小容量注射剂装量检查规程
最低装量检查法标准操作规程

最低装量检查法标准操作规程目的:建立最低装量检查法的标准操作规程,保证临床用药剂量。
2.依据:《中华人民共和国药典》2000年版二部。
3 范围:适用于固体、半固体制剂中标示装量不大于500g(ml),液体制剂标示装量为500ml以下的制剂。
4.职责:质量部QC检验员对本标准的实施负责。
5. 程序:5.1.重量法(适用于标示装量以重量计者)。
除另有规定外,取供试品5个(50g以上者3个),除去外盖和标签,容器外壁用适宜的方法清洁并干燥,分别精密称定重量,除去内容物,容器内容物用适宜的溶剂洗净并干燥,再分别精密称定空容器的重量,求出每个容器物的装量与平均装量,均应符合规定。
如有1个容器装量不符合规定,则另取5个(或3个)复试,应全部符合规定。
5.2. 容量法(适用于标示装量以容量计者)。
除另有规定外,取供试品5个(50ml以上者3个),开启时注意避免损失,将内容物分别用干燥并预经标化的注射器(包括注射针头)抽尽,50ml以上者可倾入预经标化的干燥量筒中,黏稠液体倾出后,将容倒置15分钟,尽量倾净。
读出每个容器内容物的装量,并求其平均装量,均应符合规定。
如有1个容器装量不符合规定,则另取5个(或3个)复试,应全部符合规定。
6. 附:装量控制表:注射液中不溶性微粒检查法(光阻法)标准操作规程目的:建立注射液不溶性微粒检查法(光阻法)标准操作规程。
2.依据:《中华人民共和国药典》2000年版二部。
3. 范围:适用于静脉滴注用注射液(装量为100ml以上者)经澄明度检查符合规定后的不溶性微粒的检查。
4.职责:质量部QC检验员对本标准的实施负责。
5. 程序:5.1. 原理:当液体中的微粒通过一窄小的检测区时,与流体流向垂直的入射光,由于被不溶性微粒所阻挡,从而使传感器输出的信号变化,这种信号变化与微粒的截面积成正比,光阻法检查注射液中不溶性微粒即依据此原理。
5.2. 仪器装置:仪器通常应包括取样器、传感器和数据处理三部分。
小容量注射剂

1. 最终灭菌小容量注射剂的生产流程与环境要求图2.小容量注射剂验证检查要点小容量注射剂指将配制好药淮灌入小于50ml安瓿内的注射剂。
根据药品新版GMP原则要求,结合注射剂生产影响药品质量的关键环节,提出小容量注射剂GMP检查要点,旨在督查药品生产企业质量保证体系和实际运行状态。
检查组须对如下内容按照药品GMP要求,逐一核实。
1、机构与人员a)主管生产和质量管理的企业负责人、生产管理和质量管理部门负责人均应具有医药或相关专业大专以上学历,并具有药品生产和质量管理经验,并履行其职责。
b)企业负责人和各级管理人员应定期接受药品管理法律法规培训。
c)质量检验、生产、维修保养、清洁人员应定期进行卫生和微生物学基础知识、洁净作业等方面的培训和考核,并具有实际操作技能。
2、厂房设施的管理要点及检查重点a)洁净区:我国《药品生产质量管理规范》对最终灭菌的无菌药品生产厂房洁净度级别的要求是:浓配或采用密闭系统的稀配应在100000级洁净区内进行;稀配、滤过、灌封、直接接触药品的包装材料的最终处理等操作应在10000级洁净区内进行;物料、中间品应经过物流缓冲间或传递柜进、出洁净区。
称量配料间如产尘应与洁净走廊呈相对负压,必要时设捕尘设施。
中国药典规定:微生物限度检查、无菌检查应在100级或10000级背景下的局部100级区内进行,并与生产区分开。
微生物限度检查与无菌检查用的实验室和空气净化系统最好彼此分开,以尽可能减少对无菌检查的干扰。
b)空气净化系统应能确保洁净区的洁净度级别、温湿度、压差等符合生产工艺要求并经过验证。
初效、中效过滤器应明确清洗/更换周期,高效过滤器应定期检测其完整性,如有泄漏或阻塞应及时更换。
空气净化系统应每天24小时运行,停用后再次运行应进行清洁、消毒并经过再验证,符合要求的方可开始生产。
c)与产品直接接触的压缩空气、氮气、二氧化碳等辅助设施这些气体因与产品直接接触,不得对产品带来污染。
应对系统进行验证。
任务12小容量注射剂
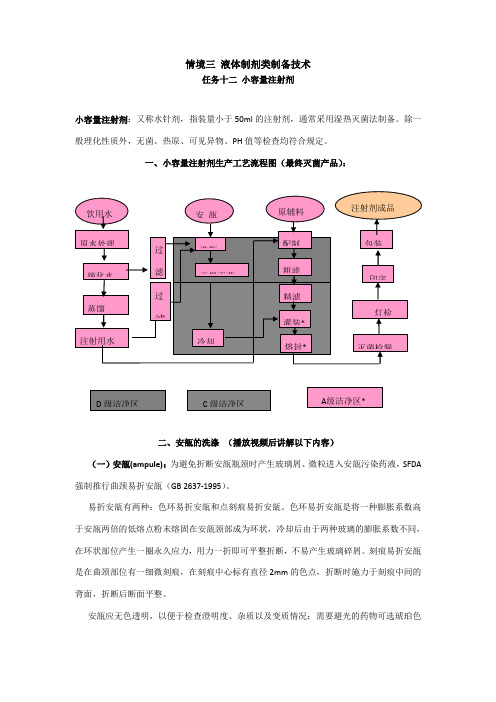
情境三 液体制剂类制备技术任务十二 小容量注射剂小容量注射剂:又称水针剂,指装量小于50ml 的注射剂,通常采用湿热灭菌法制备。
除一般理化性质外,无菌、热原、可见异物、PH 值等检查均符合规定。
一、小容量注射剂生产工艺流程图(最终灭菌产品):二、安瓿的洗涤 (播放视频后讲解以下内容)(一)安瓿(ampule):为避免折断安瓿瓶颈时产生玻璃屑、微粒进入安瓿污染药液,SFDA 强制推行曲颈易折安瓿(GB 2637-1995)。
易折安瓿有两种:色环易折安瓿和点刻痕易折安瓿。
色环易折安瓿是将一种膨胀系数高于安瓿两倍的低熔点粉末熔固在安瓿颈部成为环状,冷却后由于两种玻璃的膨胀系数不同,在环状部位产生一圈永久应力,用力一折即可平整折断,不易产生玻璃碎屑。
刻痕易折安瓿是在曲颈部位有一细微刻痕,在刻痕中心标有直径2mm 的色点,折断时施力于刻痕中间的背面,折断后断面平整。
安瓿应无色透明,以便于检查澄明度、杂质以及变质情况;需要避光的药物可选琥珀色玻璃安瓿(可滤除紫外线),但因含氧化铁,已被铁离子催化的药物不能使用。
制造安瓿用玻璃分中性玻璃(适宜中性或弱酸性药液)、含钡玻璃(耐碱性好,适宜碱性较强药液)和含锆玻璃(耐酸碱性好,适宜酸碱性较强的及对PH敏感的药液)三种。
安瓿规格:1、2、5、10、20ml。
(二)安瓿的质量要求及检查1.安瓿的质量要求:应具有低的膨胀系数,优良的耐热性。
要有足够的物理强度。
高度的化学稳定性,不改变溶液的pH,不被侵蚀。
熔点较低,易于熔封。
不得有气泡、麻点及砂粒。
2.安瓿的检查:物理检查:外观、尺寸、应力、清洁度、热稳定性等。
化学检查:耐酸、耐碱和中性检查。
尚需做装药试验,检查安瓿与药液的相容性,证明无影响后方能使用。
(三)安瓿的洗涤技术与设备1.超声洗涤法与加压气水喷射洗涤法综合洗涤方法。
2.设备:洗、烘(灭菌)、灌、封联动线。
(四)安瓿洗涤岗位洁净度要求:D级(五)安瓿洗涤操作过程(六)安瓿清洗过程的工艺管理要点和质量控制1.生产工艺管理要点:(1)清洗过程随时检查水气压力,保证水气能冲到安瓿底部。
最低装量检查标准操作规程

最低装量检查标准操作规程1简述最低装量检查法(中国药典 2005年版二部附录X F)适用于固体、半固体或液体制剂。
凡放射性药品及制剂通则中规定检查重(装)量差异的剂型不再进行最低装量检查。
2仪器与用具2.1天平感量 1mg 或 10mg 或 0.1g。
2.2注射器规格 5、 10、 20 及 50ml ,经定期检定合格(容量包括注射针头)。
2.3量筒(量入型)规格 50、100、200及500ml,经定期检定合格。
3操作方法3.1重量法(适用于标示装量以重量计者)除另有规定外,取供试品5个(标示装量为50g以上者3个),除去外盖和标签,容器外壁用适宜的方法清洁并干燥后,分别精密称定重量,除去内容物,容器内壁用适宜的溶剂洗净并干燥,再分别精密称定容器的重量,求出每个容器内容物的装量与平均装量(均取三为有效数字)。
3.2容量法(适用于标示量以容量计者)除另有规定外,取供试品 5个(50ml以上者3个0,将内容物分别用响应体积的干燥注射器抽尽; 50ml 以上者可倾入响应体积的干燥量筒(量入型)中,黏稠液体倾出后,将容器倒置 15分钟,尽量倾净。
读出每个容器内容物的装量,并求出其平均装量。
(均取三为有效数字)。
4注意事项4.1开启瓶盖时,应注意避免损失。
4.2每个供试品的两次称量中,应注意编号顺序和容器的对号。
4.3所用注射器或量筒必须洁净、干燥并经定期检定;其最大刻度值与供试品的标示装量一致,或不超过标示装量的2 倍。
5记录与计算5.1记录室温、标示装量、仪器及其规格、每个容器内容物读数(ml),或每个供试品重量及其自身空容器重量、并求算每个容器装量。
5.2每个容器装量之和除以 5(或 3),即得平均装量。
5.3按下表,求出每个容器允许的最低装量,以及黏稠液体允许的最低平均装量(均取三为有效数字)。
5.4如遇平均装量处于标示装量边缘者,计算出平均装量为标示量的百分率,再取三为有效数。
标示装量固体、半固体、液体粘稠液体(容量法)平均装量每个容器装量平均装量每个容器装量20g(ml )以下不少于标示装量不少于标示装量的93%不少于标示装量的 90%不少于标示装量的 85%20g (ml) ~50g ( ml)不少于标示装量不少于标示装量的95%不少于标示装量的 95%不少于标示装量的 90%50g (ml)以上不少于标示装量不少于标示装量的97%不少于标示装量的 95%不少于标示装量的 93%6结果判定6.1每个容器的装量不少于允许最低装量,且平均装量不少于标示装量(黏稠液体不少于允许最低平均装量),判为符合规定。
小容量注射剂

1. 最终灭菌小容量注射剂的生产流程与环境要求图2.小容量注射剂验证检查要点小容量注射剂指将配制好药淮灌入小于50ml安瓿内的注射剂。
根据药品新版GMP原则要求,结合注射剂生产影响药品质量的关键环节,提出小容量注射剂GMP检查要点,旨在督查药品生产企业质量保证体系和实际运行状态。
检查组须对如下内容按照药品GMP要求,逐一核实。
1、机构与人员a)主管生产和质量管理的企业负责人、生产管理和质量管理部门负责人均应具有医药或相关专业大专以上学历,并具有药品生产和质量管理经验,并履行其职责。
b)企业负责人和各级管理人员应定期接受药品管理法律法规培训。
c)质量检验、生产、维修保养、清洁人员应定期进行卫生和微生物学基础知识、洁净作业等方面的培训和考核,并具有实际操作技能。
2、厂房设施的管理要点及检查重点a)洁净区:我国《药品生产质量管理规范》对最终灭菌的无菌药品生产厂房洁净度级别的要求是:浓配或采用密闭系统的稀配应在100000级洁净区内进行;稀配、滤过、灌封、直接接触药品的包装材料的最终处理等操作应在10000级洁净区内进行;物料、中间品应经过物流缓冲间或传递柜进、出洁净区。
称量配料间如产尘应与洁净走廊呈相对负压,必要时设捕尘设施。
中国药典规定:微生物限度检查、无菌检查应在100级或10000级背景下的局部100级区内进行,并与生产区分开。
微生物限度检查与无菌检查用的实验室和空气净化系统最好彼此分开,以尽可能减少对无菌检查的干扰。
b)空气净化系统应能确保洁净区的洁净度级别、温湿度、压差等符合生产工艺要求并经过验证。
初效、中效过滤器应明确清洗/更换周期,高效过滤器应定期检测其完整性,如有泄漏或阻塞应及时更换。
空气净化系统应每天24小时运行,停用后再次运行应进行清洁、消毒并经过再验证,符合要求的方可开始生产。
c)与产品直接接触的压缩空气、氮气、二氧化碳等辅助设施这些气体因与产品直接接触,不得对产品带来污染。
应对系统进行验证。
小容量注射剂

1. 最终灭菌小容量注射剂的生产流程与环境要求图2.小容量注射剂验证检查要点小容量注射剂指将配制好药淮灌入小于50ml 安瓿的注射剂。
根据药品新版GMP 原则要求,结合注射剂生产影响药品质量的关键环节,提出小容量注射剂GMP 检查要点,旨在督查药品生产企业质量保证体系和实际运行状态。
检查组须对如下容按照药品GMP 要求,逐一核实。
1、 机构与人员a) 主管生产和质量管理的企业负责人、生产管理和质量管理部门负责人均应具有医药或相关专业大专以上学历,并具有药品生产和质量管理经验,并履行其职责。
b) 企业负责人和各级管理人员应定期接受药品管理法律法规培训。
c)质量检验、生产、维修保养、清洁人员应定期进行卫生和微生物学基础知识、洁净作业等方面的培训和考核,并具有实际操作技能。
2、厂房设施的管理要点及检查重点a)洁净区:我国《药品生产质量管理规》对最终灭菌的无菌药品生产厂房洁净度级别的要:浓配或采用密闭系统的稀配应在100000级洁净区进行;稀配、滤过、灌封、直接接触药品的包装材料的最终处理等操作应在10000级洁净区进行;物料、中间品应经过物流缓冲间或传递柜进、出洁净区。
称量配料间如产尘应与洁净走廊呈相对负压,必要时设捕尘设施。
中国药典规定:微生物限度检查、无菌检查应在100级或10000级背景下的局部100级区进行,并与生产区分开。
微生物限度检查与无菌检查用的实验室和空气净化系统最好彼此分开,以尽可能减少对无菌检查的干扰。
b)空气净化系统应能确保洁净区的洁净度级别、温湿度、压差等符合生产工艺要求并经过验证。
初效、中效过滤器应明确清洗/更换周期,高效过滤器应定期检测其完整性,如有泄漏或阻塞应及时更换。
空气净化系统应每天24小时运行,停用后再次运行应进行清洁、消毒并经过再验证,符合要求的方可开始生产。
c)与产品直接接触的压缩空气、氮气、二氧化碳等辅助设施这些气体因与产品直接接触,不得对产品带来污染。
应对系统进行验证。
注射剂检验标准操作规程

一、目的:建立注射剂检验标准操作规程,防止错检、漏检的发生。
二、范围:适用于注射剂的检验操作方法。
三、责任:质量部、化验室相关操作人员。
四、内容:注射剂注射剂(《中国药典》2010年版二部附录IB)系指药物与适宜的溶剂或分散介质制成的供注入体内的溶液、乳状液或混悬液,以及供临用前配制或稀释成溶液或混悬液的粉末或浓溶液的无菌制剂。
注射剂可分注射液(其中供静脉滴注用的大体积注射液也称静脉输液)、注射用无菌粉末与注射用浓溶液。
注射剂除应按药典品种项下规定的检验项目外,还应检查“装量”或“装量差异”、“可见异物”和“无菌”。
静脉用注射剂应加查“热原”或“细菌内毒素”;溶液型静脉用注射液、溶液型静脉注射用粉末及注射用浓溶液应加查“不溶性微粒”;静脉输液及插管注射用注射液应加查“渗透压摩尔浓度”。
混悬型注射液,除另有规定外,药物粒度应控制在l5um以下,含15~20um(间有个别20~50um)者,不应超过10%,若有可见沉淀,振摇时应容易分散均匀;乳状液型注射液不得有相分离现象;静脉用乳状液型注射液分散相球粒的粒度90%应在lum以下,并不得有大于5um的乳滴。
“装量”检查法1 简述1.1本法适用于50ml及50ml以下的单剂量注射液的装量检查,其目的在于保证单剂量注射液的注射用量不少于标示量,以达到临床用药剂量要求。
1.2标示装量为50ml以上的注射液和注射用浓溶液,按最低装量检查法标准操作规范检查,应符合规定。
1.3 凡规定检查含量均匀度的注射液(如塞替派注射液).可不进行“装量”检查。
2 仪器与用具2.1注射器及注射针头。
2.2量筒(量入型)规格l、2、5、1O、20及50ml的量筒,均应预经标化。
3 操作方法3.1 按下表规定取用量抽取供试品。
标示装量供试品取用量(支)2ml或2ml以下 52ml以上至50ml 33.2取供试品,擦净瓶外壁,轻弹瓶颈部使液体全部下落,小心开启,将每支内容物分别用相应体积的干燥注射器(包括注射器针头)抽尽,注入预经标化的量筒内,在室温下检视,读出每支装量。
任务12小容量注射剂

情境三 液体制剂类制备技术任务十二 小容量注射剂小容量注射剂:又称水针剂,指装量小于50ml 的注射剂,通常采用湿热灭菌法制备。
除一般理化性质外,无菌、热原、可见异物、PH 值等检查均符合规定。
一、小容量注射剂生产工艺流程图〔最终灭菌产品〕:二、安瓿的洗涤 〔播放视频后讲解以下容〕〔一〕安瓿(ampule):为防止折断安瓿瓶颈时产生玻璃屑、微粒进入安瓿污染药液,SFDA强制推行曲颈易折安瓿〔GB 2637-1995〕。
易折安瓿有两种:色环易折安瓿和点刻痕易折安瓿。
色环易折安瓿是将一种膨胀系数高于安瓿两倍的低熔点粉末熔固在安瓿颈部成为环状,冷却后由于两种玻璃的膨胀系数不同,在环状部位产生一圈永久应力,用力一折即可平整折断,不易产生玻璃碎屑。
刻痕易折安瓿是在曲颈部位有一细微刻痕,在刻痕中心标有直径2mm 的色点,折断时施力于刻痕中间的背面,折断后断面平整。
安瓿应无色透明,以便于检查澄明度、杂质以与变质情况;需要避光的药物可选琥珀色玻璃安瓿〔可滤除紫外线〕,但因含氧化铁,已被铁离子催化的药物不能使用。
制造安瓿用玻璃分中性玻璃〔适宜中性或弱酸性药液〕、含钡玻璃〔耐碱性好,适宜碱性较强药液〕和含锆玻璃〔耐酸碱性好,适宜酸碱性较强的与对PH敏感的药液〕三种。
安瓿规格:1、2、5、10、20ml。
〔二〕安瓿的质量要求与检查1.安瓿的质量要求:应具有低的膨胀系数,优良的耐热性。
要有足够的物理强度。
高度的化学稳定性,不改变溶液的pH,不被侵蚀。
熔点较低,易于熔封。
不得有气泡、麻点与砂粒。
2.安瓿的检查:物理检查:外观、尺寸、应力、清洁度、热稳定性等。
化学检查:耐酸、耐碱和中性检查。
尚需做装药试验,检查安瓿与药液的相容性,证明无影响后方能使用。
〔三〕安瓿的洗涤技术与设备1.超声洗涤法与加压气水喷射洗涤法综合洗涤方法。
2.设备:洗、烘〔灭菌〕、灌、封联动线。
〔四〕安瓿洗涤岗位洁净度要求:D级〔五〕安瓿洗涤操作过程〔六〕安瓿清洗过程的工艺管理要点和质量控制1.生产工艺管理要点:〔1〕清洗过程随时检查水气压力,保证水气能冲到安瓿底部。
最低装量检查标准操作规程

最低装量检查标准操作规程1 简述最低装量检查法(中国药典2005年版二部附录X F)适用于固体、半固体或液体制剂。
凡放射性药品及制剂通则中规定检查重(装)量差异的剂型不再进行最低装量检查。
2 仪器与用具2.1 天平感量1mg或10mg或0.1g。
2.2 注射器规格5、10、20及50ml,经定期检定合格(容量包括注射针头)。
2.3 量筒(量入型)规格50、100、200及500ml,经定期检定合格。
3 操作方法3.1 重量法(适用于标示装量以重量计者)除另有规定外,取供试品5个(标示装量为50g以上者3个),除去外盖和标签,容器外壁用适宜的方法清洁并干燥后,分别精密称定重量,除去内容物,容器内壁用适宜的溶剂洗净并干燥,再分别精密称定容器的重量,求出每个容器内容物的装量与平均装量(均取三为有效数字)。
3.2 容量法(适用于标示量以容量计者)除另有规定外,取供试品5个(50ml以上者3个0,将内容物分别用响应体积的干燥注射器抽尽;50ml以上者可倾入响应体积的干燥量筒(量入型)中,黏稠液体倾出后,将容器倒置15分钟,尽量倾净。
读出每个容器内容物的装量,并求出其平均装量。
(均取三为有效数字)。
4 注意事项4.1开启瓶盖时,应注意避免损失。
4.2 每个供试品的两次称量中,应注意编号顺序和容器的对号。
4.3 所用注射器或量筒必须洁净、干燥并经定期检定;其最大刻度值与供试品的标示装量一致,或不超过标示装量的2倍。
5 记录与计算5.1 记录室温、标示装量、仪器及其规格、每个容器内容物读数(ml),或每个供试品重量及其自身空容器重量、并求算每个容器装量。
5.2 每个容器装量之和除以5(或3),即得平均装量。
5.3按下表,求出每个容器允许的最低装量,以及黏稠液体允许的最低平均装量(均取三为有效数字)。
5.4 如遇平均装量处于标示装量边缘者,计算出平均装量为标示量的百分率,再取三为有效数。
标示装量固体、半固体、液体粘稠液体(容量法)平均装量每个容器装量平均装量每个容器装量20g(ml)以下不少于标示装量不少于标示装量的93%不少于标示装量的90%不少于标示装量的85%20g(ml)~50g(ml)不少于标示装量不少于标示装量的95%不少于标示装量的95%不少于标示装量的90% 50g(ml)以上不少于标示装量不少于标示装量的97%不少于标示装量的95%不少于标示装量的93%6 结果判定6.1 每个容器的装量不少于允许最低装量,且平均装量不少于标示装量(黏稠液体不少于允许最低平均装量),判为符合规定。
小容量注射剂
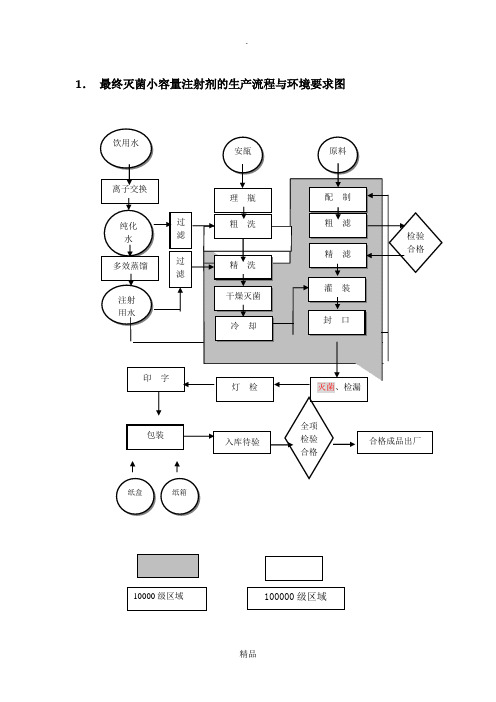
1. 最终灭菌小容量注射剂的生产流程与环境要求图2.小容量注射剂验证检查要点小容量注射剂指将配制好药淮灌入小于50ml安瓿内的注射剂。
根据药品新版GMP原则要求,结合注射剂生产影响药品质量的关键环节,提出小容量注射剂GMP检查要点,旨在督查药品生产企业质量保证体系和实际运行状态。
检查组须对如下内容按照药品GMP要求,逐一核实。
1、机构与人员a)主管生产和质量管理的企业负责人、生产管理和质量管理部门负责人均应具有医药或相关专业大专以上学历,并具有药品生产和质量管理经验,并履行其职责。
b)企业负责人和各级管理人员应定期接受药品管理法律法规培训。
c)质量检验、生产、维修保养、清洁人员应定期进行卫生和微生物学基础知识、洁净作业等方面的培训和考核,并具有实际操作技能。
2、厂房设施的管理要点及检查重点a)洁净区:我国《药品生产质量管理规范》对最终灭菌的无菌药品生产厂房洁净度级别的要求是:浓配或采用密闭系统的稀配应在100000级洁净区内进行;稀配、滤过、灌封、直接接触药品的包装材料的最终处理等操作应在10000级洁净区内进行;物料、中间品应经过物流缓冲间或传递柜进、出洁净区。
称量配料间如产尘应与洁净走廊呈相对负压,必要时设捕尘设施。
中国药典规定:微生物限度检查、无菌检查应在100级或10000级背景下的局部100级区内进行,并与生产区分开。
微生物限度检查与无菌检查用的实验室和空气净化系统最好彼此分开,以尽可能减少对无菌检查的干扰。
b)空气净化系统应能确保洁净区的洁净度级别、温湿度、压差等符合生产工艺要求并经过验证。
初效、中效过滤器应明确清洗/更换周期,高效过滤器应定期检测其完整性,如有泄漏或阻塞应及时更换。
空气净化系统应每天24小时运行,停用后再次运行应进行清洁、消毒并经过再验证,符合要求的方可开始生产。
c)与产品直接接触的压缩空气、氮气、二氧化碳等辅助设施这些气体因与产品直接接触,不得对产品带来污染。
应对系统进行验证。
注射剂检查操作规程

制药GMP管理文件一、引用标准:中华人民共和国S药典(2005年版)一部。
二、目的:本标准规定了注射剂检查法标准操作规程。
三、适用范围:适用于注射剂的检查。
四、责任者:质检人员。
五、正文注射剂注射剂系指药物与适宜的溶剂或分散介质制成的供注入体内的溶液、乳状液或混悬液及供监用前配制或稀释成溶液或混悬液的粉末或浓液的无菌制剂。
注射剂可分为为注射液、注射用无菌粉末与注射用浓溶液。
注射液系指药物制成的供注射人作内用的无菌溶液型注射液、乳状液型注射液或混悬型注射液。
可用于肌内注射、静脉注射、静脉滴注等。
其中,供静脉滴注用的大体积(除另有规定外,一般不小于100ml)注射液也称静脉输液。
注射用无菌粉末系指药物制成的供临用前用适宜的无菌溶液配制成澄清液或均匀混液的无菌粉末或无菌块状物。
可用适宜的注射用溶剂配制后注射,也可用静脉输液配制后静脉。
无菌粉末用溶剂结晶法、喷雾干燥法或冷冻干燥法等方法制得。
注射用无菌粉末系指药物制成的供临用适宜的无菌溶液配制成澄清溶液或均匀混悬液的无菌粉末或无菌块状物。
可用适宜的注射用溶剂配后注射,也可用静脉输液配制后静脉滴注。
无菌粉末用溶剂结盟法、喷雾干燥法或冷冻干燥法等方法制得。
注射用浓溶液系指药物制成的供临用前稀释供静脉滴注用的无菌浓溶液。
注射剂在生产与贮藏期间应符合下列有关规定。
一,溶液型注射液应澄明;除另有规定外,混悬型注射液药物粒度应控制在15um以下,含15~20um(间有个别20~50um)者,不应超过10%,若有可见沉淀,振摇里应容易分散均匀,不得用于静脉注射或椎管注射;乳状液型注射液应稳定,不得有相分离现象,不得用于椎管注射,静脉用乳状液型注射液分散相球粒的粒度90%应在1um 以下,不得大于5um的球粒。
静脉输液应尽可能与血液等渗。
二、注射剂所用溶剂必须安全无害,并不得影响疗效和质量。
一般分为水溶剂和非水性溶剂。
1.水性溶剂:最常用的为注射用水,也可用0.9%氯化钠溶液或其他适宜的水溶液。
注射剂检查操作规程
制药GMP管理文件一、引用标准:中华人民共和国S药典(2005年版)一部。
二、目的:本标准规定了注射剂检查法标准操作规程。
三、适用范围:适用于注射剂的检查。
四、责任者:质检人员。
五、正文注射剂注射剂系指药物与适宜的溶剂或分散介质制成的供注入体内的溶液、乳状液或混悬液及供监用前配制或稀释成溶液或混悬液的粉末或浓液的无菌制剂。
注射剂可分为为注射液、注射用无菌粉末与注射用浓溶液。
注射液系指药物制成的供注射人作内用的无菌溶液型注射液、乳状液型注射液或混悬型注射液。
可用于肌内注射、静脉注射、静脉滴注等。
其中,供静脉滴注用的大体积(除另有规定外,一般不小于100ml)注射液也称静脉输液。
注射用无菌粉末系指药物制成的供临用前用适宜的无菌溶液配制成澄清液或均匀混液的无菌粉末或无菌块状物。
可用适宜的注射用溶剂配制后注射,也可用静脉输液配制后静脉。
无菌粉末用溶剂结晶法、喷雾干燥法或冷冻干燥法等方法制得。
注射用无菌粉末系指药物制成的供临用适宜的无菌溶液配制成澄清溶液或均匀混悬液的无菌粉末或无菌块状物。
可用适宜的注射用溶剂配后注射,也可用静脉输液配制后静脉滴注。
无菌粉末用溶剂结盟法、喷雾干燥法或冷冻干燥法等方法制得。
注射用浓溶液系指药物制成的供临用前稀释供静脉滴注用的无菌浓溶液。
注射剂在生产与贮藏期间应符合下列有关规定。
一,溶液型注射液应澄明;除另有规定外,混悬型注射液药物粒度应控制在15um以下,含15~20um(间有个别20~50um)者,不应超过10%,若有可见沉淀,振摇里应容易分散均匀,不得用于静脉注射或椎管注射;乳状液型注射液应稳定,不得有相分离现象,不得用于椎管注射,静脉用乳状液型注射液分散相球粒的粒度90%应在1um 以下,不得大于5um的球粒。
静脉输液应尽可能与血液等渗。
二、注射剂所用溶剂必须安全无害,并不得影响疗效和质量。
一般分为水溶剂和非水性溶剂。
1.水性溶剂:最常用的为注射用水,也可用0.9%氯化钠溶液或其他适宜的水溶液。
小容量注射剂装量检查规程

小容量注射剂装量检查规程
目的:
建立小容量注射剂装量检查的标准操作规程,保证单剂量注射液的注射用量不少于标示量,保证临床用药剂量。
2. 依据:
《中华人民共和国药典》(2000年版二部)附录。
3. 范围:
适用于50ml及50ml以下的单剂量注射剂的装量检查。
4. 职责:
QA检查员、QC检验员对本标准的实施负责。
5. 程序:
5.1. 仪器与用具:
5.1.1. 注射器规格1、2、5、10、20及50ml。
5.1.2.与上述注射器相适应的注射针头。
5.1.3.经过标化的干燥量筒。
5.2. 操作方法:
5.2.1. 按表1规定取样抽取供试品。
表1. 注射剂装量检查取样量规定:
取供试品,擦净安瓿外壁,轻弹安瓿颈部使液体全部下落,折断安瓿颈(开启时注意避免损失),将每支内容物分别用相应体积的干燥注射器(配有适宜针头)抽尽,然后注入标化的量筒内,在室温下检视。
5.2.3.如供试品为油溶液或混悬液时,检查前应先微温摇匀,立即按“5.2.2.”项
下操作,并冷至室温后检视。
5.3. 注意事项:
5.3.1.所用注射器必须洁净、干燥。
5.3.2.所用注射器应配上适宜号数的针头,其大小与临床使用情况相近为宜。
5.4. 记录:主要记录室温、抽取供试品支数、供试品的标示装量、每支供试品实测装
量。
5.5. 结果与判定:每支注射液的装量均不得少于其标示量,如有少于其标示量者,即判为不符合规定。
0942 最低装量检查法1 - 国家药典委员会
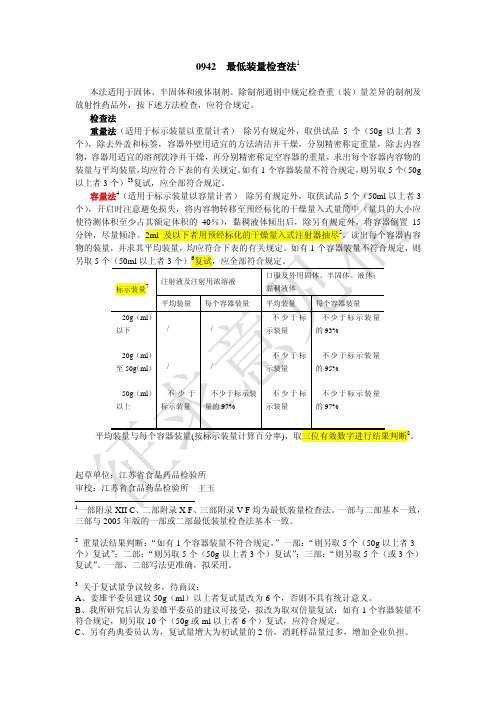
0942最低装量检查法1本法适用于固体、半固体和液体制剂。
除制剂通则中规定检查重(装)量差异的制剂及 放射性药品外,按下述方法检查,应符合规定。
检查法 重量法(适用于标示装量以重量计者) 除另有规定外,取供试品 5 个(50g 以上者 3 个) ,除去外盖和标签,容器外壁用适宜的方法清洁并干燥,分别精密称定重量,除去内容 物,容器用适宜的溶剂洗净并干燥,再分别精密称定空容器的重量,求出每个容器内容物的 装量与平均装量, 均应符合下表的有关规定。
如有 1 个容器装量不符合规定, 则另取 5 个 (50g 以上者 3 个)23复试,应全部符合规定。
容量法4(适用于标示装量以容量计者) 除另有规定外,取供试品 5 个(50ml 以上者 3 个) ,开启时注意避免损失,将内容物转移至预经标化的干燥量入式量筒中(量具的大小应 使待测体积至少占其额定体积的 40%) ,黏稠液体倾出后,除另有规定外,将容器倒置 15 分钟,尽量倾净。
2ml 及以下者用预经标化的干燥量入式注射器抽尽5。
读出每个容器内容 物的装量,并求其平均装量,均应符合下表的有关规定。
如有 1 个容器装量不符合规定,则 另取 5 个(50ml 以上者 3 个)6复试,应全部符合规定。
标示装量7注射液及注射用浓溶液 平均装量 每个容器装量 /口服及外用固体、半固体、液体; 黏稠液体 平均装量 不少于标 每个容器装量 不少于标示装量 的 93% 不少于标示装量 的 95% 不少于标示装量 的 97%20g(ml) 以下 20g(ml) 至 50g (ml) 50g(ml) 以上 / / /示装量 不少于标 示装量 不少于标 示装量不少于 标示装量不少于标示装 量的 97%平均装量与每个容器装量(按标示装量计算百分率),取三位有效数字进行结果判断8。
起草单位:江苏省食品药品检验所 审校:江苏省食品药品检验所 王玉 一部附录 XII C、 二部附录 X F、 三部附录 V F 均为最低装量检查法。
中国药典最低装量检查法

2 仪器与用具
2.1 注射器 规格5及lOml,经定期检定 合格。
2.2 注射针头
3 操作方法
取供试品5支,开启时注意避免损失,将 内容物分别用干燥并预经标化的注射器抽尽, 读取每支装量(读数精确至0.1m1),并求其 平均装量。
4 注意事项
4.1 所用注射器必须洁净,干燥并经定期 校正。
6.2 如其平均装量符合规定,但仅有1瓶的装量 不符合规定,则另取5瓶复试,应全部符合规定。
6.3 如其平均装量有2瓶(或2瓶以上)的装量不 符合规定,或在复试中不能全部符合规定者,均判 为不符合规定。
谢谢
5.2 根据每支眼膏重量与其自身空容器重 量之差求算每支装量。
5.3 5支装量之和除以5,得平均装量 (m)。保留3位有效数字。
6 结果与判定
6.1 平均装量应不少于标示装量,且每支装量 均不少于标示装量的93%者,判为符合规定。
如仅有一支的装量少于标示装量的93%,且平 均装量不少于标示装量,则另取5支复试,复试结 果全部符合规定者,仍判为符合规定。
5.2 每个容器装量之和除以5(或3),即得平均装量。 5.3 按下表规定的平均装量及每个容器装量的低限,
求出每个容器允许的最低装量,以及黏稠液体允许 的最低平均装量(均取三位有效数字)。〔附图〕
6 结果判定
6.1 每个容器的装量不少于允许最低装量,且平均装量不少于 标示装量(黏稠液体不少于允许最低平均装量)者,判为符合规 定。如仅有一个容器的装量不符合规定,则另取5个[50g(或 m1)以上者3个]复试,复试结果全部符合规定者,仍判为符合 规定。
4 注意事项
4.1 所用注射器和量筒均必须洁净、干燥并 经定期标化。
4.2 糖浆剂为粘稠液体,如标示装量为 50ml以上者,倾入量筒后,应将容器倒置 15分钟,尽量倾尽。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
小容量注射剂装量检查规程
目的:
建立小容量注射剂装量检查的标准操作规程,保证单剂量注射液的注射用量不少于标示量,保证临床用药剂量。
2. 依据:
《中华人民共和国药典》(2000年版二部)附录。
3. 范围:
适用于50ml及50ml以下的单剂量注射剂的装量检查。
4. 职责:
QA检查员、QC检验员对本标准的实施负责。
5. 程序:
5.1. 仪器与用具:
注射器规格1、2、5、10、20及50ml。
5.1.2.与上述注射器相适应的注射针头。
5.1.3.经过标化的干燥量筒。
5.2. 操作方法:
按表1规定取样抽取供试品。
表1. 注射剂装量检查取样量规定:
取供试品,擦净安瓿外壁,轻弹安瓿颈部使液体全部下落,折断安瓿颈(开启时注意避免损失),将每支内容物分别用相应体积的干燥注射器(配有适宜针头)抽尽,然后注入标化的量筒内,在室温下检视。
5.2.3.如供试品为油溶液或混悬液时,检查前应先微温摇匀,立即按“
下操作,并冷至室温后检视。
5.3. 注意事项:
5.3.1.所用注射器必须洁净、干燥。
5.3.2.所用注射器应配上适宜号数的针头,其大小与临床使用情况相近为宜。
5.4. 记录:主要记录室温、抽取供试品支数、供试品的标示装量、每支供试品实测装量。
5.5. 结果与判定:每支注射液的装量均不得少于其标示量,如有少于其标示量者,即判为不符合规定。
职工有以下表现之一者,给予罚款:
1、违反总公司规章制度;
2、给总公司造成经济损失;
3、泄露总公司内部信息、机密;
4、破坏总公司的形象、名誉。
以上处罚由部门随时将书面材料报人事本部审核,人事本部也可根据检查结果直接提出处理意见,报总公司总经理批准。
职工有上述表现者视情况给予20元以上500元以下的罚款,情节严重的也可给予工资降级。
各管理部门可在20元至500元的范围内制订本部门管辖范围内的罚款事项并将执行通知转人事本部,由人事本部通知财务本部在工资中扣除。
各管理部门可根据本部门管辖权限向人事本部提出具体处罚建议。
(二)职工有下列表现之一者,给予工资降级。
1、消极怠工、工作没有效益;
2、工作效率低,服务质量差:
3、没有达到本岗位职责要求;
4、有其它不利于公司发展的行为;
5、没有按时完成任务和计划目标;
6、总公司各部门领导决策重大失误;
7、虚报功绩,不实事求是。
上述第1至4款规定的处罚均由各部门于每季度未以书面形式报人事本部,经人事本部审核、报总公司急经理批准后给予工资降级,第三款规定的处罚由各部门于每年十二月以书面形式报人事本部,经人事本部审核、报总公司总经理批准后,给予工资降级。
(三)职工犯有以下错误之一,确属部门执行规章制度不够,管理不严不力造成的,所在单位负责人应负有领导责任:
1、严重违反劳动纪律,多次旷工或消极怠工;
2、有贪污、盗窃、流氓、打架斗殴行为;
3、违反保密制度、泄露总公司商业秘密;
4、违反外事纪律,严重损害总公司声誉和利益;
5、严重违反总公司人事管理和办公秩序制度及有关行为规范的规定。
对负有领导责任的负责人视情况予以以下处罚:
1、在全体职工大会做检查;
2、罚款500元以示教育;
3、降职、降级。
人事本部应随时对职工进行考核,发现符合工资降级的情况时,随时提出书面意见,报总公司总经理批准后给予工资降级,每年年末,人事本部对职工进行考核时,将以处罚情况作为主要依据之一,提出不称职的各级领导岗位人员和不能胜任本岗位工作人员的名单,报总公司总经理批准给予降职、降级。