The Path-A metabolic pathway prediction web server
肿瘤相关巨噬细胞的脂质代谢重编程

四川大学学报(医学版)2021,52(1) :45-49J Sichuan Univ (Med Sci)doi: 10.12182/20210160202肿瘤相关巨噬细胞的脂质代谢重编程+赵昆,时荣臣,缪洪明4中国人民解放军陆军军医大学(第三军医大学)基础医学院生物化学与分子生物学教研室(重庆400038)【摘要】肿瘤相关巨唾细胞(tumor associated macrophages, TAMs)是实质肿瘤中最常见的间质细胞类型之一,且与 肿瘤微环境的免疫抑制状态有着紧密联系,并促进肿瘤的恶性进展。
TAMs内的代谢发生了重编程,并且参与调控其自身 的极化以及相应的功能表型。
本文详细论述了 TAMs中包括三酰甘油、脂肪酸及其衍生物、胆固醇和磷脂在内的脂质代 谢重编程以及它们对肿瘤进展的调控。
然而,肿瘤细胞与肿瘤微环境间质细胞的代谢极具异质性。
肿瘤细胞与间质细胞 之间脂代谢重编程的异同点以及重编程如何调控细胞活性的机制值得深人探索。
同时,综合考虑肿瘤不同的组织类型、不同的发展阶段,精准靶向干预TAMs脂质代谢重编程,促进TAMs向M l样巨噬细胞极化,将成为代谢调节肿瘤免疫治疗 的新策略。
【关键词】肿瘤相关巨噬细胞 免疫抑制 脂质代谢重编程 肿瘤进展A Review of the Lipid Metabolism Reprogramming in Tumor Associated Macrophages Z H A O K u n, SH I R ong-chen,M IA O H o n g-m in g A. D e p a r tm e n t o f B io c h e m is try a n d M o le c u la r B io lo g y y S ch o o l o f B a sic M e d ic in e, A r m y M e d ic a lU n iversity (T h ird M ilita ry M e d ica l U n iversity)y Chongqing 400038, ChinaACorrespondingauthor,E-mail:*********************【Abstract】Tumor associated macrophages (TAMs) are one of the most common types of stromal cells in solid tumors. They are closely related to the immunosuppressive status of tumor microenvironment and potentiate the malignant progress of tumors. Studies have shown that metabolism in tumor associated macrophages has been reprogrammed and involved in the regulation of their own polarization and corresponding functions and phenotypes.Metabolic reprogramming refers to the alteration of key enzymes activity, substrate and its associated metabolites’concentration in a certain metabolic pathway, which accounts for the disorder of original metabolic states. In this paper, we mainly concentrated on the lipid metabolic reprogramming of TAMs, including triglycerides, fatty acids and their derivatives, cholesterol, phospholipids, and their regulations on tumor progression. However, the metabolism of tumor and tumor microenvironment cells is highly heterogeneous. It is worthy of further exploration on the similarities and differences of lipid metabolism reprogramming between stromal cells and tumor cells, and the mechanism of how reprogramming modulates cell activity. It will be a new strategy for immunotherapy of tumor with metabolic intervention to accurately target the lipid metabolism reprogramming of TAMs, so as to promote the polarization of TAMs to Ml like macrophages, when synthetically considering the diverse types of tumors and different stages of development.[K e y w o rd s] Tum or associated m acrophages Im m unosuppression Lipid m etabolism reprogramming Tumor progression肿瘤相关巨唾细胞(tumor associated macrophages, TAMs),—般指实体肿瘤微环境中的巨噬细胞,在实体肿 瘤内浸润的髓源细胞中占有最大比例,并与癌症患者的 不良预后密切相关。
基于转录组学探讨白屈菜生物碱促进小鼠睡眠障碍免疫修复药效学和机制

福建中医药2023 年7 月第54 卷第7期Fujian Journal of TCM July 2023,54(7)基于转录组学探讨白屈菜生物碱促进小鼠睡眠障碍免疫修复药效学和机制朱丽萍1,李景琳2,王凌1*(1.福建省立医院,福建福州 350001;2.福建医科大学药学院,福建福州 350122)摘要:目的基于转录组学探讨白屈菜生物碱促进小鼠睡眠障碍免疫修复的药效学和机制。
方法将30只昆明小鼠随机分为对照组、模型组和低、中、高剂量组,采用改良多平台水环境法每日14:00—次日10:00构建小鼠睡眠剥夺模型,共剥夺3 d。
在睡眠剥夺开始前5 d,低、中、高剂量组分别按体质量0.012 mL/(g·d)给予0.83、1.67、3.33 mg/mL白屈菜碱药液灌胃,对照组和模型组按体质量0.012 mL/(g·d)给予生理盐水灌胃,共8 d。
干预后观察小鼠的一般情况;ELISA法测定5组血清白细胞介素-6(IL-6)、肿瘤坏死因子-α(TNF-α)、白细胞介素-1β(IL-1β)、γ干扰素(IFN-γ)水平;流式细胞仪检测5组血清CD4+ T细胞、CD8+ T细胞和CD4+/CD8+。
对对照组、模型组和高剂量组进行转录组测序,采用DESeq 2.0软件包进行两两差异表达基因分析,运用GO数据库和KEGG 数据库对差异表达基因进行功能富集分析,构建蛋白互作(PPI)网络,筛选白屈菜碱促进睡眠剥夺小鼠免疫修复的关键基因,采用qPCR检测关键基因mRNA相对表达水平。
结果与对照组比较,模型组小鼠精神状态及毛发较差,饮食量减少,体质量均明显下降(P均<0.05),IL-6、TNF-α、IL-1β和IFN-γ浓度均明显升高(P均<0.05),中剂量组CD4+ T细胞、CD4+/CD8+明显升高(P均<0.05),高剂量组CD4+ T细胞、CD8+ T细胞以及CD4+/CD8+均明显升高(P均<0.05);与模型组比较,低、中、高剂量组小鼠精神状态、毛发和饮食量有所改善,体质量均明显增加(P均<0.05),IL-6、TNF-α、IL-1β和IFN-γ水平均明显降低(P均<0.05),CD4+ T细胞、CD8+ T细胞、CD4+/ CD8+均明显升高(P均<0.05);与对照组比较,模型组中有15个基因明显上调,9个基因显著下调;高剂量组15个明显上调的基因明显下调,9个明显下调的基因明显上调;高剂量组与模型组之间的差异表达基因主要富集在细胞外区域部分、细胞外空间、细胞外基质、IgA肠道免疫网络等通路;通过PPI网络图以及Degree值确定CD8A为关键基因,与模型组比较,低、中、高剂量组CD8A mRNA相对表达水平均明显上调(P均<0.05),与测序结果一致。
nmn的副作用是真实的吗,nmn的副作用整理

nmn的副作用是真实的吗,nmn的副作用整理nmn的副作用是真实的吗,nmn的副作用整理,共鸣感拉满了!NMN是合成NAD +的一种前体辅酶,而NAD+是提升各个细胞活性、修复受损DNA和线粒体、决定衰老的关键物质,但是,也有人担心NMN的安全性,服用有副作用吗?从nmn成分的属性来看,NMN真的安全NMN的安全性可以放心,首先要知道,NMN不是药、不是什么化学成分,它是一种安全的膳食补充剂,像钙片、维生素C片一样,可以帮助人体补充缺乏的营养物质。
很多我们日常食用的食物都含有NMN,比如西兰花、卷心菜、毛豆、黄瓜等,对人体来说是一种纯天然无害的物质。
人体本身中也含有NMN,甚至母乳也含有,因此NMN这种成分对人体是耐受的,服用也是非常安全的。
在日本,厚生劳动省 (卫生部) 批准NMN作为食品和药品原料添加使用,在美国,NMN已经作为新药在进行研究。
其中,在日本W+NMN25000黑金版通过长达5年的实验与观察发现:临床试验中,单剂量100mg、250mg和500mg的W+NMN被发现具有代谢和安全作用,而没有发现任何副作用。
In Japan, the Ministry of Health, Labor and Welfare (MoHW) has approved NMN for additive use as a food and pharmaceutical ingredient, and in the United States, NMN is already being investigated as a new drug. Among them, in Japan W+NMN25000 black gold Edition through 5 years of experiment and observation found: in clinical trials, single doses of 100mg, 250mg and 500mg of W+NMN were found to have metabolic and safety effects, without any side effects.NMN本身的安全性完全不需要怀疑和担心,但是这几年NMN越来越热门,市面上相关产品增多,其中不乏一些不合规、虚假夸大宣传的劣质产品,这就需要购买者提高甄别能力,选择出真正安全有效的产品。
A Balancing Act for Taxol Precursor Pathways in Ecoli

DOI: 10.1126/science.1191652, 70 (2010);330 Science , et al.Parayil Kumaran Ajikumar Escherichia coliin Isoprenoid Pathway Optimization for Taxol Precursor OverproductionThis copy is for your personal, non-commercial use only.clicking here.colleagues, clients, or customers by , you can order high-quality copies for your If you wish to distribute this article to othershere.following the guidelines can be obtained by Permission to republish or repurpose articles or portions of articles): August 4, 2011 (this infomation is current as of The following resources related to this article are available online at/content/330/6000/70.full.html version of this article at:including high-resolution figures, can be found in the online Updated information and services, /content/suppl/2010/09/27/330.6000.70.DC1.htmlcan be found at:Supporting Online Material /content/330/6000/70.full.html#related found at:can be related to this article A list of selected additional articles on the Science Web sites /content/330/6000/70.full.html#ref-list-1, 4 of which can be accessed free:cites 33 articles This article 1 article(s) on the ISI Web of Science cited by This article has been /content/330/6000/70.full.html#related-urls 1 articles hosted by HighWire Press; see:cited by This article has been/cgi/collection/chemistry Chemistrysubject collections:This article appears in the following registered trademark of AAAS.is a Science 2010 by the American Association for the Advancement of Science; all rights reserved. The title Copyright American Association for the Advancement of Science, 1200 New York Avenue NW, Washington, DC 20005. (print ISSN 0036-8075; online ISSN 1095-9203) is published weekly, except the last week in December, by the Science o n A u g u s t 4, 2011w w w .s c i e n c e m a g .o r g D o w n l o a d e d f r o mIsoprenoid Pathway Optimizationfor Taxol Precursor Overproductionin Escherichia coliParayil Kumaran Ajikumar,1,2Wen-Hai Xiao,1Keith E.J.Tyo,1Yong Wang,3Fritz Simeon,1 Effendi Leonard,1Oliver Mucha,1Too Heng Phon,2Blaine Pfeifer,3*Gregory Stephanopoulos1,2* Taxol(paclitaxel)is a potent anticancer drug first isolated from the Taxus brevifolia Pacific yew tree. Currently,cost-efficient production of Taxol and its analogs remains limited.Here,we report a multivariate-modular approach to metabolic-pathway engineering that succeeded in increasing titers of taxadiene—the first committed Taxol intermediate—approximately1gram per liter(~15,000-fold)in an engineered Escherichia coli strain.Our approach partitioned the taxadiene metabolic pathwayinto two modules:a native upstream methylerythritol-phosphate(MEP)pathway forming isopentenyl pyrophosphate and a heterologous downstream terpenoid–forming pathway.Systematic multivariate search identified conditions that optimally balance the two pathway modules so as to maximize the taxadiene production with minimal accumulation of indole,which is an inhibitory compound found here. We also engineered the next step in Taxol biosynthesis,a P450-mediated5a-oxidation of taxadieneto taxadien-5a-ol.More broadly,the modular pathway engineering approach helped to unlock the potential of the MEP pathway for the engineered production of terpenoid natural products.T axol(paclitaxel)and its structural analogs are among the most potent and commer-cially successful anticancer drugs(1).Taxol was first isolated from the bark of the Pacific yew tree(2),and early-stage production methods required sacrificing two to four fully grown trees to secure sufficient dosage for one patient(3). Taxol’s structural complexity limited its chemical synthesis to elaborate routes that required35to 51steps,with a highest yield of0.4%(4–6).Asemisynthetic route was later devised in whichthe biosynthetic intermediate baccatin III,isolatedfrom plant sources,was chemically converted toTaxol(7).Although this approach and subse-quent plant cell culture–based production effortshave decreased the need for harvesting the yewtree,production still depends on plant-based pro-cesses(8),with accompanying limitations onproductivity and scalability.These methods ofproduction also constrain the number of Taxolderivatives that can be synthesized in the searchfor more efficacious drugs(9,10).Recent developments in metabolic engineer-ing and synthetic biology offer new possibilitiesfor the overproduction of complex natural productsby optimizing more technically amenable micro-bial hosts(11,12).The metabolic pathway forTaxol consists of an upstream isoprenoid pathwaythat is native to Escherichia coli and a het-erologous downstream terpenoid pathway(fig.S1).The upstream methylerythritol-phosphate(MEP)or heterologous mevalonic acid(MV A)pathwayscan produce the two common building blocks,isopentenyl pyrophosphate(IPP)and dimethyl-allyl pyrophosphate(DMAPP),from which Taxoland other isoprenoid compounds are formed(12).Recent studies have highlighted the engi-neering of the above upstream pathways to sup-port the biosynthesis of heterologous isoprenoidssuch as lycopene(13,14),artemisinic acid(15,16),and abietadiene(17,18).The downstream taxadienepathway has been reconstructed in E.coli andSaccharomyces cerevisiae together with the over-expression of upstream pathway enzymes,but todate titers have been limited to less than10mg/liter(19,20).The above rational metabolic engineering ap-proaches examined separately either the upstreamor the downstream terpenoid pathway,implicitlyassuming that modifications are additive(a linearbehavior)(13,17,21).Although this approachcan yield moderate increases in flux,it generallyignores nonspecific effects,such as toxicity of in-termediate metabolites,adverse cellular effects ofthe vectors used for expression,and hidden path-ways and metabolites that may compete with themain pathway and inhibit the production of thedesired binatorial approaches canovercome such problems because they offer theopportunity to broadly sample the parameter spaceand bypass these complex nonlinear interactions(21–23).However,combinatorial approaches re-quire high-throughput screens,which are often notavailable for many desirable natural products(24).Considering the lack of a high-throughputscreen for taxadiene(or other Taxol pathwayintermediate),we resorted to a focused combi-1Department of Chemical Engineering,Massachusetts Institute of Technology(MIT),Cambridge,MA02139,USA.2Chemical and Pharmaceutical Engineering Program,Singapore-MIT Alli-ance,117546Singapore.3Department of Chemical and Bio-logical Engineering,Tufts University,4Colby Street,Medford, MA02155,USA.*To whom correspondence should be addressed.E-mail: gregstep@(G.S.);blaine.pfeifer@(B.P.)Upstream moduleFig.1.isoprenoid pathwaythe flux through thewe targeted reported(dxs,idi,ispD,andexpression by anTo channel theversal isoprenoidtoward Taxolsynthetic operon of downstream genes GGPP synthase(G)and taxadienesynthase(T)(37).Both pathways were placed under the control of induciblepromoters in order to control their relative gene expression.In the E.colimetabolic network,the MEP isoprenoid pathway is initiated by the con-densation of the precursors glyceraldehyde-3phosphate(G3P)and pyruvate(PYR)from glycolysis.The Taxol pathway bifurcation starts from the universalisoprenoid precursors IPP and DMAPP to form geranylgeranyl diphosphate,and then the taxadiene.The cyclic olefin taxadiene undergoes multiple roundsof stereospecific oxidations,acylations,and benzoylation to form the lateintermediate Baccatin III and side chain assembly to,ultimately,form Taxol. REPORTS1OCTOBER2010VOL330SCIENCE 70onAugust4,211www.sciencemag.orgDownloadedfromnatorial approach,which we term “multivariate-modular pathway engineering.”In this approach,the overall pathway is partitioned into smaller modules,and the modules ’expression are varied simultaneously —a multivariate search.This ap-proach can identify an optimally balanced path-way while searching a small combinatorial space.Specifically,we partition the taxadiene-forming pathway into two modules separated at IPP,which is the key intermediate in terpenoid bio-synthesis.The first module comprises an eight-gene,upstream,native (MEP)pathway of which the expression of only four genes deemed to be rate-limiting was modulated,and the second mod-ule comprises a two-gene,downstream,heterolo-gous pathway to taxadiene (Fig.1).This modular approach allowed us to efficiently sample the main parameters affecting pathway flux without the need for a high-throughput screen and to unveil the role of the metabolite indole as in-hibitor of isoprenoid pathway activity.Addition-ally,the multivariate search revealed a highly nonlinear taxadiene flux landscape with a global maximum exhibiting a 15,000-fold increase in taxadiene production over the control,yielding 1.02T 0.08g/liter (SD)taxadiene in fed-batch bioreactor fermentations.We have further engineered the P450-based oxidation chemistry in Taxol biosynthesis in E.coli to convert taxadiene to taxadien-5a -ol and provide the basis for the synthesis of sub-sequent metabolites in the pathway by means of similar cytochrome P450(CYP450)oxida-tion chemistry.Our engineered strain improved taxadiene-5a -ol production by 2400-fold over the state of the art with yeast (25).These ad-vances unlock the potential of microbial pro-cesses for the large-scale production of Taxol or its derivatives and thousands of other valuable terpenoids.The multivariate-modular approach in which various promoters and gene copy-numbers are combined to modulate diverse expression levels of upstream and downstream pathways of taxadiene synthesis is schematically described in fig.S2.A total of 16strains were constructed in order to widen the bottleneck of the MEP pathway as well as optimally balance it with the downstream tax-adiene pathway (26).The dependence of tax-adiene accumulation on the upstream pathway for constant values of the downstream pathway is shown in Fig.2A,and the dependence on the downstream pathway for constant upstream path-way strength is shown in Fig.2B (table S1,cal-culation of the upstream and downstream pathway strength from gene copy number and promoter strength).As the upstream pathway expression increases in Fig.2A from very low levels,tax-adiene production also rises initially because of increased supply of precursors to the overall path-way.However,after an intermediate value further upstream pathway increases cannot be accom-modated by the capacity of the downstream path-way.For constant upstream pathway expression (Fig.2B),a maximum in downstream expressionwas similarly observed owing to the rising edge to initial limiting of taxadiene production by low expression levels of the downstream pathway.At high (after peak)levels of downstream pathway expression,we were probably observing the neg-ative effect on cell physiology of the high copy number.These results demonstrate that dramatic changes in taxadiene accumulation can be obtained fromchanges within a narrow window of expression levels for the upstream and downstream path-ways.For example,a strain containing an ad-ditional copy of the upstream pathway on its chromosome under Trc promoter control (strain 8)(Fig.2A)produced 2000-fold more taxadiene than one expressing only the native MEP path-way (strain 1)(Fig.2A).Furthermore,changing the order of the genes in the downstreamsyn-Fig.2.Optimization of taxadiene production through regulating the expression of the upstream and downstream modular pathways.(A )Response in taxadiene accumulation to changes in upstream pathway strengths for constant values of the downstream pathway.(B )Dependence of taxadiene on the down-stream pathway for constant levels of upstream pathway strength.(C )Taxadiene response from strains (17to 24)engineered with high upstream pathway overexpressions (6to 100a.u.)at two different down-stream expressions (31a.u.and 61a.u.).(D )Modulation of a chromosomally integrated upstream pathway by using increasing promoter strength at two different downstream expressions (31a.u.and 61a.u.).(E )Genotypes of the 32strain constructs whose taxadiene phenotype is shown in Fig.2,A to D.E,E.coli K12MG1655D recA D endA ;EDE3,E.coli K12MG1655D recA D endA with DE3T7RNA polymerase gene in the chromosome;MEP,dxs-idi-ispDF operon;GT,GPPS-TS operon;TG,TS-GPPS operon;Ch1,1copy in chromosome;Trc,Trc promoter;T5,T5promoter;T7,T7promoter;p5,pSC101plasmid;p10,p15A plasmid;and p20,pBR322plasmid. SCIENCEVOL 3301OCTOBER 201071REPORTSo n A u g u s t 4, 2011w w w .s c i e n c e m a g .o r g D o w n l o a d e d f r o mthetic operon from GT (GGPS-TS)to TG (TS-GGPS)resulted in a two-to threefold increase (strains 1to 4as compared with strains 5,8,11,and 14).Altogether,the engineered strains estab-lished that the MEP pathway flux can be substan-tial if an appropriate range of expression levels for the endogenous upstream and synthetic down-stream pathway are searched simultaneously.To provide ample downstream pathway strength while minimizing the plasmid-born metabolic bur-den (27),two new sets of four strains each were engineered (strains 17to 20and 21to 24),in which the downstream pathway was placed un-der the control of a strong promoter (T7)while keeping a relatively low number of five and 10plasmid copies,respectively.The taxadiene maxi-mum was maintained at high downstream strength (strains 21to 24),whereas a monotonic response was obtained at the low downstream pathway strength (strains 17to 20)(Fig.2C).This ob-servation prompted the construction of two addi-tional sets of four strains each that maintained the same level of downstream pathway strength as before but expressed very low levels of the up-stream pathway (strains 25to 28and 29to 32)(Fig.2D).Additionally,the operon of the up-stream pathway of the latter strain set was chro-mosomally integrated (fig S3).Not only was the taxadiene maximum recovered in these strains,albeit at very low upstream pathway levels,but a much greater taxadiene maximum was attained (~300mg/liter).We believe that this significant increase can be attributed to a decrease in the cell ’s metabolic burden.We next quantified the mRNA levels of 1-deoxy-D -xylulose-5-phosphate synthase (dxs)and taxadiene synthase (TS)(representing the up-stream and downstream pathways,respectively)for the high-taxadiene-producing strains (25to 32and 17and 22)that exhibited varying up-stream and downstream pathway strengths (fig.S4,A and B)to verify our predicted expression strengths were consistent with the actual pathway levels.We found that dxs expression level cor-relates well with the upstream pathway strength.Similar correlations were found for the other genes of the upstream pathway:idi ,ispD ,and ispF (fig.S4,C and D).In downstream TS gene expres-sion,an approximately twofold improvement was quantified as the downstream pathway strength increased from 31to 61arbitrary units (a.u.)(fig.S4B).Metabolomic analysis of the previous strains led to the identification of a distinct metabolite by-product that inversely correlated with taxadiene accumulation (figs.S5and S6).The corresponding peak in the gas chromatography –mass spectrom-etry (GC-MS)chromatogram was identified as indole through GC-MS,1H,and 13C nuclear magnetic resonance (NMR)spectroscopy studies (fig.S7).We found that taxadiene synthesis by strain 26is severely inhibited by exogenous in-dole at indole levels higher than ~100mg/liter (fig.S5B).Further increasing the indole concen-tration also inhibited cell growth,with the level ofinhibition being very strain-dependent (fig.S5C).Although the biochemical mechanism of indole interaction with the isoprenoid pathway is pres-ently unclear,the results in fig.S5suggest a possible synergistic effect between indole and terpenoid compounds of the isoprenoid pathway in inhibiting cell growth.Without knowing the specific mechanism,it appears that strain 26has mitigated the indole ’s effect,which we carried forward for further study.In order to explore the taxadiene-producing potential under controlled conditions for the en-gineered strains,fed-batch cultivations of the three highest taxadiene accumulating strains (~60mg/liter from strain 22;~125mg/liter from strain 17;and ~300mg/liter from strain 26)were carried out in 1-liter bioreactors (Fig.3).The fed-batch cultivation studies were carried out as liquid-liquid two-phase fermentation using a 20%(v/v)dodecane overlay.The organic solvent was intro-duced to prevent air stripping of secreted tax-adiene from the fermentation medium,as indicated by preliminary findings (fig.S8).In defined media with controlled glycerol feeding,taxadiene pro-ductivity increased to 174T 5mg/liter (SD),210T 7mg/liter (SD),and 1020T 80mg/liter (SD)for strains 22,17,and 26,respectively (Fig.3A).Additionally,taxadiene production significantly affected the growth phenotype,acetate accumu-lation,and glycerol consumption [Fig.3,B and D,and supporting online material (SOM)text].Clearly,the high productivity and more robustgrowth of strain 26allowed very high taxadiene accumulation.Further improvements should be possible through optimizing conditions in the bio-reactor,balancing nutrients in the growth medi-um and optimizing carbon delivery.Having succeeded in engineering the bio-synthesis of the “cyclase phase ”of Taxol for high taxadiene production,we turned next to engineer-ing the oxidation-chemistry of Taxol biosynthesis.In this phase,hydroxyl groups are incorporated by oxygenation at seven positions on the taxane core structure,mediated by CYP450-dependent monooxygenases (28).The first oxygenation is the hydroxylation of the C5position,followed by seven similar reactions en route to Taxol (fig.S1)(29).Thus,a key step toward engineering Taxol-producing microbes is the development of CYP450-based oxidation chemistry in vivo.The first oxygenation step is catalyzed by a CYP450,taxadiene 5a -hydroxylase,which is an unusual monooxygenase that catalyzes the hydroxylation reaction along with double-bond migration in the diterpene precursor taxadiene (Fig.1).In general,functional expression of plant CYP450in E.coli is challenging (30)because of the inherent limitations of bacterial platforms,such as the absence of electron transfer machin-ery and CYP450-reductases (CPRs)and trans-lational incompatibility of the membrane signal modules of CYP450enzymes because of the lack of an endoplasmic reticulum.Recently,through transmembrane (TM)engineering and the gener-24487296120T a x a d i e n e (m g /L )Time (h)1234024487296120N e t g l y c e r o l a d d e d (g /L )Time (h)A BC DC e l l g r o w t h (OD 600 n m )Time (h)24487296120A c e t i c a c i d (g /L )Time (h)Fig.3.Fed-batch cultivation of engineered strains in a 1-liter bioreactor.Time courses of (A )taxadiene accumulation,(B )cell growth,(C )acetic acid accumulation,and (D )total substrate (glycerol)addition for strains 22,17,and 26during 5days of fed-batch bioreactor cultivation in 1-liter bioreactor vessels under controlled pH and oxygen conditions with minimal media and 0.5%yeast extract.After glycerol depletes to ~0.5to 1g/liter in the fermentor,3g/liter of glycerol was introduced into the bioreactor during the fermentation.Data are mean of two replicate bioreactors.1OCTOBER 2010VOL 330SCIENCE72REPORTSo n A u g u s t 4, 2011w w w .s c i e n c e m a g .o r g D o w n l o a d e d f r o mation of chimera enzymes of CYP450and CPR,some plant CYP450s have been expressed in E.coli for the biosynthesis of functional mole-cules (15,31).Still,every plant CYP450has distinct TM signal sequences and electron transfer characteristics from its reductase counterpart (32).Our initial studies were focused on optimizing the expression of codon-optimized synthetic tax-adiene 5a -hydroxylase by N-terminal TM engi-neering and generating chimera enzymes through translational fusion with the CPR redox partner from the Taxus species,Taxus CYP450reductase (TCPR)(Fig.4A)(29,31,33).One of the chi-mera enzymes generated,At24T5a OH-tTCPR,was highly efficient in carrying out the first oxi-dation step,resulting in more than 98%taxadiene conversion to taxadien-5a -ol and the byproduct 5(12)-Oxa-3(11)-cyclotaxane (OCT)(fig.S9A).Compared with the other chimeric CYP450s,At24T5a OH-tTCPR yielded twofold higher (21mg/liter)production of taxadien-5a -ol (Fig.4B).Because of the functional plasticity of taxadiene 5a -hydroxylase with its chimeric CYP450’s en-zymes (At8T5a OH-tTCPR,At24T5a OH-tTCPR,and At42T5a OH-tTCPR),the reaction also yields a complex structural rearrangement of taxadiene into the cyclic ether OCT (fig.S9)(34).The by-product accumulated in approximately equal amounts (~24mg/liter from At24T5a OH-tTCPR)to the desired product taxadien-5a -ol.The productivity of strain 26-At24T5a OH-tTCPR was significantly reduced relative to that of taxadiene production by the parent strain 26(~300mg/liter),with a concomitant increase in indole accumulation.No taxadiene accumulation was observed.Apparently,the introduction of an additional medium copy plasmid (10-copy,p10T7)bearing the At24T5a OH-tTCPR construct dis-turbed the carefully engineered balance in the up-stream and downstream pathway of strain 26(fig S10).Small-scale fermentations were carried out in bioreactors so as to quantify the alcohol production by strain 26-At24T5a OH-tTCPR.The time course profile of taxadien-5a -ol accumulation (Fig.4C)indicates alcohol production of up to 58T 3mg/liter (SD)with an equal amount of the OCT by-product produced.The observed alcohol production was approximately 2400-fold higher than previous production in S.cerevisiae (25).The MEP pathway is energetically balanced and thus overall more efficient in converting either glucose or glycerol to isoprenoids (fig.S11).Yet,during the past 10years many attempts at en-gineering the MEP pathway in E.coli in order to increase the supply of the key precursors IPP and DMAPP for carotenoid (21,35),sesquiterpenoid (16),and diterpenoid (17)overproduction met with limited success.This inefficiency was at-tributed to unknown regulatory effects associated specifically with the expression of the MEP path-way in E.coli (16).Here,we provide evidence that such limitations are correlated with the accumu-lation of the metabolite indole,owing to the non-optimal expression of the pathway,which inhibits the isoprenoid pathway activity.Taxadiene over-production (under conditions of indole-formation suppression),establishes the MEP pathway as a very efficient route for biosynthesis of pharma-ceutical and chemical products of the isoprenoid family (fig.S11).One simply needs to carefully balance the modular pathways,as suggested by our multivariate-modular pathway –engineering approach.For successful microbial production of Taxol,demonstration of the chemical decoration of the taxadiene core by means of CYP450-based oxi-dation chemistry is essential (28).Previous ef-forts to reconstitute partial Taxol pathways in yeast found CYP450activity limiting (25),making the At24T5a OH-tTCPR activity levels an im-portant step to debottleneck the late Taxol path-way.Additionally,the strategies used to create At24T5a OH-tTCPR are probably applicable for the remaining monooxygenases that will require expression in E.coli .CYP450monooxygenases constitute about one half of the 19distinct en-zymatic steps in the Taxol biosynthetic pathway.These genes show unusually high sequence sim-ilarity with each other (>70%)but low similarity (<30%)with other plant CYP450s (36),implying that these monooxygenases are amenable to similar engineering.To complete the synthesis of a suitable Taxol precursor,baccatin III,six more hydroxylation reactions and other steps (including some that have not been identified)need to be effectively engineered.Although this is certainly a daunting task,the current study shows potential by provid-ing the basis for the functional expression of two key steps,cyclization and oxygenation,in Taxol biosynthesis.Most importantly,by unlocking the potential of the MEP pathway a new more ef-ficient route to terpenoid biosynthesis is capable of providing potential commercial production of microbially derived terpenoids for use as chem-icals and fuels from renewable resources.References and Notes1.D.G.Kingston,Phytochemistry 68,1844(2007).2.M.C.Wani,H.L.Taylor,M.E.Wall,P.Coggon,A.T.McPhail,J.Am.Chem.Soc.93,2325(1971).3.M.Suffness,M.E.Wall,in Taxol:Science and Applications ,M.Suffness,Ed.(CRC,Boca Raton,FL,1995),pp.3–26.4.K.C.Nicolaou et al .,Nature 367,630(1994).5.R.A.Holton et al .,J.Am.Chem.Soc.116,1597(1994).6.A.M.Walji,D.W.C.MacMillan,Synlett 18,1477(2007).7.R.A.Holton,R.J.Biediger,P.D.Boatman,in Taxol:Science and Applications ,M.Suffness,Ed.(CRC,Boca Raton,FL,1995),pp.97–119.8.D.Frense,Appl.Microbiol.Biotechnol.73,1233(2007).9.S.C.Roberts,Nat.Chem.Biol.3,387(2007).10.J.Goodman,V.Walsh,The Story of Taxol:Nature andPolitics in the Pursuit of an Anti-Cancer Drug .(Cambridge Univ.Press,Cambridge,2001).11.K.E.Tyo,H.S.Alper,G.N.Stephanopoulos,TrendsBiotechnol.25,132(2007).12.P.K.Ajikumar et al .,Mol.Pharm.5,167(2008).510152025T a x a d i e n -5α-o l p r o d u c t i o n (m g e q u i v a l e n t o f t a x a d i e n e /L )BC048121620020406020406080100C e l l g r o w t h (OD 600n m )T a x a d i e n e -5α-o l p r o d u c t i o n (m g e q u i v a l e n t o f t a x a d i e n e /L )Time (h)Fig.4.Engineering Taxol P450oxidation chemistry in E.coli .(A )TM engineering and construction of chimera protein from taxadien-5a -ol hydroxylase (T5a OH)and Taxus cytochrome P450reductase (TCPR).The labels 1and 2represent the full-length proteins of T5a OH and TCPR identified with 42and 74amino acid TM regions,respectively,and 3represents chimera enzymes generated from three different TM en-gineered T5a OH constructs [At8T5a OH,At24T5a OH,and At42T5a OH constructed by fusing an 8-residue synthetic peptide MALLLAVF (A)to 8,24,and 42AA truncated T5a OH]through a translational fusion with 74AA truncated TCPR (tTCPR)by use of linker peptide GSTGS.(B )Functional activity of At8T5a OH-tTCPR,At24T5a OH-tTCPR,and At42T5a OH-tTCPR constructs transformed into taxadiene producing strain 26.Data are mean T SD for three replicates.(C )Time course profile of taxadien-5a -ol accumulation and growth profile of the strain 26-At24T5a OH-tTCPR fermented in a 1-liter bioreactor.Data are mean of two replicate bioreactors.SCIENCEVOL 3301OCTOBER 201073REPORTSo n A u g u s t 4, 2011w w w .s c i e n c e m a g .o r g D o w n l o a d e d f r o m13.W.R.Farmer,J.C.Liao,Nat.Biotechnol.18,533(2000).14.H.Alper,K.Miyaoku,G.Stephanopoulos,Nat.Biotechnol.23,612(2005).15.M.C.Chang,J.D.Keasling,Nat.Chem.Biol.2,674(2006).16.V.J.Martin,D.J.Pitera,S.T.Withers,J.D.Newman,J.D.Keasling,Nat.Biotechnol.21,796(2003).17.D.Morrone et al .,Appl.Microbiol.Biotechnol.85,1893(2010).18.E.Leonard et al .,Proc.Natl.Acad.Sci.U.S.A.107,13654(2010).19.Q.Huang,C.A.Roessner,R.Croteau,A.I.Scott,Bioorg.Med.Chem.9,2237(2001).20.B.Engels,P.Dahm,S.Jennewein,Metab.Eng.10,201(2008).21.L.Z.Yuan,P.E.Rouvière,rossa,W.Suh,Metab.Eng.8,79(2006).22.Y.S.Jin,G.Stephanopoulos,Metab.Eng.9,337(2007).23.H.H.Wang et al .,Nature 460,894(2009).24.D.Klein-Marcuschamer,P.K.Ajikumar,G.Stephanopoulos,Trends Biotechnol.25,417(2007).25.J.M.Dejong et al .,Biotechnol.Bioeng.93,212(2006).26.Materials and methods are available as supportingmaterial on Science Online.27.K.L.Jones,S.W.Kim,J.D.Keasling,Metab.Eng.2,328(2000).28.R.Kaspera,R.Croteau,Phytochem.Rev.5,433(2006).29.S.Jennewein,R.M.Long,R.M.Williams,R.Croteau,Chem.Biol.11,379(2004).30.M.A.Schuler,D.Werck-Reichhart,Annu.Rev.Plant Biol.54,629(2003).31.E.Leonard,M.A.Koffas,Appl.Environ.Microbiol.73,7246(2007).32.D.R.Nelson,Arch.Biochem.Biophys.369,1(1999).33.S.Jennewein et al .,Biotechnol.Bioeng.89,588(2005).34.D.Rontein et al .,J.Biol.Chem.283,6067(2008).35.W.R.Farmer,J.C.Liao,Biotechnol.Prog.17,57(2001).36.S.Jennewein,M.R.Wildung,M.Chau,K.Walker,R.Croteau,Proc.Natl.Acad.Sci.U.S.A.101,9149(2004).37.K.Walker,R.Croteau,Phytochemistry 58,1(2001).38.We thank R.Renu for extraction,purification,andcharacterization of metabolite Indole;C.Santos for providing the pACYCmelA plasmid,constructivesuggestions during the experiments,and preparation of the manuscript;D.Dugar,H.Zhou,and X.Huang for helping with experiments and suggestions;and K.Hiller for data analysis and comments on the manuscript.We gratefully acknowledge support by the Singapore-MIT Alliance (SMA-2)and NIH,grant 1-R01-GM085323-01A1.B.P.acknowledges the Milheim Foundation Grant for Cancer Research 2006-17.A patent application that is based on the results presented here has been filed by MIT.P.K.A.designed the experiments and performed the engineering and screening of the strains;W-H.X.performed screening of the strains,bioreactorexperiments,and GC-MS analysis;F.S.carried out the quantitative PCR measurements;O.M.performed the extraction and characterization of taxadiene standard;E.L.,Y.W.,and B.P.supported with cloning experiments;P.K.A.,K.E.J.T.,T.H.P.,B.P.and G.S.analyzed the data;P.K.A.,K.E.J.T.,and G.S.wrote the manuscript;G.S.supervised the research;and all of the authors contributed to discussion of the research and edited and commented on the manuscript.Supporting Online Material/cgi/content/full/330/6000/70/DC1Materials and Methods SOM TextFigs.S1to S11Tables S1to S4References29April 2010;accepted 9August 201010.1126/science.1191652Reactivity of the Gold/Water Interface During Selective Oxidation CatalysisBhushan N.Zope,David D.Hibbitts,Matthew Neurock,Robert J.Davis *The selective oxidation of alcohols in aqueous phase over supported metal catalysts is facilitated by high-pH conditions.We have studied the mechanism of ethanol and glycerol oxidation to acids over various supported gold and platinum beling experiments with 18O 2and H 218O demonstrate that oxygen atoms originating from hydroxide ions instead of molecular oxygen are incorporated into the alcohol during the oxidation reaction.Density functional theory calculations suggest that the reaction path involves both solution-mediated and metal-catalyzed elementary steps.Molecular oxygen is proposed to participate in the catalytic cycle not by dissociation to atomic oxygen but by regenerating hydroxide ions formed via the catalytic decomposition of a peroxide intermediate.The selective oxidation of alcohols with mo-lecular oxygen over gold (Au)catalysts in liquid water offers a sustainable,envi-ronmentally benign alternative to traditional pro-cesses that use expensive inorganic oxidants and harmful organic solvents (1,2).These catalytic transformations are important to the rapidly de-veloping industry based on the conversion of bio-renewable feedstocks to higher-valued chemicals (3,4)as well as the current production of petro-chemicals.Although gold is the noblest of metals (5),the water/Au interface provides a reaction en-vironment that enhances its catalytic performance.We provide here direct evidence for the predomi-nant reaction path during alcohol oxidation at high pH that includes the coupling of both solution-mediated and metal-catalyzed elementary steps.Alcohol oxidation catalyzed by Pt-group metals has been studied extensively,although the precisereaction path and extent of O 2contribution are still under debate (4,6–8).The mechanism for the selective oxidation of alcohols in liquid water over the Au catalysts remains largely un-known (6,9),despite a few recent studies with organic solvents (10–12).In general,supported Au nanoparticles are exceptionally good catalysts for the aerobic oxidation of diverse reagents ranging from simple molecules such as CO and H 2(13)to more complex substrates such as hy-drocarbons and alcohols (14).Au catalysts are also substrate-specific,highly selective,stable against metal leaching,and resistant to overoxidation by O 2(6,15,16).The active catalytic species has been suggested to be anionic Au species (17),cat-ionic Au species (18,19),and neutral Au metal particles (20).Moreover,the size and structure of Au nanoparticles (21,22)as well as the interface of these particles with the support (23)have also been claimed to be important for catalytic ac-tivity.For the well-studied CO oxidation reaction,the presence of water vapor increases the observed rate of the reaction (24–26).Large metallic Au particles and Au metal powder,which are usually considered to be catalytically inert,have consider-able oxidation activity under aqueous conditions at high pH (27,28).We provide insights into the active intermediates and the mechanism for al-cohol oxidation in aqueous media derived from experimental kinetic studies on the oxidation of glycerol and ethanol with isotopically labeled O 2and H 2O over supported Au and Pt catalysts,as well as ab initio density functional theory calcu-lations on ethanol oxidation over metal surfaces.Previous studies indicate that alcohol oxida-tion over supported metal catalysts (Au,Pt,and Pd)proceeds by dehydrogenation to an aldehyde or ketone intermediate,followed by oxidation to the acid product (Eq.1)RCH 2OH À!O 2,catalyst RCH ¼O À!O 2,catalystRCOOH(1)Hydroxide ions play an important role during oxidation;the product distribution depends on pH,and little or no activity is seen over Au cat-alysts without added base.We studied Au par-ticles of various sizes (average diameter ranging from 3.5to 10nm)on different supports (TiO 2and C)as catalysts for alcohol oxidation and com-pared them to Pt and Pd particles supported on C.The oxidation of glycerol (HOCH 2CHOHCH 2OH)to glyceric (HOCH 2CHOHCOOH)and glycolic (HOCH 2COOH)acids occurred at a turnover frequency (TOF)of 6.1and 4.9s −1on Au/C and Au/TiO 2,respectively,at high pH (>13)whereas the TOF on supported Pt and Pd (1.6and 2.2s −1,respectively)was slightly lower at otherwise iden-tical conditions (Table 1).For these Au catalysts,particle size and support composition had negligi-ble effect on the rate or selectivity.In the absence of base,the glycerol oxidation rate was much lower over the Pt and Pd catalysts and no conver-sion was observed over the Au catalysts (Table 1).Moreover,the products detected over Pt and Pd in the absence of base are primarily the intermediate aldehyde and ketone,rather than acids.Department of Chemical Engineering,University of Virginia,102Engineers ’Way,Post Office Box 400741,Charlottesville,VA,22904–4741,USA.*To whom correspondence should be addressed.E-mail:rjd4f@1OCTOBER 2010VOL 330SCIENCE74REPORTSo n A u g u s t 4, 2011w w w .s c i e n c e m a g .o r g D o w n l o a d e d f r o m。
苯丙烷代谢途径的英文

苯丙烷代谢途径的英文英文回答:Phenylpropanoid metabolism.Phenylpropanoids are a large and diverse group of plant-derived compounds that are involved in a wide range of biological processes, including defense against herbivores and microorganisms, growth and development, and reproduction. The phenylpropanoid metabolic pathway is a complex network of enzymatic reactions that converts phenylalanine into a variety of phenylpropanoid compounds, including:Monomers: Phenylalanine, cinnamic acid, p-coumaric acid, caffeic acid, ferulic acid, sinapic acid.Dimers: Lignans, neolignans.Oligomers: Coumarins, flavonoids, isoflavonoids,stilbenes.Polymers: Lignin.The phenylpropanoid metabolic pathway is divided into three main stages:Shikimate pathway: This pathway produces the phenylalanine precursor, phosphoenolpyruvate (PEP).Phenylpropanoid pathway: This pathway converts phenylalanine into cinnamic acid, the first committed intermediate in the phenylpropanoid metabolic pathway.Downstream pathways: These pathways convert cinnamic acid into a variety of phenylpropanoid compounds.The phenylpropanoid metabolic pathway is regulated by a number of factors, including:Transcription factors: These proteins bind to specific DNA sequences and regulate the expression of genes involvedin the phenylpropanoid metabolic pathway.Enzymes: These proteins catalyze the reactions of the phenylpropanoid metabolic pathway.Hormones: These chemicals can activate or repress the phenylpropanoid metabolic pathway.Environmental factors: These factors, such as light and temperature, can affect the activity of the phenylpropanoid metabolic pathway.The phenylpropanoid metabolic pathway is essential for plant growth and development. It produces a variety of compounds that are involved in a wide range of biological processes. The pathway is also regulated by a number of factors, which ensures that the production of phenylpropanoid compounds is tightly controlled.中文回答:苯丙烷代谢途径。
代谢组学

15
4
代谢组学(Metabonomics/ Metabolomics )是通过考察生 物体系(细胞、组织 或生物体)受刺激或扰动后(如将某 个特定的基因变异或环境变化后),其代谢产物的变化或其 随时间的变化,来研究生物体系的一门科学。
代谢组(metabolome)是基因组的下游产物也是最终产物, 是一些参与生物体新陈代谢、维持生物体正常生长功能 和 生长发育的小分子化合物的集合,主要是相对分子量小于 1000的内源性小分子。
Each group of samples has…,
many sample analyses are required for statistical relevance a complex raw dataset that needs to be processed differences between sample groups which need to be highlighted
修饰或环境因子的影响 4. 上述内源性化合物的知识可以被用于疾病的诊断和药
物筛选
与转录组学和蛋白组学相比,代谢组学有以下优点: 1. 基因与蛋白质表达的微小变化会在代谢物上得到放大,
从而使检测更容易 2. 代谢组学的研究不需要建立全基因测序及大量序列标
签(EST)的数据库 3. 代谢物的研究种类远小于蛋白质的数目 4. 研究中采用的技术更通用
Discrete Applied Mathematics

Discrete Applied Mathematics157(2009)2217–2220Contents lists available at ScienceDirectDiscrete Applied Mathematicsjournal homepage:/locate/damPreface$This special issue on Networks in Computational Biology is based on a workshop at Middle East Technical University in Ankara,Turkey,September10–12,2006(.tr/Networks_in_Computational_Biology/). Computational biology is one of the many currently emerging areas of applied mathematics and science.During the last century,cooperation between biology and chemistry,physics,mathematics,and other sciences increased dramatically,thus providing a solid foundation for,and initiating an enormous momentum in,many areas of the life sciences.This special issue focuses on networks,a topic that is equally important in biology and mathematics,and presents snapshots of current theoretical and methodological work in network analysis.Both discrete and continuous optimization,dynamical systems, graph theory,pertinent inverse problems,and data mining procedures are addressed.The principal goal of this special issue is to contribute to the mathematical foundation of computational biology by stressing its particular aspects relating to network theory.This special issue consists of25articles,written by65authors and rigorously reviewed by70referees.The guest editors express their cordial thanks to all of them,as well as to the Editors-in-Chief of Discrete Applied Mathematics,Prof.Dr.Endre Boros and his predecessor,Prof.Dr.Peter L.Hammer,who was one of the initiators of this special issue but left us in2006, and to Mrs.Katie D’Agosta who was at our side in each phase of preparation of this DAM special issue.The articles are ordered according to their contents.Let us briefly summarize them:In the paper of Jacek Błażewicz,Dorota Formanowicz,Piotr Formanowicz,Andrea Sackmann,and MichałSajkowski, entitled Modeling the process of human body iron homeostasis using a variant of timed Petri nets,the standard model of body iron homeostasis is enriched by including the durations of the pertinent biochemical reactions.A Petri-net variant in which, at each node,a time interval is specified is used in order to describe the time lag of the commencement of conditions that must be fulfilled before a biochemical reaction can start.Due to critical changes in the environment,switches can occur in metabolic networks that lead to systems exhibiting simultaneously discrete and continuous dynamics.Hybrid systems represent this accurately.The paper Modeling and simulation of metabolic networks for estimation of biomass-accumulation parameters by Uˇg ur Kaplan,Metin Türkay,Bülent Karasözen,and Lorenz Biegler develops a hybrid system to simulate cell-metabolism dynamics that includes the effects of extra-cellular stresses on metabolic responses.Path-finding approaches to metabolic-pathway analysis adopt a graph-theoretical approach to determine the reactions that an organism might use to transform a source compound into a target compound.In the contribution Path-finding approaches and metabolic pathways,Francisco J.Planes and John E.Beasley examine the effectiveness of using compound-node connectivities in a path-finding approach.An approach to path finding based on integer programming is also presented. Existing literature is reviewed.This paper is well illustrated and provides many examples as well as,as an extra service,some supplementary information.In A new constraint-based description of the steady-state flux cone of metabolic networks,Abdelhalim Larhlimi and Alexander Bockmayr present a new constraint-based approach to metabolic-pathway analysis.Based on sets of non-negativity constraints,it uses a description of the set of all possible flux distributions over a metabolic network at a steady state in terms of the steady-state flux cone.The constraints can be identified with irreversible reactions and,thus,allow a direct interpretation.The resulting description of the flux cone is minimal and unique.Furthermore,it satisfies a simplicity condition similar to the one for elementary flux modes.Most biological networks share some properties like being,e.g.,‘‘scale free’’.Etienne Birmeléproposes a new random-graph model in his contribution A scale-free graph model based on bipartite graphs that can be interpreted in terms of metabolic networks,and exhibits this specific feature.$Dedicated to our dear teacher and friend Prof.Dr.Peter Ladislaw Hammer(1936–2006).0166-218X/$–see front matter©2009Elsevier B.V.All rights reserved.doi:10.1016/j.dam.2009.01.0212218Preface/Discrete Applied Mathematics157(2009)2217–2220Differential equations have been established to quantitatively model the dynamic behaviour of regulatory networks representing interactions between cell components.In the paper Inference of an oscillating model for the yeast cell cycle, Nicole Radde and Lars Kaderali study differential equations within a Bayesian setting.First,an oscillating core network is learned that is to be extended,in a second step,using‘‘Bayesian’’methodology.A specifically designed hierarchical prior distribution over interaction strengths prevents overfitting and drives the solutions to sparse networks.An application to a real-world data set is provided,and its dynamical behaviour is reconstructed.The contribution An introduction to the perplex number system by Jerry L.R.Chandler derives from his approach to theoretical chemistry,and provides a universal source of diagrams.The perplex number system,a new logic for describing relationships between concrete objects and processes,provides in particular an exact notation for chemistry without invoking either chemical or‘‘alchemical’’symbols.Practical applications to concrete compounds(e.g.,isomers of ethanol and dimethyl ether)are given.In conjunction with the real number system,the relations between perplex numbers and scientific theories of concrete systems(e.g.,intermolecular dynamics,molecular biology,and individual medicine)are described.Since exact determination of haplotype blocks is usually impossible,a method is desired which can account for recombinations,especially,via phylogenetic networks or a simplified version.In their work Haplotype inferring via galled-tree networks using a hypergraph-covering problem for special genotype matrices,Arvind Gupta,Ján Maňuch,Ladislav Stacho, and Xiaohong Zhao reduce the problem via galled-tree networks to a hypergraph-covering problem for genotype matrices satisfying a certain combinatorial condition.Experiments on real data show that this condition is mostly satisfied when the minor alleles(per SNP)reach at least30%.Recently the Quartet-Net or,for short,‘‘QNet’’method was introduced by Stefan Grünewald et al.as a method for computing phylogenetic split networks from a collection of weighted quartet trees.Here,Stefan Grünewald,Vincent Moulton,and Andreas Spillner show that QNet is a‘‘consistent’’method.This key property of QNet does not only guarantee to produce a tree if the input corresponds to a tree—and an outer-labeled planar split network if the input corresponds to such a network;the proof given in their contribution Consistency of the QNet algorithm for generating planar split networks from weighted quartets also provides the main guiding principle for the design of the method.Kangal and Akbash dogs are the two well-known shepherd dog breeds in Turkey.In the article The genetic relationship between Kangal,Akbash,and other dog populations,Evren Koban,Çigdem Gökçek Saraç,Sinan Can Açan,Peter Savolainen, andİnci Togan present a comparative examination by mitochondrial DNA control region,using a consensus neighbour-joining tree with bootstrapping which is constructed from pairwise FST values between populations.This study indicates that Kangal and Akbash dogs belong to different branches of the tree,i.e.,they might have descended maternally from rather different origins created by an early branching event in the history of the domestic dogs of Eurasia.In their paper The Asian contribution to the Turkish population with respect to the Balkans:Y-chromosome perspective,Ceren Caner Berkman and inci Togan investigate historical migrations from Asia using computational approaches.The admixture method of Chikhi et al.was used to estimate the male genetic contribution of Central Asia to hybrids.The authors observed that the male contribution from Central Asia to the Turkish population with reference to the Balkans was13%.Comparison of the admixture estimate for Turkey with those of neighboring populations indicated that the Central Asian contribution was lowest in Turkey.Split-decomposition theory deals with relations between real-valued split systems and metrics.In his work Split decomposition over an Abelian group Part2:Group-valued split systems with weakly compatible support,Andreas Dress uses a general conceptual framework to study these relations from an essentially algebraic point of view.He establishes the principal results of split-decomposition theory regarding split systems with weakly compatible support within this new algebraic framework.This study contributes to computational biology by analyzing the conceptual mathematical foundations of a tool widely used in phylogenetic analysis and studies of bio-diversity.The contribution Phylogenetic graph models beyond trees of Ulrik Brandes and Sabine Cornelsen deals with methods for phylogenetic analysis,i.e.,the study of kinship relationships between species.The authors demonstrate that the phylogenetic tree model can be generalized to a cactus(i.e.,a tree all of whose2-connected components are cycles)without losing computational efficiency.A cactus can represent a quadratic rather than a linear number of splits in linear space.They show how to decide in linear time whether a set of splits can be accommodated by a cactus model and,in that case,how to construct it within the same time bounds.Finally,the authors briefly discuss further generalizations of tree models.In their paper Whole-genome prokaryotic clustering based on gene lengths,Alexander Bolshoy and Zeev Volkovich present a novel method of taxonomic analysis constructed on the basis of gene content and lengths of orthologous genes of 66completely sequenced genomes of unicellular organisms.They cluster given input data using an application of the information-bottleneck method for unsupervised clustering.This approach is not a regular distance-based method and, thus,differs from other recently published whole-genome-based clustering techniques.The results correlate well with the standard‘‘tree of life’’.For characterization of prokaryotic genomes we used clustering methods based on mean DNA curvature distributions in coding and noncoding regions.In their article Prokaryote clustering based on DNA curvature distributions,due to the extensive amount of data Limor Kozobay-Avraham,Sergey Hosida,Zeev Volkovich,and Alexander Bolshoy were able to define the external and internal factors influencing the curvature distribution in promoter and terminator regions.Prokaryotes grow in the wide temperature range from4◦C to100◦C.Each type of bacteria has an optimal temperature for growth.They found very strong correlation between arrangements of prokaryotes according to the growth temperature and clustering based on curvature excess in promoter and terminator regions.They found also that the main internal factors influencingPreface/Discrete Applied Mathematics157(2009)2217–22202219 the curvature excess are genome size and A+T composition.Two clustering methods,K-means and PAM,were applied and produced very similar clusterings that reflect the aforementioned genomic attributes and environmental conditions of the species’habitat.The paper Pattern analysis for the prediction of fungal pro-peptide cleavage sites by SüreyyaÖzöˇgür Ayzüz,John Shawe-Taylor,Gerhard-Wilhelm Weber,and Zümrüt B.Ögel applies support-vector machines to predict the pro-peptide cleavage site of fungal extra-cellular proteins displaying mostly a monobasic or dibasic processing site.A specific kernel is expressed as an application of the Gaussian kernel via feature spaces.The novel approach simultaneously performs model selection, tests the accuracy,and computes confidence levels.The results are found to be accurate and compared with the ones provided by a server.Preetam Ghosh,Samik Ghosh,Kalyan Basu,and Sajal Das adopt an‘‘in silico’’stochastic-event-based simulation methodology to determine the temporal dynamics of different molecules.In their paper Parametric modeling of protein–DNA binding kinetics:A discrete event-based simulation approach,they present a parametric model for predicting the execution time of protein–DNA binding.It considers the actual binding mechanism along with some approximated protein-and DNA-structural information using a collision-theory-based approach incorporating important biological parameters and functions into the consideration.Murat Ali Bayır,Tacettin Doˇg acan Güney,and Tolga Can propose a novel technique in their paper Integration of topological measures for eliminating non-specific interactions in protein interaction networks for removing non-specific interactions in a large-scale protein–protein interaction network.After transforming the interaction network into a line graph,they compute betweenness and other clustering coefficients for all the edges in the network.The authors use confidence estimates and validate their method by comparing the results of a test case relating to the detection of a molecular complex with reality.The article Graph spectra as a systematic tool in computational biology by Anirban Banarjee and Jürgen Jost deals with the obviously important question of how biological content can be extracted from the graphs to which biological data are often reduced.From the spectrum of the graph’s Laplacian that yields an essentially complete qualitative characterization of a graph,a spectral density plot is derived that can easily be represented graphically and,therefore,analyzed visually and compared for different classes of networks.The authors apply this method to the study of protein–protein interaction and other biological and infrastructural networks.It is detected that specific such classes of networks exhibit common features in their spectral plots that readily distinguish them from other classes.This represents a valuable complement to the currently fashionable search for universal properties that hold across networks emanating from many different contexts.Konstantin Klemm and Peter F.Stadler’s Note on fundamental,nonfundamental,and robust cycle bases investigates the mutual relationships between various classes of cycle bases in a network that have been studied in the literature.The authors show for instance that strictly fundamental bases are not necessarily cyclically robust;and that,conversely, cyclically robust bases are not necessarily fundamental.The contribution focuses on cyclically robust cycle bases whose existence for arbitrary graphs remains open despite their practical use for generating all cycles of a given2-connected graph. It presents also a class of cubic graphs for which cyclically robust bases can be constructed explicitly.Understanding the interplay and function of a system’s components also requires the study of the system’s functional response to controlled experimental perturbations.For biological systems,it is problematic with an experimental design to aim at a complete identification of the system’s mechanisms.In his contribution A refinement of the common-cause principle,Nihat Ay employs graph theory and studies the interplay between stochastic dependence and causal relations within Bayesian networks and information theory.Applying a causal information-flow measure,he provides a quantitative refinement of Reichenbach’s common-cause principle.Based on observing an appropriate collection of nodes of the network, this refinement allows one to infer a hitherto unknown lower bound for information flows within the network.In their article Discovering cis-regulatory modules by optimizing barbecues,Axel Mosig,Türker Bıyıkoˇg lu,Sonja J.Prohaska, and Peter F.Stadler ask for simultaneously stabbing a maximum number of differently coloured intervals from K arrangements of coloured intervals.A decision version of this best barbecue problem is shown to be NP-complete.Because of the relevance for complex regulatory networks on gene expression in eukaryotic cells,they propose algorithmic variations that are suitable for the analysis of real data sets comprising either many sequences or many binding sites.The optimization problem studied generalizes frequent itemset mining.The contribution A mathematical program to refine gene regulatory networks by Guglielmo Lulli and Martin Romauch proposes a methodology for making sense of large,multiple time-series data sets arising in expression analysis.It introduces a mathematical model for producing a reduced and coherent regulatory system,provided a putative regulatory network is given.Two equivalent formulations of the problem are given,and NP-completeness is established.For solving large-scale instances,the authors implemented an ant-colony optimization procedure.The proposed algorithm is validated by a computational analysis on randomly generated test instances.The practicability of the proposed methodology is also shown using real data for Saccharomyces cerevisiae.Jutta Gebert,Nicole Radde,Ulrich Faigle,Julia Strösser,and Andreas Burkovski aim in their paper Modelling and simulation of nitrogen regulation in Corynebacterium glutamicum at understanding and predicting the interactions of macromolecules inside the cell.It sets up a theoretical model for biochemical networks,and introduces a general method for parameter estimation,applicable in the case of very short time series.This approach is applied to a special system concerning nitrogen uptake.The equations are set up for its main components,the corresponding optimization problem is formulated and solved, and simulations are carried out.2220Preface/Discrete Applied Mathematics157(2009)2217–2220Gerhard-Wilhelm Weber,Ömür Uˇg ur,Pakize Taylan,and Aysun Tezel model and predict gene-expression patterns incorporating a rigorous treatment of environmental aspects,and aspects of errors and uncertainty.For this purpose,they employ Chebyshev approximation and generalized semi-infinite optimization in their paper On optimization,dynamics and uncertainty:A tutorial for gene–environment networks.Then,time-discretized dynamical systems are studied,the region of parametric stability is detected by a combinatorial algorithm and,then,the topological landscape of gene–environment networks is analyzed in terms of its‘‘structural stability’’.We are convinced that all papers selected for this special issue constitute valuable contributions to many different areas in computational biology,employing methods from discrete mathematics and related fields.We again thank all colleagues who have participated in this exciting endeavor with care,foresight,and vision,for their highly appreciated help.Guest editorsAndreas DressBülent KarasözenPeter F.StadlerGerhard-Wilhelm Weber125July2008Available online29March2009 1Assistant to the guest editors:Mrs.Cand.MSc.Bengisen Pekmen(Institute of Applied Mathematics,METU,Ankara).。
代谢组学的英语

代谢组学的英语Metabolomics: Unraveling the Complexity of Biological SystemsMetabolomics, a rapidly evolving field in the realm of systems biology, has emerged as a powerful tool for understanding the intricate workings of living organisms. This discipline focuses on the comprehensive analysis of the small molecules, known as metabolites, that are produced and consumed within biological systems. By studying the metabolome, the complete set of metabolites present in a cell, tissue, or organism, researchers can gain invaluable insights into the dynamic and interconnected processes that sustain life.The origins of metabolomics can be traced back to the early 20th century, when scientists began to recognize the importance of studying the chemical composition of living organisms. However, it was not until the advent of modern analytical technologies, such as mass spectrometry and nuclear magnetic resonance (NMR) spectroscopy, that the field truly began to flourish. These advanced techniques have enabled researchers to detect and identify a wide range of metabolites, from simple sugars and amino acids to complex lipids and secondary metabolites.One of the key advantages of metabolomics is its ability to provide a comprehensive snapshot of the physiological state of a biological system. Unlike genomics, which focuses on the genetic blueprint, or proteomics, which examines the expression of proteins, metabolomics offers a more direct and dynamic representation of the functional activities within a cell or organism. By analyzing the metabolic profiles of samples, researchers can identify biomarkers –specific metabolites or patterns of metabolites – that are associated with particular physiological or pathological conditions.The applications of metabolomics are vast and diverse, spanning a wide range of disciplines, from medicine and agriculture to environmental science and biotechnology. In the field of medicine, metabolomics has been instrumental in the early detection and diagnosis of diseases, the monitoring of disease progression, and the development of personalized treatment strategies. By identifying unique metabolic signatures associated with various health conditions, such as cancer, diabetes, and neurological disorders, clinicians can develop more targeted and effective interventions.Moreover, metabolomics has become a valuable tool in the field of drug discovery and development. By studying the metabolic responses of cells or organisms to the introduction of potential drug candidates, researchers can gain insights into the mechanisms ofaction, potential side effects, and optimal dosing regimens. This information can help streamline the drug development process and improve the chances of success for new therapeutic agents.In the realm of agriculture, metabolomics has found applications in the optimization of crop yields, the development of sustainable farming practices, and the detection of food contaminants or adulterants. By analyzing the metabolic profiles of plants, researchers can identify key metabolites that are associated with desirable traits, such as increased yield, stress tolerance, or nutritional value. This knowledge can then be used to guide breeding programs or to inform the development of more efficient agricultural practices.Beyond its applications in medicine and agriculture, metabolomics has also made significant contributions to our understanding of environmental processes and the study of microbial communities. By analyzing the metabolic signatures of environmental samples, such as soil, water, or air, researchers can gain insights into the complex interactions between living organisms and their surrounding ecosystems. This information can be used to monitor the health of natural environments, detect the presence of pollutants or toxins, and develop strategies for environmental remediation.Despite the numerous advancements in the field of metabolomics, there are still significant challenges that researchers must overcome.The complexity of biological systems, the vast diversity of metabolites, and the inherent variability in analytical techniques can all contribute to the difficulty in interpreting and integrating metabolomics data. Additionally, the development of comprehensive databases and standardized data analysis workflows remains an ongoing effort, as researchers strive to create a more unified and streamlined approach to metabolomics research.Nonetheless, the potential of metabolomics to transform our understanding of biological systems is undeniable. As the field continues to evolve, we can expect to see an increasing number of groundbreaking discoveries and innovative applications that will have far-reaching impacts on fields as diverse as medicine, agriculture, and environmental science. By unraveling the complex web of metabolic interactions within living organisms, metabolomics holds the promise of unlocking new avenues for improving human health, enhancing food production, and protecting the delicate balance of our natural world.。
Metabolic Pathways and Regulation AP Bio 2014

Aerobic Respiration Anaerobic Respiration Metabolism of Non-Sugar Molecules Regulation of Metabolism
• Ancient prokaryotes are thought to have used glycolysis long before there was oxygen in the atmosphere
• Very little O2 was available in the atmosphere until about 2.7 billion years ago, so early prokaryotes likely used only glycolysis to generate ATP
– Fats are digested to glycerol (used in glycolysis) and fatty acids are broken down by beta oxidation and yield acetyl CoA
Regulation of Respiration
• Like Lactic acid fermentation, Alcohol fermentation occurs when oxygen is not available.
• Produces Ethyl Alcohol Carbon dioxide and only 2 ATP
Anaerobes…
pyruvate or acetaldehyde – Releases only 2 ATP
Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC

Metabolic pathway analysis web service(Pathway Hunter Tool at CUBIC )S.A. Rahman*, P. Advani, R. Schunk, R. Schrader, Dietmar Schomburg*Cologne University BioInformatics Center (CUBIC ) & Institute of BiochemistryZülpicher Strasse 47, 50674 Köln, GermanyBioinfor m atics © Oxford University Press 2004; all rights reserved.Bioinformatics Advance Access published November 30, 2004ABSTRACTMotivation: Pathway Hunter Tool (PHT)(Syed Asad Rahman et al., 2004) is a fast, robust, and user friendly tool to analyse the shortest paths in metabolic pathways. The user can perform shortest path analysis for one or more organisms or can build virtual organisms (networks) using enzymes. Using PHT, the user can also calculate the average shortest path(Jungnickel, 2002), average alternate path and the top 10 hubs in the metabolic network. The comparative study of metabolic connectivity and the cross talk between metabolic pathways between various sequenced genomes is possible.Results: A new algorithm for finding the biochemically valid connectivity between metabolites in a metabolic network was developed and implemented. A predefined manual assignment of side metabolites (like ATP, ADP, Water, CO2 etc) and main metabolites is not necessary as the new concept uses chemical structure information (global and local similarity) between metabolites for identification of the shortest path.Availability:Pathway Hunter Tool (PHT) is accessible at http://www.pht.uni-koeln.de .Contact: asad.rahman@uni-koeln.de, D.Schomburg@uni-koeln.deKeywords:Pathway Hunter Tool, PHT, Shortest Path, Metabolic Pathways, Metabolic Networks, Tools, Pathway Database.INTRODUCTIONWith the advent of the "omics" era more and more system-based approaches to biological functions are being developed. Metabolome analysis and metabolomics are gaining higher attention and help to understand the complexity of the underlying cellular networks in organisms. The completion of a large number of genomes has made the comparative study of genomes possible at different levels. One way to gain a better understanding of the sequenced genomes can be achieved by analysis of the underlying metabolic network and its topology in different genomes. Several databases provide information about metabolic pathways.We have used KEGG (Kanehisa et al., 2004) as the basic database for our analysis apart from BRENDA (Schomburg et al., 2004) and PROSITE(Hulo et al., 2004). A global view of the connectivity in metabolic pathway, the contribution and usage of certain metabolite in these pathways is highly instructive. Shortest path analysis(Arita, 2004) is one of the best-defined methods to analyse a graph (Metabolic Pathways) at different levels in terms of local and global connectivity. With Pathway Hunter Tool (PHT) it is also possible to calculate statistical information(Barabasi and Oltvai, 2004) from the topology arising from the interacting molecules in order to capture the nature of connectivity.METHODWhereas a number of long-established methods exist for the analysis of shortest paths in graphs the situation in metabolic networks is a little more complicated. In the example of reactions given in figure 1 a shortest path algorithm for metabolic pathways is required to follow the path of the thick lines with the result that there exists a path between phosphate and ATP via glucose-6-phosphate (thick line) but that there is no way to produce fructose from phosphate to fructose (thin line). In the third reaction of the scheme the algorithm has to distinguish which direction to go, depending on the starting point being either glucose or phosphate.Therefore it is important to connect two metabolites in a reaction with respect to their structural similarity. We have used the fingerprint algorithm from the Chemistry Development Kit (CDK)(Steinbeck et al., 2003) to convert the 2-dimensional chemical structure information to a 1-dimesional binary stream as a fingerprint for faster similarity search (Whittle et al., 2003). Using the fingerprints, the similarity between two molecules was calculated using a normalized scoring function obtainedby combination of the atomic mass value of the metabolites and the Tanimoto algorithm (Xue et al., 2003). This allowed to avoid the false connectivity in the metabolic pathway and made the path search algorithm more robust.In order to calculate the shortest path between two metabolites, the depth first search (DFS) algorithm(Jungnickel, 2002) is used in PHT. Higher-Order Horn Logic (HOHL)(Nadathur and Miller, 1990) has been used to satisfy the constraints. Our new algorithm automatically discriminates between side metabolites (like ATP, ADP, Water, CO2 etc) and main metabolites while finding the shortest path without the need to predefine those. Predefined exclusion of small metabolites in the metabolic pathway may lead to broken links in the network or longer connectivity. This means that at each reaction step the algorithm should be able to decide, which metabolite to choose for further connectivity in the pathway and which to skip.ALGORITHMIn this section the new algorithm used in Pathway Hunter Tool (PHT) to find the shortest path in the biochemical network is described.1. Definition of the metabolite mapping scoring functionLet A be an educt and B a product metabolite and a, b the number of bits (calculated by the fingerprint algorithm from the Chemistry Development Kit (CDK)(Steinbeck et al., 2003)) “on” on A or B metabolites, respectively, c = the number of bits “on” in both A & B, d = number of bits “off” in both A & B, then we can define the equation in form of set theory (Jech and Jech, 1997).a = |A |,b = |B |,c = |A B |,d = n - | A U B |and a + b - c = | A U B |(Note: ‘| B |’ denotes cardinality of the set)where n = total number of attributes of an object (e.g, bits in a fingerprint)Once we are able to formulate the chemical structure(Whittle et al., 2003) in terms of set theory the next step was the development of a scoring scheme for the similarity between two metabolites. We have used the Tanimoto Coefficient(Willet et al., 1998)for this purpose, i.e. the structural similarity between two metabolites A and B can be defined as•Tanimoto Coefficient S A,B = | A B| / | A U B|The percentage Atomic Mass Contribution (PAMC) for two competing educt (A) and product (B) can be defined as hundred times the sum of mass for both the metabolites (A & B) divided by the total mass of the metabolites in that reaction.•Atomic Mass Contribution PAMC A,B = 100 * (M A + M B) / M RThe mapping scoring function is then defined as the product of similarity score and atomic mass contribution in each reaction between every two competing educt (A) and product (B)metabolites.Final score for top competing metabolites can be defined asScore A,B = PAMC A,B * S A,BWhere0 <= S A,B <= 1and0 <= PAMC A,B <= 1002. Local mapping metabolites in reactionsThe derived scoring function was used to find a suitable mapping between substrate molecules and product molecules. We use a slightly modified form of game theory (/) in order to map the substrate to the product metabolite. The method consists of construction of a matrix of substrates as rows and products as columns with the score defined above as matrix elements. The score between any substrate or product whose extension is smaller than three bonds is set to zero. A substrate is mapped to a product when either the score dominates all other scores in the present row or column respectively. By this procedure we keep track of the maximum structural similarity between two interacting metabolites. Fig 2 illustrates the outcome our mapping procedure when applied to a reaction.3. Shortest path between two metabolitesFor the calculation of the shortest paths the two biochemical criteria “local” and “global” structural similarity are used, where “local similarity” is defined as thesimilarity between two intermediate molecules and “global similarity”is defined as the amount of conserved structure found between the source metabolite and the destination metabolites after a series of reaction steps (Fig. 3).The only potential draw back of this method is given by the fact that not all metabolites in the metabolite databases have structures (e.g. macromolecules like proteins or nucleic acids, or generic molecules like “an alcohol”). In these cases the user may miss some connectivity due to lack of structural information. In order to cross check this result it is possible to switch off the “Atom Mapper” (Local similarity) and “Atom Tracer” (Global Similarity) options thereby performing the search on the ligand-number-based mapping obtained from the KEGG reaction database. On the other hand the power and biochemical relevance of having local similarity and global similarity is very high. In future we plan to provide non-standard structural information for these metabolites in order to allow the inclusion of such reactions.Complexity of the algorithmThe shortest path between source and destination metabolite is the minimum number of reaction steps between them(fig. 4). We consider the metabolic pathway in our system as a directed graph with all edges (reactions) sharing the same cost (here 1). Hence this does not lead us to NP-complete problem as one can calculate the k-shortest path between two metabolites using the BFS (Breadth First Search) algorithm. Higher-Order Horn Logic (HOHL)(Nadathur and Miller, 1990) has been used to satisfy the constraints (similarity) with the BFS algorithm in order to calculate k-shortest paths between two metabolites (source and destination). This means that the runtime of the tool depends on the metabolites and reactions present in an organism. We are able to generate all possible k-shortest paths between two metabolites under given criteria of global and local similarity.Program optionsPresently Pathway Hunter Tool (PHT) has four options.1.Find k-shortest path to convert one metabolite into another in a given network(organism-specific or general metabolic network).2.Find k-shortest paths from a substrate metabolite to all feasible metabolites ina given network (organism-specific or general).3.Find k-shortest path to a product metabolite from all feasible substratemetabolites in a given network (organism-specific or general).4.Statistical analysis of the metabolic pathways like average path length,diameter of the network, average node connectivity, loose ends in the network, hubs in a given network (organism-specific or general).User defined constraintsThere are sets of user-defined constraints, which can be used for an in-depth network analysis without affecting the biochemical/biological relevance.•While traversing through the metabolic pathway it is possible to set the similarity measure score (Atom Mapper) between interacting molecules and to define the amount of structure change with respect to his reference molecule at each reaction step (Atom Tracer).•By setting the Minimum path length and Maximum path length the path between two metabolites in the network can be altered. For example, if the minimum path length is set to six, then the algorithm will drop paths below it and report the next possible shortest path above or equal to six, which is the shortest possible path under the given constraint.•It is possible to choose via Metabolite, not via Metabolites and not via Enzymes options for use of a particular set of pathways.•Under Build Virtual Organism it is possible to add own set of enzymes and perform further analysis. This is very useful for identification of the missing links in the network.RESULTSWe performed a shortest path analysis (fig. 4) in Escherichia coli K-12 between beta-D-Glucose and Pyruvate, which turned out to be nine steps long. We considered global similarity and local similarity while traversing the path. The algorithm automatically identifies the correct connectivity between the metabolites at each reaction step.We also performed a comparative study between the KEGG reaction reference map, Corynebacterium glutamicum, Escherichia coli K-12 and Mycobacterium tuberculosis (fig. 5). We were interested in finding the shortest path between “D-Erythrose 4-phosphate” and“Chorismate”, which turned out to be in 7 reaction steps in all these cases. Looking closely into fig. 5 it is clear that different pathways are possible to convert “D-Erythrose 4-phosphate” to “Chorismate” in the reference map, or in Corynebacterium glutamicum, Escherichia coli K-12 and Mycobacterium tuberculosis(score 4 on the edge).Some organisms may use enzyme “1.1.99.25” (blue colour) to perform the same conversion (score 1 on the edge).OUTPUT FORMATPathway Hunter Tool (PHT) generates three kinds of output:• A Text based output can be viewed immediately in the browsers and is supplied with hyperlinks to other database like BRENDA, KEGG and PROSITE.•A Graphical view of the output is generated for “Metabolic Pathways” and “Enzyme” connectivity as Graph Modeling Language (GML) (http://infosun.fmi.uni-passau.de/Graphlet/GML/) files. These portable files can be saved on the clients system and can be viewed later in any dynamiclayout software that read the GML format (e.g. the yEd (/products/yed/ graphical editor).•Pathway Hunter Tool (PHT) also generates “Enzyme-Enzyme” connectivity matrix, which can be used for pathway alignment and other studies. The “Reaction-Organism Matrix” highlights the presence of reaction in organisms by binary 1 and 0 for absence.ACKNOWLEDGEMENTSThe authors are grateful for financial support by the German Federal Ministry for Education and Research(BMBF).REFERENCESArita, M. (2004) The metabolic world of Escherichia coli is not small. Proc Natl Acad Sci U S A, 101, 1543-1547.Barabasi, A.L. and Oltvai, Z.N. (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet, 5, 101-113.Hulo, N., Sigrist, C.J., Le Saux, V., Langendijk-Genevaux, P.S., Bordoli, L., Gattiker,A., De Castro, E., Bucher, P. and Bairoch, A. (2004) Recent improvements tothe PROSITE database. Nucleic Acids Res, 32 Database issue, D134-137. Jech, T.J. and Jech, T. (1997) Set Theory. Springer-Verlag.Jungnickel, D. (2002) Graphs, Network and Algorithm. Springer Verlag, Berlin. Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. and Hattori, M. (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res, 32 Database issue, D277-280.Nadathur, G. and Miller, D. (1990) Higher-order Horn clauses. Journal of the ACM (JACM), 37, 777-814.Schomburg, I., Chang, A., Ebeling, C., Gremse, M., Heldt, C., Huhn, G. and Schomburg, D. (2004) BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res, 32 Database issue, D431-433. Steinbeck, C., Han, Y., Kuhn, S., Horlacher, O., Luttmann, E. and Willighagen, E.(2003) The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. J Chem Inf Comput Sci, 43, 493-500.Syed Asad Rahman, Dietmar Schomburg and Schrader, R. (2004) CUBIC Metabolic Pathway Hunter Tool (CMPHT). ISMB/ECCB, Glasgow, Scotland, Vol.Poster.Whittle, M., Willett, P., Klaffke, W. and van Noort, P. (2003) Evaluation of similarity measures for searching the dictionary of natural products database. J Chem Inf Comput Sci, 43, 449-457.Willet, P., Barnard, J.M. and Downs, G.M. (1998) Chemical Similarity Searching. J Chem Inf Comput Sci, 38, 938-996.Xue, L., Godden, J.W., Stahura, F.L. and Bajorath, J. (2003) Design and evaluation ofa molecular fingerprint involving the transformation of property descriptorvalues into a binary classification scheme. J Chem Inf Comput Sci, 43, 1151-1157.FIGURES:maps to ADP (green line) and D-Glucose maps to D-Glucose-6phosphate (red line).Step1: beta-D-Glucose <=> beta-D-Glucose 6-phosphateLocal Similarity 100 %, Global Similarity 100 %Step2:ß-D-Glucose 6-phosphate <=> ß-D-Fructose 6-phosphateLocal Similarity 94%, Global Similarity 93 %Step3: ß-D-Fructose 6-phosphate <=> D-Xylulose 5-phosphate,Local Similarity 62 %, Global Similarity 45 %Figure 3: Shortest path between metabolites ß-D-Glucose to D-Xylulose 5-phosphate is in 3 steps and only 45% of the structural is common between them globally.Escherichia coli K-12is 9 reaction steps long.Figure 5: Enzyme-Enzyme connectivity map highlights the shortest path (7 reaction steps) between“D-Erythrose 4-phosphate” and “Chorismate” in the KEGG reference map and Corynebacterium glutamicum, Escherichia coli K-12 and Mycobacterium tuberculosis. The weights given at the connections reflect the number of occurrences of this step in the queried pathways. 1.1.99.25 is found only in the reference map (originating from Acinetobacter calcoaceticus).。
叶酸在治疗高同型半胱氨酸血症相关疾病中的合理应用

叶酸在治疗高同型半胱氨酸血症相关疾病中的合理应用那一凡!,谭玲#(北京医院药学部,药物临床风险与个体化应用评价北京市重点实验室,国家老年医学中心,中国医学科学院老年医学研究院,北京100730)中图分类号R977文献标志码A文章编号1672-2124(2021)04-0508-05DOI10.14009/j.issn.1672-2124.2021.04.030摘要同型半胱氨酸是蛋氨酸代谢通路上的中间代谢产物,多年来的研究结果已证明高同型半胱氨酸血症与高血压、脑卒中、抑郁、阿尔茨海默病、糖尿病周围神经病变及慢性肾脏病等疾病的发生有关"尽管叶酸可以有效降低体內同型半胱氨酸水平,但其在治疗高同型半胱氨酸血症相关疾病中可能存在超适应证、超剂量等不合理用药现象"建议临床在应用叶酸时,应采取规范的给药剂量及实施个体化用药"关键词叶酸;高同型半胱氨酸血症;剂量Rational Application of Folic Acid in the Treatment of Diseases Related to Hyperhomocysteinemia NA Yifan,TAN Ling#Dept.of Pharmacy&Beijing Hospital&National Center of Gerontology&Institute of Triatric Medicine,Chinese Academy of MedicaO Scienys,Beijing100730&China)ABSTRACT Homocysteine is an inarmediae metabolite in the metabolic pathway of methionine.Over the yeas, reseerches havv proved that hyperhomocysteinemia is related te the occurrenca of diseases such as hypeOension&stroke& depression&Alzheimer s diseasa,diabetic peripherai neuopathy and chronic kidney disasa.Although folic acid can efectiveiy reduce the I sv I of homocysteine in the body,it may have off-label indications,oyadose and other irrationai phenomena io O ic treatment of diseses related te hyperhomocysteine.It is suggested that standard dosage and individualized administration of folic aciO shoulO be adopted ip clinicct application.KEYWORDS Folic acid;Hyperhomocysteinemia;Dose同型半胱氨酸#homocysteine,Hey)是一种与半胱氨酸同系的四碳含硫氨基酸,是能量代谢和许多需甲基化反应的重要中间产物%当体内叶酸缺乏时,Hey代谢受阻导致血液中存留过多,形成高同型半胱氨酸血症(1)%过高的Hey水平具有“毒性”,其主要危害包括损伤血管内皮细胞、诱发氧化应激反应、改变脂质代谢及血栓形成等,从而导致的疾病也是多方面的(2)。
不同部位脑梗死华勒变性的特点及对神经功能的影响

[8]涂梦恬,李良平.肝细胞核因子受体4α在非酒精性脂肪性肝病中的研究现状[J].国际消化病杂志,2017,37(3):144-147.Tu MT,Li LP.Research status of hepatocyte nuclear factor receptor4αin nonalcoholic fatty liver disease[J].Int J Gastroenterol,2017,37(3):144-147.doi:10.3969/j.issn.1673-534X.2017.03.003.[9]Thayer TE,Lino Cardenas CL,Martyn T,et al.The role of bone morphogenetic protein signaling in non-alcoholic fatty liver disease [J].Sci Rep,2020,10(1):9831.doi:10.1038/s41598-020-66770-8.[10]Cariou B,Byrne CD,Loomba R,et al.NAFLD as a metabolic disease in humans:A literature review[J].Diabetes Obes Metab,2021,23(5):1069-1083.doi:10.1111/dom.14322.[11]Theofilatos D,Anestis A,Hashimoto K,et al.Transcriptional regulation of the human liver X receptorαgene by hepatocyte nuclear factor4α[J].Biochem Biophys Res Commun,2016,469(3):573-579.doi:10.1016/j.bbrc.2015.12.031.[12]Vespasiani-Gentilucci U,Dell'Unto C,De Vincentis A,et al. Combining genetic variants to improve risk prediction for NAFLD and its progression to cirrhosis:a proof of concept study[J].Can J Gastroenterol Hepatol,2018,2018:7564835.doi:10.1155/2018/ 7564835.[13]Khalid YS,Dasu NR,Suga H,et al.Increased cardiovascular events and mortality in females with NAFLD:a meta-analysis[J]. Am J Cardiovasc Dis,2020,10(3):258-271.[14]Trépo E,Valenti L.Update on NAFLD genetics:From new variants to the clinic[J].J Hepatol,2020,72(6):1196-1209.doi:10.1016/ j.jhep.2020.02.020.[15]Matsuo S,Ogawa M,Muckenthaler MU,et al.Hepatocyte nuclear factor4alpha controls iron metabolism and regulates transferrin receptor2in mouse liver[J].J Biol Chem,2015,290(52):30855-30865.doi:10.1074/jbc.M115.694414.[16]Chi X,Wei X,Gao W,et al.Dexmedetomidine ameliorates acute lung injury following orthotopic autologous liver transplantation in rats probably by inhibiting Toll-like receptor4-nuclear factor kappa B signaling[J].J Transl Med,2015,13:190.doi:10.1186/ s12967-015-0554-5.[17]Wang L,Yao M,Zheng W,et al.Insulin-like growth factor I receptor:a novel target for hepatocellular carcinoma gene therapy [J].Mini Rev Med Chem,2019,19(4):272-280.doi:10.2174/ 1389557518666181025151608.(2020-10-23收稿2021-01-17修回)(本文编辑李鹏)不同部位脑梗死华勒变性的特点及对神经功能的影响王冬梅,蔡桂淑,刘立生△摘要:目的探讨不同部位脑梗死华勒变性的特点及对神经功能的影响。
生物专业英语教学案 生物化学与代谢途径

生物专业英语教学案生物化学与代谢途径生物专业英语教学案生物化学与代谢途径I. Introduction:In the field of biology, the study of biochemistry and metabolic pathways is crucial. This lesson aims to provide students with a comprehensive understanding of the fundamental concepts and processes involved in biochemistry and metabolic pathways.II. Objectives:1. To introduce the basic principles and concepts of biochemistry.2. To familiarize students with the major metabolic pathways.3. To develop students' ability to apply biochemistry knowledge in practical scenarios.III. Lesson Plan:A. Warm-up Activity:To engage students and assess their prior knowledge, begin the lesson with a brief interactive activity. Show images related to biochemistry and metabolic pathways, such as proteins, enzymes, and chemical reactions. Ask students to identify and explain what they know about these images.B. Introduction to Biochemistry:1. Define biochemistry and its significance in the field of biology.2. Explain the fundamental components of biochemistry: biomolecules, enzymes, and metabolic pathways.3. Describe the structure and function of biomolecules, including carbohydrates, lipids, proteins, and nucleic acids.4. Discuss the role of enzymes in biochemical reactions.C. Major Metabolic Pathways:1. Glycolysis:- Explain the process of glycolysis, including the conversion of glucose into pyruvate and the production of ATP.- Discuss the importance of glycolysis in energy production and its relationship to anaerobic respiration.2. Krebs Cycle (Citric Acid Cycle):- Introduce the Krebs Cycle as a central metabolic pathway.- Explain the step-by-step process of the Krebs Cycle, highlighting the production of ATP and electron carriers.3. Electron Transport Chain (ETC):- Discuss the role of the electron transport chain in oxidative phosphorylation.- Explain the generation of ATP through the transfer of electrons and the gradient-driven synthesis of ATP.4. Photosynthesis:- Provide an overview of photosynthesis as a metabolic pathway.- Describe the light-dependent and light-independent reactions and their integral role in energy conversion.5. Protein Synthesis:- Explain the process of protein synthesis, including transcription and translation.- Discuss the importance of DNA, RNA, and ribosomes in protein synthesis.D. Application and Practical Scenarios:1. Case Study Analysis:- Present real-life scenarios where knowledge of biochemistry and metabolic pathways is crucial.- Engage students in analyzing and solving the given cases using their understanding of the topics covered.2. Laboratory Experiment:- Conduct a simple laboratory experiment related to biochemistry and metabolic pathways.- Allow students to apply theoretical concepts practically, reinforcing their understanding.E. Conclusion:Summarize the key points discussed throughout the lesson, emphasizing the importance of biochemistry and metabolic pathways in biological processes. Encourage students to explore further in the field and utilize their knowledge in future research or professional careers.IV. Assessment:Assess students' understanding through various evaluation methods:1. Class participation and engagement during discussions.2. Individual or group assignments related to biochemistry and metabolic pathways.3. Case study analysis and problem-solving activities.4. Laboratory experiment performance and reporting.V. Resources:1. Textbooks and reference materials on biochemistry and metabolism.2. Visual aids, such as diagrams and models, to enhance understanding.3. Relevant online resources (without including specific URLs).Note: The format provided above is merely a suggestion. The actual formatting and sectioning of the lesson plan can vary based on personal preferences or institutional guidelines. As the instructor, feel free to modify and adapt the content as necessary to suit your teaching style and the needs of your students.。
生物学通路英语

生物学通路英语The Biological Pathway。
A biological pathway is a series of actions or reactions that occur within a cell or organism to achieve a specific biological outcome. These pathways are crucial for the proper functioning and regulation of various biological processes. In this article, we will explore the concept of biological pathways and their significance in understanding the complexity of life.Biological pathways can be categorized into two main types: metabolic pathways and signaling pathways. Metabolic pathways involve the conversion of molecules into different forms, while signaling pathways involve the transmission of signals within and between cells.Metabolic pathways are responsible for the breakdown of nutrients and the production of energy. One well-known metabolic pathway is glycolysis, which converts glucose into pyruvate and generates ATP, the primary energy currency of cells. Another important metabolic pathway is the citric acid cycle, also known as the Krebs cycle, which generates ATP and other energy-rich molecules through the oxidation of acetyl-CoA.Signaling pathways, on the other hand, play a crucial role in cell communication and coordination. These pathways allow cells to respond to external stimuli and regulate their behavior accordingly. One example of a signaling pathway is the insulin signaling pathway, which regulates glucose uptake and metabolism in response to changes in blood sugar levels. Another well-studied signaling pathway is the MAPK pathway, which controls cell growth, proliferation, and differentiation.Biological pathways are not isolated entities but are interconnected and highly regulated. They often form complex networks that allow for the integration of multiple signals and the coordination of various cellular processes. For example, the crosstalkbetween different signaling pathways ensures a coordinated response to different stimuli and helps maintain cellular homeostasis.Understanding biological pathways is essential for unraveling the mechanisms underlying various diseases and developing effective therapeutic strategies. Many diseases, such as cancer and metabolic disorders, are characterized by dysregulation or malfunctioning of specific pathways. By studying these pathways, researchers can identify potential targets for drug development and design interventions to restore normal pathway function.Advances in technology, such as high-throughput sequencing and omics approaches, have revolutionized the field of pathway analysis. These methods allow researchers to study the expression patterns of thousands of genes or proteins simultaneously, providing a comprehensive view of pathway activity. Additionally, computational tools and databases have been developed to facilitate the analysis and interpretation of pathway data.In conclusion, biological pathways are fundamental to the understanding of complex biological processes. They play a crucial role in metabolic regulation, cell communication, and disease development. By studying these pathways, scientists can gain insights into the intricate mechanisms of life and develop innovative approaches for disease treatment and prevention. The continued exploration of biological pathways promises to uncover new knowledge and contribute to advancements in various fields, including medicine, agriculture, and biotechnology.。
卡尔文循环 糖酵解 都有的步骤

卡尔文循环糖酵解都有的步骤1.糖酵解是细胞中的生化过程,用来分解葡萄糖和其他碳水化合物,并释放能量。
Glycolysis is a biochemical process in cells used tobreak down glucose and other carbohydrates, releasing energy.2.在卡尔文循环中,植物利用光合作用将二氧化碳转化为有机物质。
In the Calvin cycle, plants use photosynthesis to convert carbon dioxide into organic compounds.3.糖酵解包括一系列酶介导的反应,最终产生乳酸或酒精。
Glycolysis involves a series of enzyme-mediated reactions, ultimately producing lactate or alcohol.4.卡尔文循环是光合成的一个重要步骤,能帮助植物固定二氧化碳。
The Calvin cycle is an important step in photosynthesis, helping plants fix carbon dioxide.5.糖酵解在细胞质中进行,不需要氧气参与。
Glycolysis takes place in the cytoplasm of cells and does not require the presence of oxygen.6.卡尔文循环是一个复杂的过程,包括多个酶催化的反应。
The Calvin cycle is a complex process involving multiple enzyme-catalyzed reactions.7.糖酵解是产生乳酸或乙醇的一种途径,同时也能生成一部分ATP。
Glycolysis is a pathway for producing lactate or ethanol, while also generating some ATP.8.卡尔文循环的目的之一是将CO2转化为三碳化合物,为接下来的有机合成提供原料。
有氧代谢与无氧代谢的关系

有氧代谢与无氧代谢的关系英文回答:Aerobic metabolism is a metabolic pathway that requires oxygen to produce energy. In aerobic metabolism, glucose is broken down into pyruvate, which is then converted into acetyl-CoA. Acetyl-CoA is then oxidized in the citric acid cycle, which generates energy in the form of ATP.Anaerobic metabolism is a metabolic pathway that does not require oxygen to produce energy. In anaerobic metabolism, glucose is broken down into pyruvate, which is then converted into lactate. Lactate can then be used to generate energy in the muscles or it can be converted back into glucose in the liver.Aerobic metabolism is more efficient than anaerobic metabolism. In aerobic metabolism, one molecule of glucose can produce up to 36 molecules of ATP. In anaerobic metabolism, one molecule of glucose can only produce 2molecules of ATP.However, anaerobic metabolism is faster than aerobic metabolism. Anaerobic metabolism can produce energy more quickly than aerobic metabolism, which is why it is used during short, intense bursts of activity.Aerobic metabolism and anaerobic metabolism are both important for human health. Aerobic metabolism is used during long, endurance activities, such as running or cycling. Anaerobic metabolism is used during short, intense bursts of activity, such as sprinting or weightlifting.中文回答:有氧代谢是一种需要氧气来产生能量的代谢途径。
食品专业英语部分翻译

P60 第一段:Alcoholic beverages are produced from a range of raw materialsbut especially from cereals, fruits and sugar crops. They include non-distilled beverages such as beers, wines, ciders, and sake. Disitlled beverages such as whisky and rum are produced from fermented cerealsand molasses, respectively, while brandy is produced by distillation of wine. Other distilled beverages, such as vodka and gin, are produced from neutral spirits obtained by distillation of fermented molasses, grain,potato or whey. A variety of fortified wines are produced by addition ofinclude sherries, port and Madeira wines.酒精饮料可由很多原料来生产,但以谷物、水果和糖料作物为主。
酒类饮料包含非蒸馏饮料,如啤酒、葡萄酒、苹果酒和日本米酒。
蒸馏饮料如威士忌和朗姆酒分别由谷物和糖浆发酵制得,而白兰地则是由葡萄酒蒸馏制得。
其余蒸馏饮料,象伏特加和杜松子酒则由发酵糖浆、谷物、马铃薯或乳清所得的白酒经蒸馏制成。
各种各种的加强葡萄酒是在葡萄酒中增添蒸馏的白酒使酒精含量提升到15 -20 %制成。
有名的产品有雪梨白葡萄酒、波尔多红葡萄酒和马德拉烈性甜酒。
信号通路12—Metabolism
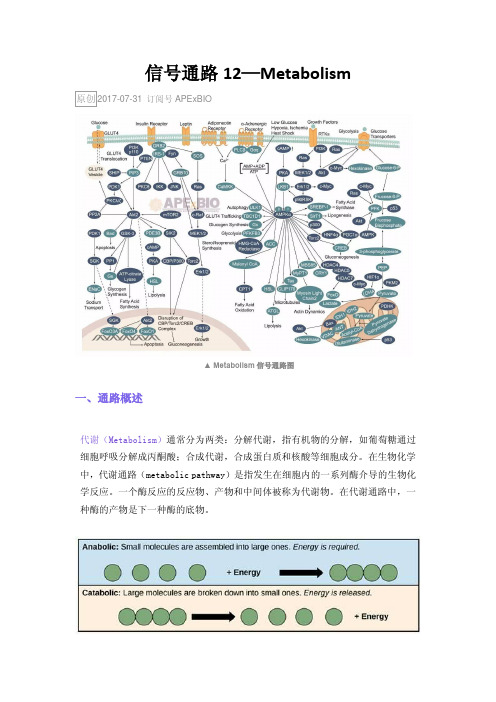
信号通路12—Metabolism订阅号APExBIO▲ Metabolism信号通路图一、通路概述代谢(Metabolism)通常分为两类:分解代谢,指有机物的分解,如葡萄糖通过细胞呼吸分解成丙酮酸;合成代谢,合成蛋白质和核酸等细胞成分。
在生物化学中,代谢通路(metabolic pathway)是指发生在细胞内的一系列酶介导的生物化学反应。
一个酶反应的反应物、产物和中间体被称为代谢物。
在代谢通路中,一种酶的产物是下一种酶的底物。
葡萄糖(glucose)是大多数细胞的主要能量来源。
进食期间升高的血糖通过葡萄糖感测通路刺激胰腺β细胞释放胰岛素(insulin)。
胰岛素通过由胰岛素受体(IR)介导的信号级联刺激血液中的葡萄糖摄取到骨骼肌和脂肪组织中。
与IR 结合的胰岛素激活胰岛素受体底物(IRS)蛋白,随后将信号传递给PI3K / Akt 和Erk1 / 2通路,导致葡萄糖转运蛋白GLUT4定位到细胞表面,增加葡萄糖摄取,细胞增殖和存活。
异常胰岛素信号传导可导致糖尿病和肥胖,也可能导致动脉粥样硬化甚至神经变性疾病。
在剥夺葡萄糖的条件下,细胞ATP水平下降时,丝氨酸/苏氨酸激酶AMPK变得活跃。
细胞和环境压力(如低葡萄糖,热休克,缺氧和局部缺血)会使AMP / ATP 比例升高并激活AMPK。
AMPK激活正调控补充细胞ATP供应的信号通路。
例如,AMPK的激活增强Glut4的转录和易位,导致胰岛素刺激的葡萄糖摄取增加。
此外,它还通过抑制ACC和激活PFK2来刺激分解代谢过程,如脂肪酸氧化和糖酵解。
AMPK 负调控ATP消耗过程的几种蛋白质如mTORC2,糖原合成酶,SREBP-1和TSC2,导致糖异生和糖原、脂质和蛋白质合成的下调或抑制。
HIF1(缺氧诱导因子1)信号通路也在调节细胞葡萄糖代谢中发挥核心作用。
HIF-1信号对低氧和生长因子作出反应来调节葡萄糖代谢。
HIF1是一种由HIF-1α和HIF-1β亚基组成的转录因子。
Metabolism Overview@生物化学精品讲义

♫Anabolic and catabolic pathways must differ in at least one step in order to be regulated
Degradation: biomolecules – building blocks – common intermediates final products
☻Nucleus (DNA replication, transcription, RNA processing) ☻ER (Rough ER: synthesis of membrane and secretory
proteins, smooth ER: lipid and steroid biosynthesis) ☻Golgi (posttranslational processing of proteins)
Anabolism: biosynthetic pathways
- energy-requiring!
Metabolic pathway
A cascade of reactions leading to the synthesis or degradation of a compound.
♪ Anabolic pathways ♪ Catabolic pathways ♪ Amphibolic pathway (无定向代谢途径) Features
万古霉素代谢

万古霉素代谢Erythromycin is a type of macrolide antibiotic that has been used for decades to treat a wide range of bacterial infections. Its broad spectrum of activity and low toxicity have made it a popular choice for healthcare providers. The mode of action of erythromycin involves inhibiting bacterial protein synthesis, which ultimately leads to bacterial cell death. The antibiotic is produced by the soil-dwelling bacterium Saccharopolyspora erythraea through a complex biosynthetic pathway. Understanding the metabolism of erythromycin is important for improving its production in industrial settings and for developing new derivatives with improved clinical efficacy. In this review, we will explore the metabolic pathway of erythromycin and the factors that influence its production.Biosynthesis of ErythromycinThe biosynthesis of erythromycin is a complex process that involves multiple enzymatic steps. The first step in the biosynthetic pathway is the condensation of malonyl-CoA and methylmalonyl-CoA to form a polyketide core, which serves as the precursor molecule for the synthesis of erythromycin. This reaction is catalyzed by the polyketide synthase (PKS) enzyme complex, which is composed of multiple subunits that work together to build the polyketide chain. The PKS enzymes are encoded by a cluster of genes known as the ery gene cluster, which is located on the chromosome of S. erythraea.Following the formation of the polyketide core, a series of enzymatic reactions are required to modify the structure of the polyketide chain to yield the final erythromycin molecule. These reactions include methylation, glycosylation, and oxidation steps, which are catalyzed by specific enzymes encoded by genes within the ery cluster. The final steps of the biosynthetic pathway involve the attachment of a macrolide ring and the formation of the deoxysugar moieties that are characteristic of erythromycin. These processes are mediated by a set of tailoring enzymes that further modify the structure of the polyketide core.Regulation of Erythromycin BiosynthesisThe production of erythromycin by S. erythraea is tightly regulated at the transcriptional and translational levels. Several regulatory proteins have been identified that control the expression of genes within the ery cluster in response to environmental cues and metabolic signals. One of the key regulators of erythromycin biosynthesis is the pathway-specific activator protein, which binds to specific DNA sequences within the ery gene cluster and stimulates the transcription of biosynthetic genes. In addition to activator proteins, global regulatory proteins that influence the overall metabolic state of the cell can also impact erythromycin production. For example, the availability of nutrients and the metabolic flux through central carbon metabolism are known to influence the synthesis of erythromycin. Improving Erythromycin ProductionEfforts to improve the production of erythromycin have focused on several strategies, including strain engineering, fermentation optimization, and bioprocess control. Strainengineering involves the manipulation of S. erythraea to enhance its ability to produce erythromycin. This can be achieved by overexpressing key biosynthetic genes, deleting negative regulators of the ery cluster, or introducing genes from related bacterial species to expand the metabolic capabilities of the host strain. Fermentation optimization aims to create an ideal growth environment for S. erythraea, including the selection of appropriate media, the optimization of fermentation conditions, and the addition of specific nutrients or inducers that stimulate erythromycin production. Bioprocess control involves monitoring and controlling the fermentation process to ensure optimal erythromycin yields, such as by regulating the pH, oxygen levels, and temperature within the fermentation vessel.Future PerspectivesThe metabolic pathway of erythromycin continues to be a topic of active research, as scientists seek to further elucidate the biosynthetic steps and regulatory mechanisms that underlie its production. In addition, efforts to engineer S. erythraea for improved erythromycin production are ongoing, with the goal of developing high-yielding strains that can be used in industrial settings. Furthermore, the development of new erythromycin derivatives with enhanced pharmacological properties, such as improved antimicrobial activity or reduced side effects, remains an important area of investigation. By gaining a deeper understanding of the metabolism of erythromycin and its biosynthetic pathway, researchers hope to advance the development and production of this important antibiotic for the treatment of bacterial infections.ConclusionIn conclusion, erythromycin is a valuable antibiotic that is produced by the bacterium S. erythraea through a complex biosynthetic pathway. Understanding the metabolic processes that govern erythromycin production is essential for improving its yield in industrial settings and for developing new derivatives with enhanced clinical efficacy. The regulatory mechanisms that control erythromycin biosynthesis and the strategies for improving its production are areas of active research. By advancing our knowledge of the metabolic pathway of erythromycin, researchers aim to enhance our ability to produce and utilize this important antibiotic for the treatment of bacterial infections.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
The Path-A metabolic pathway prediction web serverLuca Pireddu,Duane Szafron*,Paul Lu and Russell GreinerDepartment of Computing Science,University of Alberta,Edmonton,AB,Canada T6G 2E8Received February 14,2006;Revised March 6,2006;Accepted March 27,2006ABSTRACTPathway Analyst (Path-A)is a publicly available web server (http://path-a.cs.ualberta.ca)that predicts metabolic pathways.It takes a FASTA format file con-taining a set of query protein sequences from a single organism (a partial or complete proteome)and identifies those sequences that are likely to particip-ate in any of its supported metabolic pathways (currently 10).Path-A uses a number of machine-learning and sequence analysis techniques (e.g.SVM,BLAST and HMM)to predict pathways.Each machine-learned classifier exploits similarity between sequences in the pathways of its model organisms and sequences in the query set.It predicts the pathways that are present in the query organism and annotates each predicted reaction and catalyst,using the appropriate sequences from the query set.Path-A also provides a browsable and searchable database of the pathways for the model organisms that are used to make its predictions.Path-A’s pre-dictor sets (using different classifier technologies)have been evaluated using standard cross-validation techniques on a dataset of 10metabolic pathways across 13model organisms—a total of 125organism-specific pathways.The most accurate classifier tech-nology obtained a mean precision of 78.3%and a mean recall of 92.6%in predicting all catalyst proteins,of all reactions,in all pathways present in the dataset.Although Path-A currently only supports metabolic pathways,the underlying prediction techniques are general enough for other types of pathways.Consequently,it is our intent to extend Path-A to predict other types of pathways,including signalling pathways.INTRODUCTIONEach biochemical pathway describes an identifiable subset of the complex system of reactions that exist in living organisms.Understanding these pathways is essential to understanding the machinery of life.The existence of a large quantity ofproteomic sequence data,and the hard work of experimental molecular biologists,has resulted in several on-line pathway databases:KEGG PATHWAY (1),BioCarta (),aMAZE (2)and BioCyc (3).In addition,tools such as Pathway Tools (4)have appeared,providing some data-mining capabilities that try to correlate protein annotations to pathway templates so that organism-specific pathways can be derived.However,we are unaware of any freely available tools that perform protein analyses at the primary sequence level,without requiring annotations,to pre-dict organism-specific pathway variants,or pathway instances.Pathway Analyst (Path-A)is a web server that takes a set of protein sequences from a single organism as input.First,Path-A predicts which of these proteins catalyse each reaction in a set of selected metabolic pathways.Second,it assembles the predicted catalysts and reactions into an organism-specific variant of each metabolic pathway under consideration.In addition,Path-A makes it easy for users to browse and search known metabolic pathways in model organisms.Path-A is a freely available service at http://path-a.cs.ualberta.ca.PREDICTION TECHNIQUEPath-A uses a set of machine-learning techniques to predict pathway instances (5).The basic prediction algorithm is based on the realization that instances of the same pathway in dif-ferent organisms will share some degree of similarity,in both the reactions they encompass and the primary sequence struc-ture of the proteins that catalyse them.Path-A exploits this fact to predict the reactions and catalysts of pathways unknown to the system based on patterns learned from pathway instances that are already known to the system.The basic algorithm has two inputs:a set of protein sequences from the query organism and a set of model pathways,one for each target pathway.For each model pathway,the algorithm iterates through each reac-tion.For each reaction,the algorithm tries to determine which proteins from the query set are functionally compatible to any catalyst protein of the model reaction.Functional compatib-ility is predicted using one of several sequence-based meas-ures described later.If at least one protein from the query set is deemed functionally compatible to a catalyst protein for the given model reaction,the reaction is predicted to exist in the pathway for the query organism.The catalysts of the reaction*To whom correspondence should be addressed.Tel:+17804925468;Fax:+17804921071;Email:duane@cs.ualberta.ca ÓThe Author 2006.Published by Oxford University Press.All rights reserved.The online version of this article has been published under an open access ers are entitled to use,reproduce,disseminate,or display the open access version of this article for non-commercial purposes provided that:the original authorship is properly and fully attributed;the Journal and Oxford University Press are attributed as the original place of publication with the correct citation details given;if an article is subsequently reproduced or disseminated not in its entirety but only in part or as a derivative work this must be clearly indicated.For commercial re-use,please contact journals.permissions@W714–W719Nucleic Acids Research,2006,Vol.34,Web Server issue doi:10.1093/nar/gkl228in the query organism are predicted to be the proteins from the query set that were predicted to be compatible to the catalyst sequences from the model reaction.If no functionally com-patible protein is found for any catalyst of that model reaction, the reaction is predicted not to exist in the pathway of the query organism.Model pathwaysPath-A’s prediction algorithm requires a pathway model for each metabolic pathway that it supports.Typically,a single pathway instance is not a good pathway model for two reasons. First,the reactions that compose each pathway—the pathway’s structure—vary between ing only one training pathway increases the chance that the training pathway has a different structure than the target pathway that Path-A is trying to predict.If the target pathway has a different structure,predicting its true structure is impossible since the algorithm does not attempt a prediction on any reaction not found in the model pathway.Second,predictors trained using only a few positive training instances typically have poor ing more than one pathway instance to construct a model pathway increases the number of catalysts for reac-tions in the model pathway.This increase in the number of positive training instances(catalyst sequences)produces more accurate predictors.Therefore,we create a single abstract model pathway as the union of all available organism-specific instances of that pathway.The union,U,of two pathways,A and B,is a new pathway whose structure includes all the reactions occurring in either A or B.For each reaction in pathway U,if that reaction existed in both A and B,then the reaction’s protein catalyst set in U is the union of the catalyst sets from the same reaction in A and B.By using model pathways,the pathway prediction algorithm can even predict instances of a pathway with variations in structure that were never observed in the training pathway set—and perhaps never found in any physical laboratory. Such emergent structures can be computationally predicted before being observed.A Path-A user can examine each model pathway in the system.The organism-specific pathway instances that were merged to create each model can be viewed,along with each reaction in the model pathway and the pathway instances. The user can drill down to examine the catalyst proteins of each reaction,either as they appear in the abstract model pathway,or in each individual organism-specific reaction that comprises the model reaction.Path-A currently provides abstract models for10metabolic pathways,spanning 125organism-specific pathway instances.Each model reaction is annotated by information about which classifier is used to predict functional compatibility between its catalyst sequences and catalyst sequences in query organisms.As it is typically advantageous to use more training data,the model pathways in Path-A are built using all available instances of the pathway, though this is not required for the correct operation of the algorithm.In addition,since Path-A requires a model of a pathway to make a prediction,it is currently only able to make predictions on these10available metabolic pathways. We plan to support more metabolic pathways and modify existing model pathways by augmenting them with reactions and catalysts from additional organism-specific instances of these pathways.ClassifiersA machine-learned classifier is a component of Path-A’s prediction system that predicts whether a protein from a query protein set is a catalyst of a particular reaction.A single classifier is created for each reaction in a model pathway,and trained to recognize functionally compatible proteins associ-ated with this reaction.Training the classifier requires both positive and negative training sequences.We use sequences that are known to catalyse the reaction in any of the organism-specific pathways comprising the model as positive training sequences,and the rest of the sequences in those organisms as negative training sequences.To make a prediction,the trained classifier decides whether a query protein sequence is func-tionally closer to the positive or the negative class of proteins. We have evaluated several classifiers as predictors of which proteins are functionally compatible(5).A key research result, established using Path-A,is that no single classifier is best for all pathway reactions.Some are based on sequence alignment techniques such as BLAST(6).Others use machine-learning techniques such as profile hidden Markov models(HMMs)(7) and support vector machines(SVMs)(8).Some classifiers use combinations of these simple classifiplete descrip-tions of these classifiers are not germane to this paper and can be found elsewhere(5).Table1shows a sampling of accuracy measures of six different classifiers we tested. Each entry shows the mean and standard deviation of each accuracy measure for each kind of classifier computed in n-fold cross-validation tests.The tests required the prediction of125pathway instances with a total of1759reactions.There is no reason why the same classifier must be used for each reaction in a pathway.For example,the test results in the top two rows of Table1use classifiers whose parameters were tuned differently for each reaction.The current production version(v1.1)of Path-A allows the user to select a BLAST-based,HMM-based,or combination BLAST–HMM-based classifier for each path-way.This classifier choice and its parameter values are thenfixed across all reactions of that pathway.However, we are in the process of replacing these generic classifiers with reaction-specific classifiers to provide best prediction performance.We migrate our best classifiers from the experi-mental versions to the production version of Path-A as we discover them,so the user need not be concerned with the intricacies of classifier and parameter selection.Table1.Different classifiers in Path-A:mean catalyst prediction scores for each classifier type(standard deviation given in parentheses)Classifier F-measure Precision RecallOpt BLAST0.837(0.130)0.783(0.170)0.926(0.114) Opt HMM0.795(0.141)0.777(0.184)0.848(0.138) BLAST–HMM0.673(0.152)0.630(0.197)0.784(0.176) BLAST0.667(0.155)0.609(0.205)0.802(0.170) Motif SVM0.659(0.155)0.666(0.190)0.692(0.187) HMM0.654(0.164)0.704(0.190)0.671(0.221)Nucleic Acids Research,2006,Vol.34,Web Server issue W715Accessing pathway analystPath-A can be accessed at http://path-a.cs.ualberta.ca.A user can register to obtain a personal account,or login using a Guest account with no registration required.Registering on the Path-A system is free and registered users are provided with a personal space where they can store protein sets and predicted pathways.In addition,Path-A will notify registered users by email when a prediction task is complete (if desired).The Guest account has access to all services available on a regular personal account,except that data in the Guest account are only kept for five days and no personalized features,such as email notification,are available.After logging into the Path-A system,the user can select from the six services shown in the Path-A ‘Control centre’(Figure 1).The ‘Account details’service allows a registered user to change user name,password and email contact information.The ‘New analysis’and ‘View your analyses’services are used to create and view one or more analyses,and the ‘View protein sets’,‘Pathway instances’and ‘Model pathways’services allow the user to browse and search the database.Clicking the appropriate link starts the correspond-ing service.NEW ANALYSISThe user creates a new pathway analysis of a partial or com-plete proteome from a specific organism in three steps:(1)start by entering an analysis name and optional description,(2)select a protein set and (3)select pathways of interest.Step 1:Start The user starts by providing a name for the new analysis and an optional description as shown in Figure 2.The road-map at the bottom of the page indicates the current step in the context of the analysis process.Path-A uses protein sets ,where all the proteins of the set must come from the same organism.The user presses the ‘Click here to select or upload a protein set’button shown in Figure 2to begin the process of selecting a protein set.Step 2:Proteins The user may upload a new protein set,use a protein set that the user previously uploaded or use 1of the 15complete proteomes provided in the Path-A database.Figure 3shows the page used to select a protein set or upload a new one.The protein sets uploaded previously by the user are shown first,followed by the complete proteomes.Some basic information about the protein sets (name,source organism,size,description and owner)are shown for each set.For example,Figure 3shows a user uploaded protein set named Ecoli proteome (partial)on the first line of the table.It is marked with a lock to indicate that it is part of the user’s private data and that it is invisible to other Path-A users.To select a protein set,the user clicks on the corresponding selection arrow.To upload a new protein set,the user clicks on the ‘Upload new protein set’link.If the user opts to upload a new protein set,the ‘Upload a new protein set’page shown in Figure 4appears.The user enters a name for the new protein set and (optionally)a brief description that provides useful identification if the protein set is re-used for another analysis in the future.The user then provides the name of the source organism of the protein set for display purposes.This is performed by clicking on the button ‘Click here to select an organism’.A prepared list of almost 81000organisms is used.The user can quickly search through the list by typing part of the orga-nism’s name into the search field at the top of the page,as shown in Figure ing dynamic AJAX technology,the list’s contents are automatically updated to show only the matching organisms.The user clicks on the organism name to select it.The user is then asked to choose the organism’s strain from a short list (not shown).If the desired organism or its strain name is not included in the Path-A database,a new organismnameFigure 1.Path-A services in the Controlcentre.Figure 2.New analysis:Step1—Start.Figure 3.New analysis:Step 2—Proteins.W716Nucleic Acids Research,2006,Vol.34,Web Server issuecan be added by clicking on the ‘Create a new one’link at the top of the ‘Select an organism’page shown in Figure 5.After selecting the organism,the user is returned to the ‘Upload a new protein set’page of Figure 4,except that the ‘Organism:’line has its ‘Click here to select an organism’button replaced by the text,Schizosaccharomyces pombe,common (Fission yeast),and a different button labelled ‘Select a different organism’.The user can then select a local file containing the protein set by clicking on the ‘Choose File’button,and browsing to the desired local file (not shown).At this point the ‘no file selected’text in Figure 4is replaced by the file name.The user clicks on the ‘Upload’button to upload the protein set.The progress bar shown in Figure 6informs the user of the status of the file upload.After the newly uploaded protein set has been stored in the Path-A database,the user can re-use it for future analyses or browsed without the need to upload it again.At this point,Path-A returns to the ‘New analysis’page at Step 2as shown in Figure 7.The user can click on the ‘Next’button to proceed to Step 3.Step 3:Pathways The Path-A algorithm requires a model for every predicted pathway.Path-A currently provides mod-els for the 10metabolic pathways shown in Figure 8.The user can choose to predict any number of these pathways for the query protein set.Selecting a pathway results in the retrieval of the pathway’s model,classifiers and prediction parameters.The retrieved data are then used to perform the prediction.The ‘More details’button can optionally be pressed to select a non-default predictor (currently BLAST,HMM,BLAST-HMM or Opt-HMM instead of Opt-Blast)for each pathway.After selecting the desired pathways,the user clicks on the ‘Start analysis’button.After going through the three steps of the ‘New analysis’process,Path-A sends the analysis request to its computationnodes.The time required to perform an analysis depends on the number of pathways to be predicted,the type of classifier being used and the amount of activity on the system.Time to completion can range from minutes to a day.Given the unpre-dictability in the computation time,users may choose to have Path-A notify them by email when their analyses complete and become ready for viewing.Notification can be set/unset using the ‘Account details’service from the ‘Control centre’shown in Figure1.Figure 4.New analysis:Step 2—Proteins:upload a new protein setpage.Figure 5.New analysis:Step 2—Proteins:select an organismpage.Figure 6.New analysis:Step 2—Upload a new protein setrevisited.Figure 7.Protein setuploaded.Figure 8.New analysis:Step 3—Which pathways page.Nucleic Acids Research,2006,Vol.34,Web Server issue W717ANALYSIS VIEWINGA user can view the results of analyses by clicking on the ‘View your analyses’button in the Path-A control centre shown in Figure 1.This can be a new analysis that has just completed or a previously computed analysis.The ‘Your Ana-lyses’page (not shown)has two lists.One list contains the analyses that are waiting to be computed or are currently being computed.The second list contains the analyses that are com-plete.Clicking on the name of a completed analysis shows its details.For example,Figure 9shows the details of an analysis of a partial Yeast proteome.Each pathway prediction is an independent task,shown as a separate line on the page.Each line contains the name of the pathway,a link to the model used for the predictions and some information about the computation,including its status(done,computing,pendingor error).The user can click on the ‘Details link’to see more information about how a pathway prediction was carried out.The ‘Prediction details’page (not shown)explains which predictor type and parameters were used to predict each of the model pathway’s reactions,whether the reaction is predicted to exist in the query organism,based on the query protein set.Each predicted reaction also has a link to information about that reaction including a list of proteins from the query protein set that are predicted to catalyse that reaction.The user can also click on a ‘Predicted’link (e.g.148for Propanoate metabolism in Figure 9)to learn more about the predicted pathway.A dash (–)in the ‘Predicted’column indic-ates that the pathway does not exist (no reactions in that pathway exist)for the query organism,based on the protein set that was provided.For example,Figure 10shows a pre-dicted pathway in the query organism (Yeast)that is restricted to proteins from the query set.The top section of the page includes some information about the analysis that generated this pathway:the account name,the name of the analysis,the model pathway used,the organism,the number of reactions predicted and the number of catalysts found in the query set for these reactions.The following section lists each predictedreaction,along with the number of proteins predicted to cata-lyse it,as well as the predictor that was responsible for the prediction.Clicking on the reaction name (not shown)shows its details,including all the proteins predicted to be its catalysts.SEARCHING AND BROWSINGPath-A provides a text-based search engine to help locate pathways of ers are able to search through both user-provided and Path-A-provided pathway instances and model pathways.The search mechanism accepts simple text queries,as well as Boolean operators,on names and descrip-tions of pathways,organisms,reactions,molecules,genes and identifiers from external sites (e.g.UniProt,KEGG,TAIR and FlyBase).Path-A also provides browsing features to easily organize and navigate the information in different ers can quickly search a list of organisms,and then see which path-ways and protein sets associated with it exist in the database.Further,given a protein one can easily see the pathway instances that are associated with it.CONCLUSIONWe have presented Path-A,a web server for the prediction of metabolic pathways.Path-A predicts the pathways supported by arbitrary sets of proteins,using validated prediction tech-niques based on sequence alignment and machine learning.In our tests,our most accurate predictor achieved a mean preci-sion and recall of 78.3and 92.6%,respectively.Path-A can also be used as a pathway database,complete with browsing and searching functionality.Path-A is freely available for use at http://path-a.cs.ualberta.ca.ACKNOWLEDGEMENTSWe thank Jordan Patterson and Stephen Walsh for their con-tributions to the Pathway Analyst web server implementation,and the entire Proteome Analyst group at the University of Alberta for the continual discussions,advice and ideas.This work is partially funded by research or equipment grantsfromFigure 9.Viewing ananalysis.Figure 10.A predicted pathway instance.W718Nucleic Acids Research,2006,Vol.34,Web Server issuethe Canadian Protein Engineering Network of Centres of Excellence(PENCE),the Natural Sciences and Engineering Research Council of Canada(NSERC),the Informatics Circle of Research Excellence(iCORE),the Alberta Ingenuity Centre for Machine Learning(AICML),Sun Microsystems,Silicon Graphics,Inc.and the Alberta Science and Research Authority (ASRA).Funding to pay the Open Access publication charges for this article was provided by Alberta Ingenuity Centre for Machine Learning(AICML).Conflict of interest statement.None declared.REFERENCES1.Kanehisa,M.,Goto,S.,Kawashima,S.,Okuno,Y.and Hattori,M.(2004)The KEGG resource for deciphering the genome.Nucleic Acids Res.,32, D277–D280.2.Christian,L.,Antezana,E.,Couche,F.,Fays,F.,Santolaria,X.,Janky,R.,Deville,Y.,Richelle,J.and Wodak,S.J.(2004)The aMAZE lightbench:aweb interface to a relational database of cellular processes.Nucleic Acids Res.,32,D443–D448.3.Karp,P.D.,Ouzounis,C.A.,Moore-Kochlacs,C.,Goldovsky,L.,Kaipa,P.,Ahren,D.,Tsoka,S.,Darzentas,N.,Kunin,V.and Lopez-Bigas,N.(2005) Expansion of the BioCyc collection of pathway/genome databases to160 genomes.Nucleic Acids Res.,19,6083–6089.4.Karp,P.,Paley,S.and Romero,P.(2002)The pathway tools software.Bioinformatics,18,S225–S232.5.Pireddu,L.,Poulin,B.,Szafron,D.,Lu,P.and Wishart,D.S.(2005)Pathway analyst-automated metabolic pathway prediction.InProceedings of the IEEE2005Symposium on Computational Intelligence in Bioinformatics and Computational Biology.IEEE Press,La Jolla California,USA,pp.243–250.6.Altschul,S.F.,Madden,T.L.,Schaffer,A.A.,Zhang,J.,Zhang,Z.,Miller,W.and Lipman,D.J.(1997)Gapped BLAST and PSI-BLAST:a new generation of protein database search programs.Nucleic Acids Res., 25,3389–3402.7.Eddy,S.R.(1998)Profile hidden Markov models.Bioinformatics,14,755–763.8.Hastie,T.,Tibshirani,R.and Friedman,J.(2001)The Elements ofStatistical Learning:Data Mining,Inference,and Prediction.Springer Series in Statistics.Springer,NY.Nucleic Acids Research,2006,Vol.34,Web Server issue W719。