先天性肾上腺皮质增生症致女性性分化异常8例分析
什么是先天性肾上腺皮质增生

什么是先天性肾上腺皮质增生*导读:先天性肾上腺皮质增生症是我们比较常见的一种常染色体隐性遗传病,但是我们却很少听到过这个疾病名称,因此大家可能不是特别了解。
那么如果得了先天性肾上腺皮质激素会有什么症状表现呢?又应该怎么治疗呢?为了让大家更好的了解什么是是肾上腺皮质增生,小编为大家整理了以下相关资料。
……先天性肾上腺皮质增生症是我们比较常见的一种常染色体隐性遗传病,但是我们却很少听到过这个疾病名称,因此大家可能不是特别了解。
那么如果得了先天性肾上腺皮质激素会有什么症状表现呢?又应该怎么治疗呢?为了让大家更好的了解什么是是肾上腺皮质增生,小编为大家整理了以下相关资料。
先天性肾上腺皮质增生是患者的皮质激素在合成过程中所需要的酶有先天缺陷导致的。
皮质激素合成受阻,皮质醇就会合成不足,从而导致血中浓度降低,由于负反馈作用会刺激垂体分泌促肾上腺皮质激素增多,而致使患者出现一系列的临床症状。
那么这些症状到底有哪些呢?1.男性化,先天性肾上腺皮质增生会导致患者的雄心激素过多,那么女性患者往往会呈现男性化的特点。
表现为严重的痤疮、体毛过多、多囊卵巢、月经不调等。
2.失盐型,这是先天性肾上腺皮质增生最严重的一种临床表现症状,患者的孕酮的21羟化过程严重受损,导致醛固酮分泌不足,醛固酮分泌不足又会引起患者肾脏、结肠和汗腺钠的丢失。
醛固醇缺乏又会使盐皮质激素和糖皮质激素同时缺陷,这样更容易引起患者休克或者造成严重的低钠血症。
3.非经典型,非经典型患者只有轻度的雄心激素过多的临床表现,女性患者的生殖器等都不会发生异常,只是肾上腺类固醇前体物质会有轻微的升高。
上面这三点就是肾上腺皮质激素的主要症状,希望大家通过小编的文章能够对先天性肾上腺皮质增生这种疾病有进一步的了解。
如果大家还有更多的疑问,可以去正规的医院进行检查治疗,正规的医院会有专业的医师回答你的各种疑惑。
最后小编祝大家早日康复身体健康。
经新生儿筛查发现的先天性肾上腺皮质增生症的随访观察

经新生儿筛查发现的先天性肾上腺皮质增生症的随访观察陈玉林;黄美莲;张瑾;蒋涛;孙亦骏【期刊名称】《医学理论与实践》【年(卷),期】2003(016)011【摘要】目的:本组4例新生儿先天性肾上腺皮质增生症(CAH),均系经新生儿筛查确诊.通过对经新生儿筛查出的先天性肾上腺皮质增生症患者进行诊治随访观察.总结对先天性肾上腺皮质增生症诊疗经验,提高对肾上腺皮质增生症的管理质量.方法:对4例CAH患者临床资料进行回顾性分析.结果:自1993年~2002年,共筛查出4例阳性病例.1例患儿自动放弃治疗后死亡,其余3例在最后1次随访时年龄已分别为3 2/12岁,2岁,1岁,他们的身高在正常范围.南京地区先天性肾上腺皮质增生症发病率为1/35187.结论:先天性肾上腺皮质增生症是常染色体隐性遗传病,出生时常缺乏症状,易误诊,延误治疗.需要早期治疗,坚持随访.【总页数】2页(P1256-1257)【作者】陈玉林;黄美莲;张瑾;蒋涛;孙亦骏【作者单位】南京市妇幼保健院,210004;南京市妇幼保健院,210004;南京市妇幼保健院,210004;南京市妇幼保健院,210004;南京市妇幼保健院,210004【正文语种】中文【中图分类】R722.11【相关文献】1.佛山地区先天性肾上腺皮质增生症的新生儿筛查 [J], 贾德勤;刘海平;王星;简美好2.新生儿筛查发现的原发性肉碱缺乏症临床与基因分析 [J], 孙云;马定远;王彦云;程威;梁晓威;蒋涛3.先天性肾上腺皮质增生症新生儿筛查的目前进展(讲座) [J], 马燮琴4.四川省新生儿筛查中心新生儿先天性肾上腺皮质增生症筛查分析 [J], 胡琦; 欧明才; 张钰; 周婧瑶; 陈雪莲; 杨丽涓; 苏星月5.南京地区先天性肾上腺皮质增生症的新生儿筛查和随访 [J], 陈玉林;孙亦骏;等因版权原因,仅展示原文概要,查看原文内容请购买。
先天性肾上腺皮质增生症的全生命周期临床管理2024

先天性肾上腺皮质增生症的全生命周期临床管理2024先天性肾上腺皮质增生症(congenital adrenal hyperp a sia,CAH) 是肾上腺皮质激素合成途径中的酶先天缺陷,导致肾上腺皮质激素合成不足,继发下丘脑促肾上腺皮质激素释放激素(corticotropin re e asing hormone, CRH)和垂体促肾上腺皮质激素(adrenocorticotropic hormone, ACTH)代偿性分泌增加,导致肾上腺皮质增生和性腺发育异常的一组疾病,属常染色体隐性遗传病。
CA H临床表现取决千酶的阻断部位及酶活性缺失程度;大部分患者有不同程度的肾上腺皮质功能不全和性腺发育异常;伴或不伴水盐代谢紊舌店i高血压。
CAH临床表现异质性大发病年龄不一其管理贯穿整个生命周期。
生命周期不同阶段的患者需求不同,因此制定以患者为中心的全生命周期临床管理至关重要,需要多学科,包括儿科、内分泌科、妇产科、心理科等多学科联合管理,才能达到提高患者生存质量,改善患者生长、发育、婚育及延长寿命的需求。
01 CAH基因分型、临床表现及诊疗现状目前报道的有7种基因突变可引起CAH,其中5种编码合成皮质激素的生物合成酶,包括21-经化酶(CYP21入11�-轻化酶(CYP11B1入17a一经化酶/17,20裂解酶(CYP17入胆固醇侧链断裂酶(CYP11A1)和II型3P-经类固醇脱氢酶(HSD3B2);1种编码类固醇激素生成急性调节蛋白(StAR);另外一种编码细胞色素P450氧化还原酶(POR1 POR 基因突变可累及类固醇激素合成途径中多种酶,包括CYP17、CYP21、CYP11 B1和芳香化酶(CYP191 CAH的临床分型包括21-轻化酶缺乏症(21-0HD入11p轻化酶缺乏症(11p-OHD入3P-经类固醇脱氢酶缺乏症(3P-HSD)、17a一经化酶缺乏症(17a-OHD入类脂性肾上腺增生症(包括StAR缺陷症和CYP11A1缺乏症)和POR缺陷症(PORD1 21-0HD 和11P-OHD往往导致女性男性化月经紊乱或不孕;男孩同性性早熟等。
手术治疗先天性肾上腺皮质增生症致女性假两性畸形6例报告

成形 , 待青春期后行 二期 手术治疗 。
结果 : 6例患儿手术 均顺利 , 口愈合 良好 , 伤 术后 1周拔
除尿管 , 阴形 态较术前 明显改善 , 外 家长对手术效果满意。
讨论 : 素替代 治疗 是 C H 的基 本治疗 方法 , 畸形 明 激 A 但 显者需手 术矫治 。有 人认 为最佳 手术 年龄 为 2~6个月 , 也 有人认为患儿年龄 稍大 时手 术 , 阴道 损伤 和狭窄 机会 减少 , 有利于建立适 当大 小 的前 庭 和 阴蒂 阴道 间的解 剖关 系 。笔
阴道黏膜梭 形 切开 可增 加 视野 , 利 于术 中切 除并 取 出 子 有 宫。切开前后腹 膜反折时不要先切开或 同时切开前后 反折 , 可根据宫颈长短和反折 的高低决定 , 宫颈长或反 折高 的可 先
Hale Waihona Puke 手术 治疗 先 天 性 肾上 腺皮 质 增 生 症 致 女 性假 两 性 畸形 6例 报告
尿生殖窦切 V最低 点行插入缝合 , I 然后 以此为 中点 向两侧延
展缝合 , 行会 阴体成 形 、 阴道 口成 形。其 中 1例 6岁患儿 切 开尿生殖窦腹侧壁后见 阴道 开 口位 置较高 , 无法行 一期阴道
超检查可见子 宫及 双侧 附件声像 。C T检查 双侧 肾上 腺未 见 异常 。术前接受 1 2a的激素 治疗 ( 日口服醋 酸氢 化 0d~ 每
手术方法 : 常规术前准备 。术前 1d口服 醋酸氢 化可 的
松剂量加倍 , 手术 当 日静滴氢化可 的松 10 m 。术后 1d静 0 g 滴氢化 可的松 5 g 同时 按 日常治疗剂 量 V服醋 酸氢化 可 0m , I 的松 。术后第 2天 按 日常 治 疗剂 量 口服 醋酸 氢化 可 的松 。 ① 阴蒂成形术 : 患儿气管插管全麻后取 平卧位 。阴蒂头缝牵 引线 。于 阴蒂背部 正中纵行 剪开包皮 约 2 0c 切开皮 肤 、 . m, 皮下组织 , B c 筋 膜。切开 处保 留内板 约 0 5c 达 uk . m。环行 切开包 皮 , 将包皮脱 套 至阴蒂根 部 , 完全暴露 阴蒂 体。在 阴
先天性肾上腺皮质增生症

洮南 17 0 3 10 吉林 省 白城 地 区洮 南 市 医 院 ,吉林
【 摘 要 】 本 文对 3 例先天性肾上腺皮 质增生症 ( A )采用强的松及外生殖器畸形矫正手术。外 生殖 器异常按 Pae 的分型 ,其 1 CH r r d 中 I型 5例 , Ⅱ型 9例 ,Ⅲ型 l 7例 ,除 1 例无 闭经外 ,其余均为原发性 闭经 。所有病例乳房均未发育 ,呈男性乳头。治疗 前先作地塞米松 试 验,试验后 1 一K 7 s值均明显下降,根据试验前 1一 K 7 s值选择强 的松用量。全部患者第二性征得 到改善 ,月经来潮 。其 中已婚 1 例 ,6 1 例妊娠,母婴健康 。观察 中发现部分病毒停药后仍能维持月经周期 。本文就此对 C H病人是否需终生服药问题进行 了讨论 。 A 【 关键词 】 肾上腺皮质增生症 ;2 一羟化酶;尿 l 1 7酮 【 中图分类号 】R 5 . 4 8+ 3 【 文献标 识码】A 【 文章编号 】10 — 5 7 (0 0 3 0 0 — 1 0 7 8 1 2 1 )2 — 18 0
~
随访 中发现 ,有 5例 病人 月经规 律后 自行停 药 ,半年 至 1年后 ,2例 仍月经 规律 ,3例 又 出现 闭经 、皮肤 变粗 , 2 h尿 1一K 4 7 S在 8 . 5—14 1 mo之 间。2例 服药 3年 以 67 0. I l  ̄ 上 ,月经规律后不 再坚持 服药 ,每 当 出现月经 紊乱 ,甚 至 停经时再开始服药 。亦 可使月经周 期重新建 立 ,查尿 l一 7 K S亦在 正 常 范 围 。
[ ]俞霭峰 妇产科内分泌学.第 1 1 版.上海 :上海科技 出版社 19 ,2 9 98 5
~
261 .
[ ] 肖风云 男女生殖器 系畸 形 2
先天性肾上腺皮质增生症CAH的临床进展

先天性肾上腺皮质增生症CAH的临床进展先天性肾上腺增生症又称肾上腺生殖器综合征或肾上腺性变态征。
主要由于肾上腺皮质激素生物合成过程中所必需的酶存在缺陷,致使皮质激素合成不正常。
多数病例肾上腺分泌糖、盐皮质激素不足而雄性激素过多,故临床上出现不同程度的肾上腺皮质功能减退,伴有女性男性化,而男孩则表现性早熟,此外尚可有低血钠或高血压等多种症候群。
此外,由于ACTH 和促黑素细胞激素增多,患者常表现皮肤粘膜色素增深。
CAH是一个单基因突变的常染色体隐性遗传性疾病遗传上肾上腺类固醇激素合成途径中必要酶的缺乏主要有:•21-羟化酶缺陷型(占90~95%)*•11β-羟化酶缺陷型(5~8%)•3β-羟类固醇脱氢酶缺陷型•17α-羟化酶缺陷型•18-羟化酶缺陷型•胆固醇碳链酶缺陷症【临床表现】本病以女孩为多见,男与女之比约为1 : 4 。
任何一种酶(17α- OHD除外) 的缺陷皆可有肾上腺雄激素分泌过多而引起的共同症状。
按缺陷酶可分五类:①21羟化酶缺陷症(21 hydroxylase deficiency, 21-OHD) ②11β-羟化酶缺陷症(11β-hydroxylase deficiency,11β-OHD) ③3β-类固醇脱氢酶缺陷症(3β-hydroxylase dehydrogenase deficiency,3β-HSD) ,④17α-羟化酶(17α- hydroxylase deficiency,17α- OHD) / 17、20-裂解酶缺陷症(17 、20-lase deficiency,17 、20-LD),⑤胆固醇碳链酶缺陷症(cholesterol desmolase deficiency) /类脂性肾上腺增生症(adrenal lipid hyperplasia)。
一、21羟化酶缺陷症(占90~95%)1.单纯男性化型(Simple Virilism, SV)最多见,约一半男婴性早熟、早熟巨阴症、成人时体格矮小女婴阴蒂肥大,以后逐渐增大似男孩阴茎,大阴唇似男孩阴囊但无睾丸。
石家庄市新生儿先天性肾上腺皮质增生症筛查及基因突变分析

doi :10.3969/j.issn.1002-7386.2023.17.032·调查研究·石家庄市新生儿先天性肾上腺皮质增生症筛查及基因突变分析封露露 马翠霞 贾立云 马倩倩 封纪珍项目来源:河北省医学科学研究课题计划(编号:20210689)作者单位:050000 河北省石家庄市妇幼保健院遗传科(封露露、马翠霞、贾立云、封纪珍);河北省石家庄市妇产医院(马倩倩)通讯作者:封纪珍 E⁃mail:279406985@ 【摘要】 目的 分析石家庄市新生儿先天性肾上腺皮质增生症(congenital adrenal hyperplasia ,CAH )的发病率及CYP21A2基因突变情况,为患儿的个体化治疗和遗传咨询提供依据。
方法 采用免疫荧光法对石家庄市2018年9月至2021年12月活产新生儿进行筛查,对可疑阳性患儿进行基因检测,分析基因与临床表型的联系。
结果 257751例新生儿中,筛查阳性203例,召回后根据临床表现和生化检测确诊10例患儿,发病率为1/25775。
确诊患儿中,男8例,女2例;失盐型6例,单纯男性化型4例,其中3例进行了基因测序分析,均为21⁃羟化酶缺乏症。
进行基因检测的3例患儿中,纯合突变1例,杂合突变2例,共5种突变类型,突变位点主要包括c.518T>A 、c.293⁃13C>G 、c.955C>T 、c.923dupT 和外显子1~7杂合缺失。
结论 石家庄市新生儿CAH 的患病率为1/25775;男性患病率高于女性,且以失盐型为主;CYP21A2基因突变分析发现5种基因突变类型,补充和完善了石家庄市新生儿CYP21A2基因数据库。
【关键词】 先天性肾上腺皮质增生症;21羟化酶缺乏症;基因测序;CYP21A2基因【中图分类号】 R 586 【文献标识码】 A 【文章编号】 1002-7386(2023)17-2696-04Screening of neonatal congenital adrenal hyperplasia and the gene mutation analysis in Shijiazhuang City FENG Lulu ,MA Cuixia ,JIA Liyun ,et al.Department of Genetics ,Shijiazhuang Maternity &Child Healthcare Hospital ,Hebei ,Shijiazhuang 050000,China【Abstract 】 Objective To analyze the incidence of congenital adrenal hyperplasia (CAH )in neonates of Shijiazhuang City ,and the rate of the CYP21A2gene mutations in them ,thus providing references for individualized treatment and genetic counseling.Methods The living newborns in Shijiazhuang from September 2018to December 2021were screened by immunofluorescence method.Children with suspected CAH were subjected to genetic testing.The correlation between genetic findings and clinical phenotypes of CAH was analyzed.Results Among the 257,751screened neonates ,203cases of suspected CAH were screened.Finally ,10(1/25775)cases were diagnosed as CAH according to clinical and biochemical tests after the recall ,involving 8male infants and 2female infants.There were 6cases of salt⁃wasting phenotype and 4cases of simple virilizing phenotype.Three cases were tested for gene sequencing ,all of whom were 21⁃hydroxylase deficiency.There were 5types of mutations in 1child with homozygous mutation and 2with heterozygous mutations ,including the c.518T >A ,c.293⁃13C>G ,c.955C>T ,c.923dupt and heterozygous deletion of exon 1-7.Conclusion The prevalence of neonatal CAH in Shijiazhuang City is 1/25775,which is higher in males than females.The salt⁃wasting phenotype is the predominant phenotype of CAH.Five gene mutations in the CYP21A2gene are detected in CAH neonates.Our findings have supplemented and improved the profile of the CYP21A2gene mutations in neonatal CAH in Shijiazhuang City.【Key words 】 congenital adrenal hyperplasia ;21⁃hydroxylase deficiency ;gene sequencing ;CYP21A2gene 先天性肾上腺皮质增生症(congenital adrenalhyperplasia,CAH)是一种常染色体隐性遗传病[1],按照酶缺陷的不同,分为6型,21⁃羟化酶缺乏症(21⁃hydroxylase deficiency,21⁃OHD)为最常见的一种类型,患儿常出现肾上腺功能不全等症状,新生儿期表现为厌食、呕吐、体重不增、喂养困难,严重者表现为电解质紊乱等症状;儿童期会出现性早熟、皮肤色素沉着、痤疮等临床表现,根据临床表现分为三种类型:失盐型、单纯男性化型和非典型型[2]。
病例分析:一例先天性肾上腺皮质增生症((CAH))
病例
一例先天性肾上腺皮质增生症(CAH)
患者男性,31岁,结婚3年未避孕,未育入我院生殖中心检查:无精症......
基本资料:患者男性,31岁,结婚3年未避孕,未育入生殖中心检查:无精症;
生殖中心检查结果:ACTH 472 ng/L(0-46),F(皮质醇):199nmol/L,遂转来我科(内分泌科);
实验室检查:
血压正常,电解质正常
FSH:0.45IU/L(1.27-19.26),LH:0.33IU/L(1.84-2.62);
E2:204.9pmol/L(73.4-172.4),T:16.49nmol/L(6.07-27.1);
P:86.14nmol/l(1.27-3.5),PRL:239.3mIU/L(55.9-278.4);
甲状腺功能正常;
ALD:214ng/L,肾素:8.6ug/L/h;
影像学检查:肾上腺CT:两侧肾上腺占位性病变,腺瘤可能;
治疗方案:予地塞米松 7.5mg bid
预后转归:ACTH 7.64 ng/L(0-46)下降,F:3nmol/L;
FSH:6.27IU/L(1.27-19.26),LH:2.92IU/L(1.84-2.62);
E2:25.3pmol/L(73.4-172.4),T:11.08nmol/L(6.07-27.1);
P:12.28nmol/l(1.27-3.5),PRL:218.03mIU/L(55.9-278.4);
肾上腺CT:两侧肾上腺占位性病变,较之前缩小;
基因检测:确认21羟化酶缺失。
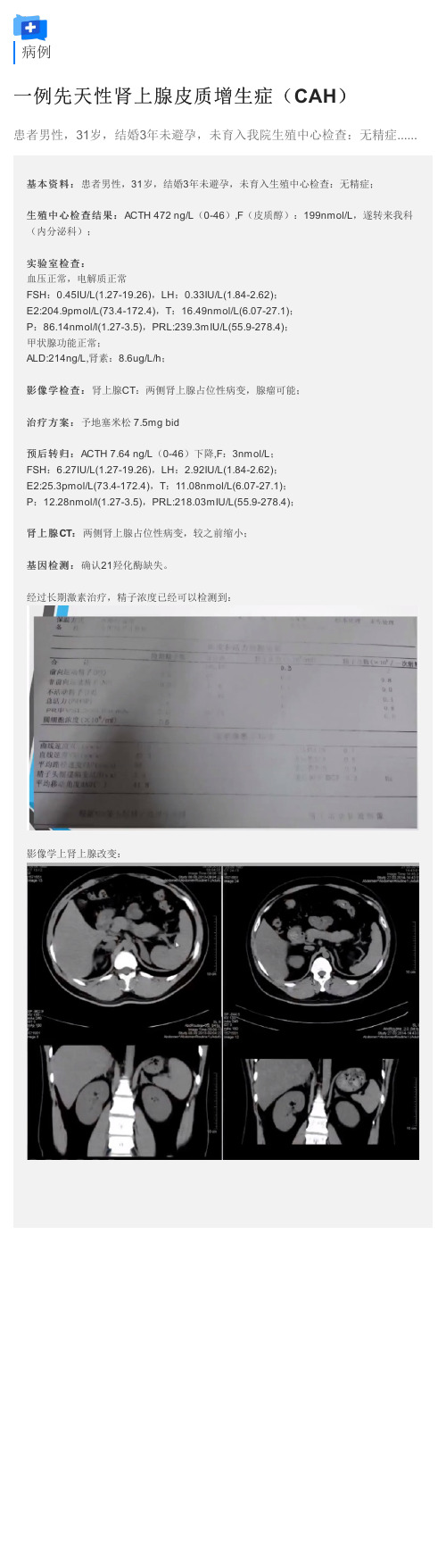
经过长期激素治疗,精子浓度已经可以检测到:
影像学上肾上腺改变:。
新生儿先天性肾上腺皮质增生症

病例1
• 体格检查:T 36.7℃,HR 144次/min, R 42次/min,Bp 72/35mmHg,体质量 3795g。反应欠佳,全身皮肤色素沉着, 以乳头(图A)、会阴处为明显(图B); 皮肤弹性欠佳。前囟门稍凹陷,约 1.0cm*1.0cm大小,张力不高。唇红, 心肺听诊未见异常。腹平软肝肋下 3.0cm,质中等,牌未触及。阴蒂及大 阴唇肥,阴蒂似男性阴茎(图B),大阴 唇似男性阴囊(图C)。四肢肌张力低下,
• 血培养(-)、梅毒筛查(一)、HV(-)。
• 染色体46,XX。
• 促肾上腺皮质激素(ACTH)22.7pgmL,尿17-羟孕酮(17-OHP)76.38ng/mL,高于正常水平。
• 皮质醇(COR)节律:下午4时330.9nmoM/L,高于正常水平。晚上12时300.39 nmol/L,上年 200.44nmol/L,睾酮(T)29.62umol/L,
• 肾上腺CT检查见双侧肾上腺稍大,提示肾上腺皮质增生可能性大。
病例1
• 临床经过:
予以3%氯化钠注射液提高血钠,碳酸氢钠注射液纠正酸中毒维持水、 电解质、酸碱平衡等对症支持治疗,尚未给予激素治疗,患儿家属签字 要求出院,出院时:Na 141.1mmo/L.,K 4.4mmol/L,Cl 114.9mmol/L, ACTH、ALD均高于正常。
先天性肾上腺皮质增生症
(congenital adrenocorticohyperplasia,CAH)
XX医院 XXX
2020/10/25
病例1
• 女,36天,发现皮肤色素沉着36天,反复呕吐10天入院。 • G2P1,胎龄40周,剖宫产,出生体重3800g。 • 出生后即发现全身皮肤色素沉着,以乳头、会阴处为明显。10余
妇科性分化异常疾病的诊治

妇科性分化异常疾病的诊治【概述】性分化异常是指性腺未分化成正常的睾丸和卵巢。
因其涉及不同的分化机制,故其临床表现不一;即使临床表现类似,背后的分化机制也可不同,所以目前尚无统一的分类。
此类疾病可给患儿的抚育、心理、学习、生活、工作和婚姻等带来诸多困扰,因此及早诊断和适时治疗对预后非常重要。
【分类】国际和国内目前尚无统一分类法(见表14及表15)。
(一)女性假两性畸形1.先天性肾上腺皮质增生(CAH)(1)分类:又称肾上腺生殖综合征,为常染色体隐性遗传性疾病,是导致女性假两性畸形的最常见类型。
其基本病变为胎儿肾上腺合成皮质醇的一些酶缺乏。
1)其中最常见的为21—羟化酶缺乏,导致17α一羟孕酮转化为皮质醇减少,当皮质醇合成量减少时,对下丘脑和腺垂体的负反馈作用消失,导致腺垂体促肾上腺皮质激素分泌量增加,刺激肾上腺增生,促使其分泌的皮质醇量趋于正常,但同时也刺激肾上腺网状带产生异常大量雄激素,致使女性胎儿外生殖器有部分男性化。
2)还有5%为11B—羟化酶缺陷致皮质酮和皮质醇合成障碍,其前体物质进入雄激素代谢途径,使雄激素升高,去氧皮质酮也增多,其有足够的盐皮质激素作用而无失盐的表现,但可造成血压升高是11B—羟化酶缺陷的特征。
基因定位于8号染色体长臂(8q22),为常染色体隐性遗传。
(2)诊断1)临床表现:患者出生时即有外生殖器畸形、高血压或呕吐、脱水、失盐等表现。
阴蒂肥大,阴唇融合遮盖阴道口和尿道口,仅在阴蒂下方见一小孔,尿液由此排出。
严重者两侧大阴唇肥厚,形成皱褶,并有程度不等的融合,状似阴囊,但其中无睾丸;子宫、输卵管、阴道均存在,但阴道下端狭窄,难以发现阴道口。
随着婴儿长大,男性化日益明显,几岁时即有阴毛和腋毛出现,至青春期乳房不发育,内生殖器发育受抑制,无月经来潮。
虽幼女期身高增长快,但因骨惭愈合早,至成年时反较正常妇女矮小。
2)辅助检查:血雄激素含量增高,尿17酮类固醇呈高值,血清ACTH及17Q一羟孕酮均显著升高。
先天性肾上腺皮质增生症一家族性21-羟化酶缺陷8例

酮皮 质类 固醇 (7 () 采用 化 学发 光法 , 用 1 一l 均 S 应
天津德 普 公 司 Imu t 00仪 测 定 。血 电解 质 m le一20 i ( 、 、 、 、 ) 用 电极 法 测 定 , 钾 钠 氯 镁 钙 采 采用 日立 一
11 一 般 资料 .
社会 性 别 女性 6例 , 外周 血 染 色
摘
要 目的 复习 8例先天性 肾上腺皮质增 生症一家族 性 2 一羟化酶缺陷患者的临床 特点 、 1 实验室 检查及 5 例行保 留阴蒂头阴蒂缩短整形 +外
治疗方法。方法 回顾性分析 8例 2 一羟化酶缺陷患者 的临床资料。结果 1
阴整 形 术 +阴 道 口成 型术 , 均行 皮质 激素 替 代 治 疗 。随 访 6 月 ~2 , 总睾 酮 、 酮 及 A r 8例 个 年 8例 孕 c H均 正 常 , 5例
别应 用 东 芝 A u i 一1 , E U I q i n 6 S Q OA一5 2 G i a l o 1 、 E S n g E CT I 器测定 。立卧位 试验 ( X IEI仪 m S 结果 采用 ) 放 射免疫法 , 应用 S N一65 9 8型智能 放射 免疫 一测
量 仪测定 。
维普资讯
22 5
Pata dcn t t ,o8 V 11。 o4 rccl i MeiieAt s 20 , o.3N . k  ̄t n
先天 性 肾上 腺 皮 质增 生症一 家 族性
2 一羟 化 酶 缺 陷 8例 1
周 小 爱 张 哲
( . 山市ห้องสมุดไป่ตู้林 医院 , 1江 浙江 江山 34 0 ; 2 10 2 浙 江大 学医学 院附属 第一 医院 , 江 杭 州 3 00 ) 浙 10 3
可治性罕见病-先天性肾上腺皮质增生症

可治性罕见病-先天性肾上腺皮质增生症展开全文一、疾病概述先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一组因肾上腺皮质激素合成途径中酶缺陷引起的疾病,又称肾上腺生殖器综合征。
是一种较少见的常染色体隐性遗传病。
新近报道其发病率约占活产婴儿的1110 000~1/20 000,与种族及地区有关[1]。
其中以21 -羟化酶缺乏症(21 -hydroxylase deficiency,21 - OHD)最常见,占本病的90%~95%。
近期筛查统计结果显示,全世界21-OHD的发生率为1:13 000,日本为1;15 000,欧洲为1:14 000~1:10 000,北美为1:15 000,我国上海地区的发生率为1:20 000[2,3]。
其次为11β -羟化酶缺乏症(11β - OHD),占3%~8% [4]。
此外尚包括3β-羟类固醇脱氢酶缺乏症(3β - HSD)、17 α-羟化酶缺乏症(17 - OHD)及先天性类脂质性肾上腺皮质增生症(CLAH)等罕见类型CAH。
21 -羟化酶由CPY21A2基因编码,也称为CYP21或P450c21,是位于肾上腺皮质内质网的一种细胞色素P450酶,能催化17 -经孕酮(17 - OHP)转化11-脱氧皮质醇(皮质醇的前体),孕酮转化为去氧皮质酮(醛固酮的前体)。
所以,21 - OHD会导致肾上腺皮质合成醛固酮及皮质醇障碍,垂体促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)增高,17 - OHP及旁路代谢产生雄激素(睾酮)增高而引起女性男性化、男性假性性早熟。
21 - OHD为CAH中可引起女性男性化的3种类型之一,其他类型的酶缺陷可引起男性假两性畸形。
二、临床特征CAH的临床表现取决于酶的阻断部位及严重程度,临床表现各异,其共同点是因皮质醇合成减少而使ACTH分泌增进而使肾上腺皮质增生。
先天性肾上腺皮质增生症外科诊治的经验总结

先天性肾上腺皮质增生症外科诊治的经验总结1. 引言1.1 疾病概述先天性肾上腺皮质增生症是一种罕见的遗传性疾病,主要特征是肾上腺皮质增生导致的激素分泌异常。
这种疾病通常在婴儿期或儿童早期就会出现,临床上表现为发育延缓、血压升高、性早熟等症状。
患者常常需要长期的激素替代治疗来维持生命。
先天性肾上腺皮质增生症是一种复杂多样的疾病,其临床表现和发病机制各不相同。
有些患者可能只表现为轻度的激素分泌异常,而有些患者则可能出现严重的合并症,如肾功能衰竭或癫痫等。
对于这种疾病的诊断和治疗需要有经验丰富的医疗团队来共同参与,以确保患者能够得到及时有效的治疗。
虽然先天性肾上腺皮质增生症是一种罕见疾病,但随着医疗水平的不断提高,对于这种疾病的认识和治疗方法也在不断改进。
希望通过本文的介绍,能够让更多的医疗人员和患者对这种疾病有更深入的了解,从而提高患者的诊疗质量,改善患者的生活质量。
1.2 研究目的研究目的是为了探讨先天性肾上腺皮质增生症外科诊治的经验,总结并分析手术治疗的有效性和安全性,为临床医生提供指导和参考。
通过对手术治疗的适应证和禁忌证、手术方式选择以及手术并发症的预防和处理等方面进行深入研究,旨在提高先天性肾上腺皮质增生症患者的治疗效果,减少手术风险,提高患者的生存率和生活质量。
我们也希望通过本研究为未来相关疾病的治疗提供经验积累和借鉴,为临床实践和学术研究提供新的思路和方法。
通过本次研究,我们期望能够为先天性肾上腺皮质增生症的外科诊治贡献新的见解和经验,为患者的健康和生命贡献力量。
1.3 研究意义先天性肾上腺皮质增生症是一种罕见的遗传疾病,其发病率较低,但病情危害性极大,容易导致严重的健康问题。
对该疾病进行外科诊治具有十分重要的意义。
外科治疗能够有效减轻患者的症状,提高其生活质量和长期存活率。
目前对该疾病的外科诊治仍存在许多不足之处,需要进一步探究和完善。
研究意义在于提高对先天性肾上腺皮质增生症外科治疗的认识和理解,为临床医生提供更为科学的诊疗方案,从而更好地服务患者。
先天性肾上腺皮质增生症外科诊治的经验总结

先天性肾上腺皮质增生症外科诊治的经验总结【摘要】先天性肾上腺皮质增生症是一种罕见的遗传性疾病,容易被误诊或漏诊。
本文将就该疾病的临床表现、影像学特征、手术指征、手术方式和术后管理进行详细讨论。
通过总结多年来的诊治经验,为临床医生提供参考。
本文也展望未来在该疾病诊治方面的挑战和发展方向。
经验总结的目的是为了更好地指导临床实践,提高患者的治疗效果。
也要感谢所有在这一领域做出贡献的专家和患者的支持与信任。
希望本文的内容能够为相关领域的医生和患者提供帮助,促进先天性肾上腺皮质增生症的治疗水平和患者生活质量的提高。
【关键词】先天性肾上腺皮质增生症, 外科诊治, 经验总结, 临床表现, 影像学特征, 手术指征, 手术方式, 术后管理, 诊治经验, 展望未来, 致谢.1. 引言1.1 疾病背景先天性肾上腺皮质增生症(Congenital Adrenal Hyperplasia,CAH)是一种常见的遗传性内分泌疾病,主要由于11β-羟化酶缺乏或其他酶缺失导致肾上腺皮质激素合成代谢障碍所致。
CAH的发病率在不同族群和地区有所不同,但全球范围内的患病率大约为1/10000-1/20000。
其中以盐皮质醇缺乏型(传统型)最为常见。
临床上,CAH患者常出现生长迟缓、早熟、骨龄提前、男性外生殖器发育不全、女性外生殖器异常等,严重者可出现低血糖、低钠血症、高钾血症、原发性肾上腺皮质功能减退等危及生命的并发症。
CAH的早期诊断和治疗非常重要,可有效避免患者出现恶性高血压、肾盂积水、青春期障碍、性腺功能坏死等严重后果。
及早发现CAH,进行规范化治疗,对于患者的生长发育和生活质量具有重要意义。
在本文中,我们将探讨CAH的外科诊治经验,以期对相关疾病的临床工作有所帮助。
1.2 治疗意义先天性肾上腺皮质增生症是一种罕见的遗传疾病,患者常常出现肾上腺皮质增生导致的激素异常,严重影响其生活质量。
针对这种情况,外科手术治疗具有重要的意义。
先天性肾上腺皮质增生症专题医学知识宣讲

糖、盐皮质激素替代治疗仍是目前CAH主要旳治 疗措施
新生儿中旳发病率为1/16000—1/20230。
误诊或晚治疗患儿,常因雄激素升高促使骨 龄加速而造成严重侏儒症,并可发生肾上 腺皮质功能减退危象而危及生命。
伴随国内新生儿CAH筛查旳逐渐开展、临 床医师对疾病辨认能力旳逐渐提升,部分 患儿在临床症状出现前或出现后就得到早 期诊治,改善了患儿旳生长发育,提升成 年底身高
(11β-OHase Deficiency)
CYP 11 A (P450SCC) HSD3β-2
CYP17 (P450C17)
CYP21 (P450C21) CYP11β (P450C 11β)
位置 15q 23-24 1p 13.1 10q 24.3
6p 21.3 8q 21-22
21-OHD旳流行病学资料
ACTH兴奋试验:适合17-OHP基础值在35ng/ml者,250μg ACTH(1-24),IV 取0,60 分钟血,测血F,17-OHP,如17-OHP 0’ ≥5ng/ml和/或17-OHP 60’ ≥10ng/ml, 21-OHD确诊。
21-OHD 治疗
㈠、糖皮质激素治疗 ⑴、激素选择:经经典最佳用氢化可旳松,血压正常 旳SV(单纯)型也可考虑用强旳松。睡前用地塞米松 也有报告。
先天性肾上腺皮质增 生症专题医学知识宣
讲
主要内容
CAH 定 义 CAH 病 理 生 理 CAH 分 类 CAH 治 疗
CAH定义
先天性肾上腺皮质增生症 (CAH) 是一组常染色体 隐性遗传病,
90%~95%是因为21-羟化酶先天缺陷 造成肾上 腺皮质合成醛固酮及皮质醇障碍,垂体促肾上腺 皮质激素 (ACTH) 增高,使酶阻断前质17羟孕酮 (17-OHP) 及旁路代谢产生雄激素(睾酮) 增高而 引起女性男性化、男性假性性早熟;
先天性肾上腺皮质增生症怎样治疗

先天性肾上腺皮质增生症怎样治疗
一、概述
大家所说的先天性肾上腺皮质增生症多数患者肾上腺分泌理糖
激素、理盐激素不足而雄性激素过多,所以出现不同程度的肾上腺皮质功能减退,伴有女孩男性化,而男孩则表现性早熟,怎样治疗先天性肾上腺皮质增生症呢,分享给大家
二、步骤/方法:
1、糖皮质激素替代治疗:GC为各种类型CAH的主要治疗手段,主要作用是抑制ACTH,减少21-OHD、11β-OHD和3β-HSD缺陷症的雄激素水平,降低11β-OHD和17α-OHD的脱氧皮质醇(DOC)水平,进而改善这些患者的骨龄、终身高或高血压,增强患者应激能力。
2、盐皮质激素替代治疗:盐皮质激素主要用于治疗失盐型21-OHD、3β-HSD缺陷症和StAR缺陷症患者,但大多数盐皮质激素缺乏的患
儿(失盐型尤其是21-OHD)“失盐”表现可以随年龄增长而缓解,
盐皮质激素治疗也可随之停止。
3、性分化和发育异常的治疗
对于性分化异常的CAH患者,应确定患者的染色体性别、性腺性别,评价外生殖器分化发育情况,尽早诊断,及时治疗可以部分消除后续的影响
4、治疗过程中的监测
CAH的治疗为终生,如果治疗及时且适当,效果较好,可获得正
常的生长、发育和生育能力。
治疗过程中的监测非常重要,。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
素。另 6 例患者表现为不同程度男性化, 假两性畸 形, 不伴高血压 、 低血钾 , 促肾上腺皮质激素增多 , 血 l一 7 羟孕酮增高 , 血睾酮增高 , 染色体核型 4 , 6 x 诊断 为 2 x, l一羟 化 酶 缺 乏 症 , 纯 男 性 化 型 。 单 治疗上 除 了补 充 糖 皮 质 激 素外 , 尚可 根 据情 况 施
・
3 8・ 7
浙江临床医学 2o 年 4月第 l 卷第生翅 09 1
注 : c H( A T 促肾上腺皮质激素) F 皮质醇) T 睾酮 )E ( ;( ;( ; 2 雌二醇 ) F H 促 卵泡激素 ) ;S ( ; L( H 黄体生成素) P 孕酮 ) 1d—O P 1a一羟孕酮) P ( ;( ;7 H (7 ;RA 肾索活性 ) A Ⅱ( ; g 血管紧 张素 Ⅱ) A D 醛固酮) z h 尿 A D 2 h尿醛 固酮 ) 2h 尿 1 一O c (4 ;L ( ;4 r 0 (4 ;4 r 7 H s 2 h尿 l 一羟 7 类固醇) 2 h 尿 l 一K (4 尿 l 酮类固醇 ) ;4 r 7 s 2 h 7一
浙江临床 医学 2 o 0 9年 4月第 l 卷第 4期 l
・
3 7・ 7
・
诊 治 分 析
・
先天性肾上腺皮质增生症致女性
性分化异常 8 例分析
徐 丹怡 沈建 国 童钟杭 道通 畅 。1 例患者 出生 时即发 现有外 阴部异 常 , 以 先天性 肾上腺皮 质增 生症 ( A 是一 组 常染 C H)
色体隐陛遗传病, 临床少见 , 是由于某些合成肾上
腺皮 质类 固醇 激 素 所 需 酶 的先 天浓密 , 8 阴蒂肥大而就诊, 服
用氢 化可 的松 2年 后 出现 乳 房异 常 增 大。另 3例 2 1一羟化酶缺 乏患者社 会性 别 为男 性 , 型 为 4 , 核 6
缺如, 原发性 闭经, 高血压 , 低血钾 , 阴呈 幼稚 外 型, 内生殖器发育不 良, 皮质醇减少 , 肾上腺皮 促 质激素增多, h 1 羟类 固醇、7一 2 尿 7一 4 l 酮类 固醇 降低或正常低值 , 染色体 核型 4 ,【 , 断符合 6) 诊 x l 一羟化 酶 缺 乏症 。治 疗 上 予 以 口服 补充 糖 皮 7【 o
。
现 报道本 院 l9 97年 1 至 20 月 06年 1 收治 2月
的 c H患者 8例 , 以此对 C H 中不 同类 型女性 A 并 A
性分 化异常 的诊断和 治疗进行讨 论 。
1 临床资料
1 1 一般资料 . 8例 c H患者外周血核型均为 A
制 , 血钾 , 谢性 碱 中毒 ,4 低 代 2h尿 1 7一羟类 固醇 、
l 酮类固醇降低或正常低值( 7一 见表 1 。6 2 ) 例 1 羟化 酶 缺乏 患 者 实 验 室 检查 : 肾上腺 皮 质 激 促
一
素增 多 , 1 血 7一羟 孕 酮 增 高 ,4 2h尿 1 7一羟 类 固 醇 、7一酮类 固醇增 高 , 睾酮增 高 , l 血 血钾 正 常 ( 见 表 2 。肾上腺 B超示 双侧 肾上 腺不 同程度增生 , ) l
一
移, 阴囊 空 虚 而 就诊 。 患者 声 音 粗 , 肉较发 达 , 肌
乳房未发育 , 两侧 阴囊空虚 , 未触及睾丸, 阴蒂类 似阴茎 , 向腹侧弯曲, 3— c 尿道外 口位于阴 长 5m, 蒂根部, 两侧大阴唇融合。 13 辅助检查 2例 l仅一羟化 酶缺 乏患 者 的辅 . 7 助检查 : 皮质醇分泌减少 , 肾上腺皮质激素分泌 促 增多 , 雄激素分泌减少 , 雌、 肾素、 血管紧张素受抑
婚 , 例未婚但有性行为 ; 例父母为近亲结婚。8 l l
例 均未生育 。
12 临床表现 2例 1仅一羟化酶缺乏患者 因第 . 7 二性 征缺如 、 原发性 闭 经就 诊 , 患者 进 入青 春 期后 无月 经来潮 , 乳 似 男 性 , 腋 毛及 阴毛 生 长 , 双 无 外
阴呈幼稚 型 , 阴道 畅 , 双侧 腹 股沟 未 及包 块 。 同时
增 生伴腺瘤 形成 , 大小 约 12m × c .c 2m。腹股 沟 B 超未 探及 睾丸 回声 。妇科 B超示 未 探 及子 宫及 双 附件 ; 始基 子 宫 , 附 件 显 示 不 清 ; 稚 子 宫 或 子 双 幼 宫偏 小 , 卵巢 内可见卵泡形 成 。 14 诊断及 治疗 . 本文 2例患 者表现 为第二性 征
1 5岁, 其余患者核型与社会性别一致 , 例患者出 1 生时即发现有外 阴部异常 , 岁时就诊 , 2例患 8 另
者就 诊年龄 为 2 5岁和 2 1岁 。8例 患 者 平 均 年龄 2. ; 高 14~l2m( 均 13m) l例 离 15岁 身 4 6c 平 5c ;
例 l 岁患者肾上腺 c l T检查示左侧肾上腺结节样
质激素 同时辅 以降压 、 钾 治疗 , 适 当补 充雌 激 补 并
伴有高血压 、 低血钾 , 血压最高达 10 l m H , 9/ l m g 0 血钾 14— .m o/ , . 25 m lL 有四肢麻木、 / 乏力等症状 , 其中 l 例伴有皮肤色素沉着。2例 2 羟化酶缺 1一 乏 患者 因原 发性 闭经 或 喉结增 大 、 嗓音增 粗 就诊 , 可见双乳未发育 , 皮肤 毛孔粗 大, 毛增 多, 体 外阴 阴毛浓密 , 呈菱形分布, 阴蒂肥大 , 长约 3— c 阴 5m,
x 因 自幼 尿道 口异位 , 能站立 排尿 或尿道 口下 x, 不
固醇激素分泌异常 , 因继 发性促 肾上腺皮质激 且
素分 泌过多而 致 肾上腺 皮质 增生 。C H 的某些 类 A
型 可引起性分 化异 常 , 如假 两性 畸 形 、 二 性 征缺 第 如 、 发 性 闭 经 等 , 性 分 化 异 常 的重 要 病 因之 原 是
4 , ) 包括 2例 l仅一 6 x( , 7 羟化酶缺乏和 6 2 一 例 l 羟 化酶 缺 乏 。2例 1 一羟化 酶 缺 乏患 者 就诊 年 龄 7
为3 8岁和 3 4岁 , 社会性 别为女性 。2 羟 化酶缺 1一 乏患 者 中有 3例 社 会性 别 为 男性 , 诊 年龄 1 就 4—