西瓜基因组文章
科技西瓜育种实验报告

一、实验背景西瓜(Citrullus lanatus)作为一种广受欢迎的水果,其品种繁多,口感和甜度各异。
随着我国西瓜产业的快速发展,对西瓜品种的多样性和品质要求日益提高。
为了满足市场需求,提高西瓜产量和品质,本实验旨在通过科技手段,结合先进的育种技术,培育出具有优良性状的新品种西瓜。
二、实验目的1. 利用基因编辑技术,对西瓜基因组进行编辑,筛选出具有优良性状的基因。
2. 通过杂交育种,培育出具有抗病性、高产、优质的新品种西瓜。
3. 对新品种西瓜进行田间试验,评估其生长性能、抗病性、产量和品质。
三、实验材料与方法1. 实验材料- 西瓜种子:选自我国多个优良品种,包括京欣系列、黑美人、甜王等。
- 基因编辑工具:CRISPR/Cas9系统。
- 抗病菌株:选取具有较强抗性的西瓜病原菌。
2. 实验方法(1)基因编辑- 利用CRISPR/Cas9系统对西瓜基因组进行编辑,针对目标基因序列设计sgRNA,并通过电穿孔法将Cas9蛋白和sgRNA导入西瓜细胞中。
- 通过PCR和测序技术检测基因编辑效果,筛选出具有编辑成功的西瓜细胞。
(2)杂交育种- 将基因编辑成功的西瓜细胞与具有优良性状的西瓜品种进行杂交,获得杂交种子。
- 对杂交种子进行种植,观察其生长性能和抗病性,筛选出优良杂交后代。
(3)田间试验- 将筛选出的优良杂交后代进行田间试验,观察其生长周期、产量、品质和抗病性。
- 对试验数据进行统计分析,评估新品种西瓜的优良性状。
四、实验结果与分析1. 基因编辑本实验成功对西瓜基因组进行编辑,共筛选出10个具有编辑成功的西瓜细胞。
通过测序分析,发现编辑成功的细胞在目标基因序列上发生了预期的突变。
2. 杂交育种通过杂交育种,共获得30个优良杂交后代。
其中,20个具有抗病性、高产、优质等优良性状。
3. 田间试验田间试验结果表明,筛选出的优良杂交后代具有以下特点:- 生长周期:从播种到收获,平均生长周期为85天。
- 产量:平均产量为每亩3000公斤。
利用西瓜愈伤组织快速检测CRISPR

收稿日期:2022-11-22;修回日期:2023-03-24基金项目:中组部青年千人计划自主科研经费(A279021801);广东省重点领域研发计划项目(2022B020*******);中央高校基本科研项目(2452022111)作者简介:彭婷,女,在读硕士研究生,主要研究方向为西瓜甜瓜分子育种。
E-mail :*******************通信作者:袁黎,男,教授,主要研究方向为西瓜甜瓜分子育种。
E-mail :***************.cn基因编辑技术是目前分子育种3.0时代的核心技术,是进行作物性状改良及新种质创制的关键技术。
其中,CRISPR/Cas9基因编辑系统因设计简单、操作方便等诸多优点成为当下应用最为广泛的基因编辑工具之一,在植物生长发育、果实品质、响应生物与非生物胁迫研究方面发挥重要作用[1]。
该工具主要是通过sgRNA 和Cas9蛋白完成对基因组DNA 双链的识别和切割,在细胞内实现基因定点修饰,激发细胞自身的修复机制,DNA 在修复的过程中造成核苷酸序列改变从而实现基因编辑[2-3]。
随着对CRISPR/Cas9系统研究的不断深入,发现该系统在工作过程中存在编辑效率低或无法正常编辑的情况,导致在转基因阳性植株筛选过程中无法获得正确编辑的突变体或突变体多为嵌合利用西瓜愈伤组织快速检测CRISPR/Cas9基因编辑效率彭婷,刘曼,刘春宇,王家发,田树娟,袁黎(旱区作物逆境生物学国家重点实验室·西北农林科技大学园艺学院陕西杨凌712100)摘要:西瓜作为重要的园艺经济作物,其CRISPR/Cas9基因编辑效率的快速检测方法仍十分匮乏。
以愈伤组织再生能力强的野生西瓜种质ZTC 为试验材料,针对西瓜ClPDS 基因构建CRISPR/Cas9双靶点敲除载体,利用农杆菌侵染西瓜幼嫩子叶,在含1.0mg·L -12,4-二氯苯氧乙酸生长素的MS 培养基中诱导愈伤组织形成,通过绿色荧光蛋白标记筛选阳性愈伤组织,检测其编辑效率。
西瓜果实长圆的遗传及候选基因定位

西瓜果实长圆的遗传及候选基因定位豆峻岭,刘文革(编译)(中国农业科学院郑州果树研究所郑州450009)目的与意义:果实形状作为一个非常重要的农艺性状,在许多作物中已经有了非常深入的研究,同时也获得了许多控制果实形状的候选基因。
随着2012年西瓜全基因组测序的完成,西瓜分子生物学的研究得到了快速发展,目前关于西瓜遗传图谱的报道有很多,根据遗传图谱也得到了许多性状的QTLs ,尤其是西瓜果实的长度、宽度、果形指数等。
然而目前还没有关于西瓜果实形状候选基因定位的报道。
因此笔者通过配制杂交组合,解析西瓜果实形状的遗传模式,同时利用现代分子生物学手段,希望能够获得控制西瓜果实形状的候选基因。
材料与方法:以长果西瓜自交系‘短125’(P 1)和圆果西瓜自交系‘郑州籽瓜’(P 2)为亲本杂交配制F 1,F 1自交配制F 2分离群体,F 1分别和P 1、P 2回交配制回交群体BC 1P 1、BC 1P 2。
采用BSA (混合群体分离分析)测序法从F 2分离群体中分别选取长瓜30株、圆瓜30株,分别采取叶片提取DNA ,然后分别将长瓜和圆瓜DNA 等量混合,送公司进行全基因组重测序。
结合GWAS (全基因组关联分析)分析得到控制西瓜果实形状的候选区间。
之后通过分析候选区间内的SNPs ,开发出298个CAPS 标记,根据这些标记对430株F 2分离群体进行酶切检测,筛选重组单株,从而缩小候选区间。
最终通过实时荧光定量PCR 技术对候选基因在不同发育时期不同组织器官中进行实时荧光定量PCR 分析。
结果与分析:(1)通过对西瓜果实形状六世代群体的遗传分析,西瓜果实长圆为单基因不完全显性遗传,其中果实长为显性纯合(OO ),果实圆为隐性纯合(oo ),而果实椭圆为杂合(Oo )。
同时对两亲本发育过程果形指数进行测定,结果发现,授粉当天果形指数最大,长瓜达到3.15,圆瓜达到1.43。
而随着果实的发育,长瓜最终维持在1.83左右,而圆瓜维持在1.02左右。
西瓜基因目录(2007)

西瓜基因目录(2007)作者:肖光辉来源:《中国瓜菜》2012年第06期译者按: 2007年的国际葫芦科遗传协会年报(CGC Report)第30期发表了最新的西瓜基因目录,对以前的西瓜基因目录进行了修改和补充,纠正了某些基因描述等方面的错误,新增加了8个基因,特别是增加了部分基因的照片。
这个西瓜基因目录包括60个基因和111个分子标记共171个基因,对从事西瓜研究特别是西瓜育种的科研人员很有用,特译出供参考。
西瓜是一种很有用的进行遗传研究的作物种类,因为它的基因组大小小,并且有许多可用的基因突变。
西瓜的基因组大小为4亿2 400万个碱基对。
DNA 序列分析显示,有用的高保守序列可用于与其他植物品种,以及葫芦科植物内其他品种进行比较基因组分析。
与其他的一些栽培瓜类作物一样,西瓜的种子和果实性状有很多遗传变异。
人们已经开展了某些性状的遗传调查,包括种子颜色、种子大小、果实形状、果皮颜色、果皮花纹和瓜瓤颜色等。
最初的西瓜基因目录由Poole[1]组织和概括总结。
西瓜基因目录后来被Robinson等[2]、葫芦科基因名录委员会(Cucurbit Gene List Committee)[3-5]、Henderson[6-7]、Rhodes 和Zhang[8]、Rhodes 和 Dane[9] 以及Guner 和 Wehner[10]进行了更新和扩展并发表。
历年来发表的西瓜基因目录如下。
Poole,1944年[1]:共15个基因。
Robinson等,1976年[2]:增加10个基因,共25个基因。
Robinson等,1979年:增加3个基因,共28个基因。
Robinson等,1982年:增加2个基因,共30个基因。
Henderson,1987年:增加3个基因,共33个基因。
Henderson,1991年[6]:增加3个基因,共36个基因(加 52个分子标记)。
Rhodes and Zhang,1995年[8]:增加3个基因,共39个基因(加 109个分子标记)。
高效的西瓜CRISPRcas9 基因敲除技术

高效的西瓜CRISPR/cas9基因敲除技术张倩倩,许勇,田守蔚(编译)(北京市农林科学院蔬菜研究中心北京100097)目的与意义:基因编辑技术在植物基因功能鉴定以及重要农艺性状突变获得方面具有重要应用价值。
CRISPR/Cas9基因编辑技术已应用于许多植物物种,成功实现了靶点突变。
然而,目前在西瓜上还没有CRISPR/Cas9基因编辑技术相关的研究报道,该技术能否成功应用于西瓜研究及育种仍未可知。
以西瓜的ClPDS 基因(编码八氢番茄红素脱氢酶,突变后植株有明显白化表型)为靶基因,尝试建立西瓜CRISPR/Cas9基因编辑技术体系,以期利用该技术在西瓜基因组实现精准编辑。
材料与方法:以西瓜自交系‘PI179878’为供试材料,利用PEG 介导的原生质体转化方法测试靶点特异性与效率,利用农杆菌介导法转化西瓜子叶建立西瓜稳定遗传转化体系,利用酶切、测序等方法检测突变效率及突变类型。
结果与分析:(1)靶点选择与载体构建:选择西瓜PDS 基因ClPDS (Cla010898)作为Cas9核酸内切酶的靶基因。
在第1和第3外显子分别选择1个靶点,BanI 和XhoI 酶切位点分别在这2个靶点预测的切割位点上,有利于识别突变。
每个靶点的sgRNA 分别被克隆到以pGreen 为骨架的PHSN401载体中,命名为PHSN1和PHSN2,用于原生质体转化。
2个靶点同时被克隆到pCAMBIA 为骨架的PHSE401载体中用于农杆菌介导的西瓜遗传转化。
(2)CRISPR/Cas9介导西瓜原生质体ClPDS 突变:利用PEG 介导的原生质体转染检测靶点的效率,采用PCR/限制性内切酶(PCR/RE )试验检测靶位点的突变并估计突变率。
PHSN1或PHSN2分别转染到西瓜原生质体细胞中,打靶效率分别为51.6和42.1%。
结果表明,构建的CRISPR/Cas9载体可在西瓜细胞中瞬时表达,Cas9/sgRNA 复合体可以高效地对这2个靶点进行打靶。
西瓜遗传图谱构建和基因定位研究进展

西瓜遗传图谱构建和基因定位研究进展作者:李兵兵刘文革来源:《中国瓜菜》2019年第02期摘 ; ;要:西瓜是一种在世界范围内广泛种植的园艺作物,中国是世界上最大的西瓜生产国和消费国,对西瓜遗传育种的研究具有重要意义。
遗传图谱是进行基因定位、图位克隆、分子标记辅助育种的重要工具,并为分子生物学的深入研究提供了基础。
近年来,随着高通量测序技术和分子标记的发展,西瓜遗传图谱研究取得了很大进展,基于不同遗传背景的西瓜遗传图谱陆续发表,极大地促进了西瓜基因组学和分子遗传育种的发展。
笔者总结了近年来国内外西瓜遗传图谱构建和基因定位的研究进展及应用,并探讨了目前西瓜遗传图谱构建方面存在的主要问题及今后的研究趋势。
关键词:西瓜; 遗传图谱; 基因定位Research progress on genetic map construction and gene mapping for watermelonLI Bingbing, LIU Wenge(Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences,Zhengzhou 450009, Henan, China)Abstract:Watermelon is a horticultural crop widely cultivated around the world. China is the largest producer and consumer of watermelon. The genetic breeding research of watermelon is of great significance. Genetic map is an important tool for gene mapping, map cloning, molecular marker-assisted breeding, and provides a basis for further research in molecular biology. In recent years, with the development of high-throughput sequencing technology and molecular markers,the research on watermelon genetic map has made great progress. Many genetic maps based on different genetic background have been constructed, which greatly promoted the development of watermelon genomics. This paper reviewed the research progress and application of watermelon genetic map construction and gene mapping in recent years, and discussed the main problems and future research trends of watermelon genetic map construction.Key words: Watermelon; Genetic map; Gene mapping西瓜(Citrullus lanatus)原产于非洲,是一种重要的葫芦科(Cucurbitaceae)作物,其果实营养丰富,甘甜多汁,具有重要经济价值,在世界主要农业产区广泛种植。
栽培西瓜与野生西瓜果实发育过程中的转录组比较分析

2019,32(8):202中国瓜菜文献编译栽培西瓜与野生西瓜果实发育过程中的转录组比较分析王准,郭绍贵,许勇(编译)(北京市农林科学院蔬菜研究中心北京100097)目的与意义:西瓜是一种重要的水果作物,但其果实发育和成熟的分子机制尚不清楚。
为了更好地了解导致其果实发育差异的遗传基础,对栽培西瓜自交系'97103'和野生种质’PI296341-FR'进行比较果实转录组分析,并对果实发育过程中特别的代谢产物进行综合分析,以便为深入了解西瓜果实品质形成和成熟过程中的基因表达调控网络提供依据。
材料与方法:以’97103'果肉和中果皮及•PI296341-FR'果肉为材料,分别在果实未成熟期(10DAP)、转色期(18 DAP)、红熟期(26DAP)和完熟期(34DAP)4个关键发育阶段取样,以便进行转录组比较分析。
采用华越洋RNA 试剂盒提取总RNA,制备链特异RNA-Seq文库.Illumina HiSeq2000平台进行单末端100bp高通量测序。
使用Trimmomatic和Bowtie处理原始RNA-Seq数据,使用Tophat将得到的高质量数据与西瓜基因组序列比对。
使用DESeq、LIMMA、Benjamini-Hochbcrg鉴定差异表达基因,GO、TermFinder、Plant MetGenMAP进行差异表达基因功能分析。
利用LightCycler480进行qRT-PCR检测,验证基因表达。
结果与分析:(1)基因表达分析:釆用Illumina平台对14个样品进行链特异RNA高通量测序,共获得3.218亿条高质量数据。
其中栽培型西瓜'97103'果肉和中果皮和野生型西瓜'PI296341-FR'果肉中分别有18198(77.6%),18 392(78.5%)和17954(76.6%)个基因表达。
组织基因差异表达分析显示,‘97103'和’PI296341-FR'的果肉和中果皮分别有2452.826和322个基因在果实发育和成熟过程中差异表达。
西瓜SSR标记中不同类型样品的DNA提取分析

西瓜SSR标记中不同类型样品的DNA提取分析DNA提取是进行分子生物学实验的基础步骤,也是研究基因组的重要手段之一。
对于不同类型的样品,如西瓜SSR标记分析,DNA提取方法也有所区别。
本文将介绍西瓜不同类型样品的DNA提取分析方法。
对于西瓜不同部位的样品,如叶片、茎、果肉等,DNA提取方法略有差异。
下面将分别介绍这些样品的DNA提取方法。
首先是西瓜叶片的DNA提取。
叶片是植物中含有较高DNA含量的部位,适合进行DNA 提取。
提取方法可按照以下步骤进行:1.取一片西瓜叶片放入研钵中,加入适量的液氮将其打碎成细碎的粉末状。
2.向打碎好的叶片粉末中加入预冷的提取缓冲液(含有高浓度的盐和洗涤剂),并用一根滴管迅速均匀搅拌。
这样可以使细胞完全裂解,并且洗涤剂可以有效去除表面的污染物。
3.将混合物转移至离心管中,进行高速离心,将细胞碎片沉淀下来。
4.将上清液转移至新的离心管中,加入等体积的冷乙醇,轻轻倒转离心管使其混匀。
5.再次进行高速离心,可使DNA沉淀到离心管底部。
6.倒掉上清液,用75%的乙醇洗涤DNA沉淀物,再次离心。
7.将离心管倾斜,将乙醇排空,然后将离心管平放,自然晾干。
8.最后用去离子水溶解DNA。
下面是西瓜茎的DNA提取。
茎是西瓜植物中的主要组织之一,也可用于DNA提取。
提取方法如下:最后是西瓜果肉的DNA提取。
西瓜果肉中的DNA含量相对较低,提取方法稍有不同。
具体步骤如下:通过以上方法,就可以从西瓜的不同部位样品中提取到高质量的DNA。
这些DNA可用于进一步的PCR扩增、序列测定、基因组分析等分子生物学实验。
利用SSR技术鉴定西瓜品种纯度

中国瓜菜
试验研究
利用 SSR 技术鉴定西瓜品种纯度
孙 波 1,邹 甜 1,王志伟 1,3,吴 琪 1,杨红波 2,王兴辉 2,孙小武 1,2
(1. 湖南农业大学 长沙 410128; 2. 邵阳市农业科学研究院 湖南邵阳 422000; 3. 湖南省农业科学院 长沙 410125)
收稿日期:2018-02-07;修回日期:2018-03-31 基金项目 :国家西甜瓜产业技术体系(CARS-25) 作者简介:孙 波,男,在读硕士研究生,主要从事瓜菜生物技术研究。E-mail:1021331329@ 通信作者 :孙小武 ,男 ,教授 ,博士生导师 ,主要从事瓜类育种及种子生产加工研究。E-mail :sunxiaowu2007@
摘 要:利用 SSR 分子标记技术对‘雪龙三号’西瓜品种及其亲本间的多态性进行了引物筛选和种子纯度研究。结果 表明,在供选的 23 对 SSR 核心引物中,有 2 对引物(BVWS02048 和 BVWS00106)在‘雪龙三号’杂交种及其亲本的带型 具有多态性。利用这 2 对引物对‘雪龙三号’进行纯度鉴定,鉴定结果均为 98%,与大田纯度检测结果吻合率达 99%, 说明 SSR 分子标记对‘雪龙三号’的纯度鉴定结果是可靠的。同时 2 对引物鉴定结果的一致也验证了品种的真实性。 关键词:西瓜;DNA 提取;SSR 标记;纯度鉴定
‘ 雪龙三号 ’西瓜品种是湖南地区主栽品种 ,耐 重茬,高抗枯萎病,笔者旨在通过对 23 对 SSR 核心 引物的筛选 ,对该品种的纯度进行快速 、准确的鉴 定 ,以缩短检测周期 、保障瓜农利益。同时为核心 SSR 标记在西瓜纯度鉴定方面的应用提供参考。
1 材料与方法
1.1 材料 ‘ 雪 龙 三 号 ’及 其 亲 本 由 湖 南 省 瓜 类 研 究 所 提
西瓜NAS_基因家族的全基因组鉴定与生物信息学分析

西瓜 NAS 家族编码的蛋白质进行三级结构预测ꎮ
AhNAS2 2 主要参与黄色条纹样蛋白 3 ( YSL3) 介导
1 3 西瓜 NAS 基因家族编码蛋白保守基序与基因结
NAS3 2 可能参与花生对镉的解毒机制ꎬ 为揭示 NAS
瓜 NAS 基因家族的启动子区含有激素应答、 光响应、 胁迫应答、 转录因子结合位点等多种顺式作用元件ꎻ 西瓜
基因组内未发现 NAS 基因的串联重复和片段复制ꎬ 但西瓜 NAS 基因与拟南芥、 番茄的 NAS 基因间分别存在 2 个
共线性基因对ꎮ 西瓜 NAS 基因家族的全基因组鉴定与生物信息学分析为进一步研究西瓜 NAS 基因的功能和分子
学分析ꎬ 共鉴定出 6 个 AhNAS 基因ꎬ 并通过对 Ah ̄
细胞定位、 顺式作用元件、 表达模式及花生响应缺铁
收稿日期: 2023-07-10
基金项目: 河南省科技攻关计划项目 ( 项目编号: 222102110447) ꎻ 河南省高等学校重点科研项目计划 ( 项目编号: 23A210032)
作者简介: 赵展 (1987-) ꎬ 女ꎬ 博士ꎬ 讲师ꎮ 研究方向: 作物遗传育种ꎮ
花生 NAS 基因家族进行了全基因组鉴定和生物信息
但与 Fe 的状态不相关ꎬ 进一步酵母单杂交和 ChIP 分
NAS 基因家族进化关系、 基因结构、 保守结构域、 亚
[4]
ꎮ NAS 基因参与植物体内多种作用机制ꎬ
在 hap5a 突变 体 中 发 现 AtNAS1 基 因 显 著 下 调 表 达ꎬ
析表明ꎬ HAP5A 直接结合到 NAS1 的启动子区域ꎬ 且
1 6 西瓜 NAS 家族的共线性分析
西瓜的基因概念

西瓜的基因概念西瓜,学名为西瓜属于葫芦科西瓜属的植物,是一种非常受欢迎的水果。
它以其甜美多汁的果肉和清凉的口感而备受喜爱。
然而,很多人对于西瓜的基因概念并不很了解。
基因是指遗传物质的基本单位,控制生物的遗传特征。
它们位于染色体上,通过DNA来传递信息。
基因决定了一个生物的各个性状,包括形态、结构、功能等。
西瓜的基因决定了它的各个特征。
例如,西瓜的颜色,可以是绿色、青色、黄色或红色。
这取决于基因是否携带了产生黄色素或红色素的信息。
一般来说,西瓜红色的品种比较受欢迎,因为它们看起来更加成熟和甜美,这是由于基因的不同导致的。
另外,西瓜的甜度也与基因有关。
甜度是由基因中携带的糖分合成与代谢相关的基因决定的。
有些基因会使西瓜中的糖分积累更多,从而使西瓜更甜。
这也解释了为什么不同品种的西瓜甜度不同。
此外,西瓜的大小和形状也与基因相关。
基因决定了西瓜的细胞分裂和扩张。
一些基因可能使细胞分裂更加频繁,有助于西瓜的生长,从而使西瓜变得更大。
另外,有些基因还会影响细胞的膨压,从而使西瓜形状不同。
除了以上特征外,西瓜的质地、口感、营养价值等也与基因有关。
基因控制了西瓜果肉中各种成分的合成和积累。
一些基因可能会使西瓜中的纤维素或碳水化合物含量增加,影响了口感和质地。
而营养价值方面,基因也会影响西瓜中维生素C和矿物质等成分的合成和积累。
通过研究基因,科学家们可以进一步了解西瓜的生长发育和性状变异的原因。
他们可以通过选择特定的基因组合来培育出更好的西瓜品种。
例如,通过选育含有更多甜度基因的品种,可以培育出更加甜美的西瓜。
通过了解基因的功能,科学家还可以进行基因编辑和转基因等技术操作,以改变西瓜的特征,使其更适合市场需求。
此外,研究西瓜的基因还有助于了解和保护遗传多样性。
通过研究不同品种的基因,可以揭示西瓜遗传多样性的范围和变异。
这有助于科学家们保护和利用这些多样性,以保证西瓜的繁育和适应性。
总之,西瓜的基因决定了它的各个特征,包括颜色、甜度、大小、形状、质地、口感和营养价值等。
西瓜高密度遗传图谱的构建及基因组Scaffolds 定位

西瓜高密度遗传图谱的构建及基因组Scaffolds 定位丁双玉,许勇,任毅(编译)(北京市农林科学院蔬菜研究中心北京100097)目的与意义:现代西瓜品种的遗传基础较窄,然而西瓜中有几个亚种具有广泛遗传多样性,它们可以成为改良西瓜品种的有用种质资源。
当前西瓜基因组资源和遗传图谱有限,并需要开发一种可用于鉴定西瓜染色体的细胞遗传学图谱,其能够划定栽培西瓜与其它相关的西瓜属植物之间存在的结构差异。
现如今,还没有任何关于西瓜基因组序列的高密度遗传连锁图谱的报道,致使西瓜在不同品种之间遗传多样性较低。
所以,构建高密度的西瓜遗传连锁图谱成为当下之需。
材料与方法:使用栽培种‘97103’和野生种‘PI 296341-FR ’杂交单粒传构建的103株系重组自交系(RILs )选择96株。
从栽培种‘97103’基因组组装scaffolds 和野生种‘PI 296341-FR ’的基因组序列读取来开发SSR (简单重复序列),InDel (插入缺失)和SV (结构变异)标记。
利用栽培种‘97103’和野生种‘PI 296341-FR ’构建的103株RIL 群体进行遗传作图。
使用JoinMap 进行图谱的构建,计算片段的重组率。
利用细菌人工染色体(BAC ),5S 和45S rDNA 探针,用于荧光原位杂交(FISH )。
结果与分析:SSR 、InDel 和SV 标记是基于栽培种‘97103’组装的353M 基因组scaffolds 序列和野生种‘PI 296341-FR ’的重测序数据开发的。
研究中共鉴定了13744个假设的SSR 位点。
在消除多种同源性的SSR 基因后,选择具有长重复基序的1877个特有SSR 用于与RIL 群体的亲本系(‘97103’和‘PI 296341-FR ’)的多态性分析。
在1877个SSR 中,787个(41.9%)在PCR 扩增后显示出栽培种‘97103’和野生种‘PI 296341-FR ’之间的多态性。
基于全基因组重测序构建西瓜高密度遗传图谱和果实相关性状的基因定位

基于全基因组重测序构建西瓜高密度遗传图谱和果实相关性状的基因定位李兵兵,刘文革(编译)(中国农业科学院郑州果树研究所郑州450009)目的与意义:遗传图谱是进行重要农艺性状QTL/基因定位、图位克隆、分子标记辅助育种等工作的重要工具。
长期的人工选择使得栽培种西瓜的遗传基础进一步狭窄,使得高效可获得的分子标记成为限制西瓜高密度遗传图谱构建的重要因素。
随着高通量测序技术的发展和西瓜参考基因组草图的组装完成,通过测序获得大量分子标记以构建高密遗传图谱成为可能。
通过全基因重测序构建高密度遗传图谱,可以更快更准确的检测重组断点,开发大量适于遗传图谱构建的分子标记。
最近几年,陆续有多张西瓜遗传图谱发表。
但这些图谱大都基于简化基因组测序,遗传图谱的饱和度相对不足;作图群体多为临时性分离群体,不能进行重复试验,影响后续基因定位工作的准确性。
笔者构建了重组自交系作为永久性作图群体,通过对其进行全基因组重测序构建了一张西瓜高密度遗传图谱,并对果皮颜色、种皮颜色和果实苦味3个性状进行了初定位,以期用于西瓜分子遗传育种等工作。
材料与方法:以具有显著性状差异的自交系材料‘9904’和‘Handel ’为亲本,经过连续7代自交,构建了含126个单株的重组自交系。
‘9904’具有墨绿色果皮、浅黄色种皮、苦味果实;‘Handel ’具有浅绿色果皮、黑色种皮、无苦味果实。
采集西瓜植株幼嫩叶片,提取植物基因组DNA ,质量检测合格的DNA 将用于测序建库。
高质量的DNA 被随机打断为200~500bp 的片段,然后经过DNA 片段末端修复、3’端加A 、添加测序接头和进行纯化、最后经PCR 扩增等完成测序文库的构建。
文库经严格的质量检测后通过Illumina HiSeqTM 测序平台进行测序。
完成标记的填补和校正后,根据子代重组情况进行bin 划分。
将各样本按照染色体物理位置排列整齐,当任何样本出现分型转变时认为出现重组断点,然后将重组断点间的SNP 化为一个bin 中。
1 197份西瓜种质资源遗传多样性和群体结构分析及核心种质构建
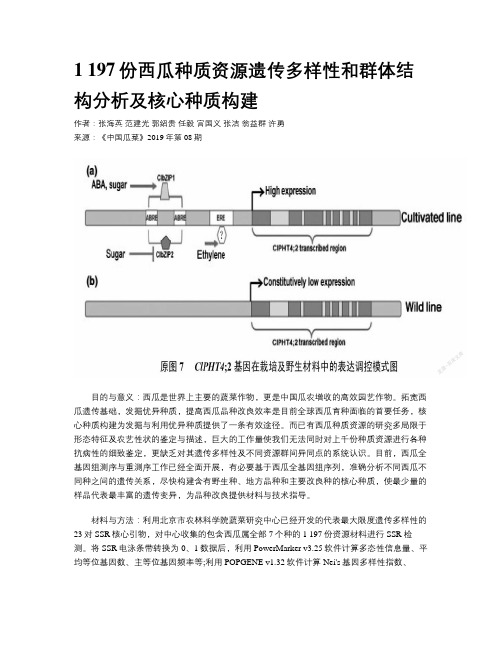
1 197份西瓜种质资源遗传多样性和群体结构分析及核心种质构建作者:张海英范建光郭绍贵任毅宫国义张洁翁益群许勇来源:《中国瓜菜》2019年第08期目的与意义:西瓜是世界上主要的蔬菜作物,更是中国瓜农增收的高效园艺作物。
拓宽西瓜遗传基础,发掘优异种质,提高西瓜品种改良效率是目前全球西瓜育种面临的首要任务,核心种质构建为发掘与利用优异种质提供了一条有效途径。
而已有西瓜种质资源的研究多局限于形态特征及农艺性状的鉴定与描述,巨大的工作量使我们无法同时对上千份种质资源进行各种抗病性的细致鉴定,更缺乏对其遗传多样性及不同资源群间异同点的系统认识。
目前,西瓜全基因组测序与重测序工作已经全面开展,有必要基于西瓜全基因组序列,准确分析不同西瓜不同种之间的遗传关系,尽快构建含有野生种、地方品种和主要改良种的核心种质,使最少量的样品代表最丰富的遗传变异,为品种改良提供材料与技术指导。
材料与方法:利用北京市农林科学院蔬菜研究中心已经开发的代表最大限度遗传多样性的23对SSR核心引物,对中心收集的包含西瓜属全部7个种的1 197份资源材料进行SSR检测。
将SSR电泳条带转换为0、1数据后,利用PowerMarker v3.25软件计算多态性信息量、平均等位基因数、主等位基因频率等;利用POPGENE v1.32软件计算Nei's基因多样性指数、Shannon信息指数、观测杂合度值、期望杂合度值等;利用STRUCTURE v.2.3软件进行群体结构分析;利用PowerCore软件进行核心种质构建。
结果与分析:(1)遗传多样性分析:采用23对SSR核心引物对1 197份资源材料进行扩增,共检测到133个等位基因,单个引物检测到的等位基因数在4~9个,平均为6.05个。
PIC值在0.35~0.70之间,平均为0.49。
1 197份材料,Nei’s基因多样性指数在0.38~0.74之间,平均为0.56。
Shannon信息指数平均为0.99。
无籽西瓜育种原理高中

无籽西瓜育种原理高中无籽西瓜是一种经过育种、选育而获得的水果品种,它的产生离不开先进的遗传学知识和育种技术。
本文将从生物学方面介绍无籽西瓜的育种原理。
无籽西瓜是如何产生的?在传统的西瓜品种中,每个果实内部都会有大量的黑色籽粒,其形成由物种基因组中的两副一样的染色体控制着,在繁殖的过程中,某些变异的基因会导致某些果实形成起来种子非常稀疏或者根本没有籽粒,这就是所谓的无籽西瓜。
但是,这种产生无籽西瓜的方法不稳定,产量也非常低,所以育种者就通过育种筛选的方法来培育无籽西瓜品种。
目前,育种者们通过选择、杂交等方法,得到了一些高产、口感好的无籽西瓜品种。
1. 无籽基因的掌握无籽西瓜的产生离不开一个特殊的基因,它被称为fs程度基因。
该基因是显性遗传的,只要母本和父本中任何一个带有该基因,那么后代就能表现出无籽的特征。
但是,这个基因具有一定的难度,可以说是育种的难点,因为要让基因充分发挥作用,获得无籽西瓜并不容易。
为了解决这个问题,育种者需要进行无籽基因的掌握和利用。
具体的方法是运用分子生物学技术,通过PCR扩增无籽基因区域,并进行基因克隆、构建表达载体等措施,进一步增强无籽基因的功能,促进无籽西瓜品种的形成。
2. 种间杂交育种种间杂交是育种者采用的一种有效的育种方法。
将两个或两个以上不同物种的优良基因进行组合杂交,从而获得优良品种。
无籽西瓜的育种就采用了这种方法。
育种者们选取了没籽瓜、长瓜、香瓜等不同的瓜类进行杂交,以期达到无籽基因的组合效应。
种间杂交育种不仅提高了产量,还能改良品质和抗病能力,同时保持物种的遗传稳定性和适应性。
3. 选用高效育种材料育种材料的选用直接关系到育种的成败。
在育种无籽西瓜中,育种者们首先要选用无籽基因丰富的育种材料,这样能减少不必要的杂交背景干扰。
而且,要选取高产、口感好、品质稳定的材料作为杂交的父本或母本,才能保证获得好的后代。
总之,无籽西瓜的育种过程离不开遗传学和育种技术。
通过对无籽基因的掌握和利用、种间杂交和选用高效育种材料等手段,进行有效育种研究,培育出品质优良、产量高的无籽西瓜品种,为人们的日常生活带来了更便捷的选择。
西瓜抗病基因同源序列的克隆与分析

西瓜 【 C i t r u l l u s l a n a t u s( T h u n b . ) ] 栽 培 历史悠 久 ,分 布 广 泛 ,经济价 值 巨大 。据联 合 国粮 农组织 ( F A O) 统计 是世 界 第五大果 品,我 国是西瓜最大的栽培和消费国 “ 】 。近年来 西瓜栽培连作现象 日趋严 重,病害逐年加 重 ,产量和品质下
0 . 5 L E x T a q D NA聚 合 酶。 反应 条 件 为 9 4℃预 变 性
5 ai r n,此 后 进行 9 4 ℃3 0 S 、4 5  ̄ C 4 0 S 、7 2 ℃1 ai r n的 3 5 个
防治 ,嫁接会对 品质产生一 定影响 ,同时需不断筛选砧木 , 而 农药使用易 引起病源抗 药性并污染环境 ,在 高温高湿地区 防治效果不佳 。培育抗病性强的新品种是解决这一 问题的最 根 本措施 ,但西瓜遗传基础狭 窄,抗病资源缺乏 ,抗病性遗
连接 p MDI 8 - T载体 ,测序。
1 . 2 . 3 西瓜 R G A s生物信 息 学分 析
西 瓜 RG As 生物 信息
学分析利用 D NA MAN 6 . 0和 B l a s t 进行 。
2 结果与分析
2 . 1 西瓜 R G A s的获得
试 验中以西瓜基 因组 D NA 为模板进行 PC R 扩增得到
CH I NE SE HOR T I CUL T UR E AB S T R AC T S
西瓜抗病基 因同源序列 的克 隆与分析
奚玉培 ,谢新 蕊 ,高 强 ,张志忠
( 福建农林大 学 园艺学院 , 福建 福州 5 5 0 0 0 2 )
摘 要 :以植 物丝氨酸 /苏氨酸蛋 白激酶 类 ( S T K ) 抗病基 因产物催化结构域 l 的保 守氨基酸序列 设计 简并引物 ,以
基于四个遗传群体的西瓜整合图谱 构建及重要经济性状QTL定位

基于四个遗传群体的西瓜整合图谱构建及重要经济性状QTL定位作者:任毅张岩宫国义张海英郭绍贵孙宏贺蔡万涛张洁许勇来源:《中国瓜菜》2019年第08期目的与意义:人们长期对于现代栽培西瓜优良性状的选育使得西瓜遗传基础变得狭隘。
野生亚种是西瓜育种中优异基因的重要来源,但将野生亚种中的优良性状基因转入到现代栽培西瓜中的成功率很低。
由于缺乏高质量的遗传图谱致使分子标记辅助选择(MAS)在西瓜中的应用尚未实现,难以在图谱之间进行整合和比较分析。
本研究的目的是整合现有的利用单核苷酸多态性(SNP)和简单重复序列(SSR)标记构建西瓜整合遗传图谱。
材料与方法:使用4种不同杂交组合的西瓜群体作为试验材料,其分别为栽培品种‘97103’和野生种‘PI 296341-FR’杂交产生的103株系重组自交系(RIL)F8群体,以及由北美栽培种‘ZWRM50’和野生种‘244019’(ZWRM × citron)杂交产生182单株F2群体,164株系 [栽培种Klondike Black Seeded ×栽培种New Hampshire Midget(KBS × NHM)] RIL群体,还有187单株[栽培种Strain II ×半野生种PI 560023 (SII×egusi)] 的F2群体。
通过将SNP标记添加到bin 来构建整合图谱。
使用不同的计算机软件来比较共线性数据、测量糖含量、计算遗传力(h2)置信区间(CI)、分析性状间的遗传相关性、联合分析单一环境和多环境互作效应。
结果与分析:作者整合了SSR(简单重复序列)标记698个,InDel(插入缺失标记)标记219个, SV(结构变异)标记36个和SNP标记386个来构建整合图谱。
该图谱包含1 339个标记,跨越798 cM,平均标记间隔为0.6 cM。
西瓜基因组的大小为425 Mbp,此处的图谱每个标记有317 Kb的平均物理区间,使其成为迄今为止最饱和的西瓜数据图谱。
西瓜遗传转化体系的优化

中国瓜菜2024,37(3):9-19西瓜(Citrullus lanatus)是最受欢迎的园艺作物之一,因果肉多汁和口感甜美而备受消费者青睐。
西瓜栽培历史悠久,地域广泛,近年来随着种植面积和产量的增加,个性化育种需求也在不断增加。
高效稳定的遗传转化技术对于改良西瓜品种具有巨大潜力[1]。
大量研究表明,西瓜是一种较难进行西瓜遗传转化体系的优化李鹏飞,陈子豪,张敏娟,豆峻岭,杨森,刘东明,牛欢欢,杨路明(河南农业大学园艺学院郑州450046)摘要:以8种基因型西瓜为试验材料,通过统计不同基因型材料的发芽率、不定芽诱导率,筛选出再生能力较强的基因型,并以其为材料对种子播种后培养时间、外植体造伤方式、菌液浓度、共培养时间及侵染方式等进行优化筛选,旨在建立高效的西瓜遗传转化体系。
结果表明,在播种2d时,YL、CYY298、CYY171及CYY137之间的种子发芽率差异不显著且均显著高于其他材料,但在不同浓度GA3诱导下,YL外植体的不定芽诱导率均最高;以YL材料为基础进行体系优化,发现播种2d后的种子状态最适于侵染;刀片划伤后的外植体荧光亮度最强,荧光率最高达74.9%;菌液浓度OD600=0.8,共培养时间为3d时,外植体分化状态最佳且荧光率最高为70.3%;真空负压方式可以显著提高侵染效率,外植体荧光率可达71.8%,愈伤组织诱导率高达60.2%;分化培养基中添加0.5mg·L-1的GA3后不定芽的诱导率最高为74%;生根培养基中添加0.5mg·L-1的IBA时生根率最高,平均生根数最多。
关键词:西瓜;遗传转化;转化效率;体系优化中图分类号:S651文献标志码:A文章编号:1673-2871(2024)03-09-11Optimization of the genetic transformation system in watermelonLI Pengfei,CHEN Zihao,ZHANG Minjuan,DOU Junling,YANG Sen,LIU Dongming,NIU Huan-huan,YANG Luming(Horticulture College of Henan Agricultural University,Zhengzhou450046,Henan,China)Abstract:In this study,eight watermelon genotypes were used as experimental materials,the germination rates and adventitious buds induction rates of different genotypes were counted and analyzed to screen out the optimal genotype with strong regeneration ability.The efficient watermelon genetic transformation system was optimized and screened through different sowing times,explant injury methods,bacterial liquid concentration,co-culture time,infection methods and other factors.The results showed that the seed germination rates of YL,CYY298,CYY171and CYY137had no dif-ference and were significantly higher than that of other materials at2days of seeding,but the germination induction rate of YL was the highest at different concentrations of GA3.The system was optimized based on YL material,and it was found that the seed germination state was the best and most suitable for infection after2days of sowing.The fluorescence brightness of the explants after scratching the blade was the strongest,and the fluorescence rate was up to74.9%.When the concentration of bacterial solution was OD600=0.8and the co-culture time was3days,the explants were in the best differentiation state and the fluorescence rate was the highest at70.3%.The infection efficiency can be significantly improved by negative pressure infection,and the fluorescence rate of explants can reach71.8%,and the callus induction rate can reach60.2%.The induction of adventitious shoots was up to74%after the addition of0.5mg·L-1GA3to the differentiation medium.The highest rooting rate and the highest average number of roots were observed when0.5mg·L-1 of IBA was added to the rooting medium.Key words:Watermelon;Genetic transformation;Transformation efficiency;System optimization收稿日期:2023-10-10;修回日期:2024-01-04基金项目:河南省科技研发计划联合基金(222103810009);河南省重大科技专项(221100110400);河南省农业良种联合攻关项目(2022010503)作者简介:李鹏飞,男,在读硕士研究生,研究方向为蔬菜遗传育种。
西瓜SSR标记中不同类型样品的DNA提取分析

西瓜SSR标记中不同类型样品的DNA提取分析DNA提取是分子生物学和遗传学研究中的重要步骤之一,它是从样品中分离和纯化DNA 分子的方法。
在蔬果中,西瓜是一种常见的水果,其DNA提取是了解西瓜的遗传特征和基因组组成的重要手段。
西瓜(SSR)标记是一种利用简单重复序列(Simple Sequence Repeat,SSR)进行基因型鉴定和遗传标记的方法。
SSR标记是通过寻找并分析基因组中的多态性重复序列来进行分析。
在DNA提取分析中,通过对不同类型的样品进行DNA提取,可以更好地了解西瓜的遗传多样性和品种分类。
在西瓜中,常见的DNA提取方法有CTAB法、硅胶膜法等。
CTAB法是一种重要且广泛应用的DNA提取方法。
该方法通过利用CTAB(十六烷基三甲基溴化铵)溶解细胞壁,并与蛋白质和其他污染物结合,最终得到纯化的DNA。
在进行DNA提取分析之前,首先需准备好实验所需的材料和试剂。
常见的实验材料包括西瓜组织样品、琼脂糖、酚/氯仿、异丙醇等。
试剂则包括CTAB提取缓冲液、TE缓冲液、RNase A酶、蛋白酶K等。
具体的实验步骤如下:1. 样品采集:从西瓜中选择不同类型的组织样品,如叶片、果肉等,保持样品新鲜且无污染。
2. 组织粉碎:将样品放入液氮中进行快速冷冻,然后用研钵和研钉将样品粉碎成细小的颗粒。
3. 去除其他污染物:加入适量的CTAB提取缓冲液,并与细胞中的蛋白质和RNA结合,形成沉淀。
4. DNA溶解:将沉淀溶解于TE缓冲液中,并加入RNase A酶进行消化,以去除RNA的污染。
5. DNA纯化:使用酚/氯仿提取法将DNA与其他溶液中的杂质分离。
6. DNA沉淀:加入等体积的异丙醇,并在低温条件下进行DNA沉淀。
7. DNA洗涤:用70%的乙醇洗涤DNA沉淀物,以去除残留的盐离子和乙醇。
8. DNA溶解:最后用TE缓冲液溶解DNA,通过测量吸光度来判断DNA的浓度和纯度。
完成以上步骤后,可以对提取的DNA进行进一步的分析,如PCR扩增、SSR标记分析等。
无籽西瓜原理

无籽西瓜原理
无籽西瓜是通过基因改良技术来实现的,其原理主要是通过对西瓜的生殖细胞进行基因操作,使其在生长过程中不产生籽粒。
具体而言,科学家们针对西瓜的籽粒形成机制进行研究,并找到关键基因。
然后,他们使用基因编辑技术,如CRISPR/Cas9,对这些关键基因进行修改或静默,以阻断籽粒的形成过程。
通过这种基因编辑技术,科学家们成功地将无籽基因引入到西瓜的基因组中。
当这些基因被转录和翻译成蛋白质时,它们会影响植物体内的发育过程,使得西瓜果实在成熟时不会形成籽粒。
此外,为了增加基因编辑的成功率,科学家们通常会选择在种植过程中使用催花剂来促使植物快速进入开花期,以便更早进行基因编辑。
通过这种基因编辑技术,无籽西瓜不仅没有籽粒,而且与传统西瓜相比保持了更多的甜度和口感。
这使得无籽西瓜成为了广受欢迎的水果之一,深受消费者的喜爱。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
©2012 N a t u r e A m e r i c a , I n c . A l l r i g h t s r e s e r v e d .A r t i c l e sWatermelon (C. lanatus ) is an important cucurbit crop, account-ing for 7% of the worldwide area devoted to vegetable production. The annual world production of watermelon isabout 90 million tons, making it among the top five most consumed fresh fruits (/). Watermelon belongs to the xerophytic genus Citrullus Schrad. ex Eckl. et Zeyh. of the botanical family Cucurbitaceae . The center of diversity and possible center of origin of Citrullus is south-ern Africa 1. C. lanatus includes three subspecies: C. lanatus subsp. lanatus , which represents a group of ancient cultigens, the ‘tsamma’ or ‘citron’ watermelon, that naturally thrives in southern Africa; C. lanatus subsp. mucosospermus , which represents the egusi watermelongroup that has large edible seeds with a fleshy pericarp 2; and C. lanatussubsp. vulgaris , which represents the sweet (dessert) watermelongroup that gave rise to the modern cultivated watermelon 3.The large edible watermelon fruits contribute to the diets of con-sumers throughout the world. Although comprised mainly of water (often over 90%), watermelon also contains important nutritional compounds, including sugars, lycopene and cardiovascular health–p romoting amino acids, such as citrulline, arginine and glutathione 4–6. Watermelon and cucurbit species in general have unique developmental mechanisms that facilitate the rapid growth and formation of giant pepo fruits 7. Fruits of modern watermelon varieties are diverse in shape, size, color, texture, flavor and nutrient composition. However, years of cultivation and selection targeting yield and desirable fruitThe draft genome of watermelon (Citrullus lanatus ) and resequencing of 20 diverse accessionsShaogui Guo 1,2,17, Jianguo Zhang 3,4,17, Honghe Sun 1,2,5,17, Jerome Salse 6,17, William J Lucas 7,17, Haiying Zhang 1, Yi Zheng 2, Linyong Mao 2, Yi Ren 1, Zhiwen Wang 3, Jiumeng Min 3, Xiaosen Guo 3, Florent Murat 6, Byung-Kook Ham 7, Zhaoliang Zhang 7, Shan Gao 2, Mingyun Huang 2, Yimin Xu 2, Silin Zhong 2,Aureliano Bombarely 2, Lukas A Mueller 2, Hong Zhao 1, Hongju He 1, Yan Zhang 1, Zhonghua Zhang 8, Sanwen Huang 8, Tao Tan 9, Erli Pang 9, Kui Lin 9, Qun Hu 10, Hanhui Kuang 10, Peixiang Ni 3,4, Bo Wang 3,Jingan Liu 1, Qinghe Kou 1, Wenju Hou 1, Xiaohua Zou 1, Jiao Jiang 1, Guoyi Gong 1, Kathrin Klee 11, Heiko Schoof 11,Ying Huang 3, Xuesong Hu 3, Shanshan Dong 3, Dequan Liang 3, Juan Wang 3, Kui Wu 3, Yang Xia 1, Xiang Zhao 3,Zequn Zheng 3, Miao Xing 3, Xinming Liang 3, Bangqing Huang 3, Tian Lv 3, Junyi Wang 3, Ye Yin 3, Hongping Yi 12, Ruiqiang Li 13, Mingzhu Wu 12, Amnon Levi 14, Xingping Zhang 1, James J Giovannoni 2,15, Jun Wang 3,16, Yunfu Li 1, Zhangjun Fei 2,15 & Yong Xu 1Watermelon, Citrullus lanatus , is an important cucurbit crop grown throughout the world. Here we report a high-quality draft genome sequence of the east Asia watermelon cultivar 97 03 (2n = 2× = 22) containing 23,440 predicted protein-coding genes. Comparative genomics analysis provided an evolutionary scenario for the origin of the watermelon chromosomes derived from a 7-chromosome paleohexaploid eudicot ancestor. Resequencing of 20 watermelon accessions representing three different C. lanatus subspecies produced numerous haplotypes and identified the extent of genetic diversity and population structure of watermelon germplasm. Genomic regions that were preferentially selected during domestication were identified. Many disease-resistance genes were also found to be lost during domestication. In addition, integrative genomic and transcriptomic analyses yielded important insights into aspects of phloem-based vascular signaling in common between watermelon and cucumber and identified genes crucial to valuable fruit-quality traits, including sugar accumulation and citrulline metabolism.1National Engineering Research Center for Vegetables, Beijing Academy of Agriculture and Forestry Sciences, Key Laboratory of Biology and Genetic Improvementof Horticultural Crops (North China), Beijing, China. 2Boyce Thompson Institute for Plant Research, Cornell University, Ithaca, New York, USA. 3Beijing Genomics Institute–Shenzhen, Shenzhen, China. 4T-Life Research Center, Fudan University, Shanghai, China. 5College of Plant Science and Technology, Beijing University of Agriculture, Beijing, China. 6Institut National de la Recherche Agrinomique, Unités Mixtes de Recherche 1095, Genetics, Diversity and Ecophysiology of Cereals, Clermont-Ferrand, France. 7Department of Plant Biology, College of Biological Sciences, University of California, Davis, California, USA. 8Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, China. 9College of Life Sciences, Beijing Normal University, Beijing, China. 10College of Horticulture and Forestry, Huazhong Agriculture University, Wuhan, China. 11Institut für Nutzpflanzenwissenschaften und Ressourcenschutz Crop Bioinformatics, University of Bonn, Bonn, Germany. 12Xinjiang Academy of Agricultural Sciences, Urumqi, China. 13Beijing Novogene Bioinformation Technology Co. Ltd, Beijing, China. 14US Department of Agriculture (USDA), Agricultural Research Service, US Vegetable Lab, Charleston, South Carolina, USA. 15USDA Robert W. Holley Center for Agriculture and Health, Ithaca, New York, USA. 16Department of Biology, University of Copenhagen, Copenhagen, Denmark. 17These authors contributed equally to this work. Correspondence should be addressed to Yong Xu (xuyong@ ), Z.F . (zf25@ ), Y.L. (liyunfu@ ) or Jun Wang (wangj@ ).Received 4 June; accepted 22 October; published online 25 November 2012; doi:10.1038/ng.2470©2012 N a t u r e A m e r i c a , I n c . A l l r i g h t s r e s e r v e d .A r t i c l e squalities have narrowed the genetic base of watermelon 8, resulting in a major bottleneck in watermelon improvement.Knowledge of genome sequences is indispensable for basic biological research and crop improvement. Here we report a high-quality genome sequence of an east Asia watermelon cultivar, 97103 (2n = 2× = 22), and resequencing of 20 watermelon accessions span-ning the genetic diversity of C. lanatus . Our comprehensive genomic and transcriptome analyses provide insights into the structure and evolution of the watermelon genome, the genetic diversity and struc-ture of watermelon populations and the molecular mechanisms of important biological processes such as fruit quality and phloem-based vascular signaling. Together, these results will assist in identifying and accessing the plethora of watermelon genetic diversity that remains to be tapped for biological discovery and crop improvement.RESULTSGenome sequencing and assemblyWe selected the Chinese elite watermelon inbred line 97103 for genome sequencing. We generated a total of 46.18 Gb of high-quality genomic sequence using Illumina sequencing technology (Supplementary Table 1), representing 108.6-fold coverage of the entire watermelon genome, which has an estimated genome size of ~425 Mb on the basis of our 17-mer depth distribution analysis of the sequenced reads (Supplementary Fig. 1) and an earlier flow cytometry analysis 9. De novo assembly of the Illumina reads resulted in a final assembly of 353.5 Mb, representing 83.2% of the water-melon genome. The assembly consists of 1,793 scaffolds (≥500 bp) with N50 lengths of 2.38 Mb and 26.38 kb for the scaffolds and con-tigs, respectively (Supplementary Table 2). A total of 234 scaffolds covering approximately 330 Mb (93.5% of the assembled genome) were anchored to the 11 watermelon chromosomes, among which 126 and 94 scaffolds accounting for 70% and 65% of the assembled genome were ordered and oriented, respectively 10.We sought to determine why 16.8% of the genome was not covered by our genome assembly by aligning unassembled reads (17.4% of the total reads) to the assembled genome with less stringent criteria (Supplementary Note and Supplementary Table 3). We found that the unassembled genome regions are composed primarily of sequences that are similar to those of the assembled regions. Distribution of the unassembled reads on the watermelon chromosomes showed the same pattern as that for transposable elements (Fig. 1a and Supplementary Fig. 2). We identified three major repeat units from the unassembled sequences on the basis of their substantial read depths and sequence similarities to centromeres, telomeres and ribosomal DNA (rDNA) clusters. We further confirmed the nature of these repeats by FISH (Fig. 1b –d ). Together these results support the notion that under-estimation of the repeat proportion has an important role in the unassembled component of de novo genome assemblies, especially those generated using next-generation sequencing technologies 11–18.We further evaluated the quality of the assembled watermelon genome using approximately one million ESTs, four completely sequenced BACs and paired-end sequences of 667 BAC clones. Our analyses supported the high quality of the watermelon genome assembly (Supplementary Note , Supplementary Tables 4–6 and Supplementary Figs. 3 and 4), which is favorably comparable to several other recently published plant genomes 11–18 using next- generation sequencing technologies (Table 1).Repeat sequence annotation and gene predictionTransposable elements are major components of eukaryotic genomes. We identified a total of 159.8 Mb (45.2%) of the assembled watermelongenome as transposable element repeats. Among these repeats,68.3% could be annotated with known repeat families. The long terminal repeat (LTR) retrotransposons, mainly Gypsy-type and Copia-type LTRs, are predominant. The distribution of transposable element divergence rates showed a peak at 32% (Supplementary Fig. 5). We further identified 920 (7.8 Mb) full-length LTR retro-transposons in the watermelon genome. We found that over the past 4.5 million years, LTR retrotransposons accumulated much faster in watermelon than in cucumber 14 (Supplementary Fig. 6) such that the overall difference in their genome sizes may reflect the differential LTR retrotransposon accumulation.We predicted 23,440 high-confidence protein-coding genes in the watermelon genome (Supplementary Table 7), which is close to the number of genes predicted in the cucumber genome 19. Approximately 85% of the watermelon predicted genes had either known homologs or could be functionally classified (Supplementary Table 8). In addition, we also identified 123 ribosomal RNA (rRNA), 789 transfer RNA, 335 small nuclear RNA and 141 microRNA genes (Supplementary Table 9).In accordance with previously reported plant genomes, the water-melon protein-coding genes showed a clear enrichment pattern within subtelomeric regions. In contrast, the transposable element–related fraction of the genome was located primarily within the pericentro-meric and centromeric regions. The short arms of chromosomes 4, 8 and 11 are highly enriched with repeat sequences (Supplementary Fig. 7). The 97103 genome contained one 5S and two 45S rDNA clusters on the short arm of chromosomes 4 and 8 (ref. 10). Using FISH, we further investigated rDNA patterns in genomes of 20 rep-resentative watermelon accessions (Supplementary Table 10). The number and location of 5S and 45S rDNA sites in the genomes of the ten modern cultivated (C. lanatus subsp. vulgaris ) and six semiwild watermelon (C. lanatus subsp. mucosospermus ) were identical to those in the 97103 genome, whereas the genomes of the four more distantly related wild watermelon (C. lanatus subsp. lanatus ) contained one 45S and two 5S rDNA sites, with the additional 5S rDNA site on the short arm of chromosome 11 (Supplementary Fig. 8). These results indicate that chromosome fusion, fission and transposition of rDNA might occur during the evolution of C. lanatus species. Our analysis alsoconfirmed the phylogenetic relationship of these three watermelonaChr1TEs100Unassembledb c dFigure 1 Distribution of unassembled reads on chromosome 1 and FISH patterns of probes from three repeat units related to the centromere, telomere and 45S rDNA clusters. (a ) Distribution of unassembled reads on chromosome 1. The distribution of unassembled reads on the other ten chromosomes is shown in supplementary Figure 2. TEs, transposable elements. (b ) FISH of watermelon chromosomes stained with4′-6-diamidino-2-phenylindole (DAPI, blue) using probes from repeat units similar to the centromere (pink). (c ) FISH using probes from repeat units similar to the telomere (red). (d ) FISH using probes from repeat units similar to the rDNAs (green, 45S; pink, 5S). Scale bars, 5 µm.©2012 N a t u r e A m e r i c a , I n c . A l l r i g h t s r e s e r v e d .A r t i c l e ssubspecies 20 and supported the hypothesis that C. lanatus subsp. mucosospermus is the recent ancestor of C. lanatus subsp. vulgaris .Cucurbit genome evolutionGenome-wide duplication in angiosperms is common and represents an important molec-ular mechanism that has shaped modern plant karyotypes. In the watermelon genome,we identified seven major triplications thatcorresponded to 302 paralogous relationshipscovering 29% of the genome (Fig. 2a ). These ancestral triplicates corresponded to theshared paleohexaploidization event (refer-enced as γ) reported for eudicots 21 that datesback to 76–130 million years ago. This would be well in advance of the cucurbit genome speciation event that occurred 15–23 million years ago (Supplementary Fig. 9).To access the nature of evolutionary events leading to modern cucurbit genome structures, we analyzed the syntenic relationships between watermelon, cucumber 19, melon 22 and grape 21. We chose grape as the reference, as it is known to be the closest relative to the eudicot ancestor structured in seven protochromosomes 23. We iden-tified a total of 3,543 orthologous relationships covering 60% of the watermelon genome. We then investigated the detailed chromosome-to-chromosome relationships within the Cucurbitaceae family and identified orthologous chromosomes between watermelon, cucumber and melon (Fig. 2b ). The complicated syntenic patterns illustrated as mosaic chromosome-to-chromosome orthologous relationships unveiled a high degree of complexity of chromosomal evolution and rearrangement among these three important crop species of the Cucurbitaceae family.Integration of independent analyses of duplications within, and syntenies between, the four eudicot genomes (watermelon, cucumber, melon and grape) led to the precise characterization in watermelon of the seven paleotriplications identified recently as the basis for the definition of seven ancestral chromosomal groups in eudicots 24. On the basis of the ancestral hexaploidization (γ) reported for the eudicots, we propose an evolutionary scenario that has shaped the 11 watermelon chromosomes from the 7-chromosome eudicot ancestors through the 21 paleohexaploid intermediates. We suggest that the transition from the 21-chromosome eudicot intermediate ancestors involved 81 fissions and 91 fusions to reach the modern 11-chromosome structure of watermelon, which is represented as a mosaic of 102 ancestral blocks (Fig. 2c ).Assessment of genetic diversity in watermelon germplasm We selected 20 representative watermelon accessions for genome rese-quencing. These included ten cultivated accessions representing the major varieties of C. lanatus subsp. vulgaris (five east Asia and five America ecotypes), six semiwild C. lanatus subsp. mucosospermus and four wild C. lanatus subsp. lanatus (Supplementary Table 10 and Supplementary Fig. 10). We sequenced these accessions to between 5× and 16× coverage and mapped the short reads to the genome of 97103 (Supplementary Table 11). We identified a total of 6,784,860 candidate SNPs and 965,006 small insertions/deletions (indels) among the 20 resequenced lines and 97103. The majorv ariations existed between C. lanatus subsp. lanatus and the other two s ubspecies, whereas the variation within the cultivated watermelon, especially C. lanatus subsp. vulgaris America ecotype, was relatively low (Supplementary Table 12). The accuracies of our SNP and indel calling were 99.3% and 98%, respectively, as indicated by Sanger sequencing (Supplementary Note and Supplementary Table 13). This extensive watermelon genome variation dataset, covering a wide spectrum of watermelon genetic diversity, represents a valuable resource for biological discovery and germplasm improvement.We evaluated the genetic diversity of the watermelon population using two common summary statistics, π and θw values 25. The esti-mated amount of diversity in watermelon (Supplementary Table 14) was substantially lower than that found in maize 26, soybean 27 and rice 28. Wild watermelon contains greater genetic diversity, indicat-ing additional genetic opportunity for watermelon improvement. We also investigated the population structure and relationships among the watermelon accessions through construction of a neighbor-joining tree (Fig. 3a ) and principal component analysis (PCA) (Fig. 3b ). Both analyses indicated the close relationship between C. lanatus subsp. vulgaris and C. lanatus subsp. mucosospermus (Supplementary Note ). Additional analysis of population structure using the FRAPPE pro-gram 29 with K (the number of populations) set from 2 to 5 identified a new subgroup within the C. lanatus subsp. mucosospermus group (when K = 5) and admixtures between C. lanatus subsp. vulgaris and C. lanatus subsp. mucosospermus (Fig. 3c and Supplementary Note ). The new subgroup shows some characteristics of the cultivated watermelon, such as soft flesh texture, pink flesh color and relatively high sugar con-tent (Supplementary Table 10 and Supplementary Fig. 10). Together these results offer further support for our proposed evolutionary scenario of C. lanatus subsp. mucosospermus to C. lanatus subsp. vulgaris derived from the FISH analysis of chromosomal rDNA distribution.We next scanned the genome for regions with the highest differ-ences of genetic diversity (πmucosospermus /πvulgaris ) between C. lanatus subsp. mucosospermus and C. lanatus subsp. vulgaris . These regionsrepresent potential selective sweeps during watermelon domesti-cation, as modern watermelon cultivars are thought to have been domesticated from C. lanatus subsp. mucosospermus . We identified a total of 108 regions (7.78 Mb in size) containing 741 candidate genes (Fig. 4 and Supplementary Table 15). Although gene complements in these regions could have been affected by genetic hitchhiking, we identified biological processes significantly enriched in candidate genes that were related to important selected traits when compared to the whole genome, including regulation of carbohydrate use, sugar-mediated signaling, carbohydrate metabolism, response to sucrose stimulus, regulation of nitrogen-compound metabolism, cellular response to nitrogen starvation and growth (Supplementary Note and Supplementary Tables 16–18).It is noteworthy that certain noncentromeric regions, especially a large region on chromosome 3 (from ~3.4 Mb to ~5.6 Mb), have particularly high nucleotide divergence only among C. lanatus subsp.table 1 comparison of watermelon genome assembly with other plant genomesSpeciesGenome assembly size (Mb)Estimated genome size (Mb)Genome covered by assembly (%)N50 scaffold (kb)N50 contig (kb)Sequencing technologiesWatermelon 353.342583.22,378.226.4Illumina Date palm38165857.930.5 6.4Illumina Pigeonpea605.8833.172.7516.122IlluminaCucumber243.536766.3226.519.8Sanger+IlluminaApple 603.9742.381.3NA 16.2Sanger+454Strawberry 201.924084.11,360NA 454+Illumina+SOLiDCacao 326.943076473.819.8Sanger+Illumina+454Chinese cabbage 283.852953.61,971.127.3Sanger+IlluminaThellungiella parvula 137.116085.75,290NA Illumina+454NA, not applicable.©2012 N a t u r e A m e r i c a , I n c . A l l r i g h t s r e s e r v e d .A r t i c l e smucosospermus accessions (Fig. 4). A previous report described a similar finding in three different rice crosses, and it was suggested that these population-specific high-divergence regions were highly associated with genes involved in reproductive barriers 30. We ana-lyzed genes in the large high-diversity region on chromosome 3 and, indeed, found that the most significantly enriched gene categories were recognition of pollen and the pollen-pistil interaction; both of these gene categories are related to reproductive barriers (Supplementary Table 19). In addition, we determined that the region contained a large cluster of 12 tandemly arrayed S-locus protein kinase genes, which are involved in reproductive barriers 31. The high nucleotide divergence of reproductive barrier genes in C. lanatus subsp. mucosospermus , the recent progenitor of modern cultivated watermelon, indicates that the domestication of water-melon could be a possible force responsible for the rapid evolution of reproductive barriers, as has been reported in rice 30. Furthermore, genes involved in plant responses to abiotic and biotic stresses were also significantly enriched in this region, in addition to genes related to several known selected traits such as carbohydrate metabolism, fruit flavor (terpene metabolism) and seed oil content (fatty acid metabolism) (Supplementary Table 19).Evolution of disease resistance genes in watermelonThe watermelon crop suffers major losses from numerous diseases. Therefore, improvement in pathogen resistance is an ongoing objec-tive of watermelon breeding programs. To investigate the molecular basis for pathogen susceptibility, we searched for three major classes of resistance genes in the watermelon genome, namely the nucleotide-binding site and leucine-rich repeat (NBS-LRR), lipoxygenase (LOX)32 and receptor-like gene families 33. We identified a total of 44 NBS-LRR genes, including 18 Toll interleukin receptor (TIR)-NBS-LRR– and 26 coiled-coil (CC)-NBS-LRR–encoding genes (Supplementary Table 20). The watermelon NBS-LRR genes evolved independently, and we detected no sequence exchanges between different homologs.Such evolutionary patterns are similar to those of type II R genes in lettuce and Arabidopsis 34, indicating that watermelon has low diver-sity of NBS-LRR genes. The number of NBS-LRR genes in the water-melon genome is similar to that in cucumber 14 and papaya 35 but is considerably fewer than that in maize 36, rice 37 and apple 12. In con-trast, the LOX gene family has undergone an expansion in the water-melon genome with 26 members, 19 of which are arranged in two tandem gene arrays (Supplementary Fig. 11). Similar findings have been reported in cucumber, with expansion of the LOX gene family having been considered as a possible complementary mechanism to cope with pathogen invasion 14. We further identified 197 receptor- like genes in the watermelon genome, among which 35 encode receptor-like proteins lacking a kinase domain and 162 encode receptor-like kinases that have an intracellular kinase domain in addition to the extracellular LRR and transmembrane domains (Supplementary Table 20). Many of these resistance genes are located on chromosomes in clusters (Supplementary Fig. 11), suggesting tandem duplications as their evolutionary basis.It has been speculated that the lack of resistance to a wide range of diseases in modern watermelon cultivars is the result of the many years of cultivation and selection that have focused on desirable fruit qualities at the expense of disease resistance 8,38. To test this notion, we performed de novo assemblies of unmapped reads pooled each from modern cultivated (C. lanatus subsp. vulgaris ) and semiwild and wild (C. lanatus subsp. mucosospermus and C. lanatus subsp. lanatus , respectively) accessions. We identified 11 and 69 genes from the cultivated and the semiwild and wild groups, respectively, that are homologous to known plant proteins (Supplementary Table 21). It is worth mentioning here that the 69 new genes identified from the semiwild and wild group were highly enriched with disease-related genes including, 6 TIR-LRR-NBS genes, 1 PR-1 gene and 3 lipoxyge-nase genes, whereas none of the 11 genes identified in the cultivated group were disease related. In addition, all of the 44 NBS-LRR genes identified in the 97103 genome were also present in the semiwild andw11w1w2w3w4w5w6w7w8w9w10aA1A4A7A10A13A16A19WatermelonCucumberMelonWatermelonChr10Chr3Chr10Chr5Chr6Chr3Chr1Chr7Chr4Chr11Chr8Chr2Chr9LG4LG6LG10LG5LG2LG12LG9LG8LG7LG3LG11LG1Chr1Chr5Chr4Chr6Chr2Chr7Chr5Chr6Chr3Chr1Chr7Chr4Chr11Chr8Chr2Chr9bDiploid ancestor n = 7A1A 1w 1w 2w 3w 4w 5w 6w 7w 8w 9w 10w 11A 2A 3A 4A 5A 6A 7A 8A 9A 10A 11A 12A 13A 14A 15A 16A 17A 18A 19A 20A 21A4A7A10A13A16A19n = 2181fissions 91fusionsTE invasionWGD Hexaploid ancestorModern watermelon speciesS t r u c t u r a l s h u f f l i n gcFigure 2 Genome synteny, duplication patterns and evolutionary history of watermelon, cucumber and melon.(a ) Schematic representation of paralogous pairs identified within the watermelongenome (chromosomes w1–w11). Each line represents a syntenic region. Different colors reflect origin from the sevenancestral eudicot chromosome karyotype (A1, A4, A7, A10, A13, A16 and A19). (b ) Schematic representation of syntenies among watermelon (chromosomes 1–11), cucumber (chromosomes 1–7) and melongenomes (linkage groups (LG) 1–12). Each line represents a syntenic region. Shared synteny between two of the three species is linked by a light gray line. (c ) Evolution of the watermelon genome (w1–w11 at the bottom) from the common eudicot genome ancestors of seven chromosomes (A1, A4, A7, A10, A13, A16 and A19) and the derived paleohexaploid n = 21 (A1–A21) ancestor intermediate. Colored blocks represent the evolution of segments from the 7- or 21-chromosome ancestors to reach the modern watermelon genome structure. The 172 chromosomal fusions and fissions are highlighted with colored arrows. TE, transposable element; WGD, whole-genome duplication.©2012 N a t u r e A m e r i c a , I n c . A l l r i g h t s r e s e r v e d .A r t i c l e swild accessions (Supplementary Note ). These findings support the hypothesis that a large portion of disease resistance genes has been lost during watermelon domestication.Analysis of cucurbit phloem sap and vascular transcriptomes The angiosperm enucleate sieve tube system contains mRNA, some of which has been shown to function as a long-distance signaling agent 39,40. Through deep transcriptome sequencing (Supplementary Table 22), we identified 13,775 and 14,242 mRNA species in water-melon and cucumber vascular bundles, respectively, and 1,519 and 1,012 transcripts in the watermelon and cucumber phloem sap, respec-tively (Supplementary Tables 23–26). Notably, we found that the gene sets in the vascular bundles between the two cucurbit species were almost identical, whereas only 50–60% of the transcripts detected in the phloem sap were common between the two species (Supplementary Note and Supplementary Table 27). Gene Ontology (GO) term enrich-ment analysis indicated that the major categories among the common phloem transcripts were response to stress or stimulus (Supplementary Table 28), which is fully consistent with the central role of the plantvascular system, and the phloem in particular, in the long-distance communication system that integrates abiotic and biotic stress signal-ing at the whole-plant level 41. In contrast, analysis of the phloem transcripts that are unique to watermelon identified macro-molecular biosynthesis process and protein metabolic process as the major GO categories (Supplementary Table 29). The unique phloem sap transcripts may reflect special-ized functions that are unique to the role ofthe phloem in these species. It is noteworthythat the watermelon phloem contained 118 transcription factors, whereas we identified only 46 transcription factors in cucumber and 32 transcription factors that were common to both (Supplementary Tables 30–32).Pumpkin (Cucurbita maxima ) has been used as a model system for phloems tudies 42,43. We developed pumpkin vascular bundle and phloem sap transcript catalogs through generation and de novo assembly of the Illumina paired-end RNA sequencing (RNA-Seq) reads. Comparative analysis of the watermelon, cucumber and pumpkin phloem transcriptomes indicated that approximately36% of their transcripts were in common (Supplementary Fig. 12). These conserved transcripts probably carry out functions that are central to the operation of the sieve tube system in most cucurbit and possibly additional species.Regulation of watermelon fruit development and qualityWatermelon fruit development is a complex process involving major changes in size, color, texture, sugar content and nutritional components. To obtain a comprehensive characterization of the genes involved in the development and quality of watermelon fruit,we performed strand-specific RNA-Seq 44 of both the flesh and rindat four crucial stages of fruit development in the inbred line 97103 (Supplementary Table 33). We identified 3,046 and 558 genes that were differentially expressed in the flesh and rind, respectively, during fruit development and 5,352 genes that were differentially expressed between the flesh and rind in at least one of the four stages (Supplementary Tables 34–36). GO term enrichment analysis indicated that during fruit development in both the flesh and rind, biological processes such as cell-wall biogenesis, flavonoid metabolism and0.6b0.40.20–0.2–0.4E i g e n v e c t o r 2–0.6–0.8–1.0–0.2–0.10.10.2Eigenvector 10.30.40.5C. lanatus subsp. vulgaris east Asia ecotype C. lanatus subsp. vulgaris America ecotype C. lanatus subsp. mucosospermus C. lanatus subsp. lanatus.cK = 2K = 3K = 4K = 5RZ-900C. lanatussubsp. vulgaris Americaecotype C. lanatussubsp. vulgaris East-AsiaecotypeC. lanatussubsp. mucosospermusC. lanatus subsp. lanatusSugarleeSy-904304Black DiamondCalhoun Gray97103JLMJX-2JXFRZ-901XHBFGMPI189317PI248178PI249010PI595203PI482271PI500301PI482276PI482303PI482326PI296341-FRS y-904304P I 189317P I 48227697103XH BF GMR Z-901J L MJ XF J X -2B la c kD ia mo n dC a l ho un Gr a yS u g a reeR Z-900P I 5952031001001001001001004910010010010010099100PI 249010P I500301PI482271P I 482326P I 482303P I 296341-F R0a0.51.01.5P I 248178Figure 3 Population structure of watermelon accessions. (a ) Neighbor-joining phylogenetic tree of watermelon accessions on the basis of SNPs. (b ) PCA analysis of watermelonaccessions using SNPs as markers. C. lanatus subsp. vulgaris east Asia and America ecotypes and C. lanatus subsp. mucosospermus accessions cluster together (arrow) and are almost indistinguishable. (c ) Populationstructure of watermelon accessions. Each color represents one population, each accession is represented by a vertical bar, and the length of each colored segment in each vertical bar represents the proportion contributed byancestral populations. Shown are representative watermelon pictures from each subspecies or ecotype.。