Bay_41-4109_less_active_enantiomer_LCMS_24381_MedChemExpress
质粒图谱查询方法

3.google scholar: / 有些质粒是经过改造的,所以通过上述方法不能查询到相应信息。这时,可以在google scholar中输入质粒名称,可以直观地看哪些学者在何文章中使用了该质粒,从而可了解到质粒的来源;或者籍此向作者咨询或索取。 4.尝试从各大生物公司,例如invitrogen网站查询. 5. 这个网站收录了大量图谱: http://www.embl-hamburg.de/~geerlof/webPP/vectordb/bact_vectors/table.html
Pe
te
rX u
file:///D|/中科院/Selective Serotonin Transporter/质粒信息/质粒图谱查询方法.txt
file:///D|/中科院/Selective Serotonin Transporter/质粒信息/质粒图谱查询方法.txt(第 2/6 页)[2011/8/4 18:39:52]
By
0099--pGE-1—Stratagene--RNAi载体 0100--pSUPER.p53—OligoEngine--RNAi载体 0101--palter-ex1--promega 0102--pACYCDuet-1--NOVAGEN 0103--pEX lox(+) Vector—NOVAGEN--原核表达 0104--质粒名称:pBACgus-8 Transfer Plasmid—NOVAGEN--CHUANSUO 0105--pSCREEN?-1b(+) Vector Map—novagen--筛选 0106--PGEX-2T--BD Co--pDsRed2--Clontech 0107--pbgal-Basic—Clontech--mammalian reporter vector 0108—pBI—Clontech--express two genes of interest from a bidirectional tet-responsive promoter 0109--质粒名称:pbgal-Control—Clontech--mammalian reporter vector 0110-- pGEX-5X-1--原核表达 0111--pBI-EGFP—Clontech--pBI-EGFP-- coexpress 0112--pBI-G—Clontech--pBI-G--express b-galactosidase 0113--pBI-GL—Clontech--pBI-GL --express luciferase and b-galactosidase 0114--pCMS-EGFP—Clontech--mammalian expression vector 0115--pd2EYFP-1—Clontech--启动子测定 0116--质粒名称--pd2EYFP-N1—Clontech--融合表达 0117--pd4EGFP-Bid—Clontech--融合表达 Bid 0118--pDNR-CMV—Clontech--pDNR-CMV 0119--pDNR-EGFP Vector—Clontech 0120--pDNR-LacZ –Clontech 0121--pECFP-Endo—Clontech--真核表达0122--pECFP-ER—Clontech--真核表达0123--pEGFP-Actin—Clontech--真核表达0124--pGAD GH--Clontech--酵母表达 0125--pGADT7-Rec –Clontech--酵母表达 0126--pGADT7-RecAB—Clontech--酵母表达 0127--pGADT7-Rec2—Clontech--酵母表达 0128--pGBKT7—Clontech--酵母表达 0129--pHAT 10/11/12—Clontech 0130--pHAT20—Clontech 0131—pHygEGFP—Clontech 0132—pLacZi—Clontech 0133—pM—Clontech--pM is used to generate a fusion of the GAL4 DNA-BD 0134--pPKCa-EGFP—Clontech 0135--pPKCb-EGFP—Clontec 0136--pSIREN-DNR Vector—Clontech--RNAi 0137--pSIREN-DNR-DsRed-Express Vector—Clontech--RNAi 0138--pSIREN-RetroQ—Clontech--RNAi 0139--pIRES-EYFP—Clontech--RNAi 0140--pSRE-Luc—Clontech--RNAi 0141--pTK-neo—novagen--原核表达 0142--pZsGreen Vector—Clontech--pZsGreen is a pUC19-derived prokaryotic expression vector 0143--pTandem-1—novagen--原核表达 0144--pZsGreen1-C1Vector—Clontech----真核表达 0145--质粒名称:M13mp18—novagen--原核表达 0146--pZsGreen1-DR Vector—Clontech--真核表达 0147--PZsGreen1-N1 Vector—Clontech --真核表达 0148--T7Select415-1b—novagen----真核表达 0149--pZsYellow Vector—Clontech --真核表达 0150—pTimer—Clontech --真核表达 0151--pTA-Luc—Clontech --真核表达 0152--pTAL-Luc—Clontech --真核表达 0153--pTA-SEAP—Clontech --真核表达 0154--pTAL-SEAP—Clontech --真核表达 0155--pTet-On—Clontech --真核表达 0156--pTet-Off—Clontech --真核表达 0157--pTet-ATF—Clontech --真核表达 0158--pTet-CREB—Clontech --真核表达
PREDICTION OF RESPONSIVENESS TO ANTI-ESTROGEN TRE

专利名称:PREDICTION OF RESPONSIVENESS TO ANTI-ESTROGEN TREATMENT IN BREAST CANCER 发明人:UMAR, Arzu,LUIDER, Theo, Marten,FOEKENS, John, A.申请号:NL2007000112申请日:20070427公开号:WO08/133493P1公开日:20081106专利内容由知识产权出版社提供摘要:The invention relates to a method for predicting the responsiveness to anti-estrogen treatment in breast cancer patients by assaying for one or more of the following proteins, which have found to be differentially expressed in responders and non-responders: - nascent polypeptide associated complex alpha subunit (NACA, HSD48) (EMBL Q13765); - splice isoform 1 of epsin 4 (CLINT1, EPN4) (EMBL Q14677-1); -adenylate kinase isoenzyme 4, mitochondrial (AK3L1, AK3, AK4) (EMBL P27144); - annexin A8 (ANXA8, ANX8) (EMBL P13928); - coronin-1B (CORO1B) (EMBL Q9BR76); - EPH receptor B2 (EPHB2, ERK, EPTH3) (EMBL P29323-3); - NADH-cytochrome B5 reductase (CYB5R3, DIA1) (EMBL P00387-1); and - splice isoform 2 of basigin precursor (BSG, EMMPRIN, TCSF, CD147) (EMBL P35613-2.申请人:UMAR, Arzu,LUIDER, Theo, Marten,FOEKENS, John, A.地址:Dr. Molewaterplein 50 NL-3015 GE Rotterdam NL,c/o Erasmus University Medical Center Rotterdam Dr. Molewaterplein 50 NL-3015 GE Rotterdam NL,c/o Erasmus University Medical Center Rotterdam Dr. Molewaterplein 50 NL-3015 GE Rotterdam NL,c/o Erasmus University Medical Center Rotterdam Dr. Molewaterplein 50 NL-3015 GERotterdam NL国籍:NL,NL,NL,NL代理机构:VAN LOON, C.J.J.更多信息请下载全文后查看。
Elabscience Annexin V-PE 7-AAD Apoptosis Kit E-CK-
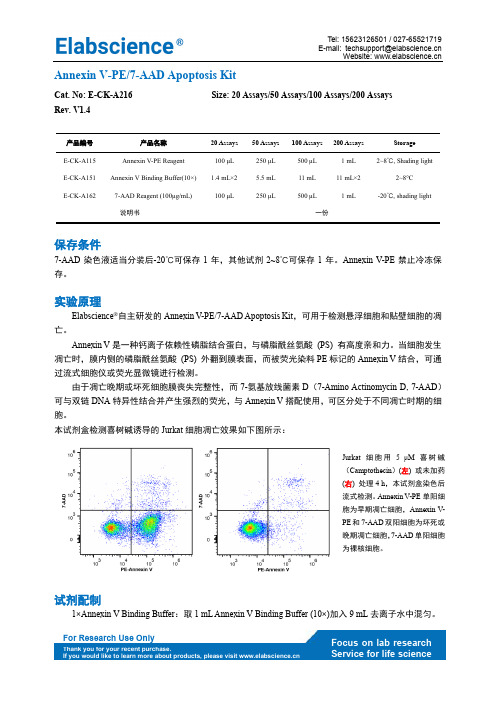
E-mail: Website: Annexin V-PE/7-AAD Apoptosis KitCat. No: E-CK-A216 Size: 20 Assays/50 Assays/100 Assays/200 Assays Rev. V1.4产品编号 产品名称 20 Assays 50 Assays 100 Assays 200 Assays Storage E-CK-A115 Annexin V-PE Reagent 100 μL 250 μL 500 μL 1 mL 2~8℃, Shading lightE-CK-A151 Annexin V Binding Buffer(10×) 1.4 mL×2 5.5 mL 11 mL 11 mL×2 2~8°C E-CK-A1627-AAD Reagent (100μg/mL) 100 μL250 μL500 μL1 mL-20℃, shading light说明书一份保存条件7-AAD 染色液适当分装后-20℃可保存1年,其他试剂2~8℃可保存1年。
Annexin V-PE 禁止冷冻保存。
实验原理Elabscience ®自主研发的Annexin V-PE/7-AAD Apoptosis Kit ,可用于检测悬浮细胞和贴壁细胞的凋亡。
Annexin V 是一种钙离子依赖性磷脂结合蛋白,与磷脂酰丝氨酸 (PS) 有高度亲和力。
当细胞发生凋亡时,膜内侧的磷脂酰丝氨酸 (PS) 外翻到膜表面,而被荧光染料PE 标记的Annexin V 结合,可通过流式细胞仪或荧光显微镜进行检测。
由于凋亡晚期或坏死细胞膜丧失完整性,而7-氨基放线菌素D (7-Amino Actinomycin D, 7-AAD )可与双链DNA 特异性结合并产生强烈的荧光,与Annexin V 搭配使用,可区分处于不同凋亡时期的细胞。
Adaptive tracking control of uncertain MIMO nonlinear systems with input constraints

article
info
abstract
In this paper, adaptive tracking control is proposed for a class of uncertain multi-input and multi-output nonlinear systems with non-symmetric input constraints. The auxiliary design system is introduced to analyze the effect of input constraints, and its states are used to adaptive tracking control design. The spectral radius of the control coefficient matrix is used to relax the nonsingular assumption of the control coefficient matrix. Subsequently, the constrained adaptive control is presented, where command filters are adopted to implement the emulate of actuator physical constraints on the control law and virtual control laws and avoid the tedious analytic computations of time derivatives of virtual control laws in the backstepping procedure. Under the proposed control techniques, the closed-loop semi-global uniformly ultimate bounded stability is achieved via Lyapunov synthesis. Finally, simulation studies are presented to illustrate the effectiveness of the proposed adaptive tracking control. © 2011 Elsevier Ltd. All rights reserved.
Shimadzu - LCMS-2020 Vacuum System Main Unit ESI P

6 month Service X
X X X
X X X X X X X X
36 month Service 48 month Service 60 month Service
X X
X X X
X X X
Note: These time frames are only suggestions and items may need to be replaced more frequently depending on sample throughput and use
X
Note: These time frames are only suggestions and items may need to be replaced more frequently depending on sample throughput and use
Shop online ~ or call Shimadzu at 800-477-1227
Shop online ~ or call Shimadzu at 800-477-1227
2
LCMS-8030/8040
Triple Quadrupole Mass Spectrometer
LCMS-8030
LCMS-8040
Vacuum System
Main Unit
SSI-LCMS-11-2015
Liquid Chromatography Mass Spectrometry
LCMS Consumables
LCMS-2020
Single Quadrupole Mass Spectrometer
Inspection/Maintenance Schedule
Octet RED384 CMI Getting Started Guide to Biolaye

Data CollectionData AnalysisShutdownData Managementoptical technique that measures macromolecular interactions by white light reflected from the surface of a biosensor tip. BLI the kinetics and affinity of molecular interactions. In a BLI ) is immobilized to a Dip and Read Biosensor and binding ) is then measured. A change in the number of molecules a , k d ) and equilibrium binding constantsof the Octet Data Acquistion software is called Octet BLI Octet Analysis Studio 13.0.welllight welllight• ForteBio Biosensors.o See table below for popular sensor types and part numbers. Go to the Sartorius/ForteBiowebsite: https:///en/products/protein-analysis/octet-bli-detection/biosensors-chips-kits , for additional sensor types, including Anti-Mouse IgG Fc, Anti-Human Fab, Anti-GST, and biosensors recommended for quantitation.• At least two black microplates per experiment (one for soaking sensors and at least one forsamples and reagents).o Only Greiner Bio-One brand, black microplates or ForteBio plates are recommended (seetable below).• An empty biosensor tray to use as a working tray. • Pipettes (recommended). OCTET Black MicroplatesPart NumberGreiner Bio-One 96-well black flat-bottom PP, 200 µL 655209 (VWR 82050-784) Greiner Bio-One 384-well black flat-bottom PP, 80-120 µL 781209 (VWR 82051-318) ForteBio 384-well black tilted-bottom PP, 60 µL18-5080Sample PreparationAssay Buffers• Many buffers are compatible with BLI. It’s usually a good idea to start with a buffer system in whichyour proteins are well behaved.• Addition of 0.05% Tween 20 (or other surfactant) is usually required to prevent non-specificbinding, which is a frequent problem in BLI experiments.o Try detergent concentrations above the CMC, typically in the range of 0.02-0.1%.• The sample used for the association phase should be in a buffer identically matched to that usedfor the baseline and dissociation phase.o Buffer match is especially important when a buffer component has a high refractive index,such as DMSO. Immobilized load sample should also be in the same buffer, if possible.• 0.1% BSA can also be used to minimize non-specific binding.o ForteBio sells a detergent-based Kinetic Buffer (PBS + 0.02 % Tween20, 0.1 % BSA, 0.05 %sodium azide) that you might consider.o NOTE: BSA is not universally beneficial and can sometimes increase non-specific binding.Popular ForteBio Dip and Read Biosensors for Kinetics Part Number Streptavidin (SA) biosensors 18-5019 (96/tray) High Precision Streptavidin (SAX) biosensors 18-5117 High Precision Streptavidin (SAX2) biosensors 18-5136 Super-Streptavidin (SSA) biosensors (for small molecules) 18-5057 anti-His (HIS1K) biosensors 18-5120 Ni-NTA (NTA) biosensors 18-5101 Anti-Human IgG Fc biosensors 18-5010•All BLI experiments are setup with one molecule fixed to the biosensor surface (the Load Sample) and a second molecule in solution (the Analyte Sample).•Concentration should be accurately measuredo Errors in Load concentration can affect signal intensityo Errors in the Analyte concentration will directly translate to errors in the K D•Protein aggregates will interfere with BLI.o Filter or centrifuge samples before use.o Assess protein heterogeneity via light scattering.o Purify protein samples with soluble aggregates by size-exclusion chromatography. •Recommended concentration ranges:o Load Sample (immobilized) 10-50 µg/ml (~µM range)o Analyte 0.01 – 100 X K D (0.1 – 10 X K D)•Sample and Reagent plate well volumeo96-well 200 µlo384-well 80 – 120 µlo384-well tilted bottom 60 µlGetting StartedAn Octet experiment involves multiple steps in which ForteBio Dip and Read Biosensor are moved between wells in microplates containing buffers, reagents and samples. The instrument can hold up to 96 sensors (one tray) for use in experiments with multiple assays, each using up to 16 sensors. Reagents and samples are placed in 96- or 384-well black microplates. Plate 1 is used in all experiments and can hold any/all sample or reagent types. Plate 2 is an optional reagent plate that cannot hold analytes use in the association phase.timeReagent sequenceResourcesAdditional resources are available at the instrument, including Data Collection and Data Analysis software manuals.Experimental Design Tips•Do not overload the immobilized molecule.•The same well containing buffer should be used for the baseline and dissociation phase, assuming inter-step correction is performed.•For small molecule work, use Super-Streptavidin sensors and quench with biocytin (biotinyl-lysine at 10 µg/mL).•Use reference subtraction (there are several types).o Reference sample well has immobilized load sample and no analyte during association.o Reference sensor is a sensor to which no load sample is immobilized and is matched for analyte concentration.o Double Reference uses both reference well and reference sensor.•Experiments should be less than 3 hr (10 % volume loss ~ 3.5 hours at 30 ˚C).Data CollectionStartup1.Book time on the PPMS calendar before you start.2.Login to the computer using your PPMS credentials (eCommons ID and password).3.The instrument should generally be left powered on at all times.a.If it is not powered on, the power supply is on the shelf above the instrument.4.Design you experiment in the Data Acquisition software before setting up your sample and reagentplates.5.Open the instrument door with the Present Stage button in the software (green eject button). DoNOT pull on the thing that looks like a handle.Experimental Data Collection1.Start the Octet BLI Discovery Software (formerly Data Acquisition Software). The current version willbe on the Desktop (older versions are also usable and found in “older versions” folder).a.Wait for initialization.b.Select New Kinetics Experiment in the Experiment Wizard, or open a method file.c.The Experiment Wizard has 5 sections tabs to guide you through experimental setup.2.Plate Definition:a.Select data acquisition mode. Read Head: 16 channel or 8 channel.i)16 channel mode uses up to 16 sensors and moves in 2 column increments.ii)8 channel mode uses up to 8 sensors and can move in 1 column increments.b.To modify plate format, select Modify Plate:i)Choose either 96-well or 384-well format for Plate 1 and Plate 2CMI Getting Started Guide C enter forM acromolecular I nteractionsSelect a well or wells to define (shift-click to select all wells in column on 96-well or alternating wells on 384-well plate).c. Right-click to pull up Sample Type Menu and choose type for the selected wells.i) Select "Set Well Data" from the Sample Type Menu to add information or fill in the PlateData tables.ii) A molar concentration for the samples is required for fitting. iii) Required Reagent Types(1) Buffer (baseline before load) (2) Load Sample (load)(3) Mock Load Buffer (for reference sensor load step) (4) Buffer (baseline before association and dissociation)(5) Sample (association phase), use a range of concentrations, include 0 for reference sample iv) Optional Reagent Types(1) Quench (e.g. biocytin for blocking)(2) Regeneration solutions (for sensor reuse or serial data collection)v) Include a zero concentration of analyte (a reference sample well) to correct for a drift in thebaseline(1) Several sensor types have significant drift. (2) A reference sample well is required for NTA. vi) Test non-specific binding with Reference Sensors d. Fill your reagent plate(s) according to the plate map. 3. Assay Definition:a. Plate 1 and Plate 2 show plate layouts (step back to Plate Definition to modify).b. Create a list of steps in the Step Data List.i) Sample steps (s):(1) Baseline 120 (60 – 300) (2) Loading 120 (120 – 600) (3) (Quench) 60 (30 – 120) optional quench (eg. Biocytin on SA or SSA) (4) Association 300 (60 – 600) (5) Dissociation 600 (60 – 600+) ii) Shake speed 1,000 RPM.c. Create an assay (a group of ordered steps with plate information).i) Select a column from a plate and select a step in Step Data List. ii) Double-click or click Add… to add a step to Assay Step List. iii) Select the sensor type for the assay. iv) Typical assay order:(1) Baseline (2) Loading (3) Baseline (4) Association (5) Dissocation d. To create a new Assays, click New Assay or select all steps in Assay 1 and click replicate. Modifythe sample column as needed.A group of “Baseline – Assocation – Dissocation” steps are required for recognition as a binding assay and can be overlayedfor kinetic analysis. Don’t forget the baseline before association.CMI Getting Started GuideC enter forM acromolecular I nteractions 4. Sensor Assignment:a. This step tells you where to put sensors in the Sensor Tray.i) You should always check the box marked Replace sensors in tray after use . This will returnsensors to the tray after your experiment (and prevent them from clogging the instrument, as there is a design flaw in the sensor discard shoot).ii) Plate 1 (and 2) indicate wells that each sensor group will enter in an assay. iii) Sensor type should match those from the Assay Definition.b. Fill a sensor tray with sensors in the marked positions and place a 96-well plate underneath withsoaking buffer (usually running buffer) in the wells with sensors.5. Review Experiment:a. Click the arrow to review each assay step in your experiment. 6. Run Experiment:a. Assign a location for your data (choose your folder inside your lab folder).b. Enter the experiment run name (avoid using the default name Experiment_1).c. Set the Run Settings:i) Check all boxes except for “Present Stage at end of experiment” which should be uncheckedunless you will be present at the end of your experiment.ii) Set experiment delay for soaking and equilibration time (10 min). iii) Set temperature (30 ˚C minimum). d. Advanced Settings:i) Set Sensor Offset: 4 mm for standard well volumes (see table below for other offsets). ii) Set Acquisition Rate: Standard Kinetics (5Hz, avg by 20). e. Save method file if using later. f. Start Run by pressing Go button. 7. During Run:a. Watch data collection in real-time.b. Avoid skipping or extending steps, especially if performing an experiment with a second assay(such as a reference assay). Only assays with identical number of steps and duration of steps can be merged for subtractions or comparison.c. When data collection is complete, close the Data Acquisition software and go to Data Analysis.Table of Recommended sensor offsets by well volume (from Octet User Manual)CMI Getting Started GuideC enter forM acromolecular I nteractionsData AnalysisOverviewThe current version of Octet Data Analysis software is called Octet Analysis Studio 13.0. The legacy software, Data Analysis and Data Analysis HT, is still available for use in the “older versions” folder.Data Analysis Studio features:• Data from multiple plates and/or experiments can be combined into one analysis. • More flexible reference subtraction options are available for kinetic analysis. • Customizable report format.• Export Results as a single file (this was not available older Data Analysis HT versions).Data Analysis Protocols1. Open the Octet Analysis Studio Software.2. Select Data:a. Click Explore in the Icon Bar to view data folders.b. Drag an Octet data folder to the Experiment Builder.i) More than one data file can be combined into one analysis by dragging additional files intothe Overlay section (must have the same number of steps and step lengths). ii) Octet data can also be Appended to the beginning or end of a data set.(1) To combine data with different step sizes.(2) For experiments with steps performed in different files (such as immobilization andassociation performed as different experiments).3. Process Data:a. Click Preprocess from the Operations Section of the Icon Bar.b. Assign Reference Sensors (sensors to which no ligand is immobilized).i) Go to the Reference Sensor Tab.ii) Click on column number or drag to select the reference sensors. iii) Right click on selection.iv) Set Sensor Type to Reference Sensor (sensor icon will change to diamond). v) Right click on plate.vi) Go to Subtract Reference and choose subtraction method.(1) In Rows (selects a reference from the same row as the sample).vii) Scroll down to see multiple plates and repeat the subtraction for each plate.viii) Check the table at the bottom to see the sensor subtraction formula used for each sensor. c. Assign Reference Samples (samples with zero concentration of analyte).i) Go to Reference Sample Tab.ii) Click (or CTRL-click) to select well(s) for reference samples (zero concentration). iii) Right click on selection.iv) Set Reference to Reference Sample Wells. v) Right click on plate.vi) Go to Subtract Reference and choose subtraction method.vii) In columns (selects a reference well from the same column as the samples).CMI Getting Started GuideC enter forM acromolecular I nteractionsviii) Scroll down to see multiple plates and repeat for each plate. ix) Check the table to see the well subtraction formula for each sensor. d. Data Corrections:i) Go to Data Correction Tab.ii) Align the Y axis to the average of baseline step (default is the last 5 seconds of the baseline). iii) Inter-step correction.(1) This step corrects for system artifacts (optical artifacts from buffer mismatch, etc.). (2) Choose a step to align (try dissociation first).(3) The baseline before association and dissociation steps must be performed from the samewell of a sample plate.(4) Should not be performed with very fast on-rates as kinetic data may be lost.iv) Noise Filtering (Savitsky-Golay Filtering, smoothing function) is recommended but optional. e. Export Processed data by clicking on Processed Data in Export Section of the Icon Bar to exportdata in a csv format for graphing in other programs.4. Kinetic Analysis:a. Fitting Parameters:i) Click on Kinetics in the Operations section of the Icon Bar. ii) Choose Steps to Analyze: Association and Dissociation. iii) Choose Model: 1:1.iv) Select Global Fitting (full).v) Group by Sample ID (or other grouping scheme) when performing a parallel experiment. vi) Group by Sensor if all concentrations are measured on the same sensor (a serial experiment). b. Examine Fitted curves.c. Steady-State Analysis (Equilibrium Fit).i) Check to include or uncheck to exclude data from the table.ii) In Steady-state dialog, choose Response as the mode of analysis (when kinetic data reachessteady-state at each concentration).iii) Choose Region of Analysis by defining Average from X to X (time interval).(1) This should be a region of the association curve that has reached equilibrium (or steady-state).(2) Default is a five sec window, five seconds from the end of the association phase.(3) For viewing, go to the graph window in the bottom right corner and select the steady-state tab.d. Save the Excel Report (for a summary of the analysis) or create a custom reporte. Export Results as a single file or multiple files for re-graphing and/or analysis in 3rd party software.f. Processing parameters are autosaved in the HTSettings.efrdi) To restart with default/no settings, you may delete the HTSettings file and reopen. ii) Save As to save processing parameters in Extended ForteBio File format (*.efrd). iii) Click on Open Workspace from the Icon Bar to reopen saved processed data.CMI Getting Started GuideC enter forM acromolecular I nteractionsShutdown 1. Remove sensor tray and reagent plates from the instrument. 2. Close the Octet door.3. Discard used biosensors with tips.4. Return borrowed empty sensor trays to the drawer under the instrument.5. Clean up in and around the instrument.6. Close the control and analysis software.7. Logoff from PPMS!Data ManagementTechnology Biolayer Interferometry InstrumentOctet RH16 (Octet Red 384) Recommended Repository Generalist RepositoryData Collection SoftwareCurrent Version Octet BLI Discovery, Version 13.0 Data Files (Type, ~size) experiment folder (contents below) 5 MB/experiment data file .frd 80 KB/measurement method file .fmf 35-45 KB/experiment plate definitions .jpg 30-90 KB/plate assay image .jpg 30-90 KB/experiment preprocessed data.xslx 200 KB/experimentData Analysis SoftwareCurrent Version Octet Analysis Studio, Version 13.0Data Files (Type, ~size) HT SettingsReadable Exports results table .xslx 5-10 KBexport report.xslxBook time and Report Problems through the PPMS system: https:///hms-cmi• rates are based on booked and real time usageContact ***************.edu with questions.last edited: 2023-05-12。
液晶弹性体

3. Actuators based on LCEs
3.1. Actuators based on thermally actuated LCEs
Fig 3. Micrometer-sized nematic LCE actuators consisting of a pillar array. (a) Experimental setup used to prepare the responsive pillars. (b) Top view (under an optical microscope) of the pillar pattern obtained by the imprint in the nematic liquid crystal elastomer. (Inset) Zoom on the structure (pillar diameter=20mm)[1]. [1 ]Buguin A, Li M H, Silberzan P, et al. Journal of the American Chemical Society, 2006, 128(4): 1088-1089.
4. Summary
1.Introduction
Smart materials:
There is a group of materials capable of responding to external stimuli with mechanical deformation.
Fig 1. The diferent kinds of actuator materials both in natural and synthetic systems
3. Actuators based on LCEs
Bay_41-4109_racemate_DataSheet_MedChemExpress

Inhibitors, Agonists, Screening Libraries Data SheetBIOLOGICAL ACTIVITY:BAY 41–4109 racemate is the racemate of BAY 41–4109. BAY 41–4109 is a potent inhibitor of human hepatitis B virus (HBV ) with an IC 50 of 53 nM.IC50 & Target: IC50: 53 nM (HBV)[1]In Vitro: BAY 41–4109 is able to both accelerate and misdirect capsid assembly in vitro . Preformed capsids are stabilized by BAY 41–4109, up to a ratio of one inhibitor molecule per two dimers [2]. BAY 41–4109 is equally effective at inhibiting HBV DNA releaseand the cytoplasmic HBcAg level, with IC 50s of 32.6 and 132 nM in HepG2.2.15 cells, respectively. HBV DNA and HBcAg are inhibited in a dose–dependent manner, indicating that the anti–HBV mechanisms are associated with and dependent on the rate of HBcAg inhibition [3].In Vivo: BAY 41–4109 reduces viral DNA in the liver and in the plasma dose–dependently with efficacy comparable to3TC. BAY 41 –4109 reduces hepatitis B virus core antigen (HBcAg) in livers of HBV–transgenic mice. Pharmacokineticstudies in mice have shown rapid absorption, a bioavailability of 30% and dose–proportional plasma concentrations,about 60% in rats and dogs [1].BAY41–4109 inhibits virus production in vivo by a mechanism that targets the viral capsid [2].PROTOCOL (Extracted from published papers and Only for reference)Cell Assay:[3]Cellular metabolism is evaluated by MTT colorimetry. HepG2.2.15 cells are plated at a density of 2 × 103 cells per well in 96–well plates. After 8 d of treatment with different concentrations of each antiviral compound, 20 μL of MTT solution (5 g/L) are added to each well and incubated at 37°C for 4 h. Next, 150 μL of DMSO is added and stirred for 10 min to dissolve the crystals.Absorbance values are recorded at 490 nm by using an ELISA reader. The MTT values are calculated using the curve regression equation [3].Animal Administration:[1]Mouse: The HBV–transgenic mice are used in the study. Compounds (BAY 41–4109) are formulated as a suspension in 0.5% Tylose and administered per os to mice two times/day for a 28 day period. The 0.5% Tylose serves as a placebo.Six hours after the last treatment, the animals are sacrificed and livers are removed and immediately frozen for subsequent analysis.Blood is obtained by cardiac puncture of the anesthesized animals [1].References:[1]. Weber O, et al. Inhibition of human hepatitis B virus (HBV) by a novel non–nucleosidic compound in a transgenic mouse model. Antiviral Res. 2002 May;54(2):69–78.[2]. Stray SJ, et al. BAY 41–4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit. 2006 Nov–Dec;19(6):542–8.[3]. Wu GY, et al. Inhibition of hepatitis B virus replication by Bay 41–4109 and its association with nucleocapsid disassembly. J Chemother. 2008 Aug;20(4):458–67.Product Name:Bay 41–4109 (racemate)Cat. No.:HY-100029A CAS No.:298708-79-9Molecular Formula:C 18H 13ClF 3N 3O 2Molecular Weight:395.76Target:HBV Pathway:Anti–infection Solubility:DMSO: ≥ 28 mg/mLCaution: Product has not been fully validated for medical applications. For research use only.Tel: 609-228-6898 Fax: 609-228-5909 E-mail: tech@Address: 1 Deer Park Dr, Suite Q, Monmouth Junction, NJ 08852, USA。
基质效应在生物样品质谱分析中的优化措施研究

基质效应在生物样品质谱分析中的优化措施研究吴文静【摘要】高效液相色谱-质谱联用法(LC - MS /MS )由于其高灵敏度和高选择性现阶段被广泛应用于食品检测、环境评估等方面的样品定量分析.然而,由于实际样品特别是复杂样品分析中基质效应的存在,样品分析进程以及检测结果的特异性、灵敏度和准确度都会受到影响.本文立足于实际的生物样品质谱分析,阐述了基质效应的产生原因、检测及评定方法,其优化措施包括四个方面,即样品前处理的优化、色谱条件的优化、质谱条件的优化以及同位素内标的选择.【期刊名称】《信阳农林学院学报》【年(卷),期】2017(027)004【总页数】5页(P115-118)【关键词】高效液相色谱一质谱联用基质效应生物样品分析【作者】吴文静【作者单位】安徽公安职业学院公安科学技术系,安徽合肥230031;【正文语种】中文【中图分类】O657.63高效液相色谱-质谱联用法(液相质谱,HPLC-MS/MS)是一种高灵敏度和高选择性的样品定量分析方法,由于其高效样品的选择性和准确的测定能力而被广泛推广应用于食品相关检测、环境风险评估、农药残留分析、药物组分以及代谢研究中[1-2]。
近几年,随着液质技术的快速发展,与检测相关的基质效应问题也开始被广泛关注。
基质效应作为质谱检测中存在的必然问题,对样品检测、分析方法和结果的特异性、灵敏度和准确度都有显著影响[3]。
目前,国外的学者已经开展了大量的与基质效应相关的工作和研究,但国内相关的研究还未能构成完整的研究体系。
本文结合国内外相关文献,对液质检测过程中基质效应的产生原因、相关作用以及目前常规的检测方法和消除或降低基质干扰的途径等问题进行阐述。
1 基质效应产生机制及影响基质效应指的是在样品检测过程中,除待测组分以外的其它物质对待测组分的分析进程产生的干扰,并影响检测结果的灵敏度和准确性。
基质效应的产生主要是源于样品中的待测组分与基质成分在离子化过程中的竞争。
CellTiter Glo Luminescent Cell Viability Assay Protocol

Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·1.Description (1)2.Product Components and Storage Conditions (4)3.Performing the CellTiter-Glo ®Assay (5)A.Reagent Preparation (5)B.Protocol for the Cell Viability Assay (6)C.Protocol for Generating an ATP Standard Curve (optional) (7)4.Appendix (7)A.Overview of the CellTiter-Glo ®Assay..............................................................7B.Additional Considerations..................................................................................8C.References............................................................................................................11D.Related Products. (12)1.DescriptionThe CellTiter-Glo ®Luminescent Cell Viability Assay (a–e)is a homogeneous method to determine the number of viable cells in culture based on quantitation of the ATP present, which signals the presence of metabolically active cells. The CellTiter-Glo ®Assay is designed for use with multiwell-plate formats, making it ideal for automated high-throughput screening (HTS) and cell proliferation and cytotoxicity assays. The homogeneous assay procedure (Figure 1) involves adding a single reagent (CellTiter-Glo ®Reagent) directly to cells cultured in serum-supplemented medium. Cell washing, removal of medium or multiple pipetting steps are not required.The homogeneous “add-mix-measure” format results in cell lysis and generation of a luminescent signal proportional to the amount of ATP present (Figure 2).The amount of ATP is directly proportional to the number of cells present in culture in agreement with previous reports (1). The CellTiter-Glo ®Assay relies on the properties of a proprietary thermostable luciferase (Ultra-Glo™ Recombinant Luciferase), which generates a stable “glow-type” luminescent signal and improves performance across a wide range of assay conditions. The luciferase reaction for this assay is shown in Figure 3. The half-life of the luminescent signal resulting from this reaction is greater than five hours (Figure 4). This extended half-life eliminates the need for reagent injectors and provides flexibility for continuous or batch-mode processing of multiple plates. The unique homogeneous format reduces pipetting errors that may be introduced during the multiple steps required by other ATP-measurement methods.CellTiter-Glo ®Luminescent Cell Viability AssayAll technical literature is available on the Internet at: /protocols/ Please visit the web site to verify that you are using the most current version of this Technical Bulletin. Please contact Promega Technical Services if you have questions on useofthissystem.E-mail:********************Figure 1. Flow diagram showing preparation and use of CellTiter-Glo ®Reagent.Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3170M A 12_0ACellTiter-Glo CellTiter-Glo MixerLuminometer®System Advantages•Homogeneous:“Add-mix-measure” format reduces the number of plate-handling steps to fewer than that required for similar ATP assays.•Fast:Data can be recorded 10 minutes after adding reagent.•Sensitive:Measures cells at numbers below the detection limits of standard colorimetric and fluorometric assays.•Flexible:Can be used with various multiwell formats. Data can be recorded by luminometer or CCD camera or imaging device.•Robust:Luminescent signal is very stable, with a half-life >5 hours,depending on cell type and culture medium used.•Able to Multiplex:Can be used with reporter gene assays or other cell-based assays from Promega (2,3).Figure 3. The luciferase reaction.Mono-oxygenation of luciferin is catalyzed byluciferase in the presence of Mg 2+, ATP and molecular oxygen.Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3171M A 12_0A L u m i n e s c e n c e (R L U )Cells per Well10,00060,00020,00030,00040,00050,0000R² = 0.9990.5 × 1061.0 × 1061.5 × 1062.0 × 1062.5 × 1063.0 × 1063.5 × 1064.0 × 106r² = 0.99020,00010,00030,00040,00050,000r² = 0.9900100200300400HO SN S N O S N S N OCOOH +ATP+O 2Ultra-Glo™ Recombinant Luciferase +AMP+PP i +CO 2+LightBeetle Luciferin OxyluciferinMg 2+0Figure 2. Cell number correlates with luminescent output.A direct relationship exists between luminescence measured with the CellTiter-Glo ®Assay and the number of cells in culture over three orders of magnitude. Serial twofold dilutions of HEK293cells were made in a 96-well plate in DMEM with 10% FBS, and assays wereperformed as described in Section 3.B. Luminescence was recorded 10minutes after reagent addition using a GloMax ®-Multi+ Detection System. Values represent the mean ± S.D. of four replicates for each cell number. The luminescent signal from 50HEK293 cells is greater than three times the background signal from serum-supplemented medium without cells. There is a linear relationship (r 2= 0.99)between the luminescent signal and the number of cells from 0to 50,000 cells per well.Figure 4. Extended luminescent half-life allows high-throughput batchprocessing.Signal stability is shown for three common cell lines. HepG2 and BHK-21cells were grown and assayed in MEM containing 10% FBS, while CHO-K1 cells were grown and assayed in DME/F-12 containing 10% FBS. CHO-K1, BHK-21 and HepG2 cells, at 25,000 cells per well, were added to a 96-well plate. After an equal volume of CellTiter-Glo ®Reagent was added, plates were shaken and luminescence monitored over time with the plates held at 22°C. The half-lives of the luminescent signals for the CHO-K1, BHK-21 and HepG2 cells were approximately 5.4, 5.2 and5.8hours, respectively.2.Product Components and Storage ConditionsProduct Size Cat.#CellTiter-Glo ®Luminescent Cell Viability Assay 10ml G7570Substrate is sufficient for 100 assays at 100µl/assay in 96-well plates or 400 assays at 25µl/assay in 384-well plates. Includes:• 1 × 10mlCellTiter-Glo ®Buffer • 1 vial CellTiter-Glo ®Substrate (lyophilized)Product Size Cat.#CellTiter-Glo ®Luminescent Cell Viability Assay 10 × 10ml G7571Each vial of substrate is sufficient for 100 assays at 100µl/assay in 96-well plates or 400 assays at 25µl/assay in 384-well plates (1,000 to 4,000 total assays). Includes:•10 × 10mlCellTiter-Glo ®Buffer •10 vials CellTiter-Glo ®Substrate (lyophilized)Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·R e l a t i v e L u m i n e s c e n c e (%)Time (minutes)CHO-K101020304050607080901003173M A 12_0AProduct Size Cat.# CellTiter-Glo®Luminescent Cell Viability Assay100ml G7572 Substrate is sufficient for 1,000 assays at 100µl/assay in 96-well plates or 4,000assays at 25µl/assay in 384-well plates. Includes:•1 × 100ml CellTiter-Glo®Buffer• 1 vial CellTiter-Glo®Substrate (lyophilized)Product Size Cat.# CellTiter-Glo®Luminescent Cell Viability Assay10 × 100ml G7573Each vial of substrate is sufficient for 1,000 assays at 100µl/assay in 96-well plates or4,000 assays at 25µl/assay in 384-well plates (10,000to 40,000 total assays). Includes:•10 × 100ml CellTiter-Glo®Buffer•10 vials CellTiter-Glo®Substrate (lyophilized)Storage Conditions:For long-term storage, store the lyophilized CellTiter-Glo®Substrate and CellTiter-Glo®Buffer at –20°C. For frequent use, the CellTiter-Glo®Buffer can be stored at 4°C or room temperature for 48hours without loss of activity. See product label for expiration date information. ReconstitutedCellTiter-Glo®Reagent (Buffer plus Substrate) can be stored at room temperaturefor up to 8hours with <10% loss of activity, at 4°C for 48hours with ~5% lossof activity, at 4°C for 4days with ~20% loss of activity or at –20°C for 21weekswith ~3% loss of activity. The reagent is stable for up to ten freeze-thaw cycles,with less than 10% loss of activity.3.Performing the CellTiter-Glo®AssayMaterials to Be Supplied by the User•opaque-walled multiwell plates adequate for cell culture•multichannel pipette or automated pipetting station for reagent delivery•device (plate shaker) for mixing multiwell plates•luminometer, CCD camera or imaging device capable of reading multiwell plates •optional:ATP for use in generating a standard curve (Section 3.C)3.A.Reagent Preparation1.Thaw the CellTiter-Glo®Buffer, and equilibrate to room temperature priorto use. For convenience the CellTiter-Glo®Buffer may be thawed andstored at room temperature for up to 48hours prior to use.2.Equilibrate the lyophilized CellTiter-Glo®Substrate to room temperatureprior to use.Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3.A.Reagent Preparation (continued)3.Transfer the appropriate volume (10ml for Cat.# G7570 and G7571, or 100mlfor Cat.# G7572 and G7573) of CellTiter-Glo ®Buffer into the amber bottlecontaining CellTiter-Glo ®Substrate to reconstitute the lyophilizedenzyme/substrate mixture. This forms the CellTiter-Glo ®Reagent.4.Mix by gently vortexing, swirling or inverting the contents to obtain ahomogeneous solution. The CellTiter-Glo ®Substrate should go intosolution easily in less than 1minute.3.B.Protocol for the Cell Viability AssayWe recommend that you perform a titration of your particular cells todetermine the optimal number and ensure that you are working within thelinear range of the CellTiter-Glo ®Assay. Figure 2 provides an example of sucha titration of HEK293 cells using 0 to 50,000 cells per well in a 96-well format.1.Prepare opaque-walled multiwell plates with mammalian cells in culturemedium, 100µl per well for 96-well plates or 25µl per well for 384-wellplates.Multiwell plates must be compatible with the luminometer used.2.Prepare control wells containing medium without cells to obtain a value forbackground luminescence.3.Add the test compound to experimental wells, and incubate according toculture protocol.4.Equilibrate the plate and its contents at room temperature forapproximately 30 minutes.5.Add a volume of CellTiter-Glo ®Reagent equal to the volume of cell culturemedium present in each well (e.g., add 100µl of reagent to 100µl of mediumcontaining cells for a 96-well plate, or add 25µl of reagent to 25µl ofmedium containing cells for a 384-well plate).6.Mix contents for 2 minutes on an orbital shaker to induce cell lysis.7.Allow the plate to incubate at room temperature for 10 minutes to stabilizeluminescent signal.Note:Uneven luminescent signal within standard plates can be caused bytemperature gradients, uneven seeding of cells or edge effects in multiwellplates.8.Record luminescence.Note:Instrument settings depend on the manufacturer. An integration timeof 0.25–1 second per well should serve as a guideline.Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3.C.Protocol for Generating an ATP Standard Curve (optional)It is a good practice to generate a standard curve using the same plate onwhich samples are assayed. We recommend ATP disodium salt (Cat.# P1132,Sigma Cat.# A7699 or GE Healthcare Cat.# 27-1006). The ATP standard curveshould be generated immediately prior to adding the CellTiter-Glo®Reagentbecause endogenous ATPase enzymes found in sera may reduce ATP levels.1.Prepare 1µM ATP in culture medium (100µl of 1µM ATP solution contains10–10moles ATP).2.Prepare serial tenfold dilutions of ATP in culture medium (1µM to 10nM;100µl contains 10–10to 10–12moles of ATP).3.Prepare a multiwell plate with varying concentrations of ATP standard in100µl medium (25µl for a 384-well plate).4.Add a volume of CellTiter-Glo®Reagent equal to the volume of ATPstandard present in each well.5.Mix contents for 2 minutes on an orbital shaker.6.Allow the plate to incubate at room temperature for 10 minutes to stabilizethe luminescent signal.7.Record luminescence.4.Appendix4.A.Overview of the CellTiter-Glo®AssayThe assay system uses the properties of a proprietary thermostable luciferase toenable reaction conditions that generate a stable “glow-type” luminescentsignal while simultaneously inhibiting endogenous enzymes released duringcell lysis (e.g., ATPases). Release of ATPases will interfere with accurate ATPmeasurement. Historically, firefly luciferase purified from Photinus pyralis(LucPpy) has been used in reagents for ATP assays (1,4–7). However, it hasonly moderate stability in vitro and is sensitive to its chemical environment,including factors such as pH and detergents, limiting its usefulness fordeveloping a robust homogeneous ATP assay. Promega has successfullydeveloped a stable form of luciferase based on the gene from another firefly,Photuris pennsylvanica(LucPpe2), using an approach to select characteristics thatimprove performance in ATP assays. The unique characteristics of this mutant(LucPpe2m) enabled design of a homogeneous single-reagent-addition approachto perform ATP assays with cultured cells. Properties of the CellTiter-Glo®Reagent overcome the problems caused by factors, such as ATPases, thatinterfere with ATP measurement in cell extracts. The reagent is physicallyrobust and provides a sensitive and stable luminescent output.Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·4.A.Overview of the CellTiter-Glo®Assay (continued)Sensitivity and Linearity:The ATP-based detection of cells is more sensitivethan other methods (8–10). In experiments performed by Promega scientists,the luminescent signal from 50HEK293 cells is greater than three standarddeviations above the background signal from serum-supplemented mediumwithout cells. There is a linear relationship (r2= 0.99) between the luminescentsignal and the number of cells from 0 to 50,000 cells per well in the 96-wellformat. The luminescence values in Figure 2 were recorded after 10minutes ofincubation at room temperature to stabilize the luminescent signal as describedin Section3.B. Incubation of the same 96-well plate used in the experimentshown in Figure 2 for 360minutes at room temperature had little effect on therelationship between luminescent signal and number of cells (r2= 0.99).Speed:The homogeneous procedure to measure ATP using the CellTiter-Glo®Assay is quicker than other ATP assay methods that require multiple steps toextract ATP and measure luminescence. The CellTiter-Glo®Assay also is fasterthan other commonly used methods to measure the number of viable cells(such as MTT, alamarBlue®or Calcein-AM) that require prolonged incubationsteps to enable the cells’ metabolic machinery to convert indicator moleculesinto a detectable signal.4.B.Additional ConsiderationsTemperature:The intensity and decay rate of the luminescent signal from theCellTiter-Glo®Assay depends on the luciferase reaction rate. Environmentalfactors that affect the luciferase reaction rate will change the intensity andstability of the luminescent signal. Temperature is one factor that affects therate of this enzymatic assay and thus the light output. For consistent results,equilibrate assay plates to a constant temperature before performing the assay.Transferring eukaryotic cells from 37°C to room temperature has little effect onATP content (5). We have demonstrated that removing cultured cells from a37°C incubator and allowing them to equilibrate to 22°C for 1–2 hours hadlittle effect on ATP content. For batch-mode processing of multiple assayplates, take precautions to ensure complete temperature equilibration. Platesremoved from a 37°C incubator and placed in tall stacks at room temperaturewill require longer equilibration than plates arranged in a single layer.Insufficient equilibration may result in a temperature gradient effect betweenwells in the center and at the edge of the plates. The temperature gradientpattern also may depend on the position of the plate in the stack.Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·Chemicals:The chemical environment of the luciferase reaction affects theenzymatic rate and thus luminescence intensity. Differences in luminescenceintensity have been observed using different types of culture media and sera.The presence of phenol red in culture medium should have little impact onluminescence output. Assaying 0.1µM ATP in RPMI medium without phenolred resulted in ~5% increase in luminescence output (in relative light units[RLU]) compared to assays in RPMI containing the standard concentration ofphenol red, whereas assays in RPMI medium containing twice the normalconcentration of phenol red showed a ~2% decrease in luminescence.Solvents for the various test compounds may interfere with the luciferasereaction and thus the light output from the assay. Interference with theluciferase reaction can be detected by assaying a parallel set of control wellscontaining medium without cells. Dimethylsulfoxide (DMSO), commonly usedas a vehicle to solubilize organic chemicals, has been tested at finalconcentrations of up to 2% in the assay and only minimally affects light output.Plate Recommendations:We recommend using standard opaque-walledmultiwell plates suitable for luminescence measurements. Opaque-walledplates with clear bottoms to allow microscopic visualization of cells also maybe used; however, these plates will have diminished signal intensity andgreater cross talk between wells. Opaque white tape may be used to decreaseluminescence loss and cross talk.Cellular ATP Content:Different cell types have different amounts of ATP,and values reported for the ATP level in cells vary considerably (1,4,11–13).Factors that affect the ATP content of cells may affect the relationship betweencell number and luminescence. Anchorage-dependent cells that undergocontact inhibition at high densities may show a change in ATP content per cellat high densities, resulting in a nonlinear relationship between cell numberand luminescence. Factors that affect the cytoplasmic volume or physiology ofcells also will affect ATP content. For example, oxygen depletion is one factorknown to cause a rapid decrease in ATP (1).Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·4.B.Additional Considerations (continued)Mixing:Optimal assay performance is achieved when the CellTiter-Glo®Reagent is mixed completely with the cultured cells. Suspension cell lines (e.g., Jurkat cells) generally require less mixing to achieve lysis and extract ATP than adherent cells (e.g., L929 cells). Tests were done to evaluate the effect ofshaking the plate after adding the CellTiter-Glo® Reagent. Suspension cellscultured in multiwell plates showed only minor differences in light outputwhether or not the plates were shaken after adding the CellTiter-Glo®Reagent.Adherent cells are more difficult to lyse and show a substantial differencebetween shaken and nonshaken plates.Several additional parameters related to reagent mixing include the force ofdelivery of CellTiter-Glo®Reagent, sample volume and dimensions of the well.All of these factors may affect assay performance. The degree of reagent mixing required may be affected by the method used to add the CellTiter-Glo®Reagent to the assay plates. Automated pipetting devices using a greater or lesser force of fluid delivery may affect the degree of subsequent mixing required.Complete reagent mixing in 96-well plates should be achieved using orbitalplate shaking devices built into many luminometers and the recommended2-minute shaking time. Special electromagnetic shaking devices that use aradius smaller than the well diameter may be required to efficiently mixcontents of 384-well plates. The depth of medium and geometry of themultiwell plates may have an effect on mixing efficiency. We recommend that you take these factors into consideration when performing the assay andempirically determine whether a mixing step is necessary for the individualapplication.LuminometersFor highly sensitive luminometric assays, the luminometer model and settings greatly affect the quality of data obtained. Luminometers from differentmanufacturers will vary in sensitivities and dynamic ranges. We recommend the GloMax®products because these instruments do not require gainadjustments to achieve optimal sensitivity and dynamic range. Additionally, GloMax®instruments are preloaded with Promega protocols for ease of use.If you are not using a GloMax®luminometer, consult the operating manual for your luminometer to determine the optimal settings. The limits should beverified on each instrument before analysis of experimental samples. The assay should be linear in some portion of the detection range of the instrument used.For an individual luminometer there may be different gain settings. Werecommend that you optimize the gain settings.4.C.References1.Crouch, S.P. et al.(1993) The use of ATP bioluminescence as a measure of cellproliferation and cytotoxicity. J. Immunol. Methods160, 81–8.2.Farfan, A.et al.(2004) Multiplexing homogeneous cell-based assays. Cell Notes10, 2–5.3.Riss, T., Moravec, R. and Niles, A. (2005) Selecting cell-based assays for drugdiscovery screening. Cell Notes13, 16–21.4.Kangas, L., Grönroos, M. and Nieminen, A.L. (1984) Bioluminescence of cellular ATP:A new method for evaluating cytotoxic agents in vitro. Med. Biol.62, 338–43.5.Lundin, A. et al.(1986) Estimation of biomass in growing cell lines by adenosinetriphosphate assay.Methods Enzymol. 133, 27–42.6.Sevin, B.U. et al.(1988) Application of an ATP-bioluminescence assay in human tumorchemosensitivity testing. Gynecol. Oncol.31, 191–204.7.Gerhardt, R.T.et al.(1991) Characterization of in vitro chemosensitivity ofperioperative human ovarian malignancies by adenosine triphosphatechemosensitivity assay. Am. J. Obstet. Gynecol. 165, 245–55.8.Petty, R.D. et al.(1995) Comparison of MTT and ATP-based assays for themeasurement of viable cell number. J. Biolumin. Chemilumin.10, 29–34.9.Cree, I.A. et al.(1995) Methotrexate chemosensitivity by ATP luminescence in humanleukemia cell lines and in breast cancer primary cultures: Comparison of the TCA-100assay with a clonogenic assay. AntiCancer Drugs6, 398–404.10.Maehara, Y. et al.(1987) The ATP assay is more sensitive than the succinatedehydrogenase inhibition test for predicting cell viability. Eur. J. Cancer Clin. Oncol.23, 273–6.11.Stanley, P.E. (1986) Extraction of adenosine triphosphate from microbial and somaticcells. Methods Enzymol.133, 14–22.12.Beckers, B. et al.(1986) Application of intracellular ATP determination in lymphocytesfor HLA-typing. J. Biolumin. Chemilumin.1, 47–51.13.Andreotti, P.E. et al.(1995) Chemosensitivity testing of human tumors using amicroplate adenosine triphosphate luminescence assay: Clinical correlation forcisplatin resistance of ovarian carcinoma. Cancer Res. 55, 5276–82.4.D.Related ProductsCell Proliferation ProductsProduct Size Cat.# ApoLive-Glo™ Multiplex Assay10ml G6410 ApoTox-Glo™ Triplex Assay10ml G6320 CellTiter-Fluor™ Cell Viability Assay (fluorescent)10ml G6080 CellTiter-Blue®Cell Viability Assay (resazurin)20ml G8080 CellTiter 96®AQ ueous One SolutionCell Proliferation Assay (MTS, colorimetric)200 assays G3582 CellTiter 96®AQ ueous Non-RadioactiveCell Proliferation Assay (MTS, colorimetric)1,000 assays G5421 CellTiter 96®AQ ueous MTS Reagent Powder1g G1111 CellTiter 96®Non-RadioactiveCell Proliferation Assay (MTT, colorimetric)1,000 assays G4000 Additional sizes available.Cytotoxicity AssaysProduct Size Cat.# CytoTox-Glo™ Cytotoxicity Assay (luminescent)*10ml G9290Mitochondrial ToxGlo™ Assay*10ml G8000 MultiTox-Glo Multiplex Cytotoxicity Assay(luminescent, fluorescent)*10ml G9270 MultiTox-Fluor Multiplex Cytotoxicity Assay(fluorescent)*10ml G9200 CytoTox-Fluor™ Cytotoxicity Assay (fluorescent)*10ml G9260 CytoTox-ONE™ Homogeneous MembraneIntegrity Assay (LDH, fluorometric)*200–800 assays G7890 CytoTox-ONE™ Homogeneous MembraneIntegrity Assay, HTP1,000–4,000 assays G7892 CytoTox 96® Non-Radioactive Cytotoxicity Assay1,000 assays G1780 (LDH, colorimetric)*GSH-Glo™ Glutathione Assay10ml V691150ml V6912 GSH/GSSG-Glo™ Assay10ml V661150ml V6612 *Additional sizes available.LuminometersProduct Size Cat.# GloMax®-Multi+ Detection System with Instinct™ Software:Base Instrument with Shaking 1 each E8032 GloMax®-Multi+ Detection System with Instinct™ Software:Base Instrument with Heating and Shaking 1 each E9032 GloMax®-Multi+ Luminescence Module 1 each E8041Apoptosis ProductsProduct Size Cat.# Caspase-Glo®2 Assay*10ml G0940 Caspase-Glo®6 Assay*10ml G0970 Caspase-Glo®3/7 Assay* 2.5ml G8090 Caspase-Glo®8 Assay* 2.5ml G8200 Caspase-Glo®9 Assay* 2.5ml G8210Apo-ONE®Homogeneous Caspase-3/7 Assay1ml G7792 DeadEnd™ Fluorometric TUNEL System60 reactions G3250 DeadEnd™ Colorimetric TUNEL System20 reactions G7360Anti-ACTIVE®Caspase-3 pAb50µl G7481Anti-PARP p85 Fragment pAb50µl G7341Anti-pS473Akt pAb40µl G7441 Caspase Inhibitor Z-VAD-FMK, 20mM50µl G7231125µl G7232*Additional sizes available.(a)U.S. Pat. Nos. 6,602,677 and 7,241,584, European Pat. No. 1131441, Japanese Pat. Nos. 4537573 and 4520084 and other patents pending(b)U.S. Pat. No. 7,741,067, Japanese Pat. No. 4485470 and other patents pending.(c)U.S. Pat. No. 7,700,310, European Pat. No. 1546374 and other patents pending.(d)U.S. Pat. Nos 7,083,911, 7,452,663 and 7,732,128, European Pat. No. 1383914 and Japanese Pat. Nos. 4125600 and 4275715.(e)The method of recombinant expression of Coleoptera luciferase is covered by U.S. Pat. Nos. 5,583,024, 5,674,713 and 5,700,673.© 2001–2012 Promega Corporation. All Rights Reserved.Anti-ACTIVE, Apo-ONE, Caspase-Glo, CellTiter 96, CellTiter-Blue, CellTiter-Glo, CytoTox 96 and GloMax are registered trademarks of Promega Corporation. ApoTox-Glo, ApoLive-Glo, CellTiter-Fluor, CytoTox-Fluor, CytoTox-Glo, CytoTox-ONE, DeadEnd, GSH-Glo, GSH/GSSG-Glo, Instinct, Mitochondrial ToxGlo and Ultra-Glo are trademarks of Promega Corporation. alamarBlue is a registered trademark of Trek Diagnostic Ssystems, Inc.Products may be covered by pending or issued patents or may have certain limitations. Please visit our Web site for more information.All prices and specifications are subject to change without prior notice.Product claims are subject to change. Please contact Promega Technical Services or access the Promega online catalog for the most up-to-date information on Promega products.。
ViralSEQ Lentivirus Physical Titer Kit说明书

ViralSEQ™ Lentivirus Physical Titer KitCatalog Numbers A52597 and A52598Pub. No. MAN0026127 Rev. A.0Note: For safety and biohazard guidelines, see the “Safety” appendix in the ViralSEQ™ Lentivirus Titer Kits User Guide(Pub. No. MAN0026126). Read the Safety Data Sheets (SDSs) and follow the handling instructions. Wear appropriate protective eyewear, clothing, and gloves.Product descriptionThe Applied Biosystems™ ViralSEQ™ Lentivirus Physical Titer Kit is a TaqMan™-based RT-qPCR kit. The kit measures viral count based on highly sensitive viral RNA quantitation from the supernatants of cell-based, bioproduction systems. Viral titers of 104 to 1011 viral particles (VP) per mL can be quantitated using a standard curve generated from the synthetic RNA control included with the kit. Lentivirus quantitation by RT-qPCR is accurate, sensitive, and reproducible.The ViralSEQ™ Lentivirus Physical Titer Kit is compatible with the PrepSEQ™ Nucleic Acid Sample Preparation Kit (Cat. A50485), which offers both a manual and automated sample preparation workflow. For real‑time PCR, the ViralSEQ™ Lentivirus Physical Titer Kit has been validated on the Applied Biosystems™ 7500 Fast Real-Time PCR System and the Applied Biosystems™ QuantStudio™ 5 Real‑Time PCR System. Data analysis is streamlined using AccuSEQ™ Real‑Time PCR Software that provides accurate quantitation and security, audit, and e-signature capabilities to help enable 21 CFR Pt 11 compliance.For more information about reagent use, see the ViralSEQ™ Lentivirus Titer Kits User Guide (Pub. No. MAN0026126).Treat samples with DNase I, RNase‑free (1 U/µL)DNase I, RNase‑free (1 U/µL) treatment is used to digest double-stranded DNA.Thaw all reagents on ice. Invert the DNase I, RNase‑free (1 U/µL) several times to mix, then centrifuge briefly. All other reagents should be vortexed, then centrifuged briefly before use.1.Set up the DNase I, RNase‑free (1 U/µL) reactions in a MicroAmp™ Optical 96-Well Reaction Plate (0.2 mL).[1]Mix gently by pipetting 3-5 times when adding.2.Mix the reactions by gently pipetting up and down 5 times, then seal the reaction plate with MicroAmp™ Clear Adhesive Film.3.Centrifuge the plate at 1,000 x g for 2 minutes.4.Load the reactions onto the VeritiPro™ 96-well Thermal Cycler, then start the DNase I treatment.Set cover temperature: 105℃Set reaction volume: 18 µL[1]Do not hold for more than 5 minutes. Proceed immediately to DNase I inactivation.5.Centrifuge the plate at 1,000 x g for 2 minutes.CAUTION! The plate is in contact with the heated lid. Remove carefully.6.Gently remove the MicroAmp™ Clear Adhesive Film, then discard.IMPORTANT! Do not touch wells when removing the MicroAmp™ Clear Adhesive Film. Contamination can lead to inaccurate results.7.Add 2 µL of 50mM EDTA to each reaction well. Mix by gently pipetting 5 times with a P10/P20 pipettor set to 10 µL.8.Seal the reaction plate with MicroAmp™ Clear Adhesive Film, then centrifuge the plate at 1,000 x g for 2 minutes.9.Load the reactions onto the VeritiPro™ 96-well Thermal Cycler, then start the DNase I inactivation.Set cover temperature: 105℃Set reaction volume: 20 µL[1]Do not hold for more than 5 minutes.10.Centrifuge the plate at 1,000 x g for 2 minutes.CAUTION! The plate is in contact with the heated lid. Remove carefully.IMPORTANT! Do not vortex.Place the plate on ice until use.Prepare the serial dilutionsThaw the Physical Titer RNA Control (2 ✕1010 copies/µL) on ice. Vortex at medium speed for 5 seconds, briefly centrifuge, then place on ice until use.bel nonstick 1.5‑mL microfuge tubes: NTC, SD1, SD2, SD3, SD4, SD5, and SD6 [used for limit of detection (LOD)].2.Add 35 µL of RNA Dilution Buffer (RDB) to the NTC (no template control) tube. Place the tube on ice.3.Perform the serial dilutions.When dispensing RNA, pipette up and down gently. After each transfer, vortex for 7 seconds, then centrifuge briefly.Table 1 Standard curve dilutions (ViralSEQ™ Lentivirus Physical Titer Kit)Store the standard curve dilution tubes at 4°C or on ice. Use the dilutions within 6 hours for RT-qPCR.Prepare the kit reagents and premix solutionThaw all kit reagents on ice. Vortex the reagents for 5 seconds, briefly centrifuge, then place the reagents on ice until use.bel a microcentrifuge tube for the Premix Solution.2.Prepare the Premix Solution according to the following tables.IMPORTANT! Use a separate pipette tip for each component.Table 2 Premix Solution[1]Includes 10% excess to compensate for pipetting loss.3.Vortex the Premix Solution for 10 seconds to mix, then briefly centrifuge.Store the Premix Solution at 4°C or on ice until use.Prepare the PCR reactionsPlace the plate containing DNase I-treated samples on a MicroAmp™ 96-Well Base, then gently remove the MicroAmp™ Clear Adhesive Film. Gently pipette up and down 3 times to mix the samples.1.Dispense the following into the appropriate wells of a MicroAmp™ Fast Optical 96-Well Reaction Plate, 0.1 mL, gently pipetting at thebottom of the well.Figure 1 Recommended plate layout2.Seal the plate with MicroAmp ™Optical Adhesive Film.3.Vortex the reaction plate for 10 seconds, then centrifuge at 1,000 x g for 2 minutes.Note: Ensure there are no bubbles in the reaction wells. If present, tap the well gently to remove bubbles, then re-centrifuge.Proceed immediately to “Start the run (QuantStudio ™ 5 Real ‑Time PCR Instrument)”.Create a ViralSEQ ™ templateCreate a new template in the (Home) screen of the AccuSEQ ™Real ‑Time PCR Software v3.1.1.ClickCreate New on the home screen.Create New ExperimentpaneFactory default/Admin Defined Templates —List of existing default or Admin Defined templates. These templates can beused as templates for new experiments.My Templates —List of templates available to the user that is signed in. These templates can be used as templates for newexperiments.Create New —Used to create an experiment or template with nopre-existing settings.Create My Template —Used to create a new template (stored locally in My Templates).Create Admin Defined Template —Used to create a new template (Administrator only).2.Select Create My Template or Create Admin Defined Template .3.Edit the Experiment Properties as required.a.In the Template Name field, modify the template name. For example, LV Titer template.b.(Optional ) Enter information in the Comments field.c.In the Setup tab, select:•Experiment Type —Quantitation-Standard Curve •Chemistry —TaqMan ® Reagents•Ramp Speed —Standard-2hrs •Block Type —96-Well 0.1mL Blockd.(Optional ) Select Is Locked to lock the template. If locked, users are unable to edit the template.4.ClickAnalysis settings to change the default C t Settings and Flag Settings.a.In the C t Settings tab, click Edit Default Settings .b.Deselect Automatic Threshold , then enter 0.200.c.Ensure that Automatic Baseline is selected.d.Click Save Changes .e.Deselect Default Settings , then click Applyto save any changes before closing the window.2C tSettingsFlag SettingsEdit Default SettingsbuttonDefault Settingscheckbox Apply buttonf.In the Flag Settings tab, deselect the following flags.•CQCONF —Low Cq confidence •EXPFAIL —Exponential algorithm failed •NOAMP —No amplification •NOSIGNAL —No signal in wellNote: Use the scrollbar on the right to scroll down the list of flags.g.Click Apply to save any changes before closing the window.5.Click Next .Template name cannot be changed after this step.The qPCR Method screen is displayed.Edit the run method and optical filter selectionThis section provides general procedures to edit the run method and optical filter selection in the qPCR Method. To edit the default run method, see the AccuSEQ™ Real‑Time PCR Software v3.1 User Guide (Pub. No. 100094287).1.Set the reaction volume to 25 µL.2.Edit Step 1 of the Hold Stage to 45℃ for 30 minutes.3.Set Step 2 of the Hold Stage to 95℃ for 10 minutes.4.Set Step 1 of the PCR Stage to 95℃ for 15 seconds.5.Edit Step 2 of the PCR Stage to 60℃ for 45 seconds.6.Set the cycle number to 40.7.Ensure that Data Collection occurs after Step 2.123Figure 2 Lentivirus Physical Titer Run MethodReaction volume- set to 25µLStageCycle number- set to 40 cycles8.(Optional) Click (Optical Filter Settings) to view the default filter settings.•The default optical filter selection is suitable for the ViralSEQ™ Lentivirus Physical Titer Kit.•The ViralSEQ™ Lentivirus Physical Titer Kit requires the QuantStudio™ 5 System to be calibrated for FAM™, VIC™, and ROX™.•For more information about system dyes and their calibration and optical filter selection, see QuantStudio™ 3 and 5 Real‑Time PCR Systems Installation, Use, and Maintenance Guide (Pub. No. MAN0010407).9.Click Next.Assign plate and well attributesNote: This section provides general procedures to set up the plate.For specific instructions for each assay type, see the corresponding chapter in this guide. Do not change Targets for default assay templates.TargetsSamplesPlate setup toolbarSelect Item to highlight (Sample, Target, or Task).Select Item. For example, Sample 1. Sample 1 replicates arehighlighted.Define& setup Standard(View Legend)(Print Preview)View (Grid View or Table View)1.In Plate Setup screen, click or click‑drag to select plate wells in the (Grid View) of the plate.2.Assign the well attributes for the selected wells. Each well should have a Sample Name under Samples, as well as the appropriateTargets under Targets. Reporters should be FAM™ dye for Lentivirus Physical Titer, and VIC™ dye for internal positive control (IPC).a.To add new Samples or Targets, click Add in the appropriate column on the left of the screen, then edit the new Name andother properties as required. The new sample or target is then selectable within the wells of the plate.b.For each sample (e.g. DNase-treated lentivirus sample, standard curve dilution, or NTC), two targets should be included.•Select the FAM™ dye reporter for Lentivirus Physical Titer detection.•Select the VIC™ dye for IPC detection.c.Select NFQ-MGB as the quencher for both targets.d.For standard curve dilution samples (SD1 to SD5), the Task for Lentivirus Physical Titer target should be indicated as “S” forStandard, with the appropriate copy number written under Quantity. For instance, the quantity for SD1 is 1E9 copies. Change the Task by clicking on the field and using the drop-down menu. Copy numbers can be indicated using scientific notation (e.g.“1E9”) and the program will convert it to numerical format.e.For DNase-treated samples and SD6, set the Task for Lentivirus Physical Titer target to U for Unknown.f.For NTC wells, set the Task for Lentivirus Physical Titer target to N for NTC.g.For IPC wells, set the Task for Lentivirus Physical Titer target to U for Unknown.h.To change sample names, click the name in the Name column, then type the new name. To change Reporters and Quenchers,click the dye, then select from the dropdown list.When a Sample or Targetname are edited, two entries are added to the Audit trail (one for Delete, and another for Create).23AddbuttonCheckbox—Select Targets and Samples to go in the selectedwell.Textbox—Click the name to edit.Scrollbar—Use to scroll to additional properties.•Use the plate setup toolbar (above the plate) to make edits to the plate.–Click View to show/hide the Sample Name, Sample Color, or Target from the view.•To add consecutive samples (with the same Target ), select a well, then click ‑drag the dark blue box to the right.3.(Optional ) Double ‑click a well to enter comments for the selected well.4.Select ROX ™dye from the Passive Reference drop-down list (bottom left of screen).5.Click Save to save the template.This template can then be used to create experiments.Start the run (QuantStudio ™ 5 Real ‑Time PCR Instrument)Ensure that the plate is loaded in the QuantStudio ™5 Real ‑Time PCR Instrument.™A message stating Run has been started successfully is displayed when the run has started.Review the resultsAfter the qPCR run is finished, use the following general procedure to analyze the results. For more detailed instructions see theAccuSEQ ™Real ‑Time PCR Software v3.1 User Guide (Pub. No. 100094287).1.In the AccuSEQ ™Real ‑Time PCR Software, open your experiment, then navigate to the Result tab.ResulttabAnalysis SettingsPlot horizontal scrollbar Analyze button2.In the Result Analysis tab, select individual targets, then review the Amplification Curve plots for amplification profiles in thecontrols, samples, and the standard curve. Ensure that threshold is set to 0.200 with an automatic baseline.3.In the Result Analysis tab, review the QC Summary for any flags in wells.4.In the Result Analysis tab, review the Standard Curve plot. Verify the values for the Slope, Y ‑intercept, R 2, and Efficiency are withinacceptable limits.Note: The Standard Curve efficiency should be between 90-110% and the R 2>0.99. If these criteria are not met, up to two points,not in the same triplicate, can be removed from the standard curve data, and the analysis repeated.5.In Table View, ensure that C t values are within the standard curve range.•Samples with C t values that exceed the upper limit of quantitation (109 copies) of the standard curve should be diluted and re-run.•Samples with C t values that exceed the lower limit of detection (LoD of 10 copies) and IPC shows no signs of PCR inhibition,suggests the absence of lentivirus.6.(Optional ) Outliers can be excluded from the results. To exclude, select the well, then click Omit/Include , then reanalyze by clickingAnalyze .7.(Optional ) Select File 4Print Report to generate a hard copy of the experiment, or click Print Preview to view and save the report asa PDF or HTML file.8.Export the results.a.Navigate to the Report tab.b.Check all boxes under Contents .c.Select Export Data in One File .d.Select the XLS format, then click Export .Calculate the titer (VP/mL)1.Download the Physical Titer Calculation Tool .a.Go to .b.Search for the ViralSEQ ™Lentivirus Physical Titer Kit.c.Download the tool from the Documents section.2.Open the tool, then follow the instructions in the tool to calculate the titer.Calculate lentivirus titers from qPCR dataTo determine the number of lentivirus RNA copies per mL in the original sample, the copy numbers obtained from the qPCR must be multiplied by the dilution factor of the sample during extraction and DNase I treatment. Since there are 2 copies of RNA/target per lentivirus particle, the number of viral particles per mL (VP/mL) is 0.5x the number of lentivirus RNA copies.Viral particles per mL =qPCR copies x sample dilution factor x 0.5Volume of sample used (mL)For help in determining the qPCR copy numbers, see the QuantStudio ™Design and Analysis Desktop Software User Guide (Pub. No. MAN0010408).For example, if the following parameters were used,•10 µL of lentivirus culture was extracted with the KingFisher ™Flex Purification System with 96 Deep-Well Head and eluted in 200 µL •10 µL of this eluate (20x dilution) was treated with DNase I, RNase ‑free (1 U/µL) in a total volume of 20 µL.• 5 µL of the DNase-treated sample (4x dilution) was used for the qPCR reaction.Lentivirus sample10 μL Sample Extraction Elute: 200 μL DNaseI Treat Total: 20 μL reaction RT-qPCRTotal: 25 μL reactionUse 10 μL (1:20 dilution)Use 5 μL (1:4 dilution)then, the calculation would be:Viral particles per mL =qPCR copies x (20x4) x 0.50.01 (mL)Note: qPCR can only determine the number of physical particles in a virus culture. To determine the numbers of infectious units,cell-based transduction experiments must be carried out. The titers of physical particles are often higher than infectious titers by 10-1000fold, depending on the purity of the lentivirus preparation and the levels of infectious particles within the culture.Limited product warrantyLife Technologies Corporation and/or its affiliate(s) warrant their products as set forth in the Life Technologies' General Terms and Conditions of Sale at /us/en/home/global/terms-and-conditions.html . If you have any questions, please contact Life Technologies at /support .Life Technologies Ltd | 7 Kingsland Grange | Woolston, Warrington WA1 4SR | United KingdomThe information in this guide is subject to change without notice.DISCLAIMER : TO THE EXTENT ALLOWED BY LAW, THERMO FISHER SCIENTIFIC INC. AND/OR ITS AFFILIATE(S) WILL NOT BE LIABLE FOR SPECIAL, INCIDENTAL, INDIRECT,PUNITIVE, MULTIPLE, OR CONSEQUENTIAL DAMAGES IN CONNECTION WITH OR ARISING FROM THIS DOCUMENT, INCLUDING YOUR USE OF IT.Important Licensing Information : These products may be covered by one or more Limited Use Label Licenses. By use of these products, you accept the terms and conditions of all applicable Limited Use Label Licenses.©2022 Thermo Fisher Scientific Inc. All rights reserved. All trademarks are the property of Thermo Fisher Scientific and its subsidiaries unless otherwise specified. Vortex-Genie is a trademark of Scientific Industries. Windows and Excel are trademarks of Microsoft Corporation. TaqMan is a registered trademark of Roche Molecular Systems, Inc., used under permission and license./support | /askaquestion 23 August 2022。
欧洲药典7.5版

INDEX
To aid users the index includes a reference to the supplement in which the latest version of a text can be found. For example : Amikacin sulfate...............................................7.5-4579 means the monograph Amikacin sulfate can be found on page 4579 of Supplement 7.5. Note that where no reference to a supplement is made, the text can be found in the principal volume.
English index ........................................................................ 4707
Latin index ................................................................................. 4739
EUROPEAN PHARMACOPபைடு நூலகம்EIA 7.5
Index
Numerics 1. General notices ................................................................... 7.5-4453 2.1.1. Droppers...................
急诊PCI的若干问题

0.6
P<0.02
0.58
0.56
0.54
0.52
0.5
0.59
n=28 0.52
0.48 n=28 Presence of plaque material
n=21 Presence of thrombus only
LVEF 16hr post-procedural
Plaque material
300 250 200 150 100 50
Tirofiban *
Angiogram Tirofiban
cont’d
Christian W. Hamm et al.
Conclusions
ON-TIME-2
• High dose tirofiban on top of clopidogrel (600mg) in the prehospital setting is safe
0 n=28 n=21
P<0.02
146
257.9
Presence of plaque material Presence of thrombus only
Peak CK-MB post-procedural
Should TA be routinely performed in TIMI 2-3 patients? Needs trials
4 Fresh
1
3 5
2
4
Formalin fixed
Formalin fixed
HE
4 HE
1. 2.
3. 4.
5 5.
Erythrocyte-rich thrombi
Platelet / fibrin-rich thrombi