol on page 13.Note: To perform SDS-PAGE with samples in Guanidinium Lysis Buffer, you need to dilute the samples, dialyze the samples, or perform TCAprecipitation prior to SDS-PAGE to prevent the precipitation of SDS.Harvesting Insect Cells For detailed protocols dealing with insect cell expression, consult the manual for your particular system. The following lysate protocols are for baculovirus-infected cells and are intended to be highly generic. They should be optimized for your cell lines.For baculovirus-infected insect cells, when the time point of maximal expression has been determined, large scale protein expression can be carried out. Generally, the large-scale expression is performed in 1 liter flasks seeded with cells at a density of 2 × 106 cells/ml in a total volume of 500 ml and infected with high titer viral stock at an MOI of 10 pfu/cell. At the point of maximal expression, harvest cells in 50 ml aliquots. Pellet the cells by centrifugation and store at -70°C until needed. Proceed to preparing cell lysates using native or denaturing conditions as described on the next page.Continued on next page4Preparing Insect Cell Lysate—Native Condition 1. Prepare 8 ml Native Binding Buffer (recipe on page 8) containingLeupeptin (a protease inhibitor) at a concentration of 0.5 µg/ml.2. After harvesting the cells (previous page), resuspend the cell pellet in8 ml Native Binding Buffer containing 0.5 µg/ml Leupeptin.3. Lyse the cells by two freeze-thaw cycles using a liquid nitrogen or dryice/ethanol bath and a 42°C water bath.4. Shear DNA by passing the preparation through an 18-gauge needle fourtimes.5. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris.Transfer the supernatant to a fresh tube.6. Remove 5 µl of the lysate for SDS-PAGE analysis. Store remaining lysateon ice or freeze at -20°C. When ready to use, proceed to the protocol on page 7.Preparing Insect Cell Lysate—Denaturing Condition 1. After harvesting insect cells (previous page), resuspend the cell pellet in8 ml Guanidinium Lysis Buffer (supplied with the system or see page 19for recipe).2. Pass the preparation through an 18-gauge needle four times.3. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.4. Remove 5 µl of the lysate for SDS-PAGE analysis. Store remaining lysateon ice or freeze at -20° C. When ready to use, proceed to the denaturingprotocol on page 11 or hybrid protocol on page 13.Note: To perform SDS-PAGE with samples in Guanidinium Lysis Buffer, you need to dilute the samples, dialyze the samples, or perform TCAprecipitation prior to SDS-PAGE to prevent the precipitation of SDS.Continued on next pagePreparing Mammalian Cell Lysate—Native Conditions For detailed protocols dealing with mammalian expression, consult the manual for your particular system. The following protocols are intended to be highly generic, and should be optimized for your cell lines.To produce recombinant protein, you need between 5 x 106and 1 x 107 cells. Seed cells and grow in the appropriate medium until they are 80–90% confluent. Harvest cells by trypsinization. You can freeze the cell pellet in liquid nitrogen and store at -70°C until use.1. Resuspend the cell pellet in 8 ml of Native Binding Buffer (page 8). Theaddition of protease inhibitors such as bestatin and leupeptin may benecessary depending on the cell line and expressed protein.2. Lyse the cells by two freeze-thaw cycles using a liquid nitrogen or dryice/ethanol bath and a 42°C water bath.3. Shear the DNA by passing the preparation through an 18-gauge needlefour times.4. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.5. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or freeze at -20° C. When ready to use, proceed to theprotocol on page 7.Preparing Mammalian Cell Lysates—Denaturing Conditions For detailed protocols dealing with mammalian expression, consult the manual for your particular system. The following protocols are intended to be highly generic, and should be optimized for your cell lines.To produce recombinant protein, you need between 5 x 106and 1 x 107 cells. Seed cells and grow in the appropriate medium until they are 80–90% confluent. Harvest cells by trypsinization. You can freeze the cell pellet in liquid nitrogen and store at -70°C until use.1. Resuspend the cell pellet in 8 ml Guanidinium Lysis Buffer (suppliedwith the system or see page 19 for recipe).2. Shear the DNA by passing the preparation through an 18-gauge needlefour times.3. Centrifuge the lysate at 3,000 ×g for 15 minutes to pellet the cellulardebris. Transfer the supernatant to a fresh tube.4. Remove 5 µl of the lysate for SDS-PAGE analysis. Store the remaininglysate on ice or freeze at -20° C until use. When ready to use, proceed to the denaturing protocol on page 11 or hybrid protocol on page 13.Note: To perform SDS-PAGE with samples in Guanidinium Lysis Buffer, you need to dilute the samples, dialyze the samples, or perform TCAprecipitation prior to SDS-PAGE to prevent the precipitation of SDS.Purification Procedure—Native ConditionsIntroduction In the following procedure, use the prepared Native Binding Buffer, NativeWash Buffer, and Native Elution Buffer, columns, and cell lysate preparedunder native conditions. Be sure to check the pH of your buffers before starting.Buffers for Native Purification All buffers for purification under native conditions are prepared from the5X Native Purification Buffer supplied with the system. Dilute and adjust the pH of the 5X Native Purification Buffer to create 1X Native Purification Buffer (page 8). From this, you can create the following buffers:• Native Binding Buffer• Native Wash Buffer• Native Elution BufferThe recipes described in this section will create sufficient buffers to perform one native purification using one kit-supplied purification column. Scale up accordingly.If you are preparing your own buffers, see page 18 for recipe.Materials Needed You will need the following items:• 5X Native Purification Buffer (supplied with the system or see page 18 forrecipe)• 3 M Imidazole (supplied with the system or see page 18 for recipe)• NaOH• HCl• Sterile distilled water• Prepared ProBond™ columns with native buffers (next page)• Lysate prepared under native conditions (page 2)Imidazole Concentration in Native Buffers Imidazole is included in the Native Wash and Elution Buffers to minimize the binding of untagged, contaminating proteins and increase the purity of the target protein with fewer wash steps. Note that, if your level of contaminating proteins is high, you may add imidazole to the Native Binding Buffer.If your protein does not bind well under these conditions, you can experiment with lowering or eliminating the imidazole in the buffers and increasing the number of wash and elution steps.Continued on next page1X Native Purification Buffer To prepare 100 ml 1X Native Purification Buffer, combine:• 80 ml of sterile distilled water• 20 ml of 5X Native Purification Buffer (supplied with the system or see page 18 for recipe)Mix well and adjust pH to 8.0 with NaOH or HCl.Native Binding Buffer Without ImidazoleUse 30 ml of the 1X Native Purification Buffer (see above for recipe) for use as the Native Binding Buffer (used for column preparation, cell lysis, and binding).With Imidazole (Optional):You can prepare the Native Binding Buffer with imidazole to reduce the binding of contaminating proteins. (Note that some His-tagged proteins may not bind under these conditions.).To prepare 30 ml Native Binding Buffer with 10 mM imidazole, combine: • 30 ml of 1X Native Purification Buffer• 100 µl of 3 M Imidazole, pH 6.0Mix well and adjust pH to 8.0 with NaOH or HCl.Native Wash Buffer To prepare 50 ml Native Wash Buffer with 20 mM imidazole, combine:• 50 ml of 1X Native Purification Buffer• 335 µl of 3 M Imidazole, pH 6.0Mix well and adjust pH to 8.0 with NaOH or HCl.Native Elution Buffer To prepare 15 ml Native Elution Buffer with 250 mM imidazole, combine:• 13.75 ml of 1X Native Purification Buffer• 1.25 ml of 3 M Imidazole, pH 6.0Mix well and adjust pH to 8.0 with NaOH or HCl.Continued on next pageDo not use strong reducing agents such as DTT with ProBond™ columns. DTTreduces the nickel ions in the resin. In addition, do not use strong chelatingagents such as EDTA or EGTA in the loading buffers or wash buffers, as thesewill strip the nickel from the columns.Be sure to check the pH of your buffers before starting.PreparingProBond™ ColumnWhen preparing a column as described below, make sure that the snap-off capat the bottom of the column remains intact. To prepare a column:1. Resuspend the ProBond™ resin in its bottle by inverting and gentlytapping the bottle repeatedly.2. Pipet or pour 2 ml of the resin into a 10-ml Purification Columnsupplied with the kit. Allow the resin to settle completely by gravity(5-10 minutes) or gently pellet it by low-speed centrifugation (1 minuteat 800 ×g). Gently aspirate the supernatant.3. Add 6 ml of sterile, distilled water and resuspend the resin byalternately inverting and gently tapping the column.4. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant.5. For purification under Native Conditions, add 6 ml Native BindingBuffer (recipe on page 8).6. Resuspend the resin by alternately inverting and gently tapping thecolumn.7. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant.8. Repeat Steps 5 through 7.Storing PreparedColumnsTo store a column containing resin, add 0.02% azide or 20% ethanol as apreservative and cap or parafilm the column. Store at room temperature.Continued on next pagePurification Under Native Conditions Using the native buffers, columns and cell lysate, follow the procedure below to purify proteins under native conditions:1. Add 8 ml of lysate prepared under native conditions to a preparedPurification Column (page 9).2. Bind for 30–60 minutes using gentle agitation to keep the resinsuspended in the lysate solution.3. Settle the resin by gravity or low speed centrifugation (800 ×g), andcarefully aspirate the supernatant. Save supernatant at 4°C forSDS-PAGE analysis.4. Wash with 8 ml Native Wash Buffer (page 8). Settle the resin by gravityor low speed centrifugation (800 ×g), and carefully aspirate thesupernatant. Save supernatant at 4°C for SDS-PAGE analysis.5. Repeat Step 4 three more times.6. Clamp the column in a vertical position and snap off the cap on thelower end. Elute the protein with 8–12 ml Native Elution Buffer (seepage 2). Collect 1 ml fractions and analyze with SDS-PAGE.Note: Store the eluted fractions at 4°C. If -20°C storage is required, addglycerol to the fractions. For long term storage, add protease inhibitors to the fractions.If you wish to reuse the resin to purify the same recombinant protein, wash the resin with 0.5 M NaOH for 30 minutes and equilibrate the resin in a suitable binding buffer. If you need to recharge the resin, see page 17.Purification Procedure—Denaturing ConditionsIntroduction Instructions to perform purification using denaturing conditions with prepareddenaturing buffers, columns, and cell lysate are described below.Materials Needed You will need the following items:• Denaturing Binding Buffer (supplied with the system or see page 19 forrecipe)• Denaturing Wash Buffer, pH 6.0 (supplied with the system or see page 19 forrecipe) and Denaturing Wash Buffer, pH 5.3 (see recipe below)• Denaturing Elution Buffer (supplied with the system or see page 20 forrecipe)• Prepared ProBond™ columns with Denaturing buffers (see below)• Lysate prepared under denaturing conditions (page 11)Preparing the Denaturing Wash Buffer pH 5.3 Using a 10 ml aliquot of the kit-supplied Denaturing Wash Buffer (pH 6.0), mix well, and adjust the pH to 5.3 using HCl. Use this for the Denaturing Wash Buffer pH 5.3 in Step 5 next page.Be sure to check the pH of your buffers before starting. Note that thedenaturing buffers containing urea will become more basic over time. PreparingProBond™ ColumnWhen preparing a column as described below, make sure that the snap-off capat the bottom of the column remains intact.If you are reusing the ProBond™ resin, see page 17 for recharging protocol.To prepare a column:1. Resuspend the ProBond™ resin in its bottle by inverting and gentlytapping the bottle repeatedly.2. Pipet or pour 2 ml of the resin into a 10-ml Purification Columnsupplied with the kit. Allow the resin to settle completely by gravity(5-10 minutes) or gently pellet it by low-speed centrifugation (1 minuteat 800 ×g). Gently aspirate the supernatant.3. Add 6 ml of sterile, distilled water and resuspend the resin byalternately inverting and gently tapping the column.4. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant.5. For purification under Denaturing Conditions, add 6 ml of DenaturingBinding Buffer.6. Resuspend the resin by alternately inverting and gently tapping thecolumn.7. Allow the resin to settle using gravity or centrifugation as described inStep 2, and gently aspirate the supernatant. Repeat Steps 5 through 7.Continued on next pagePurification Procedure—Denaturing Conditions, ContinuedPurification Under Denaturing Conditions Using the denaturing buffers, columns, and cell lysate, follow the procedure below to purify proteins under denaturing conditions:1. Add 8 ml lysate prepared under denaturing conditions to a preparedPurification Column (page 11).2. Bind for 15–30 minutes at room temperature using gentle agitation (e.g.,using a rotating wheel) to keep the resin suspended in the lysatesolution. Settle the resin by gravity or low speed centrifugation (800 ×g), and carefully aspirate the supernatant.3. Wash the column with 4 ml Denaturing Binding Buffer supplied with thekit by resuspending the resin and rocking for two minutes. Settle theresin by gravity or low speed centrifugation (800 ×g), and carefullyaspirate the supernatant. Save supernatant at 4°C for SDS-PAGEanalysis. Repeat this step one more time.4. Wash the column with 4 ml Denaturing Wash Buffer, pH 6.0 supplied inthe kit by resuspending the resin and rocking for two minutes. Settle the resin by gravity or low speed centrifugation (800 ×g), and carefullyaspirate the supernatant. Save supernatant at 4°C for SDS-PAGEanalysis. Repeat this step one more time.5. Wash the column with 4 ml Denaturing Wash Buffer pH 5.3 (see recipeon previous page) by resuspending the resin and rocking for 2 minutes.Settle the resin by gravity or low speed centrifugation (800 ×g), andcarefully aspirate the supernatant. Save supernatant at 4°C for SDS-PAGE analysis. Repeat this step once more for a total of two washes with Denaturing Wash Buffer pH 5.3.6. Clamp the column in a vertical position and snap off the cap on thelower end. Elute the protein by adding 5 ml Denaturing Elution Buffersupplied with the kit. Collect 1 ml fractions and monitor the elution bytaking OD280readings of the fractions. Pool the fractions that contain the peak absorbance and dialyze against 10 mM Tris, pH 8.0, 0.1% Triton X-100 overnight at 4°C to remove the urea. Concentrate the dialyzedmaterial by any standard method (i.e., using 10,000 MW cut-off, low-protein binding centrifugal instruments or vacuum concentrationinstruments).If you wish to reuse the resin to purify the same recombinant protein, wash the resin with 0.5 M NaOH for 30 minutes and equilibrate the resin in a suitable binding buffer. If you need to recharge the resin, see page 17.。