紫外吸收光谱1
第二章 可见紫外吸收光谱分析1

由于玻璃可吸收紫外光,所以玻璃棱镜只能用于
用于可见光域内。 石英棱镜可使用的波长范围较宽,可从185- 4000nm,即可用于紫外、可见和近红外三个光域。
光栅是利用光的衍射与干涉作用制成的。
它可用于紫外、可见及红外光域,而且
在整个波长区具有良好的、几乎均匀一 致的分辨能力。
它具有色散波长范围宽、分辨本领高、 成本低、便于保存和易于制备等优点。 缺点是各级光谱会重叠而产生干扰。
它是分光光度法定量分析的依据。
吸光系数
朗伯-比耳定律中,当c以克/升,液层厚 度b以厘米表示时,常数K以a表示,称 为吸光系数。 a的单位为升/克.厘米。 朗伯-比耳定律 :A=abc
摩尔吸光系数
朗伯-比耳定律中,浓度用摩尔/升,液 层厚度b用厘米为单位表示,则K用另一 符号ε来表示。 ε称为摩尔吸光系数(或克分子消光系数), 单位为升/摩尔.厘米。 它表示物质的浓度为1摩尔/升,液层厚 度为1厘米时溶液的吸光度。 朗伯-比耳定律 : A=εbc
72型 721型
751型 WFD-8型
760 40000
~
硅碳棒或 辉光灯
岩盐或萤 石棱镜
WFD-3型 WFD-7型
一、组成部件
光源
单色器
样品池
记录装置
检测器
(一)光源
对光源的基本要求是应在仪器操作所 需的光谱区域内能够发射连续辐射,有足 够的辐射强度和良好的稳定性,而且辐射 能量随波长的变化应尽可能小。 常用的光源有热辐射光源(如钨丝灯 和卤钨灯)和气体放电光源(如氢灯和氘 灯)两类。
1)非单色光的影响: 光吸收定律的重要前提是入射光
紫外吸收光谱(UV)
2.3 有机化合物基团分类
1)发色基团(200-400nm产生吸收的基团) n *, *
C
C, C
O ,
C
N,
O N O
*,n *
2)非发色基团(200-400nm不产生吸收的基团)
3)助色基团 本身为非发色基团,使发色基团吸收位置 移向 长波。 n *.助色能力 –F<CH3<-Cl<-Br<OH<OCH3<-NH2< -NHCH<-N(CH3)2<-NHC5H6<O-
⑴无环、非稠环二烯母体:
max= 基+nii
基-----是由非环或六环共轭二烯母体决定的基准值
max=217 nm
⑵异环(稠环)二烯母体:
max=214 nm
⑶同环(非稠环或稠环)二烯母体:
max=253 nm nii---是由双键上取代基种类和个数决定的校正项
190
210
230
250
270
290
310
330
350
370
¨¤ ² ³ nm
2)脂肪烃不饱和化合物
H
ⅰ单烯烃 C=C
H c c H
发色基团, 但
* 200nm.
max=162nm
助色基团取代
-OR 30(nm)
H
* (K带)发生红
-Cl 5(nm) CH3 5(nm)
移。
取代基 -SR -NR2 40(nm)
350
400
0.1 弱带
红位移——向长波长位移
兰位移——向短波长位移 浓色效应——摩尔吸光系数max增加 浅色效应——摩尔吸光系数max减少 强带——max≥ 104(多为允许跃迁)
紫外吸收光谱分析1

n → π*跃迁:兰移; λ↓ ;ε↑ 兰移; 兰移
π → π*跃迁:红移; λ↑;ε↓ λ↑;
21:29:56
1
1:正己烷 2:水 2
250 300 异丙叉丙酮 非极性 → 极性 兰移; 兰移 极性溶剂使精细结构消失; n → π*跃迁:兰移; λ↓ ;ε↑ π → π*跃迁:红移; λ↑;ε↓ λ↑;
[Fe3+CNS-]2+
电子接受体
电子给予体
21:29:56
分子内氧化还原反应; 分子内氧化还原反应 特点:ε > 104 ,吸收峰在紫外区 Fe2+与邻菲罗啉配合物的紫外吸收光谱属于此。
2.配体场跃迁 配体场跃迁 d-d电子跃迁和 f - f 电子跃迁 电子跃迁和
在配体的作用下,过渡金属离子 的d轨道和镧系、锕系的f 轨道裂分, 吸收辐射后,产生d一d、 f 一f 跃迁; 一 必须在配体的配位场作用下才可能 产生也称配位场跃迁 配位场跃迁; 配位场跃迁 特点:ε〈 100,吸收峰在可见区
第三节
无机化合物的紫外可见吸收光谱
电子跃迁形式: 电子跃迁形式:电荷迁移跃迁与配位场跃迁 1.电荷迁移跃迁 1.电荷迁移跃迁
吸收光辐射后,分子中原定域在配位体L轨道的电子转移 到金属M轨道上,或按相反方向转移,所产生的吸收光谱称 为荷移光谱 荷移光谱。 荷移光谱 Mn+—Lbhν hν M(n-1) +—L(b-1) [Fe2+CNS]2+
21:29:56
21:29:56
λmax(正己烷) λmax(氯仿) λmax(甲醇) λmax(水)
π→π n→π
230 329
21:29:56
238 315
有机化合物的紫外吸收光谱

直链共轭二烯基本值
217
非骈环共轭双烯
217
烷基或环残余取代
5
环外双键
5
卤素取代
17
CH3 CH2=C-C=CH2
CH3
基本值 烷基取代 计算值
测量值
217nm 2× 5nm
227nm 226nm
H3C
1
3
4
C
CH3
2
CH3
基本值 烷基取代 环外双键
计算值 测量值
217 4× 5nm
5nm 242nm 243nm
溶剂 修正值 溶剂 修正值
溶 剂
水 -8nm 乙醚 +7nm
修 正
甲醇
0 正己烷 +11nm
值
氯仿 +1nm 环己烷 +11nm
二氧六环 +5nm
(CH3)2C=CHCOCH3
计算值 甲醇中的测定值 己烷中的测定值
239nm 237nm 230nm (230+11=241nm)
B、α、β不饱和羧酸及酯吸收波长的计算方法
R2 -C6H4 -COR
R1为烷基时的基本值 R1为H时的基本值 R1为OH时的基本值 R2为下列基团时
烷基
-OH -OR
-O-
-Cl
-Br
-NH2 -NHAc
-NR2
K吸收带波长λ/nm
246 250
230 邻位 间位 对位
3
3 10
7
7
25
11
20 78
0
0
10
2
2
15
13 13 58
20 20 45 20 20 85
2、α、β不饱和羰基化合物π→π*跃迁的吸收波长计算办法
紫外吸收光谱

正己烷
CH3Cl 315nm
CH3OH
水
π→π*跃迁
n →π*跃迁
230nm
329nm
238nm
237nm
309nm
243nm
305nm
π*
Δ E n < ΔE p C O
ΔE n>Δ Ep
π*
ΔE n Δ Ep
C+
C-
ΔE n
Δ Ep
n
C+
O极性
π
C C 极性 非极性
非极性
溶剂极性效应
微粒理论(光子的量子化理论):电磁波的 能量E 可用下式表示: E=hν=hc/λ h-普朗克常数=6.625×10-34J· s E=Ee+Eν+Er Ee—电子能 1~20eV Eν—振动能 0.05~1 eV Er—转动能 10-4~0.05 eV
(3)吸收光谱的表示方法
当l以cm,c以g/L为单位,k称为吸光系数, 用 a表示。 A= a cl a的单位为L/(g.cm) 当l以cm,c以mol/L为单位,k称为摩尔吸光 系数,用 ε表示。 ε的单位为L/mol.cm,它表示物质的浓度为 1mol/L,液层厚度为1cm时,溶液的吸光度。
朗伯-比耳定律成立的前提条件: a) 入射光为单色光; b) 吸收发生在均匀的介质中; c) 在吸收过程中,吸收物质互相不发生作用。 朗伯-比耳定律偏离线性的原因:化学因素、 仪器因素 。 a) 样品浓度过高(>0.01mol/L); b) 溶液中粒子的散射; c) 入射光的非单色性; ④ 对数吸光系数lgε; ⑤ 吸光率A(%) A(%)=1-T(%)
第一章 紫外光谱

精选2021版课件
18Eσ* Nhomakorabeaπ*
π
* 4
π
* 3
n
π
π2
π1
σ
C-C C=C C=O
C=C-C=C
能级跃迁图
精选2021版课件
19
三、 分子吸收光谱的表达(紫外光谱图)
UV:A~λ;IR:T~ v 有时仅记录吸收峰的相关参数:λmax和εmax
Ultraviolet Absorption Spectrometry
3. B吸收带(Benzenoid):苯环π→π*跃迁产生,
230-270nm , 中 心 在 256nm 处 , 宽 而 弱 , 有 精 细 结
构,是苯环的特征吸收ε约220
4. E吸收带(Ethylenic):芳环中碳碳双键π→π* 跃迁产生,在184(E1)( ε约60000)和204(E2)nm 处( ε约7900)。
精选2021版课件
10
二、分子能级图
1. 分子能级
分子的总能量:
E = Et + Ee + Ev + Er 其中:Et(平动动能)是连续的,分子光谱 主要取决于Ee(电子能量)、Ev(振动能量) 和Er(转动能量) 的变化,即:
E = Ee + Ev + Er 这些能量都是不连续的、量子化的
分子能级图:
吸收带: K带; R带 含硫化合物:类似于醇、醚和羰基化合物,
吸收带λmax较大。
精选2021版课件
52
二、共轭双键化合物
跃迁类型: σ→σ*;π→π*; (n →π*)
吸收谱带: K(、R)吸收带
Woodward等人提出了一套计算此类化 合物π→π*跃迁的λmax的方法,可用于确定 此类化合物的可能结构。
紫外吸收光谱1

第九章紫外吸收光谱1. 试简述产生吸收光谱的原因.基本要点:1. 分子吸收光谱;2. 有机化合物的紫外吸收光谱;3. 无机化合物的紫外吸收光谱;4. 溶剂对紫外吸收光谱的影响 ;5. 紫外吸收光谱的应用等 .利用紫外吸收光谱进行定量分析的由来已久,公元60年古希腊已知道利用五味子浸液来估计醋中铁的含量。
这一古老的方法由于最初是运用人的眼睛来进行检测,所以叫比色法。
20世纪30年代产生了第一台光电比色计,40年代出现的BakmanUV 分光光度计, 促进了新的分光光度计的发展。
随着计算机的发展,紫外分光光度计已向着微型化﹑自动化﹑在线和多组分同时测定等方向发展。
第一节分子吸收光谱Molecular Absorption Spectroscopy一、分子内部的运动及分子能级前面讲的AAS和AES都属与原子光谱,是由原子中电子能级跃迁所产生的。
原子光谱是由一条一条的彼此分离的谱线组成的线状光谱。
分子光谱比原子光谱要复杂得多。
这是由于在分子中,除了有电子相对于原子核的运动外,还有组成分子的各原子在其平衡位置附近的振动,以及分子本身绕其重心的转动。
如果考虑三种运动形式之间的相互作用,则分子总的能量可以认为是这三种运动能量之和。
即E=E e+ E v+ E r式中E e为电子能量,E v为振动能量,E r转动能量。
这三种不同形式的运动都对应一定的能级,即:分子中除了电子能级外,还有振动能级和转动能级这三种能级都是量子化的、不连续的。
正如原子有能级图一样,分子也有其特征的能级图。
简单双原子分子的能级图如图9-1所示。
A和B表示电子能级,间距最大;每个电子能级上又有许许多多的振动能级,用V'=0,1,2,……等表示A能级上个振动能级,V"=0,1,2,……等表示B能级上各振动能级;每个振动能级上又有许许多多的转动能级,用j'=0,1,2,……等表示A能级上V'=0各转动能级,j"= 0,1,2,……等表示A能级上V'=1各振动能级等等。
紫外-可见吸收光谱

若d轨道原来是未充满的,则可以吸收电磁波, 电子由低能级的d轨道跃迁到高能级的d*轨道而产 生吸收谱带。所以这类跃迁吸收能量较小,多出现 在可见光区。
分子轨道 对称 波函数ψ 操作
符号不变 符号改变
g型轨道,ψg,对称波函数 u型轨道,ψu,反对称波函数
允许跃迁要求电子只能在对称性不同性的不同能级之间跃迁
例:g→u 允许 g→g 禁阻 u→u 禁阻
1.3.1 有机化合物的电子跃迁类型
现以羰基C=O为例来说明电子跃迁类型。
HC O n
s
Hp
碳上2个电子,氧上4个电子,形成σ、π、n、π*、σ*轨道
即 E=Ee+Ev+Er ΔΕe>ΔΕv>ΔΕr
(1) ΔEr=0.005~0.050eV λ = 250~25μm 远红外光谱
(2) ΔEv=0.05~1eV λ = 25~1.25μm 红外吸收光谱
(3) ΔEe=1~20eV λ = 0.06~1.25μm 紫外-可见光谱
分子吸收 光谱的产生
紫外-可见吸收 光谱
组员:贺小云 吕丹丹
第一节 紫外光谱基本原理
1.1 概述 1.2 紫外可见吸收光谱的产生 1.3 电子的跃迁类型 1.4 常用光谱术语及谱带分类 1.5 影响因素
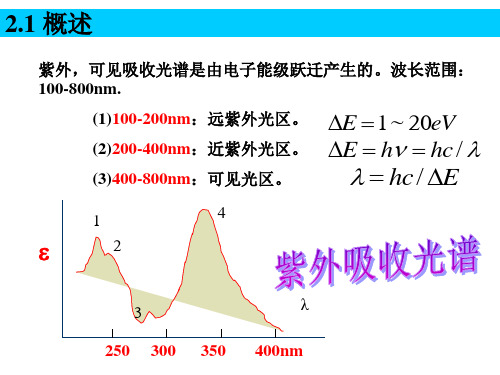
1.1 概述
分子吸收紫外-可见光区190~800nm的电磁波,使其电 子从基态跃迁到激发态,从而产生的吸收光谱称紫外-可见 吸收光谱(Ultraviolet-Visible Absorption Spectra)。简称紫 外光谱(UV-Vis)。又称为电子吸收光谱。
紫外吸收光谱

三、分子离子化的影响(pH的影响)
若化合物在不同的pH介质中能形成阳离子或阴离 子,则吸收带会随分子的离子化而改变。如苯胺在酸 性介质中会形成苯胺盐阳离子。苯胺形成阳离子之后, 氮原子的未成键电子消失,氨基的助色作用也随之消 失,因此苯胺盐的吸收带从230和280nm移到203和 254nm处。 苯酚分子中OH基团含有两对孤对电子,与苯环上 π电子形成n→π共轭,当形成酚盐阴离子时,氧原子上 孤对电子增加到三对,使n→π共轭作用进一步增强, 从而导致吸收带红移,同时吸收强度也有所增加。
表 2 环状共轭二烯π→π*跃迁的吸收波 长计算方法
π→π*跃迁λ/nm
同环二烯基本值 异环二烯基本值 每一个烷基或环残余取代 每一个环外双键 每一个烷氧基取代 –OR 每一个含硫基团取代 –SR 每一个胺基取代-NRR’ 每一个卤素取代 每一个酰基取代-COOR 增加一个共轭双键
253 214 +5 +5 +6 +30 +60 +5 +0 +30
3、醇、醚、含氮、含硫化合物及卤代物
醇、醚含有未成键电子,能产生n→ζ*跃迁,其 吸收波长都低于200nm。醇的分子间容易形成氢键而 发生缔合,吸收带波长及强度将随缔合的程度而变化。 胺是最简单的含氮有机化合物,胺中氮原子含有 未成键电子,所产生n→ζ*跃迁,其吸收波长处于 200nm附近。 硝基及亚硝基化合物中由于存在氮、氧原子,可 形成n→π共轭体系,所以能产生π→π*和n→π*跃迁, 吸收带位于近紫外区,例如硝基甲烷的吸收波长分别 为210和270nm。
气体 气体 己烷 己烷 水 己烷 水 甲醇 己烷 乙醇 甲醇 甲醇 乙醇 乙醇 己烷 己烷 甲醇 甲醇
第二节 紫外分光光度计光路图
一、单光束分光光度计
光源
光电倍增管
一 紫外可见吸收光谱法
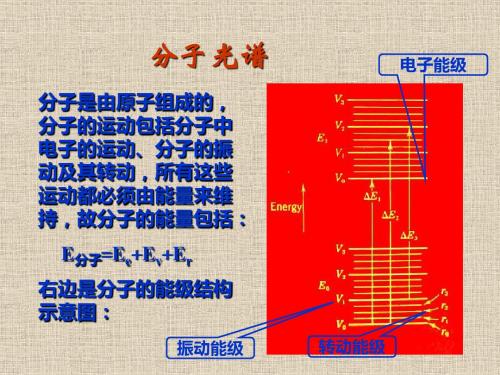
分子是由原子组成的, 分子的运动包括分子中 电子的运动、分子的振 动及其转动,所有这些 运动都必须由能量来维 持,故分子的能量包括:
E分子=Ee+Ev+Er 右边是分子的能级结构 示意图:
振动能级 转动能级
电子能级
分子吸收光谱
由于分子能量由三部分组成,当分子处于辐射中时,故可 以形成相对应的三类光谱: 转动光谱或远红外光谱,对应的波长范围在25m~12.5cm, 属于远红外区和微波区,分子的转动能级差r在0.05~ 110-4ev。 红外光谱或振-转光谱,对应的波长范围在1.25m~25m, 属于红外区,分子的振动能级差v在1~0.05ev之间,又 由于在分子振动的同时伴随有分子的转动,振-转光谱。 电子光谱或可见-紫外光谱,主要分布在可见-紫外光谱区, 对应的波长范围在100~1000nm之间,电子在分子轨道间 的跃迁能级差e在1~20ev之间。
紫外-可见分光光度计_狭缝与带宽
狭缝:一般分光光度计的出射狭缝是不变的,如 721、722型,分别为0.2nm和0.3~0.5nm,对于中、 高档仪器,狭缝是可变的,如751,狭缝的改变对 分析结果有很大的影响。
带宽:单色器出光狭缝所包含的“单色光”的波 长范围。
W或= d S(倒线色散率乘以狭缝宽度) dl
波长精度:一般3nm,751在600nm以上可达1.5nm
波长重现性:一般1.5nm
测量范围:0~100T;0~2A或 0~3A
测量精度:0.5 T%;751可达0.3 T% 杂散光:1% T。 对于中、高档仪器,还有波长分辨率,对谱线的分离程 度,一般能分开Hg三线(365.0、365.5、366.3nm),狭 缝可调范围(一般0~2nm),灵敏度等指标。
紫外-可见分子吸收光谱法

NN
溶剂与溶质之相互作用增强 C H
溶质分子的振动受到限制
水中 环己烷中
振动引起的精细结构消失
蒸汽中
500
555
对称四嗪的吸收光谱
/nm
b. 溶剂极性对π →π*跃迁谱带的影响
➢ 溶剂极性增大时,由π →π*跃迁产生的吸收 带发生红移。
c. 溶剂极性对n →π*跃迁谱带的影响
➢ 溶剂极性增大,由n →π*跃迁产生的吸收谱 带发生蓝移。
(4)多通道分光光度计
以光二极管阵列作检测器
光源
透镜
光二极管阵列
试样池
光栅
三、光吸收定律
1、朗伯-比尔定律
A lg T lg I0 bc 或 A lg T lg I0 abc
I
I
2、吸光度的加和性
当溶液中含有多种对光产生吸收的物质,且各组分之
间不存在相互作用时,则该溶液对波长λ光的总吸光度A总
➢ 根据分子轨道理论,这三种电子的能级高 低为: σ<π<n <π*<σ*
三种价电子可能产生六种形式电子跃迁:
σ→ σ*, σ→ π*, π→ σ*对应的吸收光谱处于 远紫外区,研究少。
(1) n → σ* 跃迁:
➢ 吸收光谱出现在远紫外光区和近紫外光区 ➢ 某些含有氧、氮、硫、卤素等杂原子的基 团(如—NH2、—OH、—SH、—X等)的 有机物可产生n → σ* 跃迁。 例如:CH3OH:λmax=183 nm 、CH3NH2:λmax=213 nm
② 吸收峰通常位于200~400nm之间。
(7) K带
➢ 由共轭体系的π →π*跃迁产生的吸收带。
特点:
ε ① 强度大,一般 > 104 L ·mol-1 ·cm-1 ;
课件紫外可见吸收光谱(共83张PPT)

T I I0
I 为透射光的强度
I0 为入射光的强度
A lgI0
lgT
I
1760年朗伯(Lambert)阐明了光的吸收程度和吸收层厚度的 关系,即 A∝b
1852年比耳(Beer)又提出了光的吸收程度和吸收物浓度之间 也具有类似的关系,即 A∝ c
二者的结合称为朗伯-比尔定律,其数学表达式为:
AlgTkbc
Abc
摩尔吸光系数ε的讨论:
(1)吸收物质在一定波长和溶剂条件下的特征常数; (2)不随浓度c和光程长度b的改变而改变。在温度和波长等条件一定时 ,ε仅与吸收物质本身的性质有关,与待测物浓度无关;
(3)同一吸收物质在不同波长下的ε值是不同的。在最大吸收波长λmax 处的摩尔吸光系数,常以εmax表示。εmax表明了该吸收物质最大限度的
➢ 含有杂原子的不饱和化合物可以发生n→p*跃迁, 如含有羰基、硝基、亚硝基等
➢ n→p*跃迁所产生的吸收带称为R带
常用概念
➢ 发色团(或生色团):具有π电子的不饱和基团,即 可在紫外-可见光区产生吸收的官能团。如C=C、 C≡C、 C=O、-NO2等
➢ 助色团:有一些含有n电子的基团(如-OH、-NH2、OR、-SH、-Cl、-Br、-I等),它们本身没有生色功能
第二节
紫外-可见分光 光度计
UV-Vis spectrometer
一、基本组成
二、分光光度计的 类型
一、基本组成
1. 光源
➢ 要求:提供能量,激发被测物质分子使之产生价电子的跃迁, 从而产生电子光谱;在整个紫外光区或可见光谱区可以发射连续光 谱;具有足够的辐射强度、较好的稳定性、较长的使用寿命。
2. 有机化合物的紫外可见吸收光谱
紫外吸收光谱分析法.

254
200
甲苯
261
300
含取代基时, B带简化, 间二甲苯 红移。
263
300
1,3,5-三甲苯 266
305
六甲苯
272
300
02:56:43
乙酰苯紫外光谱图
羰基双键与苯环共扼: K带强;苯的E2带与K带合 并,红移; 取代基使B带简化; 氧上的孤对电子: R带,跃迁禁阻,弱;
C H3
C
n p* ; R带
第一章 紫外吸收光谱
分析法
ultraviolet spectrometry, UV
第一节 紫外吸收 光谱分析基本原理
principles of UV
一、 紫外吸收光谱的产生 formation of UV 二、 有机物紫外吸收光谱 ultraviolet spectrometry of organic compounds
O
p p* ; K带
02:56:43
苯环上助色基团对吸收带的影响
02:56:43
苯环上发色基团对吸收带的影响
02:56:43
5. 立体结构和互变结构的影响
H C
H C
H C
C H
顺反异构: 顺式:λmax=280nm; εmax=10500 反式:λmax=295.5 nm;εmax=29000
有一些含有n电子的基团(如—OH、—OR、—NH2、— NHR、—X等),它们本身没有生色功能(不能吸收λ>200nm的 光),但当它们与生色团相连时,就会发生n—π共轭作用,增 强生色团的生色能力(吸收波长向长波方向移动,且吸收强度 增加),这样的基团称为助色团。
02:56:43
红移与蓝移
有机化合物的吸收谱带 常常因引入取代基或改变溶 剂使最大吸收波长λ max和吸 收强度发生变化:
1.紫外-可见吸收光谱

3. P- π共轭作用
含 O、S、N、Cl等杂原子的P电子与π电子产生共轭,使 λmax发生红移,吸收强度增加。
举例
E2带, λmax =203nm
OH
NH2
λmax =215.5nm
λmax =230nm
4.溶剂效应
(1 )对最大吸收波长的影响
a. n→π*跃迁所产生的吸收峰随溶剂极性的增加而向短波长方 向移动。因为具有孤对电子的分子能与极性溶剂发生氢键缔 合,使基态和激发态能级的能量均下降,但由于其作用强度 以极性较强的基态大于极性较弱的激发态,致使基态能级的 能量下降较大,而激发态能级的能量下降较小(如图a), 故两个能级间的能量差值增加。实现n→π*跃迁需要的能量 也相应增加,故使吸收峰向短波长方向位移,即蓝移。
蓝移(blue shift) :相反,使吸收带向 短波长方向移动的现象称为短移或蓝移,引 起蓝移效应的基团称为向蓝基团。
(4)增色效应和减色效应 增色效应(hyperchromic effect) :由
于取代基的变更或溶剂的影响使吸收带吸收
强度增加的现象称为浓色效应或增色效应。
减色效应(hypochromic effect) :由
图 1,3丁二烯分子轨道能级示意图
△E2
2 .超共轭效应
当烷基与共轭体系相连时,可以使波长产生红移, 因通常σ- π超 共轭作用较弱,所以红移程度也较 小。
H 举例 H C H CH CH2
λmax1>λmax(CH2=CH2)
H
H
C
CH
CH CH
CH2
λmax2
H
比较λmax2 、 λmax1、 λmax(CH2=CH2)
*几个关系式
紫外可见吸收光谱

2. 电荷迁移跃迁
——指配合物中配位体与金属离子之间,一个电子
由一方的一个轨道跃迁到另一方相关的轨道上。 ——产生电荷迁移跃迁的必要条件:一组分是电子
给予体,另一组分是电子接收体。
例: [Fe3+ (SCN-)]2+ h [Fe2+(SCN)]2+
电子接受体 电子给予体
——电荷迁移跃迁光谱的很大,一般在104以上,
——当苯环上有羟基、氨基等取代基时,吸收峰红移, 吸收强度增大.像羟基、氨基等一些助色团,至少 有一对非键n电子,这样才能与苯环上的电子相互 作用,产生助色作用.
——取代基不同,变化程度不同,可由此鉴定各种 取代基
例: 苯
λmax B带 254
λmax
E2
204
甲苯
262
208
苯酚
271
213
苯甲酸
(一)紫外可见吸收光谱 由紫外可见分光光度计获得
光源——单色器——吸收池——检测器——显示器
ΔE电 = h 光 (200—800 nm)
激发态 基态
吸收曲线
将不同波长的光透过某一固定浓度和 厚度的待测溶液,测量每一波长下待测溶 液对光的吸收程度(即吸光度),然后以 波长为横坐标,以吸光度为纵坐标作图, 可得一曲线。这曲线描述了物质对不同波 长的吸收能力,称吸收曲线或吸收光谱。
不同波长的光
L
图3-1紫外可见吸收光谱示意图
A
末端吸收
最强峰
肩 峰
次强峰 峰谷
max
min
A
分析吸收曲线 可以看到:
1.同一浓度的 待测溶液对不 同波长的光有 不同的吸光度;
max
min
各类化合物的紫外吸收光谱

在气态或非极性溶剂中,
苯及其许多同系物的B谱带有
许多的精细结构,这是由于
当苯环上有取代基时,
振动跃迁在基态电子上的跃 迁上的叠加而引起的。
在极性溶剂中,这些精 细结构消失。
苯的三个特征谱带都会发生 显著的变化,其中影响较大 的是E2带和B带。
23
稠环芳烃及杂环化合物
稠环芳烃,如萘、蒽、芘等,均显示苯的三个吸收带,但是 与苯本身相比较,这三个吸收带均发生红移,且强度增加。随 着苯环数目的增多,吸收波长红移越多,吸收强度也相应增加。
6
1、直链共轭二烯*跃迁吸收波长计算方法
母体共轭二烯基本值 双键上烷基取代 环外双键
217 nm +5 nm +5 nm
环的外向双键:指碳碳双键的两个sp2杂化的碳 原子中,其中之一是环原子的一员。
7
例1 max=217nm(基数)+2×5nm +5nm =232nm 观察值max=235nm
2
2、饱和羰基化合物
在羰基化合物中除有电 子和电子外,羰基的氧原子 上还有一对孤对电子-n电子。 因此存在着四种跃迁*、 *、n*、n*。前三 种跃迁落在测量范围之外。
孤立羰基化合物研究最 多 的 是 n* 跃 迁 , 其 吸 收 谱 带 出 现 在 2 7 0 — 3 0 0 nm 附 近,一般呈低强度吸收 (ε10~20)的宽谱带,其吸收 位置的变化对溶剂很敏感, 称之为R带。
羧酸及羧酸的衍生物虽然也有n*吸收带,但是羧酸 及羧酸的衍生物的羰基上的碳原子直接连结含有未共用电子 对的助色团,如-OH、-Cl、-OR等。
由于这些助色团上的n电子与羰基双键的电子产生n- 共轭,导致*的能级有所提高,但这种共轭作用并不能改变 n轨道的能级。
因此实现n*跃迁所需的能量变大,使n*吸现方式做保护处理对用户上传分享的文档内容本身不做任何修改或编辑并不能对任何下载内容负责
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第九章紫外吸收光谱1. 试简述产生吸收光谱的原因.基本要点:1. 分子吸收光谱;2. 有机化合物的紫外吸收光谱;3. 无机化合物的紫外吸收光谱;4. 溶剂对紫外吸收光谱的影响 ;5. 紫外吸收光谱的应用等 .利用紫外吸收光谱进行定量分析的由来已久,公元60年古希腊已知道利用五味子浸液来估计醋中铁的含量。
这一古老的方法由于最初是运用人的眼睛来进行检测,所以叫比色法。
20世纪30年代产生了第一台光电比色计,40年代出现的BakmanUV 分光光度计, 促进了新的分光光度计的发展。
随着计算机的发展,紫外分光光度计已向着微型化﹑自动化﹑在线和多组分同时测定等方向发展。
第一节分子吸收光谱Molecular Absorption Spectroscopy一、分子内部的运动及分子能级前面讲的AAS和AES都属与原子光谱,是由原子中电子能级跃迁所产生的。
原子光谱是由一条一条的彼此分离的谱线组成的线状光谱。
分子光谱比原子光谱要复杂得多。
这是由于在分子中,除了有电子相对于原子核的运动外,还有组成分子的各原子在其平衡位置附近的振动,以及分子本身绕其重心的转动。
如果考虑三种运动形式之间的相互作用,则分子总的能量可以认为是这三种运动能量之和。
即E=E e+ E v+ E r式中E e为电子能量,E v为振动能量,E r转动能量。
这三种不同形式的运动都对应一定的能级,即:分子中除了电子能级外,还有振动能级和转动能级这三种能级都是量子化的、不连续的。
正如原子有能级图一样,分子也有其特征的能级图。
简单双原子分子的能级图如图9-1所示。
A和B表示电子能级,间距最大;每个电子能级上又有许许多多的振动能级,用V'=0,1,2,……等表示A能级上个振动能级,V"=0,1,2,……等表示B能级上各振动能级;每个振动能级上又有许许多多的转动能级,用j'=0,1,2,……等表示A能级上V'=0各转动能级,j"= 0,1,2,……等表示A能级上V'=1各振动能级等等。
且ΔE e > ΔE v > ΔE r二、能级跃迁与分子吸收光谱的类型通常情况下,分子处于较低的能量状态,即基态。
分子吸收能量具有量子化特征,即分子只能吸收等于二个能级之差的能量。
如果外界给分子提供能量(如光能),分子就可能吸收能量引起能级跃迁,而由基态跃迁到激发态能级。
ΔE=E1-E2=hν=hc/λ由于三种能级跃迁所需要的能量不同,所以需要不同的波长范围的电磁辐射使其跃迁,即在不同的光学区域产生吸收光谱。
1.转动能级跃迁与远红外光谱转动能级间的能量差ΔE r约为:0.025~0.003eV。
假如是0.01 eV,可计算出:λ=hc/ΔE=6.624×10-34×2.998×108/0.01×1.6×10-19=1.24×10-5m=12400nm=124μm可见,转动能级跃迁产生吸收光谱位于远红外区(50~300 m), 称远红外光谱或分子转动光谱。
2.振动能级:振动能级间的能量差ΔE v约为:1~0.025eV。
假如是0.1 eV,可计算出:λ=hc/ΔE=6.624×10-34×2.998×108/0.1×1.6×10-19=1.24×10-5m=12400nm=12.4μm可见,振动能级跃迁产生的吸收光谱位于红外区(0.78~50μm),称红外光谱或分子振动光谱。
振动能级跃迁时不可避免地会产生转动能级间的跃迁。
即振动光谱中总包含有转动能级间跃迁,因而产生光谱也叫振动-转动光谱。
3.电子能级电子能级的能量差ΔE e : 20~1eV。
假如是5eV,可计算出:λ=hc/ΔE=6.624×10-34×2.998×108/5×1.6×10-19=2.48×10-7m=248nm可见,电子跃迁产生的吸收光谱在紫外—可见光区(200~780nm),称紫外—可见光谱或分子的电子光谱。
电子能级跃迁时不可避免地会产生振动和转动能级间的跃迁。
即电子光谱中总包含有振动能级和转动能级间跃迁,因而产生的谱线呈现宽谱带。
紫外—可见光谱实际上是电子-振动-转动光谱。
应该指出,紫外光可分为近紫外光(200 ~ 400 nm)和真空紫外光(60 ~ 200 nm)。
由于氧、氮、二氧化碳、水等在真空紫外区(60 ~ 200 nm)均有吸收,因此在测定这一范围的光谱时,必须将光学系统抽成真空,然后充以一些惰性气体,如氦、氖、氩等。
鉴于真空紫外吸收光谱的研究需要昂贵的真空紫外分光光度计,故在实际应用中受到一定的限制。
我们通常所说的紫外—可见分光光度法,实际上是指近紫外、可见分光光度法。
第二节有机化合物的紫外吸收光谱Organic Molecular Ultraviolet Absorption Spectroscopy有机化合物此外吸收光谱(电子光谱)是由分子外层电子或价电子跃迁所产生的。
按分子轨道理论,有机化合物分子中有:成键σ轨道,反键σ*轨道;成键π轨道,反键π*轨道(不饱和烃);另外还有非键轨道(杂原子存在)。
各种轨道的能级不同,如图9-2所示。
相应的外层电子和价电子有三种:σ电子、π电子和n 电子。
通常情况下,电子处于低的能级(成键轨道和非键轨道)。
当用合适能量的紫外光照射分子时,分子可能吸收光的能量,而又低能级跃迁到反键*轨道。
在紫外可见光区,主要有下列几种跃迁类型:①.N→V跃迁:电子又成键轨道跃迁到反键轨道,包括σ→σ*;π→π*跃迁。
②. N→Q跃迁:分子中未成键的n 电子跃迁到反键轨道,包括n→σ*;n→π*跃迁。
③. N→R跃迁:σ电子逐级跃迁到各高能级,最后脱离分子,使分子成为分子离子的跃迁。
(光致电离)④.电荷迁移跃迁:当分子形成配合物或分子内的两个大π体系相互接近时,外来辐射照射后,电荷可以由一部分转移到另一部分,而产生电荷转移吸收光谱。
可见,有机化合物一般主要有4种类型的跃迁:n→π*、π→π*、n→σ*和σ→σ*。
各种跃迁所对应的能量大小为n→π*< π→π*< n→σ*< σ→σ*[讨论]:①σ→σ*跃迁所需能量最大。
σ电子只有吸收远紫外光的能量才能发生跃迁饱和烷烃的分子吸收光谱出现在远紫外区,吸收波长λ<200 nm,甲烷的λmax 为125nm , 乙烷λmax为135nm,只能被真空紫外分光光度计检测到;作为溶剂使用。
②.n→σ*跃迁所需能量较大。
吸收波长为150~250nm,大部分在远紫外区,近紫外区仍不易观察到。
含非键电子的饱和烃衍生物(含N、O、S和卤素等杂原子)均呈现n→σ*跃迁。
③. π→π*跃迁所需能量较小。
吸收波长处于远紫外区的近紫外端或近紫外区,εmax一般在104L·mol-1·cm-1以上,属于强吸收。
1.饱和烃饱和烃类分子中只含有σ键,因此只能产生σ→σ*跃迁,即σ电子从成键轨道(σ)跃迁到反键轨道(σ*),所需能量最大。
饱和烷烃的分子吸收光谱出现在远紫外区,吸收波长λ10~200nm,已超出紫外、可见分光光度计的测量范围,只能被真空紫外分光光度计检测到(空气中的氧吸收波长< 160nm的紫外光)。
如甲烷的λmax为125nm,乙烷λmax为135nm。
这类物质在紫外光谱分析中常用作溶剂。
当饱和烷烃的分子中的氢被氧、氮、卤素、硫等杂原子取代时,因有n 电子存在,而产生n→σ*跃迁,所需能量减小。
吸收波长向长波方向移动,这种现象称之为红移。
例如,CH3Cl、CH3Br和CH3I的n→σ* 跃迁分别出现在173、204和258nm 处。
又如,CH4跃迁范围125~135nm(σ→σ*),CH3I跃迁范围150~210nm(σ→σ*)和259nm (n→σ*);CH2I2吸收峰292nm(n→σ*);CHI3吸收峰349nm(n→σ*)。
这些数据不仅说明氯、溴和碘原子引入甲烷后,其相应的吸收波长发生了红移,显示了助色团的助色作用。
而且说明,虽杂原子半径增加,n→σ*跃迁向长波方向移动。
直接用烷烃和卤代烃的紫外吸收光谱分析这些化合物的实用价值不大。
但是它们是测定紫外和(或)可见吸收光谱(200~1000nm)的良好溶剂。
2.不饱和脂肪烃在不饱和烃类分子中,除含有σ键外,还含有π键,它们可以产生σ→σ*和π→π*两种跃迁。
π→π*跃迁的能量小于σ→σ*跃迁。
例如,在乙烯分子中,π→π*跃迁最大吸收波长为180nm。
这种含有不饱和键的基团称为生色团。
See Table 9-3。
在不饱和烃类分子中,当有两个以上的双键共轭形成大π键时,随着共轭系统的延长,π→π*跃迁的吸收带将明显向长波方向移动,吸收强度也随之增强[原因:轭系效应使单键具有双键的性质(加强),双键具有单键的性质(削弱),即平均化。
电子易激发] 。
例如,C2H4(孤立单键)λmax=171nm,εmax=1.553×104;CH2=CH-CH=CH2λmax=217nm,εmax=2.1×104。
[See Table 9-4]。
在共轭体系中,π→π*跃迁产生的吸收带又称为K(Konjugation)带。
K带(π→π*)的特点:强度大,εmax›104;位置一般在217~280nm;λmax 和εmax的大小共轭链的长短及取代基的位置有关。
根据K带是否出现,可判断分子中共轭体系的存在的情况。
在紫外光谱分析中有重要应用。
乙酰苯紫外光谱图(See Power Point):羰基双键与苯环共扼:K带强;苯的E2带与K带合并,红移;取代基使B带简化;氧上的孤对电子:R带,n→π*跃迁,跃迁禁阻,弱。
3.芳香烃Fig. 9-5为苯的紫外光谱图(乙醇溶剂)。
苯有三个吸收带,它们都是由π→π*跃迁引起的。
E1带出现在185nm(εMAX = 47,000),E2带出现在204nm(εMAX = 79,00 ),强吸收带。
它们是由苯环结构中,三个乙烯的环状共轭系统的跃迁所产生的,是芳香族化合的特征吸收。
B带出现在255nm (εMAX = 200)。
这是由π→π*跃迁的振动重叠引起的。
在气态或非极性溶剂中,苯及其许多同系物的B谱带有许多的精细结构,这是由于振动跃迁在基态电子跃迁上的叠加而引起的。
在极性溶剂中,这些精细结构消失。
当苯环上有取代基时,苯的三个特征谱带都会发生显著的变化,其中影响较大的是E2带和B谱带。
当苯环与生色团连结时,有B和K两种吸收带,有时还有R吸收带,其中R吸收带的波长最长。
稠环芳烃,如萘、蒽、芘等,均显示苯的三个吸收带,但是与苯本身相比较,这三个吸收带均发生红移,且强度增加。
随着苯环数目的增多,吸收波长红移越多,吸收强度也相应增加。