纤维素降解混合培养物的16S rRNA基因序列分析
16s rrna报告解读

16s rrna报告解读摘要:1.16s rRNA简介2.16s rRNA报告解读方法3.报告结果分析与应用4.报告的局限性与未来发展方向正文:随着分子生物学技术的发展,16s rRNA报告已成为微生物学、生态学等领域的重要研究手段。
16s rRNA是细菌核糖体的一部分,具有种属特异性,可通过高通量测序技术对微生物群落进行定性和定量分析。
本文将介绍16s rRNA报告的解读方法、结果分析与应用,以及报告的局限性与未来发展方向。
一、16s rRNA简介16s rRNA存在于细菌细胞中,负责蛋白质生物合成。
由于不同细菌物种的16s rRNA序列存在差异,可通过测定该序列对微生物进行分类和鉴定。
目前,16s rRNA测序已成为微生物多样性研究的主要手段。
二、16s rRNA报告解读方法1.序列分析:将测序得到的原始数据进行质控、比对、组装等处理,得到序列。
然后,通过序列之间的相似性分析,对微生物物种进行分类和鉴定。
2.生物信息学分析:基于序列数据,进行物种多样性、群落结构、代谢途径等分析。
常用的生物信息学工具包括MetaPhlAn、Kraken等。
3.统计分析:对生物信息学结果进行统计,评估样本间的差异性和相似性,为实验结果提供依据。
三、报告结果分析与应用1.物种多样性分析:通过统计不同物种的序列数量,评估样本中的微生物多样性。
2.群落结构分析:分析不同物种在样本中的相对丰度,揭示微生物群落的组成和变化。
3.功能基因分析:基于代谢途径和基因功能,分析微生物群落的代谢活性。
4.应用:16s rRNA报告可用于医学、环境、农业等领域的微生物学研究,为疾病诊断、环境保护、农业生产等提供科学依据。
四、报告的局限性与未来发展方向1.局限性:16s rRNA报告无法区分共生和病原微生物,且受样本制备和测序深度等因素影响。
2.未来发展方向:发展更高效的测序技术、生物信息学方法,以及整合多组学数据进行综合分析,提高报告的准确性和应用价值。
用16S rRNA基因序列鉴定细菌

Ezbiocloud比对,下载同源序列
培养细菌 提取基因组DNA PCR扩增16S rRNA基因 产物电泳检测,测序 Ezbiocloud比对,下载同源序列 MEGA比对和分析
1. 测序结果的分析和处理 一个测序反应结果包括两个文件,序列文件用记事本
打开,图形文件用Chromas软件打开观察。打开图形文件, 确认测序结果良好,并去除序列文件中两端不准确的部分, 得到最终的测序结果。 2. 同源序列下载
CVV
C
VC V C V
C VC V C V C V C
Prmier 1492R
细胞生物共有,功能重要,多拷贝,保守区和多变区组成,适合扩增和序列相似性分析。 ➢ 研究物种间进化和分类。 ➢ 根据16S rRNA基因序列鉴定待测菌株。 ① 同种细菌序列相似性≥98.7%,但序列相似性≥98.7%≠同种; ② 用待测菌株及相似性较高的菌株16S rRNA基因序列构建系统发育树(Phylogenetic
1. 构建系统发育树 系统发育分析后的结果文件,用mega 6软件中的邻接法(Neighbor-Joining)构建系统发育树。 2. 整理系统发育树,确定待测菌株属名
产物电泳检测,测序
Ezbiocloud比对,下载同源序列 MEGA分析
构建系统发育树,确定种属
用Neighbor-Joining法构建系统发育树
用16S rRNA鉴定细菌流程
培养细菌 提取基因组DNA PCR扩增16S rRNA基因 产物电泳检测,测序 Ezbiocloud比对,下载同源序列 MEGA比对和分析 构建系统发育树,确定种属
PCR扩增16S rRNA基因
1. PCR体系(50 μL)
引物27F (20 mmol/L) 引物1492R(20 mmol/L) 基因组DNA 双蒸水 2×PCR SuperMax(+dye)
纤维素菌分离和鉴定的方法

纤维素菌分离和鉴定的方法一、概述纤维素由于其特殊的生化性质,一直是微生物学和食品工业研究的关注焦点。
特别是纤维素的分解,除了传统的化学方法,生物法也是目前广泛采用的技术之一。
对纤维素酶活性和产酶微生物的分离和鉴定,是纤维素生物技术研究的重要内容。
纤维素分解酶是产生于微生物体内的一类酶,广泛存在于真菌和细菌中,可划分为纤维素酶和半纤维素酶两大类,规律的利用这些微生物菌种,开发新型的纤维素分解酶。
纤维素的微生物分解菌种较多,其中许多菌种能分泌出多种细胞外酶,如单糖的转化酶、纤维素酶、半纤维素酶、葡萄糖氧化酶、木糖酶、果糖酶和葡聚糖酶等。
多种新型有机物的产生依赖于工业生产中微生物的应用技术,目前各国纤维素降解的菌株和发酵产物分离和鉴定方法越来越多。
纤维素菌的分离可采用筛选、稀释和富集等方法。
实际应用中,常用的分离方法有:(一)厌氧富集法:根据纤维素菌能够在厌氧条件下进行生长和代谢的特点,采用富集培养方法,利用耐氧性微生物限制氧气的供应,引起产生厌氧性纤维素分解菌类,然后推广领域固定化、发酵和应用。
可根据繁殖时间将厌氧富集法分为短时间富集法和长时间富集法。
(二)筛选法:在纤维素富含的自然环境或人工培养环境中进行筛选。
先用一些微生物菌株做为预培养菌种,加入富含纤维素的培养基,通过短期采取接种、稀释、摇动等处理方式,寻找纤维素分解酶活力最高的培养物,然后进行分离纯化、性质鉴定。
(三)稀释法:将样品依一定比例进行稀释,将稀释后的液体均匀地均匀的加入纤维素培养基中,用深层培养的方式进行发酵,进行分离鉴定纯化。
稀释法适用于富含纤维素且菌株较多的培养基的菌群筛选。
(一)形态学特征鉴定法:根据菌株的形态学特征进行鉴定。
此方法是最基本也是最重要的鉴定方法之一。
菌株的形态学特征包括形状、结构、颜色和大小等,进一步对分离的菌株进行正确定义。
常用的形态学特征包括:形态特征、结构特征、色素特征和大肠杆菌。
(二)生理生化特征鉴定法:通过菌株的生长特性在不同培养基中的表现,或菌体在不同生长条件下表现的生化过程,如碳源利用情况,氮源利用情况,温度和pH值的影响等,进行鉴定。
16S rRNA、16S-23S rRNA基因测序分析检测主要血流感染病原菌比较

16S rRNA、16S-23S rRNA基因测序分析检测主要血流感染病原菌比较金中淦;葛平;徐蓉;陈蓉;宣瑛;刘学杰;王庆忠【摘要】目的比较细菌16S rRNA、16S-23S rRNA基因测序分析在血流感染病原菌检测中的作用.方法提取临床上血流感染常见的金黄色葡萄菌、表皮葡萄球菌、大肠埃希菌、粪肠球菌、肺炎链球菌、铜绿假单胞菌、阴沟肠杆菌、鲍曼不动杆菌、洛菲不动杆菌、肺炎克雷伯杆菌、化脓性链球菌、奇异变形杆菌、潘尼变形杆菌、屎肠球菌、粘质沙雷菌、宋内志贺菌、产气肠杆菌、小肠结肠炎耶尔森菌、腐生葡萄球菌基因组DNA,运用16S rRNA、16S-23S rRNA基因进行PCR扩增.扩增产物经测序后在美国国家生物技术中心( NCBI)上进行比对分析,确定菌种.结果在所分析的19种临床血流感染常见细菌中,16S rRNA基因测序分析可将除粘质沙雷菌外的细菌鉴定到种的水平,但无法完全区分近缘种属;16S-23SrRNA成功鉴定17种细菌,除大肠埃希菌、宋内志贺菌外所有细菌均成功鉴定到单一种的水平.结论16S-23S rRNA基因可作为血流感染细菌检测较好的分子靶标.【期刊名称】《分子诊断与治疗杂志》【年(卷),期】2012(004)003【总页数】5页(P181-185)【关键词】16SrRNA;16S-23SrRNA【作者】金中淦;葛平;徐蓉;陈蓉;宣瑛;刘学杰;王庆忠【作者单位】上海市临床检验中心,上海,200126;上海市临床检验中心,上海,200126;上海市临床检验中心,上海,200126;上海市临床检验中心,上海,200126;上海市临床检验中心,上海,200126;上海市临床检验中心,上海,200126;上海市临床检验中心,上海,200126【正文语种】中文以菌血症和败血症为主的血流感染具有低发生率、高危险性的特点[1]。
据报道,美国血流感染的总体病死率可高达14%~63%,但在明确病原菌后采用针对性治疗,其死亡率可降至5%~17%[2~4]。
16S+rRNA序列分析在多杀巴斯德氏菌鉴定中的应用

万方数据 万方数据第6期高正琴等:16SrRNA序列分析在多杀巴斯德氏菌鉴定中的应用‘3‘润(图IB)裹1分离菌株生化试验结果注:+,阳性;一阴性表2分离菌株药敏试验结果注:s,敏感;M,中等敏感;R,耐药.圈1PMr0901感染家兔肝脏和肺脏病理变化A:多杀巴斯德氏菌感染家兔肝脏组织切片,HE染色.肝脏中央静脉周围的肝细胞索紊乱,肝脏局灶性坏死,肝细胞完整结构丧失,崩解。
胞核碎裂,有大量炎性细胞浸润;B:多杀巴斯德氏菌感染家兔肺脏组织切片,HE染色.肺小叶问质增宽,有浆液纤维素性渗出物,肺泡结构不完整,大量炎性细胞浸润.2.416SrRNA序列分析结果PMr0901分离菌株的16SrRNA的扩增长度为l500bp(图2),将获得的核苷酸序列与GenBank中已知序列进行同源性比较分析,结果显示,PMr0901与巴斯德氏菌属成员的16SrRNA序列具有98%以上的高度同源性,其中与Pasteurellamultocidasubsp.multocidastr.Pm70(GenBank登录号:NC002663.1)的同源性最高,达到99.9%。
16SrRNA序列分析结果进一步表明本研究分离的PMr0901菌株确实是多杀巴斯德氏菌。
图2PCR扩增多杀巴斯德氏西PMr0901的16srRNA基因泳道1:多杀巴斯德氏菌PMr0901的161rRNA基因PCR扩增条带;M:200bpDNAladderInarker.2.5人工感染的致病作用将分离鉴定的多杀巴斯德氏菌PMr0901,按1.7节中所述方法对小鼠进行人工感染试验,结果6只小鼠于感染后24h内全部死亡,对照组6只小鼠在14d观察期内均正常存活。
对感染死亡小鼠进行剖解,发现其肺脏严重充血、出血。
取肺脏触片进行革兰染色,结果发现大量与感染菌相同的革兰阴性短球杆菌。
同时,分离到大量纯一的原感染菌的菌落,对其纯化培养后进行形态特征及理化性状的复合鉴定,结果与PMr0901一致。
16s rdna序列 16s rrna基因序列

16s rDNA序列是细菌和古细菌特有的一种特征序列,是通过测定16s rDNA基因所编码的16s rRNA的序列而得到的。
在分子生物学和微生物学领域,16s rDNA序列被广泛应用于微生物分类、微生物多样性研究和微生物系统进化研究中。
本文将从以下几个方面对16s rDNA 序列进行介绍和分析。
一、成因和结构16s rDNA序列是细菌和古细菌特有的一种特征序列,它是细菌和古细菌核糖体小亚基rRNA基因的一部分,通常有约1500个核苷酸碱基对,可由16s rRNA基因编码。
这一序列在细菌和古细菌的核糖体RNA中起着重要的作用,它能够稳定地与核糖体蛋白结合,形成核糖体的小亚基,并参与到细菌和古细菌的蛋白质合成过程中。
二、意义和应用1. 微生物分类16s rDNA序列在微生物分类中具有重要意义,通过对16s rDNA序列进行测序分析可以鉴定和分类细菌和古细菌的种属和亚属。
这是因为16s rDNA序列在不同种属和不同亚属的细菌和古细菌之间存在一定的变异,可以作为分子生物学特征用于分类鉴定。
2. 微生物多样性研究通过对环境样品中的16s rDNA序列进行测序分析,可以了解微生物裙落的组成和结构,揭示微生物在自然界的分布和多样性。
这对于研究微生物的生态学、环境适应性和生态功能具有重要意义。
3. 微生物系统进化研究利用16s rDNA序列进行系统进化研究,可以揭示细菌和古细菌的系统发育关系和演化过程,为了解细菌和古细菌的起源、多样性和进化提供重要的分子学证据。
三、研究方法1. PCR扩增通常情况下,从细菌或古细菌的DNA中提取16s rDNA序列,然后利用PCR技术进行扩增。
通过选择适当的引物和反应条件,可以特异性地扩增出16s rDNA序列,为后续的测序分析做准备。
2. 测序分析测序是获取16s rDNA序列信息的关键步骤,目前常用的测序方法包括Sanger测序和高通量测序。
通过测序分析,可以获得16s rDNA 序列的具体碱基序列信息,用于后续的分类鉴定和系统进化研究。
16s rrna基因测序原理

16s rrna基因测序原理
16S rRNA基因测序是一种常用的微生物分析方法,用于研究
和鉴定微生物的种类和数量。
16S rRNA是细菌和古细菌的核
糖体RNA的一个组成部分,它在不同的微生物中存在一定的
变异性,这种变异性可以用来区分不同的微生物。
16S rRNA基因测序的原理是通过提取样品中的总DNA或总RNA,然后使用聚合酶链式反应(PCR)扩增16S rRNA基因。
PCR反应使用一对通用引物,能扩增大多数细菌和古细菌的
16S rRNA基因片段。
扩增获得的DNA片段可以通过电泳或
其他方法进行分离和纯化。
得到纯化的DNA片段后,可以使用Sanger测序技术或高通量
测序技术对其进行测序。
Sanger测序技术是一种经典的测序方法,通过反复合成和分离DNA链的方式逐个测序碱基。
高通
量测序技术,如Illumina平台,使用一种并行测序原理,可以
同时测序大量的DNA片段。
通过测序获得的16S rRNA序列可以通过比对已知的16S
rRNA数据库进行比对分析,以确定样品中微生物的种类和亲
缘关系。
也可以通过对多个样品的测序结果进行比较,进行微生物群落结构和多样性分析。
总的来说,16S rRNA基因测序是一种通过扩增和测序16S rRNA基因来分析微生物组成和多样性的方法,能够对微生物
进行定性和定量分析。
16srRNA序列同源性分析与细菌系统分类鉴定(论文资料)

004 16s rRNA 序列同源性分析与细菌系统分类鉴定中国预防医学科学院营养与食品卫生研究所 (北京 100050)焦振泉 刘秀梅综述 孟昭赫审校 摘要 本文介绍了16s rRNA 序列测定及同源性分析的方法,并阐述了其在细菌系统分类鉴定中的重要作用。
关键词 16s rRNA 序列同源性分析 细菌 分类鉴定 近10多年来,随着分子生物学理论和技术的迅速发展,特别是作为生物技术里程碑的聚合酶链反应(PCR )技术的出现及核酸测序技术的不断完善,产生了许多新的分类方法,如:质粒图谱、限制性片段长度多态性分析、脉冲场凝胶电泳、PCR 指纹图、r DNA 指纹图、16s rRNA 序列分析等。
它们主要是对细菌染色体进行直接的DNA 分析或对染色体外的DNA 片段进行分析,从遗传进化的角度去认识细菌,从分子水平进行分类与鉴定,使细菌的分类越来越科学和精确,特别是16s rRNA 序列分析方法的出现使细菌进化可以通过试验研究来证实。
这是细菌分类史上的一次革命,必将使人们对生物进化及其与其它生物学科关系的认识更加深入。
1 16s rRNA 的结构与性质16s rRNA 为原核生物核糖体中一种核糖体RNA 。
目前,在细菌的系统分类学研究中最有用的和最常用的分子钟是rRNA ,其种类少,含量大(约占细菌RNA 含量的80%),分子大小适中,存在于所有的生物中,特别是其进化具有良好的时钟性质,在结构与功能上具有高度的保守性,素有“细菌化石”之称。
rRNA 在大多数原核生物中都具有多个拷贝[1],5s 、16s 和23s rRNA 的拷贝数相同[2],16s rRNA 由于大小适中,约115kb 左右,既能体现不同菌属之间的差异,又能利用测序技术来较容易地得到其序列,故被细菌学家及分类学家所接受[3]。
所以,“细菌系统学研究特设委员会”建议依据系统发育关系分类。
通过对其序列的分析,可以判定不同菌属、菌种间遗传关系的远近。
微生物分离纤维素降解菌的筛选与分离

微生物分离纤维素降解菌的筛选与分离纤维素是一种广泛存在于自然界中的有机化合物,它是植物细胞壁的主要组成部分。
纤维素具有高度的生物降解性,然而,其高度结晶性和复杂的结构使其难以被常规的酶解系统降解。
在生物领域中,微生物分解是一种有效且环保的方法,因此,筛选和分离纤维素降解菌对于提高纤维素降解效率具有重要意义。
一、筛选纤维素降解菌的方法1.1 培养基的选择筛选纤维素降解菌的第一步是选择合适的培养基。
常用的纤维素降解培养基包括CMC(羧甲基纤维素钠)、Avicel(微晶纤维素)、Whatman No.1滤纸等。
这些培养基能够提供纤维素降解菌所需的碳源和营养物质,有利于菌群的生长和繁殖。
1.2 筛选方法传统的筛选方法是利用纤维素作为唯一的碳源,在培养基中培养环境中的微生物,通过测定产酶能力来判断纤维素降解菌的存在。
常用的方法有:(1)红色亚甲基纤维素(RAC)将纤维素培养基添加亚甲基蓝等指示剂,在纤维素降解区域由蓝色转变为红色,表明纤维素被降解。
(2)半定量筛选利用葡萄糖法测定纤维素降解能力。
在培养基中添加不同浓度的纤维素,观察菌落的生长情况和菌液中的葡萄糖含量,评估纤维素降解能力。
(3)放射标记纤维素将放射性同位素标记在纤维素分子上,通过测定纤维素的解脱率来评估菌株的降解能力。
二、纤维素降解菌的分离与鉴定2.1 分离方法从自然环境中分离纤维素降解菌是筛选过程的关键步骤之一。
常用的分离方法包括:(1)稀释平板法将适当稀释的样品在纤维素培养基上均匀涂布,经过一段时间后,将生长的菌落分离并培养纯种。
(2)可溶性物质包埋法将样品与纤维素培养基搅拌均匀,接种到含有纤维素的胶状物上,培养一段时间后,可分离出纤维素降解菌。
2.2 鉴定方法为了确定分离的菌株是否为具有纤维素降解能力的菌株,需要进行鉴定。
常用的鉴定方法包括:(1)形态学鉴定观察菌落的形态、颜色和菌落边缘等特征,使用显微镜观察细胞的形状和结构。
(2)生理生化特性鉴定测定菌株的氧耗、氧释等生理特征,通过测定菌株对不同碳源和氮源的利用情况来判断其代谢特性。
高效纤维素降解菌的筛选鉴定及特性研究

高效纤维素降解菌的筛选鉴定及特性研究1. 本文概述随着生物技术的迅速发展,微生物在环保、能源等领域的应用备受关注。
高效纤维素降解菌作为其中之一,在解决全球气候变化、生物质能源开发等方面具有重要意义。
本文将围绕高效纤维素降解菌的筛选鉴定及特性研究展开论述。
高效纤维素降解菌的筛选鉴定是研究其特性的前提。
这一过程包括从自然界中采集样品,如土壤、腐木等,然后在选择性培养基上进行富集培养,初步筛选出能够降解纤维素的细菌。
通过形态学、生理学和分子生物学方法进行鉴定和纯化。
高效纤维素降解菌的特性研究包括生理学特性、代谢特性和营养需求等方面。
生理学特性方面,高效纤维素降解菌为好氧菌,最适生长温度范围为2535,最适pH值为0。
代谢特性方面,高效纤维素降解菌主要通过分泌纤维素酶进行纤维素的降解,这些酶在细胞内合成,然后分泌至细胞外,作用于纤维素分子不同位置,将纤维素降解为可溶性糖。
营养需求方面,高效纤维素降解菌生长需要碳源、氮源、磷源、钾源等,其中碳源主要为纤维素,氮源主要为有机氮,磷源和钾源则通过无机盐形式补充。
环境影响也是高效纤维素降解菌特性研究的一个重要方面。
在降解纤维素过程中,高效纤维素降解菌会产生二氧化碳气体,其释放量与纤维素的降解量呈正相关。
同时,高效纤维素降解菌在生长过程中可能会对培养基产生一定程度的污染,但可通过无菌操作等措施加以控制。
高效纤维素降解菌的筛选鉴定及特性研究具有重要的实践意义。
通过深入了解高效纤维素降解菌的生理学、代谢特性和营养需求等方面的特性,以及其对环境的影响,将为今后更好地开发利用纤维素资源、解决全球气候变化等问题提供理论支持和实践指导。
目前对于高效纤维素降解菌的研究仍存在一些不足之处,如不同种类的高效纤维素降解菌之间的协同作用机制尚不明确,以及如何提高纤维素降解菌的降解效率及降低生产成本等方面仍需进一步探讨。
未来,可以从这些方面展开深入研究,以推动高效纤维素降解菌的应用和发展。
16Srna序列分析比对

16Srna序列分析比对研究目的:实验工具:利用点阵分析方法可以找到两个序列之间的所有可能的残基匹配,以及两序列之间的遗传重组现象,用Vector NTI 8 软件进行双序列局部比对,设计窗口值为5,阈值为100进行点阵分析实验材料:16S中的"S"是一个沉降系数,亦即反映生物大分子在离心场中向下沉降速度的一个指标,值越高,说明分子越大大小界门纲目科属种16S RNA1 849 细菌厚壁菌门乳酸杆菌目链球菌科链球菌属变形链球菌16S RNA2 924 细菌变形菌门变形菌纲鞘脂单胞菌目鞘脂单胞菌科发酵单胞菌运动发酵单胞菌16S RNA3 762 细菌变形菌门丙型变形细菌纲弧菌目弧菌科弧菌霍乱弧菌16S RNA4 910 细菌厚壁菌门芽孢杆菌目芽孢杆菌科土芽孢杆菌属土芽孢杆菌16S RNA5 1007 细菌厚壁菌门乳酸杆菌目肠球菌科肠球菌属海氏肠球菌melmttrna 807 真核生物后生动物门线虫纲垫刃线虫目垫刃总科根结线虫属爪哇根结线虫sulfolobus 1496 古生菌泉古菌门热变形菌纲硫化叶菌目好热好酸菌科硫化叶菌属硫磺矿硫化叶菌其中变形链球菌、运动发酵单胞菌、霍乱弧菌、嗜热脂肪芽孢杆菌、海氏肠球菌属于细菌界,而硫叶菌属于古生菌,爪哇根结线虫属于真核生物。
一、原核生物和古生菌的比对:1.变形链球菌和硫磺矿硫化叶菌比对结果:2.运动发酵单胞菌和硫磺矿硫化叶菌比对结果:118136154172184511452894335777218651009115312971492变形链球菌118136154172192011452894335777218651009115312971492运动发酵单胞菌3.霍乱弧菌和硫磺矿硫化叶菌比对结果:4.土芽孢杆菌和硫磺矿硫化叶菌比对结果:118136154175811452894335777218651009115312971492霍乱弧菌11773535297059051143285427569711853995113712791491土芽孢杆菌5.海氏肠球菌和硫磺矿硫化叶菌比对结果:1181361541721901100211452894335777218651009115312971491海氏肠球菌二、原核生物和真核生物的比对:1.变形链球菌和爪哇根结线虫比对结果:2.运动发酵单胞菌和爪哇根结线虫比对结果:199197295393491589687785845179157235313391469547625703803变形链球菌199197295393491589687785883920179157235313391469547625703803运动发酵单胞菌3.霍乱弧菌和爪哇根结线虫比对结果:4.土芽孢杆菌和爪哇根结线虫比对结果:199197295393491589687758179157235313391469547625703803霍乱弧菌199197295393491589687785906179157235313391469547625703803土芽孢杆菌5.海氏肠球菌和爪哇根结线虫比对结果:1991972953934915896877858831003179157235313391469547625703803海氏肠球菌三、古生菌和真核生物比对:硫磺矿硫化叶菌和爪哇根结线虫比对分析结果:1631251872493113734354975596216837458031143285427569711853995113712791492m四、1、变形链球菌和运动发酵单胞菌比对:1113225337449561673785845191181271361451541631721811920变形链球菌531539547555563571579587597588594600606612618624630636642649变形链球菌613614615616617618619620621ATACCCTGG665666667668669670671672673ATACCCTGG变形链球菌2、土芽孢杆菌和运动发酵单胞菌比对:1113225337449561673785906191181271361451541631721811920土芽孢杆菌420430440450460470480485394402410418426434442450458466土芽孢杆菌688690692694696ATACCCTGG664665666667668669670671672673GATACCCTGG土芽孢杆菌3、海氏肠球菌和运动发酵单胞菌比对:11132253374495616737858971003191181271361451541631721811920海氏肠球菌463475487499511523535547559569390400410420430440450460470484海氏肠球菌744746748750752753GATACCCTGG664665666667668669670671672673GATACCCTGG海氏肠球菌4、变形链球菌和霍乱弧菌比对:193185277369461553645737845175149223297371445519593667758变形链球菌527539551563575587599611588598608618628638648658668675变形链球菌623624625626627628629630AGTCCACG676677678679680681682683AGTCCACG变形链球菌5、土芽孢杆菌和霍乱弧菌比对:193185277369461553645737829906175149223297371445519593667758土芽孢杆菌187191195199203207211176180184188192198土芽孢杆菌698699700701702703704705AGTCCACG676677678679680681682683AGTCCACG土芽孢杆菌6、海氏肠球菌和霍乱弧菌比对:1931852773694615536457378299211003175149223297371445519593667758海氏肠球菌484492500508516524532540409415421427433439445451457461海氏肠球菌755756757758759760761762AGTCCACG676677678679680681682683AGTCCACG海氏肠球菌7、运动发酵单胞菌和霍乱弧菌的比对:193185277369461553645737829920175149223297371445519593667758运动发酵单胞菌175181187193199205211217223227156162168174180186192198203运动发酵单胞菌8、变形链球菌和土芽孢杆菌的比对:1111221331441551661771845189177265353441529617705793906变形链球菌512530548566584602620629575589603617631645659673687701709变形链球菌504506508*********515GTGTAGCGGTGA580581582583584585586587588589590TGTAGCGGTGA变形链球菌9、海氏肠球菌和土芽孢的比对:11112213314415516617718811003189177265353441529617705793906海氏肠球菌6678901021141261381436162636465666768693海氏肠球菌636638640642644646648649GTGTAGCGGTGAAA580581582583584585586587588589590TGTAGCGGTGA海氏肠球菌10、变形链球菌和海氏肠球菌的比对:11232453674896117338451991972953934915896877858831003变形链球菌534542550558566574582590669675681687693699705711717723729变形链球菌。
16SrRNA基因序列分析在原核生物种的分类中的应用

请老师同学批评指正!
因此最适合于揭示各原核生物的亲缘关系,确定各原 核生物彼此系统发育相关性或进化距离。16SrRNA 基因 序列也因此被公认为是一把好的谱分析的“分子尺”。
【基本步骤】
细菌基因组的提取 特异引物扩增 16SrDNA序列 验证PCR产物 扩增是否成功
选取近似菌种序列 构建系统发育树
与数据库中已知 细菌比较获得样品 种属信息
再将得到的DNA序列输入MEGA软件进 行建树。
16S rRNA 基因序列分析技术的问题:
1、首先是数据库的完整性,尽管数据库收录的细菌 种类在不断增加, 但还会遇到不能确定的情况;早期 所测定的结果也不尽正确。
2、数据库中有些序列中还存在很多不清楚的位点。 其序列中存在着突变速度很快的“热点”区域和 高度保守的区域 , 因此其确切的进化速度目前还不列
判断16SrDNA序列扩增是否成功:
经琼脂糖凝胶电泳在1500bp附近出现条带, 说明16s rDNA已经扩增成功.由于16s rDNA序列 长度一般1500bp左右。
DNA测序获得16SrRNA处理后得到的结果
GCATGCGGCGTGCTATACATGCAAGTCGAACGAACTCTGGTATTGATTGGTGCTTGCATCATGATTTACATTTGAGTGAGTGGCGA ACTGTGAGTAACACGTGGGAAACCTGCCCAGAAGCGGGGGATAACACCTGGAAACAGATGCTAATACCGCATAACAACTTGGACCG CATGGTCCGAGTTTGAAAGATGGCTTCGGCTATCACTTTGGATGGTCCCGCGGCGTATTAGCTAGATGGTGGGGTAACGGCTCACC ATGGCAATGATACGTAGCCGACCTGAGAGGGTAATCGGCCACATTGGGACTGAGACACGGCCCAAACTCCTACGGGAGGCAGCAGT AGGGAATCTTCCACAATGGACGAAAGTCTGATGGAGCAACGCCGCGTGAGTGAAGAAGGGTTTCGGCTCGTAAAACTCTGTTGTTA AAGAAGAACATATCTGAGAGTAACTGTTCAGGTATTGACGGTATTTAACCAGAAAGCCACGGCTAACTACGTGCCAGCAGCCGCGG TAATACGTAGGTGGCAAGCGTTGTCCGGATTTATTGGGCGTAAGCGAGCGCAGGCGGTTTTTTAAGTCTGATGTGAAAGCCTTCGG CTCAACCGAAGAAGTGCATCGGAAACTGGGAAACTTGAGTGCAGAAGAGGACAGTGGAACTCCATGTGTAGCGGTGAAATGCGTAG ATATATGGAAGAACACCAGTGGCGAAGGCGGCTGTCTGGTCTGTAACTGACGCTGAGGCTCGAAAGTATGGGTAGCAAACAGGATT AGATACCCTGGTAGTCCATACCGTAAACGATGAATGCTAAGTGTTGGAGGGTTTCCGCCCTTCAGTGCTGCAGCTAACGCATTAAG CATTCCGCCTGGGGAGTACGGCCGCAAGGCTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTAAT TCGAAGCTACGCGAAGAACCTTACCAGGTCTTGACATACTATGCAAATCTAAGAGATTAGACGTTCCCTTCGGGGACATGGATACA GGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTATTATCAGTTGCCAGC ATTAAGTTGGGCACTCTGGTGAGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTATGACCT GGGCTACACACGTGCTACAATGGATGGTACAACGAGTTGCGAACTCGCGAGAGTAAGCTAATCTCTTAAAGCCATTCTCAGTTCGG ATTGTAGGCTGCAACTCGCCTACATGAAGTCGGAATCGCTAGTAATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTT GTACACACCGCCCGTCACACCTGAGAGTTTGTAACACCCAAAGTCGGTGGGGTAACCTTTTAGGAACCAGCCGCCTAAGGTGACAG AGATGG
土壤中纤维素分解菌的分离鉴定

土壤中纤维素分解菌的分离鉴定纤维素是植物细胞壁中最主要的组成成分之一,其分解可以释放丰富的能量和营养物质,因此纤维素分解菌对土壤生态系统具有重要作用。
因此,对于土壤中纤维素分解菌的分离鉴定,有助于深入了解其功能和生态学意义。
一、实验材料和方法1.实验样品本实验采集自我国典型红壤中的一些样品,土壤样本为表层土壤(0-15cm)。
2.分离方法对样品进行拔草、松散、天然干燥、筛分等处理,然后进行分离。
1)直接培养法:将半固态纤维素酵母提取培养基(CMC-Na:1.5g/L;NHa2PO4:0.5g/L;KH2PO4:0.5g/L;MgSO4:0.5g/L;FeSO4:0.01g/L;NaCl:0.5g/L;以及0.5%的琼脂)均匀涂布于含有纤维素的平板上。
在37℃恒温箱中孵育培养。
2)稀释培养法:起初将样品按比例加入到同等体积的0.85%氯化钠溶液中。
接着对稀释液进行梯度稀释,将每1ml的第-1-7次稀释到含有纤维素的琼脂平板上。
在37℃恒温箱中孵育培养集落。
3)筛选方法在进行分离后,筛选出7个形态和功能不同的菌落,进行细菌纯化。
然后,利用革兰染色法、生理生化实验和16S rRNA序列分析等方法进行鉴定。
二、结果及讨论1.分离菌株的特征我们通过实验分离出来7个不同的纤维素分解菌株,它们分别是:AT1、AT2、AT3、AT4、AT5、AT6和AT7。
在进行鉴定时,我们根据它们的形态特征、生理生化性质及16S rRNA基因序列,对其进行了系统的分类和鉴定。
菌株AT1,为革兰阴性杆菌,能够在CMC-Na离子互济培养基中发生较强的纤维素降解。
我们对其进行16S rRNA测序,发现与Salmonella sp. ATCC 43971的16S rRNA序列高度同源(99.7%)。
因此,我们认为其为一株Salmonella sp.的分解菌株。
2.纤维素分解菌株的多样性我们通过对红壤样品中纤维素分解菌株的分离和鉴定,发现其中的菌株分属于不同的细菌属,如Salmonella、Microbacterium、Pseudomonas、Bacillus、Acinetobacter、Streptomyces、Rhodococcus等,这表明纤维素分解菌株在细菌属的多样性方面具有很高的潜力。
菌群16s文献解读

菌群16s文献解读引言概述:菌群16s文献是指通过16s rRNA基因测序技术对菌群进行研究并发表的科学文献。
16s rRNA基因是细菌和古菌中高度保守的基因,通过对其序列进行分析可以了解菌群的多样性、组成以及功能。
本文将从菌群16s文献的优势、技术原理、分析方法、应用领域和未来发展等五个大点进行阐述。
正文内容:1. 优势1.1 高度保守的16s rRNA基因使得菌群16s文献具有较高的可靠性和稳定性。
1.2 菌群16s文献可以对不同样本中的菌群进行比较研究,揭示它们的差异和相似性。
1.3 通过菌群16s文献可以发现新的菌群种类,丰富了我们对微生物世界的认识。
2. 技术原理2.1 菌群16s文献的核心技术是16s rRNA基因的测序,通过对该基因序列的测定和比对,可以确定菌群的种类和数量。
2.2 常用的16s rRNA测序技术包括Sanger测序和高通量测序(如Illumina测序),后者具有更高的测序深度和准确性。
2.3 通过对测序结果的生物信息学分析,可以得到菌群的多样性指数、物种组成、功能预测等信息。
3. 分析方法3.1 菌群16s文献的分析方法包括OTU聚类分析、物种多样性分析、功能预测等。
3.2 OTU聚类分析是将相似的序列聚类为一个OTU(操作税单元),用于评估样本中的物种多样性。
3.3 物种多样性分析可以通过计算Shannon指数、Simpson指数等来评估样本中物种的多样性和均匀度。
3.4 功能预测可以通过基于16s rRNA序列的功能预测软件(如PICRUSt)来预测菌群的功能组成。
4. 应用领域4.1 菌群16s文献在医学领域中被广泛应用,如研究肠道菌群与肠炎、肠道疾病等的关系。
4.2 在环境科学领域,菌群16s文献可以用于研究不同环境中的微生物组成和功能。
4.3 农业领域中,菌群16s文献可以用于研究土壤菌群与作物生长、土壤肥力等的关系。
5. 未来发展5.1 随着高通量测序技术的发展,菌群16s文献将变得更加高效和准确。
菌群16s测序解读

菌群16s测序解读菌群16s测序,是一种广泛应用于微生物群落研究的技术。
通过对16s rRNA基因进行测序,并对其序列进行分析,可以对群落中存在的细菌进行分类和鉴定。
这项技术能够帮助我们了解细菌的多样性、丰度和功能,对于研究环境微生物群落结构、人体菌群与健康之间的关系等具有重要意义。
在进行菌群16s测序时,首先需要从样品中提取细菌的DNA,并进行PCR扩增。
常用的扩增方法是利用16s rRNA的保守序列设计引物,将目标序列扩增出来。
扩增后的DNA片段进一步纯化和测序,最终得到的测序数据即为16s rRNA的序列信息。
在得到16s测序数据后,需要对数据进行处理和解读。
首先是序列质量控制,去除低质量的序列。
接下来,对序列进行去噪声处理,去除PCR扩增产生的噪声和测序错误。
然后,利用聚类算法对序列进行分割,将相似的序列分为同一个OTU(操作分类单元)。
最后,通过比对数据库,将OTU与已知菌种进行分类和鉴定。
通过菌群16s测序,我们可以获得菌群的多样性指数。
多样性指数反映了菌群的物种丰富度和均一性。
常用的多样性指数包括Shannon指数、Simpson指数等。
这些指数能够告诉我们菌群内的物种多样性和优势菌种。
除了菌群的多样性,16s测序还可以帮助我们了解菌群的功能。
通过比对菌群16s测序数据和已知的功能基因数据库,我们可以推断菌群在代谢、信号传导、抗性等方面的功能。
这对于研究菌群与宿主的相互作用机制、菌群在环境中的重要功能等具有重要意义。
菌群16s测序的应用十分广泛。
在环境科学方面,可以通过菌群16s测序了解不同环境样品中细菌的组成和功能,帮助了解细菌在环境生物地球化学循环中的作用。
在医学研究中,菌群16s测序可以用于研究肠道菌群与人体健康之间的关系,探索微生物在疾病发生发展中的作用。
在食品安全检测中,通过菌群16s测序可以追踪食品中可能存在的致病菌,提供相关的病原菌检测和风险评估。
尽管菌群16s测序在微生物群落研究中具有广泛应用,但也存在一些限制。
一株芽孢杆菌一株芽孢杆菌16SrRNA的基因序列测定和系统进化分析16SrRNA的基因序列测定和系统进化分析

2.1 基因组 DNA 扩增产物鉴定结果 将 以 BS501 菌 株 的 基 因 组 DNA 为 模 板 扩 增 的 PCR
产物进行 1%琼脂糖凝胶电泳。 由图 1 可知,扩增产物大 小约为 1 500 bp 左右。 2.2 阳性重组 T 克隆鉴定结果
用琼脂糖凝胶电泳检测重组质粒是否发生转化,白斑 为重组阳性质粒,蓝斑为未重组质粒(图 2)。
LI Rui-fang, ZHAO Yu-feng, XUE Wen-wen, TIAN Yang-yuan, ZHANG Chang-fu (College of Bioengineering, Henan University of Technology, Zhengzhou 450001, China)
122
淀即为BS501 基因组 DNA;(10)用 400 μL 70%的乙醇洗 涤沉淀 2 次,自然干燥 后,用 30 μL 的 TE 或 超 纯 水 溶 解 DNA,置于-20℃冰箱备用。 1.2.2 引物合成 参照文献[9]介绍的方法,并根据芽孢杆 菌 16S rRNA 基因序列的保守区,用 Primer 5.0 软件设计 引物,由上海生工生物工程公司合成。
李瑞芳, 赵玉峰, 薛雯雯, 田泱源, 张长付 (河南工业大学生物工程学院,河南 郑州 450001)
摘 要: 从河南黄河稻区土壤中分离出一株具有较强稻瘟病菌拮 抗 活性 的芽 孢 杆菌 菌株 BS501,经 过形 态观 察 、生理 生化 特
性检测和电镜技术,确定其为芽孢杆菌属细菌。 为进一步确定其分类学地位,扩增了 BS501 基因组的 16S rDNA,并将测序结果提
Key words:Bacillus strain; 16S rRNA gene; sequencing; phylogenetic analysis
代谢组与16S rDNA测序整合分析
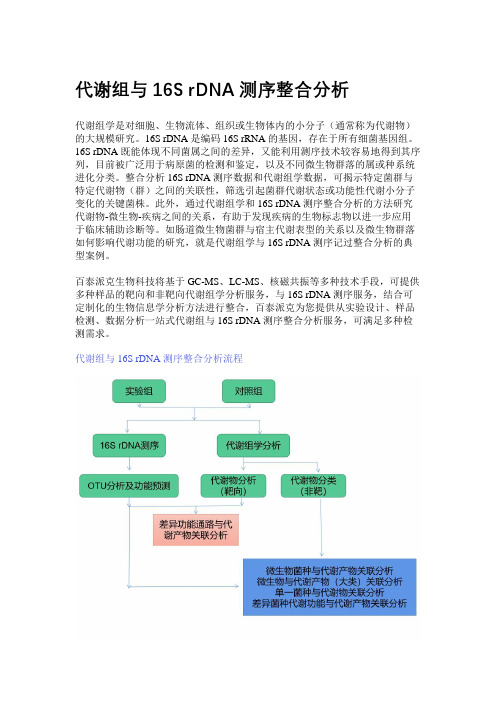
代谢组与16S rDNA测序整合分析代谢组学是对细胞、生物流体、组织或生物体内的小分子(通常称为代谢物)的大规模研究。
16S rDNA是编码16S rRNA的基因,存在于所有细菌基因组。
16S rDNA既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列,目前被广泛用于病原菌的检测和鉴定,以及不同微生物群落的属或种系统进化分类。
整合分析16S rDNA测序数据和代谢组学数据,可揭示特定菌群与特定代谢物(群)之间的关联性,筛选引起菌群代谢状态或功能性代谢小分子变化的关键菌株。
此外,通过代谢组学和16S rDNA测序整合分析的方法研究代谢物-微生物-疾病之间的关系,有助于发现疾病的生物标志物以进一步应用于临床辅助诊断等。
如肠道微生物菌群与宿主代谢表型的关系以及微生物群落如何影响代谢功能的研究,就是代谢组学与16S rDNA测序记过整合分析的典型案例。
百泰派克生物科技将基于GC-MS、LC-MS、核磁共振等多种技术手段,可提供多种样品的靶向和非靶向代谢组学分析服务,与16S rDNA测序服务,结合可定制化的生物信息学分析方法进行整合,百泰派克为您提供从实验设计、样品检测、数据分析一站式代谢组与16S rDNA测序整合分析服务,可满足多种检测需求。
代谢组与16S rDNA测序整合分析流程应用领域农林领域:抗逆胁迫机制,物种保护研究等;畜牧业:致病机理研究,肉类及乳制品品质研究等;海洋水产:渔业环境与水产品安全等;微生物:致病机理,耐药机制,病原体-宿主相互作用研究等;生物医药:生物标志物,疾病机理机制,疾病分型,药物开发,个性化治疗等;环境科学:发酵过程优化,生物燃料生产,环境危害风险评估研究等;食品科学:食品储藏及加工条件优化,食品组分及品质鉴定,食品安全监检测等。
中/英文项目报告在技术报告中,百泰派克会为您提供详细的中/英文双语版技术报告,报告包括:1. 实验步骤(中英文)2. 相关的实验参数(中英文)3. 质谱图片4. 原始数据5. 代谢组与16S rDNA测序结果How to order?关于百泰派克北京百泰派克生物科技有限公司(Beijing Bio-Tech Pack Technology Company Ltd. 简称BTP)成立于2015年,是国家级高新技术企业,业务范围主要围绕蛋白和小分子代谢物检测两大板块,从事蛋白质和小分子代谢物的理化性质分析及结构解析等相关技术服务,为客户提供高性价比、高效率的技术服务。
16s嵌合体序列

16s嵌合体序列16s嵌合体序列是指由16s rRNA基因和其相邻区域组成的DNA序列,可以用于研究微生物的分类和进化。
本文将围绕16s嵌合体序列展开,探讨其在微生物研究中的重要性和应用。
第一部分:16s嵌合体序列的基本概念和特点16s嵌合体序列是由16s rRNA基因和嵌合体序列两部分组成的,其中16s rRNA基因是一种小分子RNA,在细菌和古菌中广泛存在,具有高度保守性和变异性。
嵌合体序列是16s rRNA基因与其相邻区域的连接部分,包含了一些特定的变异位点。
第二部分:16s嵌合体序列在微生物分类中的应用由于16s rRNA基因的高度保守性,可以通过对16s嵌合体序列进行测序和比对,快速准确地鉴定和分类微生物。
通过比对16s嵌合体序列与已知序列数据库中的序列,可以确定微生物的系统发育位置,从而推断其分类和进化关系。
第三部分:16s嵌合体序列在微生物进化研究中的应用16s嵌合体序列的变异位点可以反映微生物在进化过程中的分化和演化。
通过对不同物种的16s嵌合体序列进行比较和分析,可以揭示微生物的进化历史和亲缘关系。
同时,通过比对16s嵌合体序列与已知进化树的序列,可以推测微生物的进化速度和分化模式。
第四部分:16s嵌合体序列在微生物生态学研究中的应用16s嵌合体序列可以用于研究微生物在不同环境中的分布和多样性。
通过对不同环境样品中的16s嵌合体序列进行测序和分析,可以了解微生物在不同环境中的组成和功能。
同时,通过比对不同样品中的16s嵌合体序列,可以揭示微生物在不同环境中的适应性和竞争关系。
第五部分:16s嵌合体序列在临床微生物学中的应用16s嵌合体序列可以用于鉴定和分类临床中的微生物病原体。
通过对患者样品中的16s嵌合体序列进行测序和比对,可以快速准确地确定病原体的种类和亚种。
同时,通过分析不同病原体的16s嵌合体序列,可以了解其耐药性和毒力相关基因的变异情况。
总结:16s嵌合体序列作为微生物研究的重要工具,在微生物分类、进化、生态和临床中都有广泛应用。
16srna序列分析

1 6 s R N A 序列分析前言点阵分析(dot-matrix analysis),是指将两条以上核酸或氨基酸序列分别列示于纵横坐标,在同一位置上出现相同符号并形成连线,以揭示序列中重复片段或两条序列同源性的方法。
16sRNA是原核生物核糖体小亚基上的RNA,这段RNA序列在不同的细菌中的结构、功能都具有高度保守性,完成测序后通过点阵分析可以确定其同源性比较、用于细菌的系统分类鉴定、多样性和亲缘关系分析等。
虽然相对保守,但在不同的微生物中,16sRNA基因序列还是存在差异的,一般来说,16sRNA序列越接近,菌种关系越近,同源性越大;反之,16sRNA序列差异越大,同源性越小。
而本文主要利用点阵分析的方法对不同菌种的16SrRNA基因进行局部比对,通过比较两个序列之间的相似区域和保守性位点,寻找两者可能的分子亲缘关系,以及进化途径的关系,从而为生物进化的研究提供理论基础。
内容在窗口值为50,阈值为80的情况下,对细菌(变形链球菌、运动发酵单胞菌、霍乱弧菌、嗜热脂肪土芽孢杆菌、海氏肠球菌)、古生菌(硫磺矿硫化叶菌)以及真核生物线粒体(爪哇根结线虫线粒体)16sRNA两两序列间的点阵分析如下:1. 运动发酵单胞菌subsp mobilis 部分的16srRNA序列1105209313417521625729800185169253337421505589673757875链球菌mutans16SrRNA(Streptococcus mutans 与Zymomonas mobilis subsp.mobilis )1-22. 霍乱弧菌局部的16srRNA 基因序列 18516925333742150558967375780016913720527334140947754561371316s r n a 1链球菌mutans 16SrRNA(Streptococcus mutans 与Viibrio cholerae)1-33. 嗜热脂肪土芽孢杆菌1103205307409511613715800183165247329411493575657739861链球菌mutans 16SrRNA(Streptococcus mutans 与Geobacillus srearothermophilus )1-44. 肠球菌种局部的16srRNA 基因序列 110921732543354164975318717325934543151760368977586191116s r n a链球菌mutans16SrRNA(Streptococcus mutans 与Enterococcus hirae )1-55. 爪哇根结线虫线粒体1651291932573213854495135776417057698451771532293053814575336096857618031链球菌mutans16SrRNA(Streptococcus mutans 与mitochonddrion meloidogyne javanica )1-m6. 硫磺矿硫化叶菌 16512919325732138544951357764170576984511432854275697118539951137127914921链球菌mutans16SrRNA(Streptococcus mutans 与Sulfolobus solfataricus )1-s7. 硫磺矿硫化叶菌1631251872493113734354975596216837458031143285427569711853995113712791492m爪哇根结线虫线粒体(mitochonddrion meloidogyne javanica 与Sulfolobus solfataricus)m-s结果变形链球菌-远动发酵单胞菌: 变形链球菌129~190、580~640、710~770处的碱基分别于运动发酵单胞菌180~255、630~705、800~860处的碱基大致相同;相似性较高,亲缘关系较近,分子进化关系较近;变形链球菌-霍乱弧菌:相似性一般,亲缘关系一般,分子进化关系一般;有相似序列如变形链球菌160左右、600~641处的碱基分别与霍乱弧菌217左右、649~679处的碱基大致相同;此部分序列可能对基本生命活动来说不可缺少或者有相似的功能。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2 3 2 0 0 1 ) ( 淮南师范学 院 生命科学 系 , 安徽 淮南
摘 要 : 以牦 牛瘤 胃内容物为接种物 , 以滤 纸为唯一碳 源富集到一个降解纤维素 的混合培养物. 构建混合 培7个 O T U. 其中1 9个序列与 已培养细菌 的 1 6 S r R N A基 因相 似性> 9 7 %, 占总序列 的 3 8 . 8 %; 2个 古菌的序列与 甲烷菌 的 1 6 S r R N A基 因相 似性分别 为 9 9 %和 9 8 %, 占总 序列 的 4 . 1 %; 2 8个序列属 于未培养细菌 , 占总序列 的 5 7 . 1 %. 未培养 的序列形成 4个独立 的系统发育分支 ,
t a x o n o my u n i t s( O T U s )a t t h e s i m i l a i r t y l e v e l o f 9 7 % s e q u e n c e s i mi l a r i t y .1 9 s e q u e n c e s w e r e r e l a t e d
2 0 1 3年 7月 第3 7卷 第 4期
安徽 大 学 学 报 ( 自然 科 学 版 ) J o u r n a l o f A n h u i U n i v e r s i t y( N a t u r a l S c i e n c e E d i t i o n )
Abs t r a c t : By us i ng il f t e r p a p e r a s t he s o l e c a r bo n s o u r c e,we o b t a i n e d a c e l l u l o s e-d e g r a d i n g mi x- c u l t u r e e n ic r h me n t wi t h y a k l x l me n c o n t e n t a s t h e i n o c u l a n t . T he 1 6S r RNA g e n e l i b r a r y wa s c o ns t r uc t e d a nd a t o t a l o f 49 c l o n e s we r e o b t a i n e d a nd t h e y we r e c l us t e r e d a s s e v e n o p e r a t i o n a l
t h e t o t a l c l o n e s . Th e r e ma i n i n g 2 8 c l o ne s we r e a f il f i a t e d t o t h e u nc u l t u r e d g r o u p s.a c c o u n t i n g f 0 r 5 7. 1 % o f t he t o t a l c l o ne s .Ba s e d o n t h e p h y l o g e ne t i c a n a l y s i s.t h r e e u n c u l t u r e d OTUs we r e r e l a t e d t o
J u l y 2 0 1 3
0 . 3 9 6 9 / j . i s s n . 1 0 0 0 - 2 1 6 2 . 2 0 1 3 . 0 4 . 0 1 6
纤维 素降 解 混 合培 养物 的 1 6 S r R N A基 因序 列 分析
s e q u e n c e s we r e r e l a t e d t o Me t h a n o b r e v i b a c t e r o l l e y a e a n d
r u mi n a n t i u m .a c c o u n t i n g or f 4 . 1 % 0 f
中 图分 类 号 : Q 9 3 文献标志码 : A 文章编号 : 1 0 0 0 — 2 1 6 2 ( 2 0 1 3) 0 4 — 0 0 9 3 — 0 6
An a l y s i s o f 1 6 S r RNA g e ne s e qu e n c e s o f a c e l l u l o l y t i c mi x— — c u l t u r e e nr i c h me nt
t o Ac t i n o my c e s r u mi n i c o l a a nd L ac t o b a c i l l u s v i t u l i n u s .a c c o u n t i n g f o r 38 .8% o f t he t o t a l c l o n e s .2
Z HANG Ke - g u i ,YU Di e ,YANG J i n g — y a n,W ANG S h u n — c h a n g ( D e p a r t me n t o f L i f e S c i e n c e ,H u a i n a n N o r m l a U n i v e r s i t y , H u a i n a n 2 3 2 0 0 1 ,C h i n a )
其 中 3个未培养分支 与在其 他环境中的黏附在纤 维素上 的瘤 胃细菌 的 1 6 S r R N A基 因序列相似 性> 9 7 %, 推
测 这 3类 微 生 物可 能 与 纤 维 素 降 解 相 关 .
关键词 : 牦牛瘤 胃; 纤维 素降