基因序列分析软件DNAStar简介
常用分子生物学软件简介

常用分子生物学软件简介常用分子生物学软件一、基因芯片:1、基因芯片综合分析软件。
ArrayVision 7.0一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。
Arraypro 4.0Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。
phoretix?Array Nonlinear Dynamics公司的基因片综合分析软件。
J-express挪威Bergen大学编写,是一个用JAVA语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JAVA运行环境JRE1.2后(5.1M)后,才能运行。
2、基因芯片阅读图像分析软件ScanAlyze 2.44,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。
输出为分隔的文本格式,可很容易地转化为任何数据库。
3、基因芯片数据分析软件Cluster斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。
SAMSignificance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。
4.基因芯片聚类图形显示TreeView 1.5斯坦福开发的用来显示Cluster软件分析的图形化结果。
现已和Cluster成为了基因芯片处理的标准软件。
FreeView是基于JAVA语言的系统树生成软件,接收Cluster生成的数据,比Treeview增强了某些功能。
5.基因芯片引物设计Array Designer 2.00DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具二、RNA二级结构。
DNAStar中文使用说明书概述

DNAstar---综合性序列分析平台生物谷提供下载,感谢原整理作者GETTING STARTED Introductory Tour of the LASERGENE SystemMAY 2001DNASTAR, Inc.1228 South Park StreetMadison, Wisconsin 53715(608) 258-7420Copyright . 2001 by DNASTAR, Inc.All rights reserved. Reproduction, adaptation, or translation without prior written permission is prohibited,except as allowed under the copyright laws or with the permission of DNASTAR, Inc.Sixth Edition, May 2001Printed in Madison, Wisconsin, USATrademark InformationDNASTAR, Lasergene, Lasergene99, SeqEasy, SeqMan, SeqMan II, EditSeq, MegAlign, GeneMan, Protean,MapDraw, PrimerSelect, GeneQuest, GeneFont , and the Method Curtain are trademarks or registered trademarks of DNASTAR, Inc. Macintosh is a trademark of Apple Computers, Inc.MacVertor. and GCG. are registered trademarks of Pharmacopeia, Inc.Disclaimer & LiabilityDNASTAR, Inc. makes no warranties, expressed or implied, including without limitation the implied warranties of merchantability and fitness for a particular purpose, regarding the software. DNASTAR does not warrant, guaranty, or make any representation regarding the use or the results of the use of the software in terms of correctness, accuracy, reliability, currentness, or otherwise. The entire risk as to the results andperformance of the software is assumed by you. The exclusion of implied warranties is not permitted by some states. The above exclusion may not apply to you.In no event will DNASTAR, Inc. and their directors, officers, employees, or agents (collectively DNASTAR) be liable to you for any consequential, incidental or indirect damages (including damages for loss of business profits, business interruption, loss of business information and the like) arising out of the use of, or the inability to use the software even if DNASTAR Inc. has been advised of the possibility of such damages.Because some states do not allow the exclusion or limitation of liability for consequential or incidental damages, the above limitations may not apply to you.DNASTAR, Inc. reserves the right to revise this publication and to make changes to it from time to time without obligation of DNASTAR, Inc. to notify any person or organization of such revision or changes. The screen and other illustrations in this publication are meant to be representative of those that appear on your monitor or printer.目录在苹果机(Macintosh)上的安装与升级05通过因特网升级06 软件安装 07 网络安装 08 疑难解答 12在PC机(Windows)上安装与升级17通过因特网升级18 软件安装 19 从EditSeq 开始 21 从GeneQuest开始 31 从MapDraw 开始 44 从MegAlign开始 54 从PrimerSelect开始 65 从Protean 开始 78 从SeqMan II开始 89L A S E R G E N Ef o rWi n d o w s & M a c i n t o s hDNASTA R I n c . ( 6 0 8 ) 2 5 8 - 7 4 2 0 f a x : ( 6 0 8 ) 2 5 8 - 7 4 3 9e m a i l : s u p p o r t @ d n a s t a r. c o m在苹果机(Macintosh)上的安装与升级通过因特网升级必备条件 6 下载升级程序6软件安装必备条件7 从CD安装Lasergene7网络安装必备条件8 定义8 Dongle安装10 服务器安装10 终端机安装11疑难解答系统配置冲突及网络问题12 解决网络问题的其他工具13 授权错误报告14 程序使用及更多的授权信息15通过英特网升级如果您以前已经安装了Lasergene 而且目前有升级和服务联系,您就可以通过英特网来升级您现有的版本,各种模块(module)都是以自解压形式存储的,你可以选择性的下载安装。
初中生物软件知识点归纳总结

初中生物软件知识点归纳总结软件在生物学的研究中扮演着重要角色,它们为科学家和学生提供了学习、研究和实践的工具。
在初中生物学的学习中,了解常用的生物软件知识点是非常重要的。
本文将对初中生物软件知识点进行归纳总结,帮助初中生更好地理解和运用这些软件。
1. 基因编辑软件基因编辑软件是帮助科学家编辑基因序列的工具,其中最著名的软件是CRISPR-Cas9。
CRISPR-Cas9软件可以帮助科学家准确地定位和编辑基因组中的特定基因,可以用于研究基因功能、治疗疾病等等。
初中生可以了解到CRISPR-Cas9软件的基本应用和原理,了解基因编辑的概念和意义。
2. 生物信息学软件生物信息学软件是处理和分析生物学数据的工具,其中一些最常用的软件有BLAST、NCBI、DNAStar等。
BLAST软件可以用于比对和分析DNA、蛋白质序列,帮助科学家找到相似的序列并进行进一步的研究。
NCBI是一个包含大量生物学数据库的在线平台,可以帮助科学家在数据库中搜索并浏览生物学信息。
DNAStar是一款用于DNA序列分析的软件,可以进行DNA序列的比对、注释和可视化等。
3. 模拟和建模软件初中生可以了解一些常用的生物模拟和建模软件,如Stellarium、BIOZONE和Stem cells等。
Stellarium是天文学软件,可以模拟出夜空中的星星和行星运动情况,帮助初中生了解天文学知识。
BIOZONE是一款模拟生物学实验的软件,可以帮助初中生进行虚拟实验并观察实验现象。
Stem cells软件则是帮助初中生学习干细胞的分化和发育过程。
4. 数据可视化软件数据可视化软件可以将生物学实验结果和数据转化为图表或图形,帮助科学家更好地理解和展示数据。
在初中生物学中,初中生可以了解一些简单的数据可视化软件,如Excel和GraphPad Prism。
Excel软件可以用于绘制图表、创建数据表格和计算简单统计量。
GraphPad Prism软件则更专业,可以进行复杂的数据分析、绘制高质量的图表和进行统计检验等。
DNAstar蛋白分析入门
DNAstar蛋白分析入门准备介绍一下DNAstar里面的蛋白分析功能,不过只是入门级别的介绍,我是做生化实验的,对于生物软件,只关心他们对我的实验有什么帮助,不关心后面的算法、程序等等生物信息学的东西。
DNAstar蛋白分析功能特点:1、蛋白一级和二级结构分析在同类软件里面算比较全面;2、用户设定功能少,所以比较傻,容易上手;3、结果报告图形漂亮。
缺点:1、绝大多数分析功能的报告都不能保存且无法输出,只能拷屏,简直就象demo软件;2、因为用户设定功能太少,结果常常不让人满意也无法修改,比如氨基酸颜色无法改变;3、没有基于结构的 alignment 分析;4、功能模块分割,做一个工作要进入不同模块,就象完成一个任务要打开几个软件,这是DNAstar最为愚蠢的地方。
当然这主要是因为原来的版本从unix发展而来,是历史遗留问题。
同样也因为是基于 unix 的原因,让我得到一个好处,老版本安装以后,直接拷贝目录,完全可以成为绿色免安装软件。
我到现在仍然在用这最早的版本,好像没有感觉到新版本有任何提高(除了网络查询功能,而DNAstar的网络功能跟CM7比,简直就不如没有),所以安装了新版本以后马上也就删除了。
―――――――――――――――――――――――――――――――――――――――第一步:editseq 建立蛋白序列和初步分析蛋白生化特性进入 editseq 模块后,由于默认序列文件是核酸文件,所以必须关闭默认窗口,重新建立一个序列文件,选择蛋白序列文件类型。
建立新序列的方式非常多,包括:手工输入、剪贴板拷贝、直接导入序列以及通过网络查询导入,这里不深入讨论,举例用手工输入一个短序列。
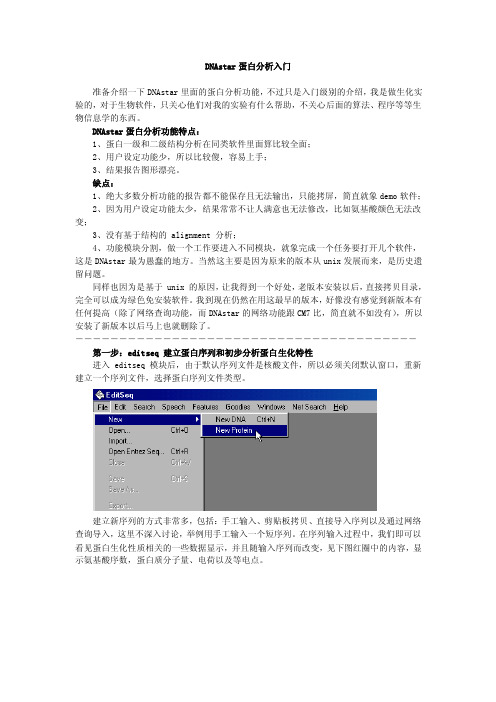
在序列输入过程中,我们即可以看见蛋白生化性质相关的一些数据显示,并且随输入序列而改变,见下图红圈中的内容,显示氨基酸序数,蛋白质分子量、电荷以及等电点。
通过蛋白统计数据显示,可以看见更多的蛋白质序列组成及生化特性的详细分析,注意分析前必须选中一段序列,这种方法可以用于分析蛋白序列中某区段的序列。
star 转录组流程

STAR转录组流程1. 简介STAR(Spliced Transcripts Alignment to a Reference)是一种广泛应用于转录组测序数据分析的软件工具。
它能够高效准确地将测序reads比对到参考基因组上,并根据比对结果计算基因表达水平。
本文将详细介绍STAR转录组流程的各个步骤和流程。
2. 数据准备在进行STAR转录组流程之前,需要准备以下数据: - 参考基因组序列文件(fasta格式) - 参考基因组注释文件(GTF/GFF格式) - 测序reads(fastq格式)3. 安装STAR首先需要在计算机上安装STAR软件。
可以从其官方网站()下载最新版本的STAR,并按照官方提供的安装说明进行安装。
4. 索引构建在进行比对之前,需要先对参考基因组进行索引构建。
索引构建只需要进行一次,之后可以多次重复使用。
索引构建的命令如下:STAR --runMode genomeGenerate --genomeDir /path/to/genomeDir --genomeFastaFiles /path/to/genome.fa --sjdbGTFfile /path/to/annotations.gtf其中,--genomeDir指定索引文件的输出目录,--genomeFastaFiles指定参考基因组序列文件,--sjdbGTFfile指定参考基因组注释文件。
5. 比对在进行比对之前,需要先将fastq格式的测序reads进行质量控制和预处理。
常见的质量控制工具有FastQC和Trimmomatic等。
质量控制和预处理的具体步骤不在本文的讨论范围内。
进行比对的命令如下:STAR --genomeDir /path/to/genomeDir --readFilesIn /path/to/reads.fastq --outFi leNamePrefix /path/to/output其中,--genomeDir指定参考基因组索引文件所在的目录,--readFilesIn指定测序reads的文件路径,--outFileNamePrefix指定输出文件的前缀。
DNAstar软件的使用(一)Megalign序列比对

DNAstar软件的使⽤(⼀)Megalign序列⽐对DNAstar软件共有七个⼩程序(见下图),各⾃执⾏不同的功能,Editseq⽤于序列编辑,Seqman可以去除载体序列和拼接序列,MegAlign则主要执⾏序列⽐对的功能.开启MegAlign软件⾸先需要点击File---New 新建⼀个⼯作⽂件再点击File---Entersequeces 添加需要⽐对的序列,⽀持多种格式的⽂件(.seq;.abi;.pro;.fas等),点Add可添加多个⽂件,点Done导⼊选中的⽂件.这是⽂件导⼊后的界⾯点Align选择序列⽐对的算法,其中多序列⽐对有三种算法:The Jotun Hein method,ClustalV和Clustal W;⼀般选择Clustal W即可。
三种算法的差异如下:“The Jotun Hein method was devised to align sequences that are previously known to be related by descent. The ClustalV and ClustalW alignment methods do not make this assumption. ClustalW is an advancement over ClustalV, and was designed to create more accurate alignments for highly diverged sequences”。
“CLUSTALW 是⼀种渐进的多序列⽐对⽅法,先将多个序列两两⽐对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产⽣系统进化指导树,对关系密切的序列进⾏加权;然后从最紧密的两条序列开始,逐步引⼊临近的序列并不断重新构建⽐对,直到所有序列都被加⼊为⽌”。
红⾊区域表⽰的是相似性最⾼的区域,蓝⾊和绿⾊是同源性较低的区域;多序列⽐对的⼀个缺点就是不会⾃动对序列反向互补后再⽐对,所以反向互补后同源性⾼的序列检测不出来。
DNAstar Megalign入门篇

先看一下megalign的简单介绍MegAlign 提供 6 列队(aligment)方法,进行DNA 和蛋白质序列的配对和多序列比较(multiple aligment) 。
多序列比较(multiple aligment)可以在MegAlign 的worktable 进行查看和编辑。
可以根据队列(aligment)的结果制作进化树(Phylogenetic trees),并且,有关序列距离的数据和残基替代可以容易地作成表格。
一般多序列比较(multiple aligment)的结果展示于队列(aligment)窗口,相似性和差异用彩色的直方图展示。
打开方法与editseq一样,只不过点选megalign图标,然后进入其界面选择File-Enter Sequences首先进行2个序列比对,选中所需序列1和2,点击add,使从左侧添加到右边的框中,单击Done出现如图所示界面,选中1与2(可按control点选),之后选择Align-One Pair-ByWilbur-Lipman method出现如图所示界面,即为blast结果,但画面不美观,可对其进行调整,点击鼠标所处位置按钮出现此对话框,里面可进行一系列设置,可根据自己喜好进行,使界面更美观形象设置后可看到错配碱基,如下,还是比较直观吧比对之后可对其进行结果查看,点选View-Alignment report即可结果如图对于多序列的比对,添加序列与一对序列一样,不过选择的Align-Clustal或者Jotun Hein命令如点了之后出现点选Jotun He 2次变成4条后点选View-P现如下结果,ein 后,出现如条了 )Phylogenetic因我用序列如图界面,图 Tree 进行系列太少,体现图中红线部分系统树分析不出很好效果分代表同源序果,欢迎大家序列(偷懒了家自己尝试了,2个序列添加简单的把Alignment report参数设置介绍下关于Alignment report,参考图8,之后选择options-Alignment Report Contents出现如图对话框,为使Alignment report界面更美观一点,首先调整其界面,点选break alignment,填入适当的碱基数,我一般选择80个,可填满整个界面,点击OK出现如图所示界面,是不是更好看些?继续选择上面对话框中的show consensus disagreement,会标示出错配碱基(绿色部分)如继续选择show consensus strength,则出现如图所示结果,一致序列用红线代表谢谢,正需要这些,不过请问Align‐Clustal或者Jotun Hein命令有什么区别呢?Clustal还有V、W之分,有什么区别?在一对比对是,One Pair‐By Wilbur‐Lipman method时要输入一些参数,那些参数怎么填,有什么作用呢?谢谢楼主关于第一个问题,见附件关于第二个问题,见下面介绍,希望对你有所帮助1 ) K‐TUPLE SIZE: This is the size of exactly matching fragment that is used.2 )GAP PENALTY: This is a penalty for each gap in the fast alignments. It has little affect on the speed or sensitivity except for extreme values.3 )WINDOW SIZE: This is the number of diagonals around each of the 'best' diagonals that will be used. Decrease for speed; increase for sensitivity.。
DNA序列分析软件介绍

DNA序列分析软件介绍Antheprot:蛋白质序列分析软件包ANTHEPROT 4.5是位于法国的蛋白质生物与化学研究院(Institute of Biology and Chemistry of Proteins)用十多年时间开发出的蛋白质研究软件包。
软件包包括了蛋白质研究领域所包括的大多数内容,功能非常强大。
应用此软件包,使用个人电脑,便能进行各种蛋白序列分析与特性预测。
更重要的是该软件能够提供蛋白序列的一些二级结构信息,使用户有可能模拟出未知蛋白的高级结构。
Applied Biosystems Primer Express:这是ABI公司销售附送的软件,可用于设计引物和探针,尤其适用于荧光PCR探针的设计,可以精确计算寡核苷酸与荧光基团鳌合后的Tm值。
可以预测引物与引物之间与模板之间等的二级结构。
Artemis R5:A DNA sequence viewer and annotation tools,一个DNA序列查看器与注释工具,可以以图形形式查看序列的各种分析结果与特性,程序读取EMBL与GENBANK格式的序列与纯DNA序列。
以Java写成,需要安装JRE1.2。
BioEdit是一个序列编辑器与分析工具软件,功能非常强大,使用十分容易。
功能包括:序列编辑、外挂分析程序、RNA分析、寻找特征序列、支持超过20000个序列的多序列文件、基本序列处理功能、质粒图绘制等等。
BLAST (Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。
BLAST程序能迅速与公开数据库进行相似性序列比较。
BLAST结果中的得分是对一种对相似性的统计说明。
BLAST对一条或多条序列(可以是任何形式的序列)在一个或多个核酸或蛋白序列库中进行比对。
BLAST还能发现具有缺口的能比对上的序列。
BLAST是基于Altschul等人在J.Mol.Biol上发表的方法(J.Mol.Biol.215:403-410(1990)),在序列数据库中对查询序列进行同源性比对工作。
DNAStar中文使用说明书

DNAStar中文使用说明书一、EditSeq打开已有序列我们从用苹果计算机打开“TETHIS21MA”和用W indows 打开“tethis21.seq”开始。
假设序列的末尾有载体序列污染。
我们在用EditSeq 打开序列的同时,用Set Ends命令去除5’和3 ’污染序列。
z 从文件菜单(),选择Open。
z 打开文件夹“D e m o S e q u e n c e s ”单击选定单击位于对话框右下角的序列“T E T H I S 2 1 ”。
z Set Ends 按z ,点钮。
Set Ends被打开(如右)。
在5 ’框和3’框中键入50和8 5 0击OK。
单击Open打开序列。
当EditSeq 窗口打开时,序列长度显示在右上角。
通过“setting ends,”现在你只有最初序列中的80 1 b p 的片段。
Se t En d s 选择在全部Lasergene 应用程序中都可以使用。
2寻找开放读框在这入门的一部分中,我们将确定序列中最大的ORF,并翻译它。
z 从S EARCH MENU找到O RF,点击打开会出现右边的对话框。
z 单击Find N ext 寻找第一个ORF 的位置。
z 继续点击Find N ext 直到你把O RF的位置选定在位置183-455。
ORF 的坐标会出现在EditSeq 窗口的顶端附近。
DNA 序列翻译这一节中我们介绍如何翻译我们的ORF,不过任何序列中的读框内部分翻译。
如果你的选择是在三联码的读框内,三联码指示棒显示为实心黑线(如左图)。
如果都可以用下面的方法进行3你的选择是不在三联码的读框内,左边的箭头和右面的箭头显示向左或向右移动一个bp,以使所选序列成为三的倍数。
z 选定ORF ,从GOODIES MENUr a n s l a t e )。
z 翻译的蛋白菜单中选择翻译(T质序列出现在一个新的未命名窗口中(如右图)。
它是使用标准的遗传密码翻译的。
4使用其它遗传密码根据你的序列的来源,你可以选择使用非标准的遗传密码进行翻译等操作。
DNASTAR中文使用说明书

从EditSeq开始EditSeq是能够迅速、正确地输入,并且修改DNA或蛋白质序列工具。
每个EditSeq文件都可以分为三个可编辑的部分,上边的一部分为序列文件,中间的一部分里是评论,底部是序列的注释。
EditSeq能读取大部分的序列格式——包括FASTA,GenBank,ABI、GCG和ASCII格式。
你可以使用菜单命令或拖拽方式输入序列文件。
另外,序列也许通过使用键盘输入,或者从其他地方复制、粘贴得到。
经Entrez或BLAST检索得到的序列可以直接从因特网或企业内部互联网服务器下载。
序列被打开后,EditSeq能使用标准或者指定的遗传密码进行翻译,或者反翻译,寻找开放读框,还可以进行阅读校对。
另外,EditSeq能以GenBank,FASTA和GCG格式输出序列。
如果在使用这软件中需要帮助,可以和DNASTAR联络。
电话:(608)258-7420,传真:(608)258-7439,电子信件:support@,或者经.内容打开已有序列23寻找开放读框24DNA序列翻译24遗传密码选择使用25遗传密码修改25序列的反向互补及反向转换26BLAST检索27序列信息查看28序列校读29序列的保存与输出29打开已有序列我们从用苹果计算机打开“TETHIS21MA”和用Windows打开“tethis21.seq”开始。
假设序列的末尾有载体序列污染。
我们在用EditSeq打开序列的同时,用Set Ends命令去除5’和3’污染序列。
l 从文件菜单(FILE MENU),选择Open。
l 打开文件夹“Demo Sequences”单击选定序列“TETHIS21”。
l 单击位于对话框右下角的Set Ends按钮。
Set Ends被打开(如右)。
l 在5’框和3’框中键入50和850,点击OK。
单击Open打开序列。
当EditSeq窗口打开时,序列长度显示在右上角。
通过“setting ends,”现在你只有最初序列中的801 bp的片段。
DNAStar详细中文使用说明书

Sequence Analysis Software for Macintosh and WindowsGETTING STARTED Introductory Tour of the LASERGENE System MAY 2001DNASTAR, Inc.1228 South Park StreetMadison, Wisconsin 53715(608) 258-7420Copyright . 2001 by DNASTAR, Inc.All rights reserved. Reproduction, adaptation, or translation without prior written permission is prohibited,except as allowed under the copyright laws or with the permission of DNASTAR, Inc.Sixth Edition, May 2001Printed in Madison, Wisconsin, USATrademark InformationDNASTAR, Lasergene, Lasergene99, SeqEasy, SeqMan, SeqMan II, EditSeq, MegAlign, GeneMan, Protean,MapDraw, PrimerSelect, GeneQuest, GeneFont , and the Method Curtain are trademarks or registered trademarks of DNASTAR, Inc. Macintosh is a trademark of Apple Computers, Inc. Windows is a trademark of Microsoft Corp. ABI Prism 373, 377 and 3700 are registered trademarks of Applera Corp., ALF is a registered trademark of Pharmacia Biotech ., MacVertor. and GCG. are registered trademarks of Pharmacopeia, Inc.Disclaimer & LiabilityDNASTAR, Inc. makes no warranties, expressed or implied, including without limitation the implied warranties of merchantability and fitness for a particular purpose, regarding the software. DNASTAR does not warrant, guaranty, or make any representation regarding the use or the results of the use of the software in terms of correctness, accuracy, reliability, currentness, or otherwise. The entire risk as to the results andperformance of the software is assumed by you. The exclusion of implied warranties is not permitted by some states. The above exclusion may not apply to you.In no event will DNASTAR, Inc. and their directors, officers, employees, or agents (collectively DNASTAR) be liable to you for any consequential, incidental or indirect damages (including damages for loss of business profits, business interruption, loss of business information and the like) arising out of the use of, or the inability to use the software even if DNASTAR Inc. has been advised of the possibility of such damages. Because some states do not allow the exclusion or limitation of liability for consequential or incidental damages, the above limitations may not apply to you.DNASTAR, Inc. reserves the right to revise this publication and to make changes to it from time to time without obligation of DNASTAR, Inc. to notify any person or organization of such revision or changes. The screen and other illustrations in this publication are meant to be representative of those that appear on your monitor or printer.目录在苹果机(Macintosh)上的安装与升级05通过因特网升级06 软件安装 07 网络安装 08 疑难解答 12在PC机(Windows)上安装与升级17通过因特网升级18 软件安装 19 从EditSeq 开始 21 从GeneQuest开始 31从MapDraw 开始 44从MegAlign开始 54从PrimerSelect开始 65从Protean 开始 78从SeqMan II开始 89L A S E R G E N Ef o rWi n d o w s & M a c i n t o s hDNASTA R I n c . ( 6 0 8 ) 2 5 8 - 7 4 2 0 f a x : ( 6 0 8 ) 2 5 8 - 7 43 9e m a i l : s u p p o r t @ d n a s t a r. c o m在苹果机(Macintosh)上的安装与升级通过因特网升级必备条件 6下载升级程序 6软件安装必备条件 7从CD安装Lasergene7网络安装必备条件 8定义 8 Dongle安装 10服务器安装 10终端机安装 11疑难解答系统配置冲突及网络问题 12解决网络问题的其他工具 13授权错误报告 14程序使用及更多的授权信息 15通过英特网升级如果您以前已经安装了Lasergene 而且目前有升级和服务联系,您就可以通过英特网来升级您现有的版本,各种模块(module)都是以自解压形式存储的,你可以选择性的下载安装。
DNASTAR使用说明

DNASTAR使用说明1.序列比对DNASTAR提供了强大的序列比对功能,可以将已知序列与未知序列进行比对,找到匹配的片段。
在DNASTAR中,用户可以将目标序列文件导入软件,然后选择合适的比对算法,如BLAST或Clustal Omega,进行比对分析。
比对结果将以可视化方式呈现,用户可以很方便地查看序列的相似性和差异性。
2.基因注释基因注释是生物学研究中非常重要的一项工作。
在DNASTAR中,用户可以将已知的基因序列导入软件,然后使用基因注释功能进行分析。
用户可以获取基因的名称、序列、结构、功能等信息。
此外,DNASTAR还提供了一些附加信息,如突变位点、剪接异常等,帮助用户更好地理解基因组的结构和功能。
3.构建进化树伴随着生物进化的研究,构建进化树成为了一种重要的方法。
DNASTAR提供了多种构建进化树的方法,如UPGMA、Neighbor-Joining等。
用户可以将感兴趣的序列导入软件,然后选择合适的构建方法进行分析。
生成的进化树将以图形化方式展示,用户可以直观地了解序列之间的进化关系。
4.引物设计DNASTAR还提供了引物设计功能,帮助用户设计用于PCR、RT-PCR等实验的引物。
用户可以选择特定的目标序列,然后设置引物的一些参数,如长度、碱基组成、Tm值等。
软件将根据用户设置的参数自动设计出合适的引物。
引物设计完毕后,用户可以查看引物的详细信息,并进行必要的调整。
5.DNA互补配对和翻译DNASTAR还包含了DNA互补配对和翻译功能。
用户可以将DNA序列输入软件,然后选择互补配对功能,软件将自动为用户生成DNA的互补序列。
此外,用户还可以将DNA序列翻译成蛋白质序列,并获取相应的氨基酸序列。
6.基于图形界面的操作DNASTAR提供了基于图形界面的操作方式,用户可以通过鼠标点击实现所需操作。
例如,用户可以通过拖拽文件到软件界面上进行导入,通过鼠标点击控制比对、注释、进化树构建等流程的进行。
DNAstar软件包的使用1

Founded 1982
DNASTAR was founded by Professor of Genetics Frederick Blattner and his Computer Scientist colleague John Schroeder 26 years ago. DNASTAR's mission then was the same as it is today: to provide life scientists with the computing tools they need for sequence analysis.
核苷酸序列分析
蛋白质序列分析
蛋白质二级结构 蛋白质超二级结构 蛋白质三级结构 序列比对 系统发育分析
DNAstar® Products
Lasergene - Sequence Analysis (1982) GenVision - Publication Quality Graphics (2000) ArrayStar - Microarray Gene Expression Analysis (2005)
TCATTTTCTCTTGCCGCCACCATGCTTCTTCCTCATTTTCTCT CCACCATGCCGCCACCACGCCACCATGCTTCTTCCTCATCTC GCTTTCTTGCCGCCACCATGCCGCCACCGCTTCTTCCtTCTCT…
现代生物技术
基因
片断) (决定某种性状的DNA片断) 决定某种性状的 片断
GC含量 含量/Codon bias 含量 基因编码区组分分析 引物设计 限制性核酸内切酶位点预测 基因结构分析 基因编码区结构分析 选择性剪切分析/SNP分析 分析 选择性剪切分析 基因调控区域分析 蛋白质一级序列 蛋白质理化性质分析 蛋白质二级结构预测 蛋白质序列信号位点分析 蛋白质结构域分析 蛋白质三维结构模拟 序列比对注释 多序列比对 系统发育分析
常用生物信息学软件介绍

常用生物学软件简介1. Oligo 6是目前使用最为广泛的一款引物设计软件,除了可以简单快捷地完成各种引物和探针的设计与分析外,还具有很多其他同类软件所不具有的高级功能: a) 已知一个PCR引物的序列,搜寻和设计另一个引物的序列。
b) 按照不同的物种对MM子的偏好性设计简并引物。
c) 对环型DNA片段,设计反向PCR引物。
d) 设计多重PCR引物。
e) 为LCR反应设计探针,以检测某个突变是否出现。
f) 分析和评价用其他途径设计的引物是否合理。
g) 同源序列查找,并根据同源区设计引物。
h) 增强了的引物/探针搜寻手段。
设计引物过程中,可以“Lock”每个参数,如Tm 值范围和引物3’端的稳定性等。
i) 以多种形式存储结果;支持多用户,每个用户可保存自己的特殊设置。
网址:/2. Vector NTI Suite是一套功能最全,而且界面最美观,最友好的分子生物学应用软件包。
主要包括四个大型软件,它们分别可以对DNA、RNA、蛋白质分子进行各种分析和操作。
Vector⑴ NTI:作为Vector NTI Suite的核心组成部分,它可以在生物研究的全过程中提供数据组织和序列编辑的软件支持。
Vector NTI 是以一种窗口形式,且支持项目组织的数据库来完成这一功能的;通过这个数据库,可以保存和组织大部分的实验数据,比如:基因结构、载体、序列片断、引物、蛋白质、多肽、电泳Markers和限制性内切酶等。
实际上,该数据库还支持对Vector NTI Suite 中各种小型的绘图和结果展示工具的管理。
Vector NTI 可以按照用户要求设计克隆策略。
用户只需提供克隆载体,外源片断序列,明确载体克隆的大致位置或酶切位点,其它工作由软件完成。
设计结果以图文形式输出到屏幕;最后根据客户定制的条件进行模拟电泳。
Vector NTI 还具有强大的设计和评估PCR引物、测序引物和杂交探针功能。
BioPlot⑵:BioPlot是一个对蛋白质和核酸序列进行各种理化特性分析的综合性工具,它是一种方便的桌面程序。
DNA star 简介

DNA序列翻译
任何序列中的读框内部分都可以用下面的方法进行翻译。选定ORF,从 GOODIES MENU菜单中选择翻译(Translate)。翻译的蛋白质序列出现 在一个新的未命名窗口中(如下图)。它是使用标准的遗传密码翻译的。 根据你的序列的来源,你可以选择使用非标准的遗传密
码进行翻译等操作。
如果你的选择是在三联 码的读框内,三联码指 示棒显示为实心黑线( 如下图箭头)。如果你 的选择是不在三联码的 读框内,左边的箭头和 右面的箭头显示向左或 向右移动一个bp,以 使所选序列成为三的倍 数。
选定序列的一部分。如果你是希望全选序列, 从EDIT MENU 菜单,选择Select All。 从GOODIES MENU 菜单,选DNA Statistics,就 会出现下面的窗口,显示序列信息。
碱基总数、A/T/G/C百分比等
这功能能帮助你校正测序序列中的错读。 选定序列。
•创建PrimerSelect文件 •定义引物特点 •查找引物对 •浏览其他的引物信息 •按特征对引物分类 •引物长度改变 •在引物中引入突变 •设计新引物 •使用寡核苷酸订购表格 •保存PrimerSelect文件
DNA star 组件
分析和预测蛋白质结构,提供各种分析方法并以图形的格式输出 结果,显示蛋白质分子的各种理化特性以及例如抗原决定族等功 能区的预测功能。
•创建蛋白质分析文件 •Protean‘s蛋白质分析方法 •应用分析方法 •方法参数改变 •优化结果显示 •使用蛋白酶消化与SDS •Feature注释 •BLAST检索
PAGE
(局部比对) •二级结构模拟 •展示滴定曲线 •保存分析文件
DNA star 组件
多序列拼接。最多支持64000条序列的同时拼接。在拼接前 可以对序列进行修正,对自动测序的序列结果可除去污染序 列或载体序列。整个拼装过程即时显示,并提示可能的完成 时间。拼装结果采用序列、策略等方式显示。
DNASTAR中文使用说明

DNAstar中文使用说明书目录DNAStar的安装与升级 (6)EditSeq的使用方法 (7)打开已有序列寻找开放读框DNA序列翻译遗传密码选择使用遗传密码修改序列的反向互补及反向转换BLAST检索序列信息查看序列校读序列的保存与输出GeneQuest的使用方法 (12)打开已有分析文件GeneQuest的DNA分析方法用分析方法操作方法参数改变结果展示优化Feature注释BLAST检索Entrez Database检索GeneQuest的其他特点保存分析文件MapDraw的使用方法 (18)新酶切图制作过泸器类型应用频率过泸器应用手动过泸器使用过泸器一览表使用Must Cut Here/Don’t Cut Here调色板工具酶信息显示环形展示ORF图显示择选保存,退出MegAlign的使用方法 (23)创建队列文件序列设置Pairwise Alignment使用 Dot Plot Method多序列比较Phylogenetic Tree查看查看队列报告Decorations/Consensi MegAlign文件保存PrimerSelect的使用方法 (28)创建PrimerSelect文件定义引物特点查找引物对浏览其他的引物信息按特征对引物分类引物长度改变在引物中引入突变设计新引物使用寡核苷酸订购表格保存PrimerSelect文件Protean的使用方法 (34)创建蛋白质分析文件Protean’s蛋白质分析方法应用分析方法方法参数改变优化结果显示使用蛋白酶消化与SDS PAGE Feature注释BLAST检索二级结构模拟展示滴定曲线保存分析文件SeqManII的使用方法 (39)输入序列片段Pre-Assembly Options操作检查修整的数据序列装配查看范围和构建结果去除矛盾碱基和缺口手动修改序列末端文件保存与序列输出EditSeq的使用方法EditSeq是能够迅速、正确地输入,并且修改DNA或蛋白质序列工具。
DNAStar软件包的使用
上机四实验报告【实验名称】DNAStar软件包的使用。
【实验目的】了解基因组序列分析的主要内容和DNAStar程序包各软件的基本功能,掌握Web logo 和 ORF finder的使用,以及EditSeq软件和GeneQuest软件的使用方法。
【实验原理】利用上述软件以及相关网络平台,对基因组进行分析、酶切电泳模拟、标注等。
【实验内容】试对文件contig3.seq(已提供)中的序列进行如下分析:1、分析整条序列的碱基组成和GC含量,以及GC含量在序列上的分布情况(窗口长度为10)。
2、标注序列中的起始密码子、终止密码子及长度大于100个密码子的ORF。
3、在反向互补序列上找到一个长度大于1000个密码子的的ORF,将其翻译成蛋白质,分析密码子使用偏好。
4、选择一个感兴趣的酶,分析序列酶切位点分布情况,给出酶切后,凝胶电泳模拟图。
【实验步骤】1、打开EditSeq,将文件contig3.seq导入。
如图,将文件在EditSeq中打开。
全选后,点击DNA statistic进行碱基统计如图,可获得详细的碱基统计信息。
打开GeneQuest,将文件contig3.seq导入,下拉左边的Base contents-Base distrubution,将G+C contents 和G+C contents region拖出,则可得到GC含量和分布,设置窗口长度为10.如图,所示结果为GC含量及分布情况(将序列信息放大后的显示)。
2、标注序列中的起始密码子、终止密码子及长度大于100个密码子的ORF点击More Methods中Coding Prediction - Starts Stops ORFs,将其拖入分析界面(该方法下的各个子方法将全部应用),即可以看到起始密码子、中止密码子、ORF在序列中的分布情况。
设置ORFs的最小长度为100如图,这是长度大于100的ORFs在序列中的分布(缩小后的图)。
(生物类研究生必学)DNAMAN、DNAstar、primer5.0、Endnote常用功能介绍

1.引物最好在模板cDNA的保守区内设计。
专题讲座(一)
2.引物长度一般在15~30碱基之间。
引物长度常用的是18-27 bp,但不应大于38, 因为过长会导致其延伸温度大于74℃,不适于Taq 酶进 行反应。 3.引物GC含量在40%~60%之间,Tm值在55-80℃之间,最好 接近72℃。 4.引物3′端要避开密码子的第3位。 如扩增编码区域,引物 3′端不要终止于密码 子的第3位,因密码子的第3位易发生简并,会影响扩增 的特异性与效率。
专题讲座(一)
DNAMAN功能很多,这里我们主要讲以下几个常用功能: 序列形式的变换 DNA序列的限制性酶切位点分析 序列比对分析
专题讲座(一)
主菜单栏
打开DNAMAN会出现如下界面
浏览器栏
工具栏
专题讲座(一)
① ② ③
载入序列的方法: 主菜单栏⇒文件⇒打开⇒选择要载入的序列 主菜单栏⇒文件⇒新建⇒直接将序列复制过来 主菜单栏 ⇒序列 ⇒载入序列 ⇒选择要打开的文件类型 ⇒ 选择相应的要打开的文件
专题讲座(一)
引物设计完之后我们再来看一下引物内部的稳定性:
主菜单栏⇒Report ⇒Internal Stability
专题讲座(一)
运行EndNote后,出现的第一个界面如下:
37
专题讲座(一)
我们可以新建一个数据库:
专题讲座(一)
如何将文献导入数据库
1. 电脑里的本地文献 选择Import File出现以下界面:
专题讲座(一)
DNAstar DNAstar软件,即著名的Lasergene Suite,功能主要有: 序列的格式转换,序列拼接和重叠克隆群的处理;基因寻 找;蛋白质结构域的查找;多重序列的比较和两两序列比 较;寡核苷酸设计( PCR 引物,测序引物,探针)。由 EditSeq 、 MegAlign、GeneQuest 、 MapDraw 、 PrimerSelect 、Protean 、SeqMan II七个模块组成。
DNASTAR中文使用说明

DNASTAR中⽂使⽤说明DNAstar中⽂使⽤说明书⽬录DNAStar的安装与升级 (6)EditSeq的使⽤⽅法 (7)打开已有序列寻找开放读框DNA序列翻译遗传密码选择使⽤遗传密码修改序列的反向互补及反向转换BLAST检索序列信息查看序列校读序列的保存与输出GeneQuest的使⽤⽅法 (12)打开已有分析⽂件GeneQuest的DNA分析⽅法⽤分析⽅法操作⽅法参数改变结果展⽰优化Feature注释BLAST检索Entrez Database检索GeneQuest的其他特点保存分析⽂件MapDraw的使⽤⽅法 (18)新酶切图制作过泸器类型应⽤频率过泸器应⽤⼿动过泸器使⽤过泸器⼀览表使⽤Must Cut Here/Don’t Cut Here调⾊板⼯具酶信息显⽰环形展⽰ORF图显⽰择选保存,退出MegAlign的使⽤⽅法 (23)创建队列⽂件序列设置Pairwise Alignment使⽤ Dot Plot Method多序列⽐较Phylogenetic Tree查看查看队列报告Decorations/Consensi MegAlign⽂件保存PrimerSelect的使⽤⽅法 (28)创建PrimerSelect⽂件定义引物特点查找引物对浏览其他的引物信息按特征对引物分类引物长度改变在引物中引⼊突变设计新引物使⽤寡核苷酸订购表格保存PrimerSelect⽂件Protean的使⽤⽅法 (34)创建蛋⽩质分析⽂件Protean’s蛋⽩质分析⽅法应⽤分析⽅法⽅法参数改变优化结果显⽰使⽤蛋⽩酶消化与SDS PAGE Feature注释BLAST检索⼆级结构模拟展⽰滴定曲线保存分析⽂件SeqManII的使⽤⽅法 (39)输⼊序列⽚段Pre-Assembly Options操作检查修整的数据序列装配查看范围和构建结果去除⽭盾碱基和缺⼝⼿动修改序列末端⽂件保存与序列输出EditSeq的使⽤⽅法EditSeq是能够迅速、正确地输⼊,并且修改DNA或蛋⽩质序列⼯具。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
生物信息基因序列分析软件DNAStar简介郑伟文,林营志,刘波,曹宜,苏明星,朱育菁,蓝江林,车建美,郑斯平,陈坚(福建省农科院生物技术中心)1.设计公司Sequence Analysis Software for Macintosh and Windows,GETTING STARTED,Introductory Tour of the LASERGENE System,MAY 2001,L A S E R G E N E f o r W i n d o w s & M a c i n t o s h,DNASTAR, Inc.,1228 South Park Street,Madison, Wisconsin 53715,(608) 258-7420,Copyright . 2001 by DNASTAR, Inc.,All rights reserved. Reproduction, adaptation, or translation without prior written permission is,prohibited,except as allowed under the copyright laws or with the permission of DNASTAR, Inc.,Sixth Edition, May 2001,Printed in Madison, Wisconsin, USA,Trademark Information。
2.应用程序在安装Lasergene网络系统之前要熟悉以下术语:应用程序:指EditSeq, GeneMan, GeneQuest, MapDraw,MegAlign, PrimerSelect, Protean, and SeqMan II。
应用程序服务器:是指存储应用程序的电脑,通常与dongle 服务,器是同一个服务器,但也可以不同,当在局部硬盘上安装网络程序,时,也可以在同一个网络系统中同时存在多个不同的应用程序服务,器,而且应用程序服务器不一定是苹果机,储存应用程序的机器也不一定必须能够运行该程序,仅仅是储存而已。
3.安装方式3.1通过英特网升级如果您以前已经安装了Lasergene 而且目前有升级和服务联系,您就可以通过英特网来升级您现有的版本,各种模块(module)都是以自解压形式存储的,你可以选择性的下载安装。
必备条件您的用户名和会员号是必需的,可以在安装盘上找到。
3.2程序升级备份您已有的Lasergene,找到您要升级的执行程序,并把它转移到备份的文件夹中。
连接到DNAstar 网站的主页(),从菜单中的Customers中点击Lasergene Updates点,安提示输入密码和用户名(与会员名相同),这样就会打开下载页面。
找到windows软件(Windows 95/98/NT Software.),就可以下载您想要的模块了。
模块下载完毕以后,双击文件将其解压缩完毕。
看到“Application name”has been updated.说明升级完毕。
3.3软件安装从CD在PC机(Windows)上安装Lasergene。
注意安装是尽量关闭所有其它程序以保证安装顺利进行。
必备条件,一张个人的Lasergene安装盘;一张Lasergene软件光碟;足够的硬盘空间和内存:至少30Mb的硬盘,32Mb的RAM。
从光盘安装Lasergene,插入安装盘和安装光盘,双击安装图标,则出现下面的窗口,点击继续,则出现安装窗口。
随后一次出现下面窗口,请按照提示做出选择然后点击Next,直至完成安装(图1)。
图1 软件安装基因序列编辑软件EditSeq的使用技术刘波,郑伟文,林营志,曹宜,苏明星,朱育菁,蓝江林,车建美,郑斯平,陈坚(福建省农科院生物技术中心)1.EditSeq功能简介EditSeq 是能够迅速、正确地输入,并且修改DNA 或蛋白质序列工具。
每个EditSeq 文件都可以分为三个可编辑的部分,上边的一部分为序列文件,中间的一部分里是评论,底部是序列的注释。
EditSeq 能读取大部分的序列格式——包括FASTA,GenBank,ABI、GCG 和ASCII 格式。
你可以使用菜单命令或拖拽方式输入序列文件。
另外,序列也许通过使用键盘输入,或者从其他地方复制、粘贴得到。
经Entrez 或BLAST 检索得到的序列可以直接从因特网或企业内部互联网服务器下载。
序列被打开后,EditSeq 能使用标准或者指定的遗传密码进行翻译,或者反翻译,寻找开放读框,还可以进行阅读校对。
另外, EditSeq 能以GenBank,FASTA 和GCG 格式输出序列。
如果在使用这软件中需要帮助,可以和DNASTAR 联络。
电话:(608)258-7420,传真:(608)258-7439,电子信件:support@,或者经。
2.打开已有序列用Windows 打开“tethis21.seq”开始。假设序列的末尾有载体序列污染。我们在用EditSeq 打开序列的同时,用Set Ends 命令去除5’和3’污染序列。从文件菜单(FILE MENU),选择Open。打开文件夹“Demo Sequences”单击选定序列“TETHIS21”。单击位于对话框右下角的Set Ends 按钮。Set Ends 被打开(如右)。在5’框和3’框中键入50 和850,点击OK。单击Open 打开序列。当EditSeq 窗口打开时,序列长度显示在右上角。通过“setting ends,”现在你只有最初序列中的801 bp 的片段。Set Ends 选择在全部Lasergene 应用程序中都可以使用(图1)。
图1 选择Open。打开文件夹“Demo Sequences”单击选定序列“TETHIS21”3.寻找开放读框在这入门的一部分中,我们将确定序列中最大的ORF,并翻译它。
从SEARCH MENU 找到ORF,点击打开会出现右边的对话框。
单击Find Next 寻找第一个ORF 的位置。
继续点击Find Next 直到你把ORF 的位置选定在位置183-455。
ORF的坐标会出现在EditSeq 窗口的顶端附近(图2)。
图2 单击Find Next 寻找第一个ORF 的位置4.DNA序列翻译这一节中我们介绍如何翻译我们的ORF,不过任何序列中的读框内部分都可以用下面的方法进行翻译。
如果你的选择是在三联码的读框内,三联码指示棒显示为实心黑线(如左图)。
如果你的选择是不在三联码的读框内,左边的箭头和右面的箭头显示向左或向右移动一个bp,以使所选序列成为三的倍数。
选定ORF,从GOODIES MENU 菜单中选择翻译(Translate)。
翻译的蛋白质序列出现在一个新的未命名窗口中(如右图)。
它是使用标准的遗传密码翻译的。
图3 图45.使用其它遗传密码根据你的序列的来源,你可以选择使用非标准的遗传密码进行翻译等操作。
在这节中,我们将标准的遗传密码转换成Ciliate Macronuclear密码。
从GOODIES MENU 菜单选择Genetic Codes打开,子菜单显示如左。
单击“Ciliate Macronuclear”就实现了遗传密码的转换,EditSeq 现在使用的就是CiliateMacronuclear 的遗传密码。
同样可以将遗传密码转换为其它类型。
图56.遗传密码的编辑这一节中我们修改CiliateMacronuclear 的遗传密码。
从GOODIESMENU 菜单选择Edit Selected Code。
这将打开右面的窗口,窗口显示遗传密码是怎样翻译DNA 和RNA 序列的。
如以DNA形式展示密码,点击DNA按钮。
编辑时,单击任何要编辑的密码,从其目前的位置拖到新氨基酸对应的位置则可。
如使用不同的启始密码子,单击Set Starts 按钮。
第二的遗传密码窗就会被打开,可以进行起始密码子选择。
单击任何氨基酸(或者codon 位置),该密码子就会变成绿色,而且旁边出现一个箭头,它就被设定为起始密码子了。
如要去除,只需单击它即可。
图6如不保存,则单击取消;如要保存,单击保存为。
7.序列的反向互补及反向转换下面的步骤可以用于反向测定的序列的正确输入。
选定序列。
从GOODIES MENU 菜单,选反向互补序列(Reverse Complement),或者把序列颠倒过来(Reverse Sequence)命令,则被选定的序列就被翻转互补或翻转过来了。
8.BLAST 检索下面我们将在NCBI 的BLAST 服务器上对TETHIS21 序列进行相似性比较。
注意为了进行BLAST 查找必须保证因特网的连接。
如果你没有连接因特网,跳过这部分,继续下一部分。
选定序,或者从EDIT 菜单中选择Select All。
从网络检索菜单(NET SEARCH MENU),选择BLAST 查找。
BLAST对话框就会出现。
程序默认为blastn,数据库默认是nr,参数转换请参照帮助。
单击OK 开始查找。
寻找结果显示为两部分(如下)。
上边的部分是按可能性的顺序显示检索到的序列的名字,下面的部分显示上面部分选定序列与提交序列(上边序列)比较的具体结果。
图7 图8有关“score”和“expectation”的详细的信息在NCBI’s 网点http:// www.ncbi. nlm.nih. gov/ BLAST.可以找到。
一般来说,更主要的score 和更低的expectation 提示较好的相似性。
BLAST 结果窗最上边的3 钮被用来打开,或者保存检索到的序列,或者让你了解更多的有关信息。
下面我们从用“Create Document”钮来打开评分最高的5 序列开始:单击Create Document。
一个小的对话框出现。
在左上角有一个下拉菜单显示默认(default)。
单击下拉菜单,选顶端(Top)。
并在右面的文本框中写入5。
单击OK,EditSeq 自动查对多余序列。
如果EditSeq 提示至少2 序列是同一个,请点击OK。
EditSeq 将从因特网数据库下在单一的序列,并分别打开一个单独的EditSeq 窗口。
下面我们用“Batch Save”钮将3-10 序列保存为EditSeq 文件:选定从顶端起第3 个序列。
单击Batch Save。
小的灰色的对话框出现。
图9 图10单击下拉菜单,选Next。
并在右面的文本框中写入8。
点击Set Location,显示文件夹对话框。
选定你要保存序列的位置。
单击OK 回到灰色的对话框。
单击OK 保存序列,文件扩展为“.seq”。
下载过程期间,EditSeq自动查对重复的序列。