欧洲药典附录3.1.3.-推荐下载
欧洲药典identification鉴别-概述说明以及解释

欧洲药典identification鉴别-概述说明以及解释1.引言1.1 概述欧洲药典identification鉴别是药品质量控制中的一项重要工作。
药品的identification鉴别可以理解为确定药品的真实性和纯度的过程,通过分析样品的特征和性质,与标准或已知样品进行比对,从而确保药品的质量和安全。
在欧洲药典中,identification鉴别是确定药品真实性的一种重要方法。
欧洲药典作为一个权威的药典体系,旨在保障欧洲地区药品的质量、安全和疗效。
其中,identification鉴别是其质量控制的核心内容之一。
通过进行identification鉴别,可以判断药品中的成分和杂质,并与标准进行比对,从而确保药品的质量符合规定标准。
此外,identification鉴别还能够帮助识别药品中可能存在的伪劣药品和假药,起到保护公众健康的重要作用。
在市场上,药品的质量状况千差万别,有些药品可能存在掺假、掺杂或不合格等情况。
通过进行identification鉴别,可以及时发现并排除这些潜在的风险,确保消费者使用的药品安全可靠。
此外,identification鉴别也有助于验证药品的纯度和稳定性。
药品的纯度和稳定性对于药物的疗效和安全性至关重要。
通过进行identification 鉴别,可以确定药品中各成分的含量和比例,以及检测药物是否受到环境因素的影响而发生变化。
这对于药品的生产和使用过程中的质量控制非常重要,可以保证药品的治疗效果和安全性。
综上所述,欧洲药典的identification鉴别是一项非常重要的工作。
它不仅可以确保药品的质量和安全,还可以帮助识别伪劣药品,保护公众健康。
随着科学技术的不断进步,我们对identification鉴别的研究和应用也将得到进一步的发展,为药品监管提供更加可靠和有效的手段。
1.2 文章结构文章结构部分的内容可以是对整篇文章的框架和组织方式进行说明。
这一部分旨在向读者介绍本文的结构及各部分的主要内容,以便读者能够更好地理解和阅读文章。
欧洲药典附录

欧洲药典附录 Prepared on 22 November 2020第二部分、附录附录1 溶液的澄清度在内径15~25mm,平底,无色、透明、中性玻璃管中,加入等量的供试溶液与浊度标准液,使液位的深度都为40mm,按如下所述方法进行比较。
浊度标准液制备5分钟后,以色散自然光照射浊度标准溶液和供试溶液,在黑色背景下从垂直方向观察、比较澄清度或浑浊程度。
色散自然光必须较容易区分浊度标准溶液Ⅰ与水,浊度标准溶液Ⅱ与浊度标准溶液Ⅰ。
如果供试溶液的澄清、透明程度与水相同,或者与所用溶剂相同,或者其澄清度不超过Ⅰ号浊度标准溶液,那么可判定该溶液为澄清。
试剂:硫酸肼溶液:取硫酸肼溶于水,加水稀释至,静置4~6小时。
乌洛托品(六亚甲基四胺)溶液:在100ml容量平中,以水溶解乌洛托品。
浊度标准贮备液:在存放乌洛托品溶液的100ml容量瓶中,加的硫酸肼溶液。
混合,静置24小时,贮存在无表面要求的玻璃容器中,可在2个月内使用。
该浊度液不得黏附玻璃,用前必须充分摇匀。
浊度标准原液:取浊度标准贮备液15ml,加水稀释、定容至1000ml。
该液临用前制备,至多保存24小时。
浊度标准液:由浊度标准原液与水按表1-1配制,即得。
本液应临用前配制。
附录2 溶液颜色检查按本药典规定,用下面两种方法之一可以检出溶液在棕色-黄色-红色范围内的颜色。
如果溶液A的外观与水或所用溶剂相同,或者颜色浅于标准比色液B9,则可判定溶液A为无色。
方法I用外径为12mm的无色、透明中性玻璃管取2ml的供试溶液,与相同玻璃管中的2ml的水,或2ml本文所规定的标准比色液(见标准比色液表)进行比较。
在散射自然光,白色的背景下,水平观察比较颜色。
方法Ⅱ用同样平底、内径为15~25mm的无色透明中性玻璃管,液位的深度为40mm,将供试溶液与水或溶剂或本文中规定的标准比色液(见标准比色液表)对比。
在散射自然光,白色的背景下,垂直地观察比较颜色。
贮备液黄色液称取46克氯化铁,加大约900ml盐酸溶液(25ml浓盐酸和975ml水混和)溶解,继续添加,并定容。
欧洲药典适用性证书的变更更新的管理程序 中英对照 2013.07.13

Procedures for management of revisions/renewals of certificates of suitability to the European Pharmacopoeia monographs Certification of suitability to Monographs of the European Pharmacopoeia欧洲药典适用性证书PROCEDURES FOR MANAGEMENT OF REVISIONS/RENEWALS OF CERTIFICATES OF SUITABILITY TO THE EUROPEAN PHARMACOPOEIAMONOGRAPHS欧洲药典适用性证书的变更/更新的管理程序Introduction:介绍This document should be read in conjunction with the EDQM “Guideline on Requirements on Revision/Renewal of Certificates of Suitability to the European Pharmacopoeia monographs”(PA/PH/CEP (04) 2, as amended), which describes the conditions to be fulfilled as well as the documentation to be submitted for each request for revision.此文件应该与EDQM的“欧洲药典适用性证书修订与更新规定指南” (PA/PH/CEP (04) 2)联合起来阅读,后者描述了每个变更所要求满足的条件,以及要提供的文件资料。
The procedures for the management of revisions of certificates of suitability (CEPs) are described below and have been revised according to the revised European Regulation for Variations to Marketing Authorisation Applications.对于CEP证书变更管理的程序,在下面进行了描述,并且按照新修订的欧洲市场授权申请的有关法规进行了修订。
eu无菌附录 解读

eu无菌附录解读1.引言概述部分的内容可以简要介绍无菌附录的背景和重要性。
以下是一个示例:1.1 概述无菌附录是指在欧洲联盟(EU)法规体系下对医疗器械生产领域中的无菌产品制造过程提出的一系列要求和规范。
无菌产品制造过程的合规性对于确保产品质量和安全至关重要。
因此,EU无菌附录对于制药和医疗器械行业的从业人员来说具有重要的解读和意义。
在医疗器械生产过程中,无菌产品扮演着至关重要的角色,尤其是在手术等关键操作中。
一旦无菌产品受到污染或制造过程中存在缺陷,就会导致严重的医疗事故和患者感染。
因此,制定一套科学合理的无菌产品制造要求和标准非常重要。
EU无菌附录作为欧盟法规的一部分,旨在确保医疗器械制造企业在生产无菌产品时遵守规范,从而保证产品的质量和安全。
该附录包含了一系列关于无菌产品生产中必须遵循的要求和指导原则,涵盖了生产环境、工作人员培训、设备验证和监测等方面。
理解和解读EU无菌附录对于从事医疗器械生产的企业和从业人员至关重要。
它可以帮助制造企业建立清晰的目标和标准,确保生产过程的合规性,并最终提供高品质的无菌产品给医疗领域使用。
从长远来看,对EU 无菌附录的解读和应用也可以促进医疗器械行业的发展,推动技术创新和质量管理体系的不断完善。
在本文接下来的部分,我们将深入研究EU无菌附录的定义和背景,明确其要求和标准,并探讨其对医疗器械生产行业的解读和意义。
我们还将探讨未来发展趋势并提出建议,以促进无菌附录的进一步完善和实施。
让我们开始吧。
1.2文章结构1.2 文章结构本文将按照以下结构进行论述和解读EU无菌附录的相关内容:1. 引言:在本节中,将对文章的背景和引言进行概述,介绍EU无菌附录的重要性和应用领域。
2. 正文:本节主要分为两个部分,分别是无菌附录的定义和背景以及EU无菌附录的具体要求和标准。
2.1 无菌附录的定义和背景:首先,将对无菌附录的定义进行详细解释,包括无菌状态的概念和其在医疗和制药行业中的重要作用。
欧洲药典对无水物及干燥品的定义

欧洲药典对无水物及干燥品的定义1. 引言1.1 欧洲药典的重要性欧洲药典是欧洲医药行业的核心指导性文献,为确保药品的质量、安全和有效性提供了重要的参考依据。
欧洲药典作为药品质量管理的标准,对无水物及干燥品的定义和要求起着至关重要的作用。
欧洲药典不仅规定了无水物和干燥品的标准化定义,还明确定义了制备、储存和贮运过程中对其质量的要求。
无水物在欧洲药典中被定义为完全不含水分的固体药品或物质,其中水分含量不得超过特定的限制。
而干燥品则被描述为含有适量水分的物质,需符合特定的含水量标准。
欧洲药典对无水物及干燥品的规定严格,要求药品的含水量、纯度、稳定性等方面达到一定标准。
1.2 无水物及干燥品的定义引言欧洲药典作为制定和维护欧洲药品质量标准的权威性组织,对药品的无水物及干燥品提出了严格的要求。
在实际生产过程中,无水物及干燥品是药品制备过程中不可或缺的一环。
无水物是指不含水的固体或溶液,通常是通过干燥技术从水解物中去除水分而得到的。
而干燥品则是指含有水分的固体或溶液,需要通过干燥过程来去除其中的水分,使其达到一定的干燥程度。
在药品生产中,无水物及干燥品的质量直接影响到药品的稳定性、有效性和安全性。
欧洲药典对无水物及干燥品的要求十分严格,包括其化学成分、物理性质、纯度和稳定性等方面都有详细的规定。
欧洲药典还规定了对无水物及干燥品的检测方法,以确保药品质量符合标准。
欧洲药典对无水物及干燥品的定义和要求,为药品生产提供了明确的指导,保障了药品质量和安全性。
在日常生活中,我们可以通过选择合格的药品来保障自身健康和安全。
2. 正文2.1 无水物的定义无水物是指在物质中不存在任何水分的固体或液体化合物。
在药学领域,无水物通常是指没有结晶水或水合物的纯净化合物。
无水物的制备通常需要严格控制环境中的水分含量,以保证最终产品的纯度和稳定性。
无水物的特点包括在室温下不会吸收空气中的水蒸气,不会因水的存在而导致化学反应的发生。
在药品生产过程中,选择纯净的无水物作为原料可以提高产品的质量和稳定性,避免因水分含量导致药品的失效或变质。
欧洲药典在线查询全文
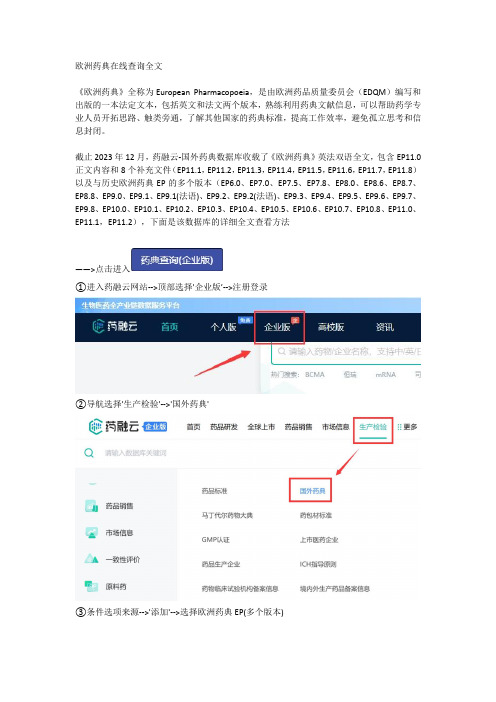
欧洲药典在线查询全文《欧洲药典》全称为European Pharmacopoeia,是由欧洲药品质量委员会(EDQM)编写和出版的一本法定文本,包括英文和法文两个版本,熟练利用药典文献信息,可以帮助药学专业人员开拓思路、触类旁通,了解其他国家的药典标准,提高工作效率,避免孤立思考和信息封闭。
截止2023年12月,药融云-国外药典数据库收载了《欧洲药典》英法双语全文,包含EP11.0正文内容和8个补充文件(EP11.1,EP11.2,EP11.3,EP11.4,EP11.5,EP11.6,EP11.7,EP11.8)以及与历史欧洲药典EP的多个版本(EP6.0、EP7.0、EP7.5、EP7.8、EP8.0、EP8.6、EP8.7、EP8.8、EP9.0、EP9.1、EP9.1(法语)、EP9.2、EP9.2(法语)、EP9.3、EP9.4、EP9.5、EP9.6、EP9.7、EP9.8、EP10.0、EP10.1、EP10.2、EP10.3、EP10.4、EP10.5、EP10.6、EP10.7、EP10.8、EP11.0、EP11.1,EP11.2),下面是该数据库的详细全文查看方法——>点击进入①进入药融云网站-->顶部选择'企业版’-->注册登录②导航选择'生产检验'-->'国外药典'③条件选项来源-->'添加'-->选择欧洲药典EP(多个版本)-----最后说下关于欧洲药典新版本EP11---《欧洲药典》EP11.0于2023年1月1日开始实施,是欧洲药品行业的权威参考书,EP11.0版本对之前版本的内容进行了全面更新和重新组织,新增和修改了600多个单体和试剂。
因此欧洲药典2024版将涵盖超过4000个全新的认证试剂,并提供7000多个化学物质的简要概述。
EP11.0版本的欧洲药典引入了一些重要的改进。
欧洲药典附录3.1.3.大全

3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
–二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3% –1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基 )- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%–二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3% –二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;–didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;–dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;–三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;–增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
欧洲药品GMP检查指南及附件(中英文)

GUIDE TO GOOD MANUFACTURINGPRACTICE FOR MEDICINAL PRODUCTS药品GMP检查指南.PIC/S July 2004Reproduction prohibited for commercial purposes.Reproduction for internal use is authorised,provided that the source is acknowledged.Editor: PIC/S SecretariatP.O. Box 5695CH-1211 Geneva 11e-mail: daniel.brunner@web site: :// 1 July 2004 PE 009-2TABLE OF CONTENT目录INTRODUCTION介绍 (1)CHAPTER 1 QUALITY MANAGEMENT 质量管理 (4)PRINCIPLE 原则 (4)QUALITY ASSURANCE 质量保证 (4)GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS (GMP) 药品GMP (6)QUALITY CONTROL 质量控制 (7)CHAPTER 2 PERSONNEL 人员 (10)PRINCIPLE 原则 (10)GENERAL 通则 (10)KEY PERSONNEL 关键人员 (10)TRAINING 培训 (13)PERSONAL HYGIENE 个人卫生 (14)CHAPTER 3 PREMISES AND EQUIPMENT 厂房和设备 (16)PRINCIPLE 原则 (16)PREMISES General总则 (16)Production Area 生产区域 (17)Storage Areas 储存区域 (19)Quality Control Areas 质量控制区域 (20)Ancillary Areas 辅助区域 (20)EQUIPMENT 设备 (21)CHAPTER 4 DOCUMENTATION 文件 (23)PRINCIPLE 原则 (23)GENERAL 总则 (23)DOCUMENTS REQUIRED 必需的文件 (25)MANUFACTURING FORMULA AND PROCESSING INSTRUCTIONS 生产方法和加工指示 (27)PACKAGING INSTRUCTIONS 包装指示 (28)BA TCH PROCESSING RECORDS 批加工记录 (29)BA TCH PACKAGING RECORDS 批包装记录 (30)PROCEDURES AND RECORDS 程序和记录 (32)CHAPTER 5 PRODUCTION 生产 (36)PRINCIPLE 原则 (36)GENERAL 通则 (36)PREVENTION OF CROSS-CONTAMINATION IN PRODUCTION 生产过程中防止交叉污染 (38)V ALIDATION 验证 (39)STARTING MA TERIALS 起始物料 (40)PROCESSING OPERA TIONS - INTERMEDIATE AND BULK PRODUCTS 加工操作:中间体和散装产品 (42)PACKAGING MATERIALS 包装材料 (42)PACKAGING OPERATIONS 包装操作 (43)FINISHED PRODUCTS 最终成品 (45)REJECTED, RECOVERED AND RETURNED MATERIALS 拒绝的,回收的和退回的物料46CHAPTER 6 QUALITY CONTROL 质量控制 (48)PRINCIPLE 原则 (48)GENERAL 通则 (48)GOOD QUALITY CONTROL LABORATORY PRACTICE 优良质量控制实验室实践 (49)DOCUMENTATION 文件 (49)SAMPLING 取样 (50)TESTING 检测 (52)CHAPTER 7 CONTRACT MANUFACTURE AND ANAL YSIS 合同加工和分析 (55)PRINCIPLE 原则 (55)GENERAL 通则 (55)THE CONTRACT GIVER 合同提供人 (55)THE CONTRACT ACCEPTOR 合同接受人 (56)THE CONTRACT 合同 (57)CHAPTER 8 COMPLAINTS AND PRODUCT RECALL 抱怨和产品召回 (59)PRINCIPLE 原则 (59)COMPLAINTS 抱怨 (59)RECALLS 召回 (60)CHAPTER 9 SELF INSPECTION 自检 (61)PRINCIPLE 原则 (61)ANNEX 1 MANUFACTURE OF STERILE MEDICINAL PRODUCTS无菌药品的生产 (63)PRINCIPLE (63)GENERAL (63)BLOW/FILL/SEAL TECHNOLOGY (67)TERMINALL Y STERILISED PRODUCTS (67)ASEPTIC PREPARA TION (68)PERSONNEL (68)PREMISES (70)EQUIPMENT (71)SANITATION (71)PROCESSING (71)STERILISATION (73)STERILISATION BY HEA T (74)MOIST HEAT (75)DRY HEAT (75)STERILISATION BY RADIATION (75)STERILISATION WITH ETHYLENE OXIDE (76)FILTRATION OF MEDICINAL PRODUCTS WHICH CANNOT BE STERILISED IN THEIR FINAL CONTAINER (77)FINISHING OF STERILE PRODUCTS (77)QUALITY CONTROL (78)ANNEX 2 MANUFACTURE OF BIOLOGICAL MEDICINAL PRODUCTS FOR HUMAN USE人用生物药品的生产 (79)SCOPE (79)PRINCIPLE (79)PERSONNEL (80)PREMISES AND EQUIPMENT (81)ANIMAL QUARTERS AND CARE (82)DOCUMENTATION (82)PRODUCTION (83)QUALITY CONTROL (84)ANNEX 3 MANUFACTURE OF RADIOPHARMACEUTICALS 放射性药品的生产 (85)PRINCIPLE (85)PERSONNEL (85)PREMISES AND EQUIPMENT (85)PRODUCTION (86)QUALITY CONTROL (86)DISTRIBUTION AND RECALLS (86)ANNEX 4 MANUFACTURE OF VETERINARY MEDICINAL PRODUCTS OTHER THAN IMMUNOLOGICALS MANUFACTURE OF PREMIXES FOR MEDICATED FEEDING STUFFS 除为预混合加药饲料原料生产的免疫产品以外的,兽药产品的生产 (87)THE MANUFACTURE OF ECTOPARASITICIDES (88)THE MANUFACTURE OF VETERINARY MEDICINAL PRODUCTS CONTAINING PENICILLINS (88)RETENTION OF SAMPLES (point 1.4. viii and point 6.14.) (88)STERILE VETERINARY MEDICINAL PRODUCTS (88)ANNEX 5 MANUFACTURE OF IMMUNOLOGICAL VETERINARY MEDICAL PRODUCTS免疫兽药产品的生产 (89)PRINCIPLE (89)PERSONNEL (89)PREMISES (90)EQUIPMENT (93)ANIMALS AND ANIMAL HOUSES (94)DISINFECTION - WASTE DISPOSAL (94)PRODUCTION (95)STARTING MA TERIALS (95)QUALITY CONTROL (98)ANNEX 6 MANUFACTURE OF MEDICINAL GASES药用气体的生产 (99)1. PRINCIPLE (99)2. PERSONNEL (99)3. PREMISES AND EQUIPMENT (99)4. DOCUMENTA TION (100)5. PRODUCTION (101)6. QUALITY CONTROL (104)7. STORAGE AND RELEASE (105)ANNEX 7 MANUFACTURE OF HERBAL MEDICINAL PRODUCTS草药产品的生产 (108)PRINCIPLE (108)PREMISES (108)DOCUMENTATION (108)SAMPLING (109)QUALITY CONTROL (110)ANNEX 8 SAMPLING OF STARTING AND PACKAGING MA TERIALS起始物料和包装材料的取样 (111)PRINCIPLE (111)PERSONNEL (111)STARTING MA TERIALS (111)PACKAGING MATERIAL (112)ANNEX 9 MANUFACTURE OF LIQUIDS, CREAMS AND OINTMENTS流体,霜体和膏体药品的生产 (113)PRINCIPLE (113)PRODUCTION (113)ANNEX 10 MANUFACTURE OF PRESSURISED METERED DOSE AEROSOL PREPARATIONS FOR INHALATION吸入式剂量仪的气雾剂的生产 (115)PRINCIPLE (115)GENERAL (115)PREMISES AND EQUIPMENT (115)PRODUCTION AND QUALITY CONTROL (116)ANNEX 11 COMPUTERISED SYSTEMS 计算机化系统 (117)PRINCIPLE (117)PERSONNEL (117)V ALIDATION (117)ANNEX 12 USE OF IONISING RADIATION IN THE MANUFACTURE OF MEDICINAL PRODUCTS使用离子放射生产药品 (120)INTRODUCTION (120)RESPONSIBILITIES (120)DOSIMETRY (121)V ALIDATION OF THE PROCESS (121)COMMISSIONING OF THE PLANT (122)PREMISES (124)PROCESSING (124)DOCUMENTATION (126)MICROBIOLOGICAL MONITORING (126)ANNEX 13 MANUFACTURE OF INVESTIGA TIONAL MEDICINAL PRODUCTS观察期药品的生产 (127)PRINCIPLE (127)GLOSSARY (128)QUALITY MANAGEMENT (130)PERSONNEL (130)PREMISES AND EQUIPMENT (130)DOCUMENT A TION (131)PRODUCTION (132)QUALITY CONTROL (136)RELEASE OF BATCHES (137)SHIPPING (139)COMPLAINTS (139)RECALLS AND RETURNS (139)DESTRUCTION (140)ANNEX 14 MANUFACTURE OF PRODUCTS DERIVED FROM HUMAN BLOOD OR HUMAN PLASMA生产自人类血液或人体组织分离的产品 (143)PRINCIPLE (143)GLOSSARY (144)QUALITY MANAGEMENT (144)PREMISES AND EQUIPMENT (145)BLOOD AND PLASMA COLLECTION (145)TRACEABILITY AND POST COLLECTION MEASURES (146)PRODUCTION AND QUALITY CONTROL (147)RETENTION OF SAMPLES (148)DISPOSAL OF REJECTED BLOOD, PLASMA OR INTERMEDIATES (148)ANNEX 15 QUALIFICATION AND V ALIDATION 确认和验证 (149)PRINCIPLE (149)PLANNING FOR V ALIDATION (149)DOCUMENTATION (150)QUALIFICATION (150)PROCESS V ALIDATION (151)CLEANING VALIDATION (153)CHANGE CONTROL (154)REV ALIDATION (154)GLOSSARY (154)[ANNEX 16] [QUALIFIED PERSON AND BA TCH RELEASE]*经授权的人员和批放行 (157)ANNEX 17 PARAMETRIC RELEASE参数放行 (158)1. PRINCIPLE (158)2. PARAMETRIC RELEASE (158)3. PARAMETRIC RELEASE FOR STERILE PRODUCTS (158)4. GLOSSARY (160)[ANNEX 18] [GMP GUIDE FOR ACTIVE PHARMACEUTICAL INGREDIENTS] 17原料药GMP 指南 (161)GLOSSARY术语表 (162)GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS药品GMP指南INTRODUCTION介绍为进一步消除药品贸易壁垒,促进许可证的一致性,以及确保整个欧洲在研发,生产和控制药品中保持高标准的质量保证,根据药品检查协会(PIC)同意,药品检查使用一致的GMP原则,和药品检查合作计划表中的欧洲药品GMP及其附录。
欧洲药典3.1.4-无添加剂的PET包装材料的检测

EUROPEAN PHARMACOPOEIA 8.0 3.1.4.Polyethylene without additives forcontainersReference solution (n).Dissolve 60mg of plastic additive 16CRS in 10mL of methylene chloride R .Dilute 2mL of this solution to 10mL with acidified methylene chloride R .Reference solution (o).Dissolve 60mg of plastic additive 17CRS in 10mL of methylene chloride R .Dilute 2mL of this solution to 10mL with acidified methylene chloride R .Reference solution (p).Dissolve 60mg of plastic additive 16CRS and 60mg of plastic additive 17CRS in 10mL of methylene chloride R .Dilute 2mL of this solution to 10mL with acidified methylene chloride R .Plate :TLC silica gel GF 254plate R .Mobile phase A :hexane R .Mobile phase B :methylene chloride R .Application :20μL of the test solution S24,the reference solution (p)and the reference solutions corresponding to all the phenolic and non-phenolic antioxidants mentioned in the type composition of the material to be examined.Development A :over a path of 18cm with mobile phase A.Drying A :in air.Development B :over a path of 17cm with mobile phase B.Drying B :in air.Detection :examine in ultraviolet light at 254nm;spray with alcoholic iodine solution R and examine in ultraviolet light at 254nm after 10-15min.System suitability :reference solution (p):–the chromatogram shows 2clearly separated spots.Limit :any spots in the chromatogram obtained with test solution S24are not more intense than the spots in thecorresponding positions in the chromatograms obtained with the reference solutions.Plastic additive 22.Liquid chromatography (2.2.29).Test solution .Evaporate 25mL of solution S2to dryness in vacuo at 45°C.Dissolve the residue in 10mL of toluene R and 10mL of a 10g/L solution of tetrabutylammonium hydroxide R in a mixture of 35volumes of toluene R and 65volumes of anhydrous ethanol R .Boil under a reflux condenser for 3h.Allow to cool and filter if necessary.Reference solution .Dissolve 30mg of plastic additive 22CRS in 50mL of toluene R .Add 1mL of this solution to 25mL of blank solution S2and evaporate to dryness in vacuo at 45°C.Dissolve the residue in 10mL of toluene R and 10mL of a 10g/L solution of tetrabutylammonium hydroxide R in a mixture of 35volumes of toluene R and 65volumes of anhydrous ethanol R .Boil under a reflux condenser for 3h.Allow to cool and filter if necessary.Column :–size :l =0.25m,Ø=4.6mm;–stationary phase :aminopropylsilyl silica gel for chromatography R (5μm).Mobile phase :anhydrous ethanol R ,hexane R (11:89V/V ).Flow rate :2mL/min.Detection :spectrophotometer at 227nm.Injection :20μL.Run time :10min.System suitability :–resolution :minimum 7between the peaks due to the “diol”component and to the diluent of the reference solution.Limit :the area of the peak due to the “diol”component from plastic additive 22in the chromatogram obtained with the test solution is less than the corresponding peak in the chromatogram obtained with the reference solution.Amides and stearates .Thin-layer chromatography (2.2.27).Test solution .Use test solution S24described in the test for non-phenolic antioxidants.Reference solution (q).Dissolve 20mg of stearic acid (plastic additive 19CRS )in 10mL of methylene chloride R .Reference solution (r).Dissolve 40mg of oleamide (plastic additive 20CRS )in 20mL of methylene chloride R .Reference solution (s).Dissolve 40mg of erucamide (plastic additive 21CRS )in 20mL of methylene chloride R .Plate :TLC silica gel GF 254plate R (2plates).A.Mobile phase :anhydrous ethanol R ,trimethylpentane R (25:75V/V ).Application :10μL of the test solution S24and reference solution (q).Development :over a path of 10cm.Drying :in air.Detection :spray with a 2g/L solution ofdichlorophenolindophenol,sodium salt R in anhydrous ethanol R and heat in an oven at 120°C for a few minutes to intensify the spots.Limit :any spot corresponding to plastic additive 19in the chromatogram obtained with test solution S24is identical in position to (R F =about 0.5)but not more intense than the spot in the chromatogram obtained with reference solution (q).B.Mobile phase A :hexane R .Mobile phase B :methanol R ,methylene chloride R (5:95V/V ).Application :10μL of the test solution S24and the reference solutions (r)and (s).Development A :over a path of 13cm with mobile phase A.Drying A :in air.Development B :over a path of 10cm with mobile phase B.Drying B :in air.Detection :spray with a 40g/L solution of phosphomolybdic acid R in anhydrous ethanol R .Heat in an oven at 120°C until spots appear.Limit :any spots corresponding to plastic additive 20or plastic additive 21in the chromatogram obtained with test solution S24are identical in position to (R F =about 0.2)but not more intense than the corresponding spots in the chromatograms obtained with reference solutions (r)and (s).01/2008:30104corrected 6.03.1.4.POLYETHYLENE WITHOUT ADDITIVES FOR CONTAINERS FOR PARENTERAL PREPARATIONS AND FOR OPHTHALMIC PREPARATIONSDEFINITIONPolyethylene without additives is obtained by thepolymerisation of ethylene under high pressure in the presence of oxygen or free-radical-forming initiators as catalyst.CHARACTERSAppearance :beads,granules,powder or,after transformation,translucent sheets of varying thickness or containers.Solubility :practically insoluble in water,soluble in hot aromatic hydrocarbons,practically insoluble in anhydrous ethanol,in hexane and in methanol.It softens at temperatures beginning at 65°C.Relative density:0.910to 0.937.IDENTIFICATIONIf necessary,cut the samples of the material to be examined into pieces of maximum dimension on a side of not greater than 1cm .A.Infrared absorption spectrophotometry (2.2.24).General Notices (1)apply to all monographs and other texts3833.1.5.Polyethylene with additives for containers EUROPEAN PHARMACOPOEIA8.0Preparation:to0.25g add10mL of toluene R and boilunder a reflux condenser for about15min.Place a fewdrops of the solution on a sodium chloride disc andevaporate the solvent in an oven at80°C.Absorption maxima:at2920cm−1,2850cm−1,1465cm−1, 730cm−1and720cm−1.The spectrum obtained is identical to that obtained with the material selected for the type sample.If the material to be examined is in the form of sheets,the identification may be performed directly on a cut piece of suitable size.B.Additives(see Tests).TESTSIf necessary,cut the samples of the material to be examined into pieces of maximum dimension on a side of not greater than1cm.Solution S1.Place25g in a borosilicate-glassflask with a ground-glass neck.Add500mL of water for injections R and heat under a reflux condenser for5h.Allow to cool and decant.Keep part of the solution for the test for appearance of solution.Filter the rest through a sintered glassfilter(16) (2.1.2).Use solution S1within4h of preparation.Solution S2.Place2.0g in a conical borosilicate-glassflask with a ground-glass neck.Add80mL of toluene R and boil under a reflux condenser with constant stirring for1h30min. Allow to cool to60°C and add with continued stirring120mL of methanol R.Filter the solution through a sintered-glassfilter(16)(2.1.2).Rinse theflask and thefilter with25mL of a mixture of40mL of toluene R and60mL of methanol R,add the rinsings to thefiltrate and dilute to250mL with the same mixture of solvents.Prepare a blank solution.Solution S3.Place100g in a conical borosilicate-glassflask with a ground-glass neck.Add250mL of0.1M hydrochloric acid and boil under a reflux condenser with constant stirring for1h.Allow to cool and decant the solution. Appearance of solution.Solution S1is clear(2.2.1)and colourless(2.2.2,Method II).Acidity or alkalinity.To100mL of solution S1add0.15mL of BRP indicator solution R.Not more than1.5mL of0.01M sodium hydroxide is required to change the colour of the indicator to blue.To100mL of solution S1add0.2mL of methyl orange solution R.Not more than1.0mL of0.01M hydrochloric acid is required to reach the beginning of the colour change of the indicator from yellow to orange. Absorbance(2.2.25):maximum0.2,determined between wavelengths of220nm and340nm on solution S1. Reducing substances.To20mL of solution S1add1mLof dilute sulfuric acid R and20mL of0.002M potassium permanganate.Boil under a reflux condenser for3min and cool immediately.Add l g of potassium iodide R and titrate immediately with0.01M sodium thiosulfate,using0.25mLof starch solution R as indicator.Carry out a blank titration. The difference between the titration volumes is not more than 0.5mL.Substances soluble in hexane.Place10g in a250mL conical borosilicate-glassflask with a ground-glass neck.Add100mL of hexane R and boil under a reflux condenser for4h,stirring constantly.Cool in iced water andfilter rapidly through a sintered-glassfilter(16)(2.1.2)maintaining the solution at 0°C(thefiltration time must be less than5min;if necessary thefiltration may be accelerated by applying pressure to the solution).Evaporate20mL of thefiltrate in a tared glass dish on a water-bath.Dry the residue in an oven at100-105°C for1h.The mass of the residue obtained is within10per cent of the residue obtained with the type sample and does not exceed5per cent.Additives.Thin-layer chromatography(2.2.27).Test solution.Evaporate50mL of solution S2to dryness in vacuo at45°C.Dissolve the evaporation residue with5mL of methylene chloride R.Prepare a blank solution from the blank solution corresponding to solution S2.Reference solution.Dissolve20mg of plastic additive15CRS and20mg of plastic additive08CRS in methylene chloride R and dilute to10mL with the same solvent.Plate:TLC silica gel G plate R.Mobile phase A:hexane R.Mobile phase B:methanol R,methylene chloride R(5:95V/V). Application:10μL.Development A:over a path of13cm using mobile phase A. Drying A:in air.Development B:over a path of10cm using mobile phase B. Drying B:in air.Detection:spray with a40g/L solution of phosphomolybdic acid R in ethanol(96per cent)R and heat at120°C until the spots appear in the chromatogram obtained with the reference solution.System suitability:reference solution:–the chromatogram shows2separated spots.Limit:no spot appears in the chromatogram obtained with the test solution,except for a spot which may be at the solvent front from thefirst development and which correspondsto oligomers.Disregard any spots corresponding to those obtained in the chromatogram with the blank solution. Extractable heavy metals(2.4.8):maximum2.5ppm. Evaporate50mL of solution S3to about5mL on a water-bath and dilute to20mL with water R.12mL of solution complies with test A.Prepare the reference solution using2.5mL of lead standard solution(10ppm Pb)R.Sulfated ash(2.4.14):maximum0.02per cent,determined on5.0g.01/2008:30105corrected7.5 3.1.5.POLYETHYLENE WITH ADDITIVES FOR CONTAINERS FOR PARENTERAL PREPARATIONS AND FOR OPHTHALMIC PREPARATIONS DEFINITIONPolyethylene with additives is obtained by the polymerisation of ethylene under pressure in the presence of a catalyst or by copolymerisation of ethylene with not more than25per cent of higher alkene homologues(C3to C10).PRODUCTIONA certain number of additives are added to the polymer in order to optimise their chemical,physical and mechanical properties in order to adapt them for the intended use.All these additives are chosen from the appended list which specifies for each product the maximum allowable content. They may contain at most3antioxidants,1or several lubricants or antiblocking agents as well as titanium dioxide as an opacifying agent when the material must provide protection from light.–butylhydroxytoluene(plastic additive07):maximum0.125per cent;–pentaerythrityl tetrakis[3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate](plastic additive09):maximum0.3per cent;–1,3,5-tris(3,5-di-tert-butyl-4-hydroxybenzyl)-S-triazine-2,4,6(1H,3H,5H)-trione(plastic additive13):maximum0.3per cent;–octadecyl3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionate, (plastic additive11):maximum0.3per cent;384See the information section on general monographs(cover pages)。
propoxycarbazone

EUROPEAN COMMISSIONHEALTH & CONSUMER PROTECTION DIRECTORATE-GENERALDirectorate E – Food Safety: plant health, animal health and welfare, international questionsE1 - Plant healthPropoxycarbozoneSANCO/4067/2001-Final30 September 2003COMMISSION WORKING DOCUMENT - DOES NOT NECESSARILY REPRESENTTHE VIEWS OF THE COMMISSION SERVICESReview report for the active substance propoxycarbazoneFinalised in the Standing Committee on the Food Chain and Animal Health at its meeting on3 October 2003 in view of the inclusion of propoxycarbazone in Annex I of Directive91/414/EEC.1. Procedure followed for the evaluation processThis review report has been established as a result of the evaluation of the new active substancepropoxycarbazone, made in the context of the work provided for in Articles 5 and 6 of Directive91/414/EEC concerning the placing of plant protection products on the market, with a view tothe possible inclusion of this substance in Annex I to the Directive.In accordance with the provisions of Article 6(2) of Directive 91/414/EEC, the Germanauthorities received on25 January 2000 an application from Bayer AG (now BayerCropScience), hereafter referred to as the applicant, for the inclusion of the active substancepropoxycarbazone in Annex I to the Directive. The German authorities indicated to theCommission on 10 March 2000 the results of a first examination of the completeness of thedossier, with regard to the data and information requirements provided for in Annex II and, for atleast one plant protection product containing the active substance concerned, in Annex III to theDirective. Subsequently, and in accordance with the requirements of Article 6(2), a dossier onpropoxycarbazone was distributed to the Member States and the Commission.The Commission referred the dossier to the Standing Committee on the Food Chain andAnimal Health in the meeting of the working group ‘legislation’ thereof on10 March 2000,during which the Member States confirmed the receipt of the dossier.In accordance with the provisions of Article 6(3), which requires the confirmation at Communitylevel that the dossier is to be considered as satisfying, in principle, the data and informationrequirements provided for in Annex II and, for at least one plant protection product containingthe active substance concerned, in Annex III to the Directive and in accordance with theprocedure laid down in Article 20 of the Directive, the Commission confirmed in its Decision 2000/463/EC 1 of 17 July 2000 that these requirements were satisfied.Within the framework of that decision and with a view to the further organisation of the works related to the detailed examination of the dossier provided for in Article 6(2) and (4) of Directive 91/414/EEC, it was agreed between the Member States and the Commission that Germany would, as rapporteur Member State, carry out the detailed examination of the dossier and report the conclusions of its examination accompanied by any recommendations on the inclusion or non-inclusion and any conditions relating thereto, to the Commission as soon as possible and at the latest within a period of one year.Germany submitted to the Commission on26 March 2001 the report of its detailed scientific examination, hereafter referred to as the draft assessment report, including, as required, a recommendation concerning the possible inclusion of propoxycarbazone in Annex I to the Directive.On receipt of the draft assessment report, the Commission forwarded it for consultation to all the Member States as well as to Bayer AG being the sole applicant on4 May 2001.The Commission organised further an intensive consultation of specialised scientific experts from a representative number of Member States, to review the draft assessment report and the comments received thereon (peer review), in particular on each of the following disciplines :- identity and physical /chemical properties ;- fate and behaviour in the environment ;;- ecotoxicology- mammalian toxicology ;- residues and analytical methods ;questions.- regulatoryThe meetings for this consultation were organised on behalf of the Commission by the Pesticide Safety Directorate (PSD) in York, United Kingdom, from November 2001 to July 2002.The report of the peer review (i.e. full report) was circulated, for further consultation, to Member States and the sole applicant on 11 October 2002.The dossier, draft assessment report and the peer review report (i.e. full report) including in particular an outline resumé of the remaining technical questions, were referred to the Standing Committee on the Food Chain and Animal Health, and specialised working groups of this Committee, for final examination, with participation of experts from the 15 Member States. This final examination took place from March to October 2003, and was finalised in the meeting of the Standing Committee on 3 October 2003.The present review report contains the conclusions of this final examination; given the importance of the draft assessment report, the peer review report (i.e. full report) and the comments and clarifications submitted after the peer review as basic information for the final examination process, these documents are considered respectively as background documents A, B and C to this review report and are part of it.These review did not reveal any open question, which would have required the consultation of the Scientific Committee on Plants.2. Purposes of this review reportThis review report, including the background documents and appendices thereto, have been developed and finalised in support of the Directive 2003/119/EC2 concerning the inclusion of propoxycarbazone in Annex I to Directive 91/414/EEC, and to assist the Member States in decisions on individual plant protection products containing propoxycarbazone they have to take in accordance with the provisions of that Directive, and in particular the provisions of article 4(1) and the uniform principles laid down in Annex VI.This review report provides also for the evaluation required under Section A.2.(b) of the above mentioned uniform principles, as well as under several specific sections of part B of these principles. In these sections it is provided that Member States, in evaluating applications and granting authorisations, shall take into account the information concerning the active substance in Annex II of the directive, submitted for the purpose of inclusion of the active substance in Annex I, as well as the result of the evaluation of those data.In parallel with the provisions of Article 7(6) of Regulation 3600/92 for existing active substances, the Commission and the Member States will keep available or make available this review report for consultation by any interested parties or will make it available to them on their specific request. Moreover the Commission will send a copy of this review report (not including the background documents) to the applicant.The information in this review report is, at least partly, based on information which is confidential and/or protected under the provisions of Directive 91/414/EEC. It is therefore recommended that this review report would not be accepted to support any registration outside the context of Directive 91/414/EEC, e.g. in third countries, for which the applicant has not demonstrated possession of regulatory access to the information on which this review report is based.3. Overall conclusion in the context of Directive 91/414/EECThe overall conclusion from the evaluation is that it may be expected that plant protection products containing propoxycarbazone will fulfil the safety requirements laid down in Article 5(1)(a) and (b) of Directive 91/414/EEC. This conclusion is however subject to compliance with the particular requirements in sections 4, 5, 6 and 7 of this report, as well as to the implementation of the provisions of Article 4(1) and the uniform principles laid down in Annex VI of Directive 91/414/EEC, for each propoxycarbazone containing plant protection product for which Member States will grant or review the authorisation.Furthermore, these conclusions were reached within the framework of the uses which were proposed and supported by the main data submitter and mentioned in the list of uses supported by available data (attached as Appendix IV to this Review Report).Extension of the use pattern beyond those described above will require an evaluation at Member State level in order to establish whether the proposed extensions of use can satisfy the requirements of Article 4(1) and of the uniform principles laid down in Annex VI of Directive 91/414/EEC.4. Specific conclusions which are highlighted in this evaluation4.1 Residues of propoxycarbazone in foodstuffsThe review has established that the residues arising from the proposed uses, consequent on application consistent with good plant protection practice, have no harmful effects on human or animal health. The Theoretical Maximum Daily Intake (TMDI) for a 60 kg adult is0,26% of the Acceptable Daily Intake (ADI), based on the FAO/WHO European Diet (August 1994). This low intake value reflects the current limited use pattern for this active substance.4.2 Exposure of operators, workers and bystandersThe review has identified acceptable exposure scenarios for operators, workers and bystanders, which require, however, confirmation for each plant protection product in accordance with the relevant sections of the above mentioned uniform principles.Ecotoxicology4.3The review has also concluded that under the proposed and supported conditions of use there are no unacceptable effects on the environment, as provided for in Article 4 (1) (b) (iv) and (v) of Directive 91/414/EEC, provided that certain conditions are taken into account as detailed in section 7 of this report.5. Identity and Physical/chemical propertiesThe main identity and the physical/chemical properties of propoxycarbazone are given in Appendix I.The active substance shall have a minimum purity of974g/kg technical product (based on a pilot plant production).The review has established that for the active substance notified by the applicant (Bayer AG), none of the manufacturing impurities considered are, on the basis of information currently available, of toxicological or environmental concern.6. Endpoints and related informationIn order to facilitate Member States, in granting or reviewing authorisations, to apply adequately the provisions of Article 4(1) of Directive 91/414/EEC and the uniform principles laid down in Annex VI of that Directive, the most important endpoints as identified during the evaluation process are listed in Appendix II.7. Particular conditions to be taken into account on short term basis by Member States in relation to the granting of authorisations of plant protection products containing propoxycarbazoneOn the basis of the proposed and supported uses, the following particular issues have been identified as requiring particular and short term (within 12 months at the latest) attention from the Member States, in the framework of any authorisations to be granted, varied or withdrawn, as appropriate:In this overall assessment Member States- should pay particular attention to the potential of propoxycarbazone and its metabolites for groundwater contamination, when the active substance is applied in regions with vulnerable soil and/or climate conditions;- should pay particular attention to the protection of aquatic ecosystems, especially of aquatic plants.Risk mitigation measures should be applied where appropriate.8. List of studies to be generatedNo further studies were identified which were considered at this stage, and under the current inclusion conditions necessary in relation to the inclusion of propoxycarbazone in Annex I.9. Updating of this review reportThe technical information in this report may require periodic updating to take account of technical and scientific developments as well as of the results of the examination of any information referred to the Commission in the framework of Articles 7, 10 or 11 of Directive 91/414/EEC. Such adaptations will be examined and finalised in the Standing Committee on the Food Chain and Animal Health, in connection with any amendment of the inclusion conditions for propoxycarbazone in Annex I of the Directive.APPENDIX IIdentity, physical and chemical propertiesPROPOXYCARBAZONECommon name (ISO) Propoxycarbazone (The given data belong to thesodium salt Propoxycarbazone-sodium if not specifiedotherwise)MKH 65 61Development Code (for new activesonly)Chemical name (IUPAC) parent2-(4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-yl)carboxamidosulfonylbenzoicacid-methylestersodium saltsodium (4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-ylcarbonyl)(2-methoxycarbonylphenylsulfonyl)azanideChemical name (CA) parentmethyl 2-[[[(4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-yl)carbonyl]amino]sulfonyl]benzoatesodium saltbenzoic acid, 2-[[[4,5-dihydro-4-methyl-5-oxo-3-propoxy-1H-1,2,4-triazol-1-yl)carbonyl]amino] sulfonyl]-, methyl ester,sodium saltCIPAC No 655 (parent); 655.011 (sodium salt)CAS No 145026-81-9 (parent); 181274-15-7 (sodium salt)EEC No not assignedFAO SPECIFICATION not availableMinimum purity 974 g/kgMolecular formula C15H18N4O7S (parent); C15H17N4NaO7S (sodium salt) Molecular mass 398.4 g/mol (parent); 420.4 g/mol (sodium salt)Structural formulaMelting point 230 – 240 °C,the melting occurs under decompositionBoiling point Not measurable, decomposition at 230 – 240 °C Appearance odourless and colourless crystalline powder (PAS, TAS) Relative density 1.42 g/cm³ at 20°CVapour pressure Cannot be determined directly due to its extreme low value;upper limit: < 9 ⋅ 10-8 Pa at 70 °C< 1 ⋅ 10-8 Pa at 20 °C (extrapolated)Henry's law constant < 1 ⋅ 10-10 Pa m3 mol-1 at 20 °C (calculated)Solubility in water (g/L) pH 8.8: 42 (unbuffered)pH 9.0: 42pH 7.2: 42pH 4.5: 2.9Solubility in organic solvents (g/L) n-heptane < 0.1xylene < 0.10.11-octanol <2-propanol <0.1ethyl acetate < 0.1polyethylen glycol 5.2acetonitrile 0.90acetone 0.50dichloromethane 1.5dimethylsulfoxide 190Partition co-efficient (log P ow) pH 9: - 1.59pH 7: - 1.55pH 4: - 0.30Hydrolytic stability (DT50) Stable at 25 ° C at pH 4 –9Dissociation constant The free acid of the sodium salt is produced by protonationof the active substance under acidic conditions.It has a pKa-value of 2.1 in aqueous solutions.Quantum yield of direct photo-Φ = 0.75transformation in water at λ >290nmFlammability Not highly flammable in the sense of EU guideline A.10.Does not undergo spontaneous combustion in the sense ofEU guideline A.16 and in 1L BCC-Test.Explosive properties Not explosive in the sense of EU guideline A.14UV/VIS absorption (max.) 230 nmPhotostability in water (DT50) DT50 18.1 days (phenyl-labelled)DT50 40.9 days (triazolinone-labelled)23 June 2003APPENDIX IIEND POINTS AND RELATED INFORMATIONPROPOXYCARBAZONE1 Toxicology and metabolismAbsorption, distribution, excretion and metabolism in mammalsRate and extent of absorption: Rapid but incomplete absorption (25 – 30 %) within48 hoursDistribution: Widely distributed, highest residues in the liverPotential for accumulation: No evidence for accumulationRate and extent of excretion: Rapid and nearly complete excretion (>88 % of theadministered dose within 48 hours) primarily via faeces(>66 %)Toxicologically significant compounds: Parent compound, plant metabolite 2-hydroxypropoxyMKH 6561 (M01) was not of greater toxicologicalsignificance than parentMetabolism in animals: Limited metabolism: unchanged parent compound in urine(>90 %) and faeces (>80 %); metabolism via cleavage ofthe amide bond, sulfonamide methylester (M05),sulfonamide acid (M06) and saccharin (M07)Acute toxicityRat LD50 oral: > 5000 mg/kg bwRat LD50 dermal: > 5000 mg/kg bwRat LC50 inhalation: > 5.03 mg/l airSkin irritation: Non-irritantEye irritation: Non-irritantSkin sensitization (test method used andNot sensitising (M & K)result):Short term toxicityTarget / critical effect: Decreased body weight gain, increased water intake;irritation of forestomach epithelium (rats); decreased foodconsumption and relative heart weights (dog)Lowest relevant oral NOAEL / NOEL: 1yr dog: 2000 ppm (56 mg/kg bw/d)Lowest relevant dermal NOAEL / NOEL: >1000 mg/kg bw/d (rat)Lowest relevant inhalation NOAEL / NOEL:No data – not requiredGenotoxicity No genotoxic potential23 June 2003Long term toxicity and carcinogenicityTarget / critical effect: Decreased body weight gain; increased urine pH value andrenal pelvic mineralization in rats onlyLowest relevant NOAEL: 2yr rat: 1000 ppm (43 mg/kg bw/d)Carcinogenicity: No carcinogenic potentialReproductive toxicityTarget / critical effect - Reproduction: No reproductive toxicity at limit dose level (rat)Lowest relevant reproductive NOAEL /NOEL:>16000 ppm (>1000 mg/kg bw/d; rat)Target / critical effect - Developmental toxicity: Embryotxic effects (reduced fetus number, reduced fetal and placental weights, increased post-implantation loss, retarded skeletal ossification) at a maternally toxic dose (1000 mg/kg bw/d) in rabbitsLowest relevant developmental NOAEL / NOEL: Rabbit:500 mg/kg bw/d (developmental toxicity) 100 mg/kg bw/d (maternal toxicity)Delayed neurotoxicity No acute or subchronic (90-day) neurotoxicity in rats(NOAEL acute: >2000 mg/kg bw; NOAEL subchronic:>1321 mg/kg bw/d)Other toxicological studies Plant metabolite M01 has a very low acute oral toxicity(LD50: >5000 mg/kg bw), was not genotoxic (Ames-Test, invitro chromosome aberration test Chinese hamster V79cells), and caused no effects in the rat after subacutefeeding of 10000 ppmMedical data Limited data (new compound); no human health problemsobservedSummaryValueStudySafetyfactor ADI: 0.4 mg/kg bw Rat, 2yr study 100AOEL systemic: 0.3 mg/kg bw/day Rabbit,developmentalstudy (approx. oralabsorption rate25%)100ARfD (acute reference dose): Not allocated Not necessaryDermal absorption10 % default value (due to physical and chemicalproperties)14 April 2003 2 Fate and behaviour in the environment2.1 Fate and behaviour in soilRoute of degradationAerobic:Mineralization after 100 days: phenyl-label:9.1 to 41.9 % (88-98d); 21.7 to 49.0% (180-361d)(n=4)triazolinone-label:1.3 to 8.9 % (93-117d);2.6 to 12.6 % (182 –365d)(n=4)Non-extractable residues after 100 days: phenyl-label:6.5 to 29.5 % (88-98d); 8.2 to 28.3 % (180-361d)(n=4)triazolinone-label:8.9 to 64.9 %(93-117d); 17.9 to 65.7 % (182-365d)(n=4)Major metabolites above 10 % of applied active substance: name and/or code% of applied rate (range and maximum) sulfonamide methyl ester (M05):Range: <10 % at day 100, max. 20.9 % (day 6) saccharin (M07):Range: 17.3 % at day 100 in 1 soil, max. 26.7 % (day 14)4-Hydroxy saccharin (M08):Range: 9.3 to 18.1 % at day 100, max. 19.5 % (day 36)N-methyl propoxy triazolinone amide (M09): Range: <10 % at day 100, max. 13.2 % (day 253)N-methyl propoxy triazolinone (M10):Range: 12.6 to 34.3 % at day 100, max. 55.2 % (day 182)Supplemental studiesAnaerobic:not required, application in springSoil photolysis:stable to photolysisRemarks:noneRate of degradationLaboratory studiesDT50lab (20 °C, aerobic): DT50lab (20°C, aerobic): propoxycarbazone-sodium:14 April 2003range: 22.8 – 98.8 (220.4*) d (n = 8), 1st order,median: 60.6 d, r²: 0.818 – 0.997,DT50calc (20°C, aerobic): sulfonamide methyl ester(M05):3.0 - 22.3 d(n = 3), 1st order, median: 3.1, r²: 0.898 - 1DT50calc (20°C, aerobic): saccharin (M07):5 - 57 d(n = 3), 1st order, median: 27.3, r²: 0.909 - 1DT50lab (20°C, aerobic): 4-Hydroxy saccharin (M08):178.4 d (r²:0.899), 185.5 d (r²:0.838), 953.8* d(r²:0.227) 1st order (n = 3),DT50lab (20°C, aerobic): N-methyl propoxy triazolinoneamide (M09):83.8 d (r²:0.777), 90.1 d (r²:0.855), >>1 year*(r²:0.025), 1st order (n = 3)DT50lab+calc (20°C, aerobic): N-methyl propoxytriazolinone (M10):38.7 - 75.6 d, 1st order (n = 3)* soil with low microbial activity (not representative) DT90lab (20 °C, aerobic): DT90lab (20°C, aerobic): propoxycarbazone-sodium:range: 76 – 328 (733*) d [n = 8], r²: 0.818 -.0.997,1st order, median: 202 d* soil with low microbial activity (not representative) DT50lab (10 °C, aerobic): DT50lab (10°C, aerobic): calculated from lab (20 °C):50.2 – 217.4 d (485* d), median 133 d (n = 8), 1storder* soil with low microbial activity (not representative)3 field trials with average temperatures ~10 °C in the firweeks after application showed DT50 between 4.9 and20 daysDT50lab (20 °C, anaerobic): DT50lab (20°C, anaerobic): not requiredField studies (country or region)DT50f from soil dissipation studies: DT50f: Southern Europe (France): 2.7 / 9.1d, √1st order[n = 2], recalc. 1st order: 12 and 37 dNorthern Europe (Germany, France, UK): 6.6 - 21.0 d,1st order [n = 4] and 4.9 d √1st order [n = 1], recalc. 1storder: 9.1 dDT50 (total residues):DT50f: Southern Europe (France): 15–29 dDT50f: Northern Europe (Germany, France, UK):12–56 dDT90f from soil dissipation studies: DT90f: Southern Europe (France): 30 / 101 d, √1storder [n = 2], recalc. 1st order: 40 and 123 dNorthern Europe (Germany, France, UK): 22 - 71 d, 1st4] and 54 d √1st order [n = 1], recalc. 1st order: 49 d14 April 2003Soil accumulation studies: not requiredSoil residue studies: Not requiredRemarks:e.g. effect of soil pH on degradation ratenoneAdsorption/desorptionK f / K oc:K d:pH dependence: K oc: propoxycarbazone-sodium:12.9 –106.2 L/kg [n=5], median: 28.8 L/kg sulfonamide methyl ester (M05):K oc: 35 L/kg (column leaching study), 71.1 L/kg (calculated, PCKOCWIN)saccharin (M07):K oc: 4.6 to 15.5 L/kg [n = 5], median: 5.2 L/kg4-Hydroxy saccharin (M08):K oc: 456.9 to 2872.7 L/kg [n = 5], median: 2033.8 L/kg N-methyl propoxy triazolinone amide (M09):K oc: 10.4 to 551.5 L/kg [n = 5], median: 99.9 L/kgN-methyl propoxy triazolinone (M10):K oc: 8.9 to 75.5 L/kg [n = 5], median: 20.6 L/kgpropoxycarbazone-sodium:K d: 0.22 – 1.7. L/kg [n = 5]sulfonamide methyl ester (M05):0.161 L/kg (column leaching study)saccharin (M07):K d: 0.02 to 0.25 L/kg [n = 5]4-Hydroxy saccharin (M08):K d: 7.5 to 46.3 L/kg [n = 5]N-methyl propoxy triazolinone amide (M09):K d: 0.26 to 3.90 L/kg [n = 5]N-methyl propoxy triazolinone (M10):K d: 0.22 to 1.22 L/kg [n = 5]propoxycarbazone-sodium, M05, M07, M08, M09,M10: noMobilityLaboratory studies:Column leaching: not requiredAged residue leaching: Loamy sand (80 % sand, 4 % clay, 0.5 % C org), ageingtime 28 d (phenyl-label) and 29 d (triazolinone-label),irrigation 508 mm, incubation at 20 °C and FC in the14 April 2003darkleachates contained 85.8 % (phenyl-label) and 89.0 %(triazolinone-label) of the applied radioactivity, containin76.5 % propoxycarbazone-sodium, 3.1 - 3.6 % carboxyacid (M04), 0.8 % sulfonamide acid (M06), 4.3 % sacch(M07) and 7.9 % N-methyl propoxy triazolinone (M10).Sulfonamide methyl ester (M05) and N-methyl propoxytriazolinone amide (M09) were not detectedField studies:Lysimeter/Field leaching studies: 70 g as/ha, spring application, 2 lysimeters over 3years, single application in year 1 and 2annual average concentration in leachate (µg/l):propoxycarbazone-sodium: 0.009, 0.004, 0.002N-methyl propoxy triazolinone(M10):0.002,0.018,0007total radioactive residue: 0.061, 0.057, 0.049maximum concentration in leachate (µg/l):propoxycarbazone-sodium: 0.02N-methyl propoxy triazolinone(M10): 0.04total radioactive residue: 0.099Remarks: under vulnerable conditions,propoxycarbazone-sodiumand N-methyl propoxy triazolinone (M 10) might leachinto ground water14 April 2003 2.2 Fate and behaviour in waterAbiotic degradationHydrolytic degradation: pH 4: stable (25°C)pH 7: stable (25°C)pH 9: stable (25°C)Major metabolites: nonePhotolytic degradation: phenyl label: propoxycarbazone-sodium: DT50: 18.1 dtriazolinone label: propoxycarbazone-sodium: DT50: 40. Major metabolites: metabolite saccharin (M07): 22 % of TAR (day 19, endof study)metabolite N-methyl propoxy triazolinone (M10): 13.6 %TAR (day 19, end of study)Biological degradationReadily biodegradable: not required (see water sediment study)Water/sediment study:DT50 water:DT90 water:DT50 whole system:DT90 whole system:Distribution in water / sediment systems (active substance)Distribution in water / sediment systems (metabolites) 10.6 and 90.8 d35.4 and 302.0 d12.0 and 189.0 d39.9 and 627.0 dwater: 97 - 97.9 % AR (0 d)0.7 - 47.2 % AR after 100 dsediment: max. 18.2 % AR (3 d) - 21.4 % AR (100 d) 0.2 - 21.4 % AR after 100 dcarboxylic acid (M04)water: max. 0.1 % AR (30 d) - 50.2 % AR (30 d)0.1 - 34.6 % AR after 100 dsediment: max. 0.0 % AR (0-100 d) - 19.3 % AR (62d)0.0 - 13.2 % AR after 100 dsulfonamide acid (M06)water: max. 1.6 % AR (100d) - 16.2 % AR (100d) sediment: max. 0 (0-100 d)- 3.2 % AR (100d)N-methyl propoxy triazolinone (M10)water: max. 3.1 % AR (100 d) - 21.2 % AR (100 d) 3.1 - 21.2 % AR after 100 dsediment: max. 3.8 % AR (100 d) - 13.2 % AR (100 d) 3.8 - 13.2 % AR after 100 d14 April 2003 Degradation in the saturated zone degradation in the saturated zone: not required Remarks: none14 April 2003 2.3 Fate and behaviour in airVolatilityVapour pressure: Cannot be determined directly due to its extreme lowvalue; upper limit: < 9 ⋅ 10-8 Pa at 70 °C< 1 ⋅ 10-8 Pa at 20 °C (extrapolated)Henry's law constant: < 1 ⋅ 10-10 Pa m3 mol-1 at 20 °C (calculated)Photolytic degradationDirect photolysis in air: no dataPhotochemical oxidative degradation in air DT50: Calculations according to Atkinson (AOPWin 1.75): t½: 4.5 h (c OH = 0.5 · 106 cm-3, 24 h day)Volatilisation: from plant surfaces: < 10 % within 24 hoursfrom soil: < 10 % within 24 hours Remarks: no short or long range transport expected14 April 20033 EcotoxicologyTerrestrial VertebratesAcute toxicity to mammals: LD 50 >5000 mg/kg bw (rat)Acute toxicity to birds: LD 50 >2000 mg/kg bw (bobwhite quail) Dietary toxicity to birds: LC 50 >10000 ppm (bobwhite quail)Reproductive toxicity to birds: NOEL 1250 ppm (bobwhite quail and mallard duck)Aquatic OrganismsGroup Test sub-stanceTime-scaleEnd-pointToxicity (mg/L)Laboratory testsAcute toxicity fish (LC50):O. mykiss a.s. acute (static) mortality>77.6 Long term toxicity fish (NOEC):P.promelasa.s. long-term (flow-through)Mortalit y,hatch, growth, beha-viour 105Bioaccumulation fish:Not relevant (Log P OW : 0.03) D. magna a.s. acute(stat.) mortalit y >107 D. magnaMKH 7017 (M10) acute (stat.) mortalit y >100D. magna MKH 7018 (M04) acute (stat.) mortalit y >100Acute toxicity invertebrate (EC50)D. magna STJ 4934 (M05)acute (stat.)mortalit y>100Chronic toxicity invertebrate (NOEC): D. magnaa.s. chronic (flow-through) Mortalit y,growth,reprodu ction110Acute toxicity algae (EC50):Selenastr umcapricorn utuma.s. chronic biomass 1.57。
欧洲药典索引版3

EUROPEAN PHARMACOPOEIA5.5INDEXTo aid users the index includes a reference to the supplement where the latest version of a text can be found.For example:Acetone...............................................5.1-2875means the monograph Acetone can be found on page2875of Supplement5.1.Note that where no reference for a supplement is made,the text can be found in the principal volume.Monographs deleted from the5th edition are not included in the index;the list of deleted texts is found in the Contents of this supplement,page xxx.EUROPEAN PHARMACOPOEIA5.5Numerics1.1.General statements (5)1.2.Other provisions applying to general chapters and monographs (5)1.3.General chapters (6)1.4.Monographs (7)1.5.Abbreviations and symbols (9)1.6.Units of the International System(SI)used in the Pharmacopoeia and equivalence with other units (10)1.General notices (5)2.1.1.Droppers (17)parative table of porosity of sintered-glass filters (17)2.1.3.Ultraviolet ray lamps for analytical purposes (17)2.1.4.Sieves (18)2.1.5.Tubes for comparative tests (19)2.1.6.Gas detector tubes (19)2.1.Apparatus (17)2.2.10.Viscosity-Rotating viscometer method.........5.3-3337 2.2.11.Distillation range (30)2.2.12.Boiling point (31)2.2.13.Determination of water by distillation (32)2.2.14.Melting point-capillary method (32)2.2.15.Melting point-open capillary method (33)2.2.16.Melting point-instantaneous method (33)2.2.17.Drop point (33)2.2.18.Freezing point (34)2.2.19.Amperometric titration (34)2.2.1.Clarity and degree of opalescence of liquids (23)2.2.20.Potentiometric titration (35)2.2.21.Fluorimetry (35)2.2.22.Atomic emission spectrometry (35)2.2.23.Atomic absorption spectrometry (36)2.2.24.Absorption spectrophotometry,infrared (37)2.2.25.Absorption spectrophotometry,ultraviolet and visible.................................................................................5.2-3089 2.2.26.Paper chromatography. (40)2.2.27.Thin-layer chromatography...............................5.2-3090 2.2.28.Gas chromatography.. (42)2.2.29.Liquid chromatography (43)2.2.2.Degree of coloration of liquids (24)2.2.30.Size-exclusion chromatography (45)2.2.31.Electrophoresis (45)2.2.32.Loss on drying (50)2.2.33.Nuclear magnetic resonance spectrometry (51)2.2.34.Thermal analysis (52)2.2.35.Osmolality (54)2.2.36.Potentiometric determination of ionic concentration using ion-selective electrodes (55)2.2.37.X-ray fluorescence spectrometry (56)2.2.38.Conductivity.........................................................5.1-2783 2.2.39.Molecular mass distribution in dextrans (57)2.2.3.Potentiometric determination of pH (26)2.2.40.Near-infrared spectrophotometry (59)2.2.41.Circular dichroism (63)2.2.42.Density of solids (64)2.2.43.Mass spectrometry (65)2.2.44.Total organic carbon in water for pharmaceutical use (68)2.2.45.Supercritical fluid chromatography (68)2.2.46.Chromatographic separation techniques (69)2.2.47.Capillary electrophoresis (74)2.2.48.Raman spectrometry (79)2.2.49.Falling ball viscometer method (80)2.2.4.Relationship between reaction of solution, approximate pH and colour of certain indicators (27)2.2.54.Isoelectric focusing (81)2.2.55.Peptide mapping (82)2.2.56.Amino acid analysis.......................................................862.2.5.Relative density.. (27)2.2.6.Refractive index (28)2.2.7.Optical rotation......................................................5.4-3695 2.2.8.Viscosity (29)2.2.9.Capillary viscometer method (29)2.2.Physical and physicochemical methods (23)2.3.1.Identification reactions of ions and functional groups...............................................................................5.5-4101 2.3.2.Identification of fatty oils by thin-layer chromatography. (98)2.3.3.Identification of phenothiazines by thin-layer chromatography (99)2.3.4.Odour (99)2.3.Identification (95)2.4.10.Lead in sugars (107)2.4.11.Phosphates (108)2.4.12.Potassium (108)2.4.13.Sulphates (108)2.4.14.Sulphated ash......................................................5.3-3341 2.4.15.Nickel in polyols.. (108)2.4.16.Total ash (108)2.4.17.Aluminium (108)2.4.18.Free formaldehyde (109)2.4.19.Alkaline impurities in fatty oils (109)2.4.1.Ammonium (103)2.4.21.Foreign oils in fatty oils by thin-layer chromatography (109)position of fatty acids by gas chroma-tography (110)2.4.23.Sterols in fatty oils..............................................5.1-2787 2.4.24.Identification and control of residual solvents (113)2.4.25.Ethylene oxide and dioxan (118)2.4.26.N,N-Dimethylaniline (119)2.4.27.Heavy metals in herbal drugs and fatty oils (119)2.4.28.2-Ethylhexanoic acid (120)position of fatty acids in oils rich inomega-3-acids...................................................................5.5-4107 2.4.2.Arsenic (103)2.4.30.Ethylene glycol and diethylene glycol in ethoxylated substances........................................................................5.2-3095 2.4.3.Calcium.. (103)2.4.4.Chlorides (104)2.4.5.Fluorides (104)2.4.6.Magnesium (104)2.4.7.Magnesium and alkaline-earth metals (104)2.4.8.Heavy metals (104)2.4.9.Iron (107)2.4.Limit tests (103)2.5.10.Oxygen-flask method (130)plexometric titrations (130)2.5.12.Water:semi-micro determination (130)2.5.13.Aluminium in adsorbed vaccines (131)2.5.14.Calcium in adsorbed vaccines (131)2.5.15.Phenol in immunosera and vaccines (131)2.5.16.Protein in polysaccharide vaccines (131)2.5.17.Nucleic acids in polysaccharide vaccines (132)2.5.18.Phosphorus in polysaccharide vaccines (132)2.5.19.O-Acetyl in polysaccharide vaccines (132)2.5.1.Acid value................................................................5.2-3099 2.5.20.Hexosamines in polysaccharide vaccines. (132)2.5.21.Methylpentoses in polysaccharide vaccines (133)2.5.22.Uronic acids in polysaccharide vaccines (133)2.5.23.Sialic acid in polysaccharide vaccines (133)2.5.24.Carbon dioxide in gases (134)2.5.25.Carbon monoxide in gases (134)2.5.26.Nitrogen monoxide and nitrogen dioxide in gases (135)2.5.27.Oxygen in gases (136)2.5.28.Water in gases (136)2.5.29.Sulphur dioxide (136)2.5.2.Ester value (127)2.5.30.Oxidising substances (137)2.5.31.Ribose in polysaccharide vaccines (137)2.5.32.Water:micro determination (137)2.5.33.Total protein (138)2.5.34.Acetic acid in synthetic peptides (141)2.5.35.Nitrous oxide in gases (141)2.5.36.Anisidine value (142)2.5.3.Hydroxyl value (127)2.5.4.Iodine value (127)2.5.5.Peroxide value (128)2.5.6.Saponification value (129)2.5.7.Unsaponifiable matter (129)2.5.8.Determination of primary aromaticamino-nitrogen (129)2.5.9.Determination of nitrogen by sulphuric acid digestion (129)2.5.Assays (127)2.6.10.Histamine (153)2.6.11.Depressor substances (153)2.6.12.Microbiological examination of non-sterile products (total viable aerobic count) (154)2.6.13.Microbiological examination of non-sterile products (test for specified micro-organisms) (156)2.6.14.Bacterial endotoxins (161)2.6.15.Prekallikrein activator........................................5.5-4111 2.6.16.Tests for extraneous agents in viral vaccines for human use (169)2.6.17.Test for anticomplementary activity of immunoglobulin (170)2.6.18.Test for neurovirulence of live virus vaccines (172)2.6.19.Test for neurovirulence of poliomyelitis vaccine (oral) (172)2.6.1.Sterility (145)2.6.20.Anti-A and anti-B haemagglutinins(indirect method) (174)2.6.21.Nucleic acid amplification techniques............5.5-4111 2.6.22.Activated coagulation factors...........................5.5-4115 2.6.24.Avian viral vaccines:tests for extraneous agents in seed lots............................................................................5.4-3699 2.6.25.Avian live virus vaccines:tests for extraneous agents in batches of finished product.....................................5.3-3345 2.6.26.Test for anti-D antibodies in human immunoglobulin for intravenous administration....................................5.3-3348 2.6.2.Mycobacteria. (149)2.6.7.Mycoplasmas (149)2.6.8.Pyrogens (152)2.6.9.Abnormal toxicity (153)2.6.Biological tests (145)2.7.10.Assay of human coagulation factor VII (203)2.7.11.Assay of human coagulation factor IX............5.5-4120 2.7.12.Assay of heparin in coagulation factors (204)2.7.13.Assay of human anti-D immunoglobulin (205)2.7.14.Assay of hepatitis A vaccine..............................5.1-2795 2.7.15.Assay of hepatitis B vaccine(rDNA). (207)2.7.16.Assay of pertussis vaccine(acellular) (208)2.7.17.Assay of human antithrombin III (209)2.7.18.Assay of human coagulation factor II (209)2.7.19.Assay of human coagulation factor X (210)2.7.1.Immunochemical methods (187)2.7.20.In vivo assay of poliomyelitis vaccine (inactivated) (210)2.7.21.Assay of human von Willebrand factor...........5.5-4120 2.7.22.Assay of human coagulation factor XI............5.5-4121 2.7.2.Microbiological assay of antibiotics. (188)2.7.4.Assay of human coagulation factor VIII...........5.5-4119 2.7.5.Assay of heparin.. (195)2.7.6.Assay of diphtheria vaccine(adsorbed).....................1962.7.7.Assay of pertussis vaccine (197)2.7.8.Assay of tetanus vaccine(adsorbed)..................5.1-2791 2.7.9.Test for Fc function of immunoglobulin. (202)2.7.Biological assays (187)2.8.10.Solubility in alcohol of essential oils (216)2.8.11.Assay of1,8-cineole in essential oils (216)2.8.12.Determination of essential oils in vegetable drugs (217)2.8.13.Pesticide residues (218)2.8.14.Determination of tannins in herbal drugs (221)2.8.15.Bitterness value (221)2.8.16.Dry residue of extracts (222)2.8.17.Loss on drying of extracts (222)2.8.1.Ash insoluble in hydrochloric acid (215)2.8.2.Foreign matter (215)2.8.3.Stomata and stomatal index (215)2.8.4.Swelling index (215)2.8.5.Water in essential oils (216)2.8.6.Foreign esters in essential oils (216)2.8.7.Fatty oils and resinified essential oils in essential oils (216)2.8.8.Odour and taste of essential oils (216)2.8.9.Residue on evaporation of essential oils (216)2.8.Methods in pharmacognosy (215)2.9.10.Ethanol content and alcoholimetric tables (237)2.9.11.Test for methanol and2-propanol...................5.3-3362 2.9.12.Sieve test (239)2.9.13.Limit test of particle size by microscopy (239)2.9.14.Specific surface area by air permeability (239)2.9.15.Apparent volume (241)2.9.16.Flowability (242)2.9.17.Test for extractable volume of parenteral preparations.....................................................................5.3-3363 2.9.18.Preparations for inhalation:aerodynamic assessment of fine particles...............................................................5.2-3103 2.9.19.Particulate contamination:sub-visible particles (253)2.9.1.Disintegration of tablets and capsules..............5.3-3351 2.9.20.Particulate contamination:visible particles. (255)2.9.22.Softening time determination of lipophilic suppositories (256)2.9.23.Pycnometric density of solids (257)2.9.24.Resistance to rupture of suppositories and pessaries (258)2.9.25.Dissolution test for medicated chewing gums..................................................................................5.2-3116 2.9.26.Specific surface area by gas adsorption.........5.1-2811 2.9.27.Uniformity of mass of delivered doses from multidose containers. (263)2.9.28.Test for deliverable mass or volume of liquid and semi-solid preparations (263)2.9.29.Intrinsic dissolution............................................5.4-3705 2.9.2.Disintegration of suppositories and pessaries (227)2.9.36.Powder flow..........................................................5.3-3363 2.9.37.Optical microscopy..............................................5.3-3366 2.9.38.Particle-size distribution estimation by analytical sieving...............................................................................5.3-3368 2.9.3.Dissolution test for solid dosage forms............5.3-3353 2.9.40.Uniformity of dosage units................................5.3-3370 2.9.42.Dissolution test for lipophilic solid dosage forms..................................................................................5.3-3373 2.9.4.Dissolution test for transdermal patches (231)2.9.5.Uniformity of mass of single-dose preparations (233)2.9.6.Uniformity of content of single-dose preparations..234 2.9.7.Friability of uncoated tablets..............................5.2-3103 2.9.8.Resistance to crushing of tablets.. (235)2.9.9.Measurement of consistency by penetrometry (235)2.9.Pharmaceutical technical procedures (225)3.1.10.Materials based on non-plasticised poly(vinyl chloride) for containers for non-injectable,aqueous solutions (289)3.1.11.Materials based on non-plasticised poly(vinyl chloride)for containers for dry dosage forms for oral administration..........................................................................2913.1.1.1.Materials based on plasticised poly(vinyl chloride)for containers for human blood and blood components. (269)3.1.1.2.Materials based on plasticised poly(vinyl chloride)for tubing used in sets for the transfusion of blood andblood components (272)3.1.13.Plastic additives (293)3.1.14.Materials based on plasticised poly(vinyl chloride)for containers for aqueous solutions for intravenous infusion......................................................................................2963.1.15.Polyethyleneterephthalatefor containers forpreparations not for parenteral use.....................................2983.1.1.Materials for containers for human blood and blood components. (269)3.1.3.Polyolefines (274)3.1.4.Polyethylene without additives for containers for parenteral preparations and for ophthalmic preparations..............................................................................2783.1.5.Polyethylene with additives for containers for parenteral preparations and for ophthalmicpreparations..............................................................................2793.1.6.Polypropylene for containers and closures for parenteral preparationsand ophthalmic preparations (282)3.1.7.Poly(ethylene -vinyl acetate)for containers and tubing for total parenteral nutrition preparations........................2853.1.8.Silicone oilused as a lubricant (287)3.1.9.Silicone elastomer for closures and tubing..............2883.1.Materials used for the manufacture of containers.....2693.2.1.Glass containers for pharmaceutical use..................3033.2.2.1.Plastic containers for aqueous solutions for parenteral infusion..................................................................3093.2.2.Plastic containers and closures for pharmaceuticaluse...............................................................................................3083.2.3.Sterile plastic containers for human blood and bloodcomponents...............................................................................3093.2.4.Empty sterile containers of plasticised poly(vinylchloride)forhuman blood and blood components...........3113.2.5.Sterile containers of plasticisedpoly (vinylchloride)for human blood containing anticoagulant solution.......3123.2.6.Sets for the transfusion of blood and blood components................................................................................3133.2.8.Sterile single-use plastic syringes................................3143.2.9.Rubber closures for containers for aqueous parenteral preparations,for powders and for freeze-dried powders..3163.2.Containers...........................................................................3034.1.1.Reagents..................................................................5.4-37094.1.1.Reagents..................................................................5.5-41254.1.2.Standard solutions for limit tests.......................5.4-38174.1.2.Standard solutions for limit tests.......................5.5-41264.1.3.Buffer solutions.....................................................5.4-38214.1.3.Buffer solutions.....................................................5.5-41264.1.Reagents,standard solutions,buffer solutions..5.4-37094.2.1.Primary standards for volumetric solutions....5.4-38274.2.2.Volumetric solutions.............................................5.4-38274.2.2.Volumetric solutions.............................................5.5-41274.2.Volumetric analysis...................................................5.4-38274.Reagents.........................................................................5.4-37095.10.Control of impurities in substances for pharmaceuticaluse......................................................................................5.5-41455.11.Characters section in monographs (565)5.1.1.Methods of preparation of sterile products..............4455.1.2.Biological indicators of sterilisation (447)5.1.3.Efficacy of antimicrobial preservation.......................4475.1.4.Microbiological quality of pharmaceuticalpreparations (449)5.1.5.Application of the F 0concept to steam sterilisation of aqueous preparations....................................................5.1-2821 5.1.6.Alternative methods for control of microbiological quality................................................................................5.5-41315.1.Generaltexts onsterility..................................................4455.2.1.Terminology used in monographs on vaccines (453)5.2.2.Chicken flocks free from specified pathogens for the production and quality control of vaccines...............5.1-28255.2.3.Cell substrates for the production of vaccines for human use.................................................................................4555.2.4.Cell cultures for the production of veterinaryvaccines (458)5.2.5.Substances of animal origin for the production ofveterinary vaccines (460)5.2.6.Evaluation of safety of veterinary vaccines andimmunosera ....................................................................5.1-28275.2.7.Evaluation of efficacy of veterinary vaccines and immunosera.....................................................................5.1-28295.2.8.Minimising the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products (463)5.2.9.Evaluation of safety of each batch of veterinary vaccines and immunosera.............................................5.1-28305.2.General texts on vaccines (453)5.3.Statistical analysis of results of biological assays andtests (475)5.4.Residual solvents...............................................................5075.5.Alcoholimetric tables.........................................................5195.6.Assay of interferons..................................................5.3-33815.7.Table of physical characteristics of radionuclidesmentioned in the European Pharmacopoeia.....................5395.8.Pharmacopoeial harmonisation.....................................5515.9.Polymorphism (555)AAbbreviationsand symbols (1.5.) (9)Abnormal toxicity (2.6.9.) (153)Absinthiiherba ........................................................................2710Absorption spectrophotometry,infrared (2.2.24.). (37)Absorption spectrophotometry,ultraviolet and visible (2.2.25.).............................................................................5.2-3089Acacia (905)Acaciae gummi (905)Acaciae gummi dispersione desiccatum .............................905Acacia,spray-dried (905)Acamprosate calcium................................................................906Acamprosatum calcicum (906)Acarbose..............................................................................5.1-2873Acarbosum .........................................................................5.1-2873Acebutololhydrochloride................................................5.4-3889Acebutololi hydrochloridum .........................................5.4-3889Aceclofenac (909)Aceclofenacum (909)Acesulfame potassium.....................................................5.4-3890Acesulfamum kalicum ....................................................5.4-3890Acetazolamide (912)Acetazolamidum (912)Acetic acid,glacial (913)Acetic acid in synthetic peptides (2.5.34.) (141)Acetone................................................................................5.1-2875Acetonum ...........................................................................5.1-2875Acetylcholine chloride...............................................................914Acetylcholini chloridum .. (914)Acetylcysteine..............................................................................915Acetylcysteinum (915)β-Acetyldigoxin..................................................................5.5-4185β-Acetyldigoxinum ...........................................................5.5-4185Acetylsalicylic acid (917)Acetyltryptophan,N - (918)Acetyltyrosine,N - (920)Aciclovir..............................................................................5.3-3436Aciclovirum.......................................................................5.3-3436 Acidum4-aminobenzoicum (973)Acidum aceticum glaciale (913)Acidum acetylsalicylicum (917)Acidum adipicum (926)Acidum alginicum (942)Acidum amidotrizoicum dihydricum (967)Acidum aminocaproicum (974)Acidum ascorbicum (1025)Acidum asparticum (1029)Acidum benzoicum (1072)Acidum boricum (1117)Acidum caprylicum (1172)Acidum chenodeoxycholicum (1247)Acidum citricum anhydricum (1306)Acidum citricum monohydricum (1307)Acidum edeticum.............................................................5.4-3933 Acidum etacrynicum.. (1542)Acidum folicum (1630)Acidum fusidicum (1645)Acidum glutamicum (1670)Acidum hydrochloridum concentratum (1755)Acidum hydrochloridum dilutum (1756)Acidum iopanoicum (1824)Acidum iotalamicum (1825)Acidum ioxaglicum (1826)Acidum lacticum..............................................................5.2-3227 Acidum lactobionicum.. (1885)Acidum maleicum (1966)Acidum malicum (1966)Acidum mefenamicum (1984)Acidum methacrylicum et ethylis acrylas polymerisatum 1:1 (2005)Acidum methacrylicum et ethylis acrylas polymerisatum 1:1dispersio30per centum (2005)Acidum methacrylicum et methylis methacrylas polymerisatum1:1 (2006)Acidum methacrylicum et methylis methacrylas polymerisatum1:2 (2007)Acidum nalidixicum (2080)Acidum nicotinicum (2097)Acidum nitricum (2105)Acidum oleicum (2132)Acidum oxolinicum (2165)Acidum palmiticum (2179)Acidum phosphoricum concentratum (2237)Acidum phosphoricum dilutum (2238)Acidum pipemidicum trihydricum (2249)Acidum salicylicum.........................................................5.1-3007 Acidum(S)-lacticum........................................................5.2-3227 Acidum sorbicum.. (2467)Acidum stearicum (2490)Acidum sulfuricum (2520)Acidum tartaricum (2534)Acidum thiocticum...........................................................5.5-4312 Acidum tiaprofenicum.. (2578)Acidum tolfenamicum (2601)Acidum tranexamicum (2609)Acidum trichloraceticum (2620)Acidum undecylenicum (2658)Acidum ursodeoxycholicum (2662)Acidum valproicum (2669)Acid value(2.5.1.)..............................................................5.2-3099 Acitretin. (922)Acitretinum (922)Acriflavinii monochloridum (924)Acriflavinium monochloride (924)Actinobacillosis vaccine(inactivated),porcine (784)Activated charcoal....................................................................1246Activated coagulation factors(2.6.22.).........................5.5-4115 Additives,plastic(3.1.13.) (293)Adenine (924)Adeninum (924)Adenosine (925)Adenosinum (925)Adeps lanae.......................................................................5.2-3285 Adeps lanae cum aqua.. (2709)Adeps lanae hydrogenatus (2708)Adeps solidus (1711)Adipic acid (926)Adrenaline tartrate (927)Adrenalini tartras (927)Aer medicinalis (929)Aer medicinalis artificiosus (932)Aerodynamic assessment of fine particles in preparations for inhalation(2.9.18.).........................................................5.2-3103 Aether.. (1548)Aether anaestheticus (1549)Agar (928)Agni casti fructus.............................................................5.4-3892 Agnus castus fruit.............................................................5.4-3892 Agrimoniae herba (929)Agrimony (929)Air,medicinal (929)Air,synthetic medicinal (932)Alanine (933)Alaninum (933)Albendazole (934)Albendazolum (934)Albumini humani solutio...............................................5.3-3511 Albumin solution,human................................................5.3-3511 Alchemilla (935)Alchemillae herba (935)Alcohol benzylicus...........................................................5.5-4197 Alcohol cetylicus...............................................................5.3-3475 Alcohol cetylicus et stearylicus....................................5.3-3474 Alcohol cetylicus et stearylicus emulsificans A.. (1239)Alcohol cetylicus et stearylicus emulsificans B (1241)Alcoholes adipis lanae (2703)Alcoholimetric tables(2.9.10.) (237)Alcoholimetric tables(5.5.) (519)Alcohol isopropylicus (1841)Alcohol oleicus (2134)Alcohol stearylicus...........................................................5.3-3621 Alcuronii chloridum.. (935)Alcuronium chloride (935)Alexandrian senna pods (2404)Alfacalcidol (937)Alfacalcidolum (937)Alfadex (938)Alfadexum (938)Alfentanil hydrochloride (939)Alfentanili hydrochloridum (939)Alfuzosin hydrochloride (941)Alfuzosini hydrochloridum (941)Alginic acid (942)Alkaline-earth metals and magnesium(2.4.7.) (104)Alkaline impurities in fatty oils(2.4.19.) (109)Allantoin (942)Allantoinum (942)Allergen products (569)Allii sativi bulbi pulvis (1651)Allium sativum ad praeparationes homoeopathicas (897)Allopurinol (943)Allopurinolum (943)all-rac-α-Tocopherol..........................................................5.5-4313 all-rac-α-Tocopheryl acetate...........................................5.5-4314 Almagate.............................................................................5.2-3169Almagatum.........................................................................5.2-3169 Almond oil,refined...........................................................5.4-3893 Almond oil,virgin.............................................................5.3-3437 Aloe barbadensis.. (947)Aloe capensis (948)Aloes,barbados (947)Aloes,Cape (948)Aloes dry extract,standardised (949)Aloes extractum siccum normatum (949)Alphacyclodextrin (938)Alprazolam (950)Alprazolamum (950)Alprenolol hydrochloride (952)Alprenololi hydrochloridum (952)Alprostadil (953)Alprostadilum (953)Alteplase for injection (956)Alteplasum ad iniectabile (956)Alternative methods for control of microbiological quality (5.1.6.)................................................................................5.5-4131 Althaeae folium (1974)Althaeae radix...................................................................5.2-3232 Alum. (959)Alumen (959)Aluminii chloridum hexahydricum (960)Aluminii hydroxidum hydricum ad adsorptionem..5.5-4186 Aluminii magnesii silicas (961)Aluminii oxidum hydricum (962)Aluminii phosphas hydricus (963)Aluminii phosphatis liquamen.....................................5.3-3438 Aluminii sulfas (964)Aluminium(2.4.17.) (108)Aluminium chloride hexahydrate (960)Aluminium hydroxide,hydrated,for adsorption........5.5-4186 Aluminium in adsorbed vaccines(2.5.13.).. (131)Aluminium magnesium silicate (961)Aluminium oxide,hydrated (962)Aluminium phosphate gel...............................................5.3-3438 Aluminium phosphate,hydrated.. (963)Aluminium sulphate (964)Amantadine hydrochloride (964)Amantadini hydrochloridum (964)Ambroxol hydrochloride (965)Ambroxoli hydrochloridum (965)Amfetamine sulphate (966)Amfetamini sulfas (966)Amidotrizoic acid dihydrate (967)Amikacin (968)Amikacini sulfas...............................................................5.4-3894 Amikacin sulphate............................................................5.4-3894 Amikacinum. (968)Amiloride hydrochloride..................................................5.3-3439 Amiloridi hydrochloridum.............................................5.3-3439 Amino acid analysis(2.2.56.).. (86)Aminobenzoic acid,4- (973)Aminocaproic acid (974)Aminoglutethimide (975)Aminoglutethimidum (975)Amiodarone hydrochloride (977)Amiodaroni hydrochloridum (977)Amisulpride (978)Amisulpridum (978)Amitriptyline hydrochloride (980)Amitriptylini hydrochloridum (980)Amlodipine besilate (981)Amlodipini besilas (981)Ammonia(13N)injection (817)Ammoniae(13N)solutio iniectabilis (817)Ammoniae solutio concentrata.............................................983Ammonia solution,concentrated. (983)Ammonii bromidum (985)Ammonii chloridum (986)Ammonii glycyrrhizas....................................................5.1-2876 Ammonii hydrogenocarbonas.. (988)Ammonio methacrylate copolymer(type A) (983)Ammonio methacrylate copolymer(type B) (984)Ammonio methacrylatis copolymerum A (983)Ammonio methacrylatis copolymerum B (984)Ammonium(2.4.1.) (103)Ammonium bromide (985)Ammonium chloride (986)Ammonium glycyrrhizate................................................5.1-2876 Ammonium hydrogen carbonate.. (988)Amobarbital (988)Amobarbital sodium (989)Amobarbitalum (988)Amobarbitalum natricum (989)Amoxicillin sodium (990)Amoxicillin trihydrate......................................................5.3-3440 Amoxicillinum natricum (990)Amoxicillinum trihydricum...........................................5.3-3440 Amperometric titration(2.2.19.).. (34)Amphotericin B (995)Amphotericinum B (995)Ampicillin,anhydrous (996)Ampicillin sodium (998)Ampicillin trihydrate (1001)Ampicillinum anhydricum (996)Ampicillinum natricum (998)Ampicillinum trihydricum (1001)Amygdalae oleum raffinatum.......................................5.4-3893 Amygdalae oleum virginale..........................................5.3-3437 Amylum pregelificatum (2490)Anaesthetic ether (1549)Analysis,thermal(2.2.34.) (52)Analytical sieving,particle-size distribution estimation by (2.9.38.).............................................................................5.3-3368 Angelicae radix (1003)Angelica root (1003)Anhydrous silica,hydrophobic colloidal......................5.5-4297 Animal anti-T lymphocyte immunoglobulin for human use (1010)Animal spongiform encephalopathies,products with risk of transmitting agents of (577)Animal spongiform encephalopathy agents,minimising the risk of transmitting via human and veterinary medicinal products(5.2.8.) (463)Aniseed (1006)Anise oil (1004)Anisi aetheroleum (1004)Anisidine value(2.5.36.) (142)Anisi fructus (1006)Anisi stellati aetheroleum (2488)Anisi stellati fructus.........................................................5.5-4297 Antazoline hydrochloride. (1006)Antazolini hydrochloridum (1006)Anthrax spore vaccine(live)for veterinary use (715)Anti-A and anti-B haemagglutinins(indirect method)(2.6.20.) (174)Antibiotics,microbiological assay of(2.7.2.) (188)Anticoagulant and preservative solutions for human blood (1007)Anticomplementary activity of immunoglobulin(2.6.17.)..170 Anticorpora monoclonalia ad usum humanum......5.2-3127 Anti-D antibodies in human immunoglobulins for intravenous administration,test for(2.6.26.)..................................5.3-3348 Anti-D immunoglobulin,human. (1732)Anti-D immunoglobulin,human,assay of(2.7.13.) (205)。
欧盟GMP附录

欧洲共同体:European Communities (EC)。
欧洲联盟:European Union (EU),简称欧盟。
人用药品注册技术标准国际协调会:ICH欧盟GMP附录1无菌药品的生产注:冻干瓶轧盖的条款自2010年3月1日开始实施。
原则为降低微生物、微粒和热原污染的风险,无菌药品的生产应有各种特殊要求。
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度。
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法和规程进行。
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验。
注:本指南没有对微粒、浮游菌和表面微生物等测试方法详细进行阐述,可参阅欧洲标准或国际标准(CEN/ISO)及药典资料。
总则1.无菌药品的生产必须在洁净区内进行,人员和(或)设备以及物料必须通过缓冲进入洁净区。
洁净区应当保持适当的洁净度,洁净区的送风须经具有一定过滤效率过滤器的过滤。
2.原料配制、产品加工和灌装等不同操作必须在洁净去内彼此分开的单独区域内进行。
生产工艺可分为两类:一类是最终灭菌工艺;第二类是部分或全部工序为无菌操作的工艺。
3.应按所需环境的特点确定无菌产品的洁净级别。
每一步生产操作都应达到适当的动态洁净度,以尽可能降低产品(或原料)被微粒或微生物污染。
洁净区的设计必须符合相应的“静态”标准,以达到“动态”的洁净要求。
“静态”是指安装已经完成并已运行,但没有操作人员在场的状态。
“动态”是指生产设施按预定的工艺模式运行并有规定数量的操作人员进行现场操作的状态。
应确定每一洁净室或每组洁净间的“动态”及“静态”标准。
无菌药品生产所需的洁净区一般可分为4个级别:A级:高风险操作区,如:灌装区,放置胶塞桶、敞口安瓿瓶、敞口西林瓶的区域及无菌装配/连接操作的区域。
通常用单向流操作台/罩来维护该区的环境状态。
单向流系统在其工作区域必须均匀送风,风速为0.36-0.54m/s(指导值)。
欧洲药典译文(药品玻璃容器)

药品玻璃容器药品玻璃容器用于直接与药品接触.现有的几种药品玻璃容器例如:安瓿: 这是一种薄壁玻璃容器,在被填装后,以熔合的方式式封装.容器中的药物在容器破碎后被抽取.针剂瓶仅为一次性使用各种规格的玻璃瓶:这是一些规格不一的厚壁玻璃容器,以玻璃或其他材料封装,例如塑料,人造橡胶等.容器中的药物可以被一次或多次按任意的量取用.盛装人类血液或人类血液中的提取物的容器:这是一种圆柱形的壁厚,容积不一的厚壁玻璃容器,由无色,透明的中性玻璃制成。
玻璃的质量:无色玻璃指人类肉眼看上去高度透明的玻璃。
有色玻璃是指添加了少量氧化金属物质的玻璃,根据需要的吸光率选材制成.中性玻璃指一种硼硅酸盐玻璃,其中含有大量硅氧化物,铝或碱土金属氧化物. 因为中性玻璃上述的成分构成,它具有很高的抗热冲击性和抗水解性.钠钙硅玻璃指一种硅玻璃含有碱金属氧化物--主要是钠氧化物;和碱土金属氧化物--主要是钙氧化物. 钠钙硅玻璃的化学成份决定它只具有中等的抗水解性. 药品玻璃容器的化学稳定性是由它的抗水解性表现出来的. 比如在一个特定的水和容器内表面接触的条件下或粉末状玻璃和水接触时,玻璃所表现出来的抗溶解性. 抗水解性以滴定法被测溶液碱度来衡量. 以下是根据其抗水解性对玻璃容器的分类以下斜体字部分是对不同的药品所应采用的玻璃容器类型的推荐. 药品生产厂家必须负责保证选用合适的容器来包装药品.一类玻璃容器通常适用于所有的药品(口服或非口服药品及人类血液或人类血液的提取物). 二类玻璃容器通常适用于酸性和中性及水性药品(非口服).三类玻璃容器通常适用于:非水性的药品(非口服)、粉末剂(非口服)以及口服类药品.四类玻璃容器通常适用于一些固体药品(口服)和一些液体或半固体药品(口服)某些特殊药品的玻璃容器的抗水解性可能高于以上介绍的玻璃容器的水解性.对于口服类药品,有色和无色玻璃容器都适用,非口服类药品通常用无色玻璃容器包装,但有色玻璃容器亦可用于某些对光不敏感的药品.建议所有包装液体和粉末状(非口服)药品的玻璃容器可以让人凭肉眼就能观察其盛装的物品.可以对玻璃容器的内表面进行特殊处理以获得或提高抗水解性. 玻璃容器的外表面也可以被处理以减少磨擦和提高抗磨损性. 对玻璃容器的外表面处理必须不能污染其内表面.除了一类玻璃容器, 药品玻璃容器不可被重复使用. 盛装人类血液或人类血液提取物的玻璃容器不可被重复使用.药品玻璃容器必须符合关于抗水解性的测试标准或其他相关标准. 对于有非玻璃部件的玻璃容器, 上述测试标准只适用于玻璃部件.测试以下是必须的测试用以评定不同用途的玻璃容器的质量.抗水解性设备和试剂—研钵、杵(如图3.2.1.-1)、锤子、磁铁—一套不锈钢方网格筛子,装在不锈钢的框架上,筛子的规格号码为:a) 筛子no. 710b) 筛子no. 425c) 筛子no. 250—一块永久磁铁—一个旧的中性玻璃制成的烧瓶和盖子(即曾被使用于同样试验的烧瓶和盖子或者是一个烧瓶被装入水试剂后保持在121°C的高压下至少一个小时)—一片惰性金属(例如铝)的箔.—一个可以使温度保持在121 °C ± 1 °C的高压锅,这个高压锅必须装有压力计、温度计、旋钮气阀和一个托盘,高压锅的水平面以上必须有足够的空间来容纳用被测试的容器。
欧洲药典重金属及溶残方法

01/2005:504005.4.RESIDUAL SOLVENTSLIMITING RESIDUAL SOLVENT LEVELS IN ACTIVE SUBSTANCES,EXCIPIENTS AND MEDICINAL PRODUCTSThe International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use(ICH)has adopted Impurities Guidelines for Residual Solvents which prescribes limits for the content of solvents which may remain in active substances,excipients and medicinal products after processing.This guideline, the text of which is reproduced below,excludes existing marketed products.The European Pharmacopoeia is, however,applying the same principles enshrined in the guideline to existing active substances,excipients and medicinal products whether or not they are the subject of a monograph of the Pharmacopoeia.All substances and products are to be tested for the content of solvents likely to be present in a substance or product.Where the limits to be applied comply with those given below,tests for residual solvents are not generally mentioned in specific monographs since the solvents employedmay vary from one manufacturer to another and the requirements of this general chapter are applied via the general monograph on Substances for Pharmaceutical Use(2034).The competent authority is to be informedof the solvents employed during the production process. This information is also given in the dossier submitted for a certificate of suitability of the monographs of the European Pharmacopoeia and is mentioned on the certificate. Where only Class3solvents are used,a test for loss on drying may be applied or a specific determination of the solvent may be made.If for a Class3solvent a justified and authorised limit higher than0.5per cent is applied,a specific determination of the solvent is required.When Class1residual solvents or Class2residual solvents (or Class3residual solvents which exceed the0.5per cent) are used,the methodology described in the general method (2.4.24)is to be applied wherever possible.Otherwise an appropriate validated method is to be employed.When a quantitative determination of a residual solventis carried out,the result is taken into account for the calculation of the content of the substance except where a test for drying is carried out.IMPURITIES:GUIDELINESFOR RESIDUAL SOLVENTS(CPMP/ICH/283/95)1.INTRODUCTION2.SCOPE OF THE GUIDELINE3.GENERAL PRINCIPLES3.1.CLASSIFICATION OF RESIDUAL SOLVENTS BY RISK ASSESSMENT3.2.METHODS FOR ESTABLISHING EXPOSURE LIMITS 3.3.OPTIONS FOR DESCRIBING LIMITS OF CLASS2 SOLVENTS3.4.ANALYTICAL PROCEDURES3.5.REPORTING LEVELS OF RESIDUAL SOLVENTS4.LIMITS OF RESIDUAL SOLVENTS4.1.SOLVENTS TO BE AVOIDED4.2.SOLVENTS TO BE LIMITED4.3.SOLVENTS WITH LOW TOXIC POTENTIAL4.4.SOLVENTS FOR WHICH NO ADEQUATE TOXICOLOGICAL DATA WAS FOUNDGLOSSARYAPPENDIX1.LIST OF SOLVENTS INCLUDED IN THE GUIDELINEAPPENDIX2.ADDITIONAL BACKGROUNDA2.1:ENVIRONMENTAL REGULATION OF ORGANIC VOLATILE SOLVENTSA2.2:RESIDUAL SOLVENTS IN PHARMACEUTICALS APPENDIX3.METHODS FOR ESTABLISHING EXPOSURE LIMITS1.INTRODUCTIONThe objective of this guideline is to recommend acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient.The guideline recommends the useof less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents. Residual solvents in pharmaceuticals are defined here as organic volatile chemicals that are used or produced in the manufacture of active substances or excipients,or in the preparation of medicinal products.The solvents are not completely removed by practical manufacturing techniques. Appropriate selection of the solvent for the synthesis of active substance may enhance the yield,or determine characteristics such as crystal form,purity,and solubility. Therefore,the solvent may sometimes be a critical parameter in the synthetic process.This guideline does not address solvents deliberately used as excipients nor does it address solvates.However,the content of solvents in such products should be evaluated and justified.Since there is no therapeutic benefit from residual solvents, all residual solvents should be removed to the extent possible to meet product specifications,good manufacturing practices,or other quality-based requirements.Medicinal products should contain no higher levels of residual solvents than can be supported by safety data.Some solvents that are known to cause unacceptable toxicities(Class1,Table1) should be avoided in the production of active substances, excipients,or medicinal products unless their use can be strongly justified in a risk-benefit assessment.Some solvents associated with less severe toxicity(Class2,Table2)should be limited in order to protect patients from potential adverse effects.Ideally,less toxic solvents(Class3,Table3)should be used where practical.The complete list of solvents included in this guideline is given in Appendix1.The lists are not exhaustive and other solvents can be used and later added to the lists.Recommended limits of Class1 and2solvents or classification of solvents may change as new safety data becomes available.Supporting safety data in a marketing application for a new medicinal product containing a new solvent may be based on concepts inthis guideline or the concept of qualification of impurities as expressed in the guideline for active substances(Q3A,Impurities in New Active Substances)or medicinal products (Q3B,Impurities in New Medicinal Products),or all three guidelines.2.SCOPE OF THE GUIDELINEResidual solvents in active substances,excipients,and in medicinal products are within the scope of this guideline. Therefore,testing should be performed for residual solvents when production or purification processes are known to result in the presence of such solvents.It is only necessary to test for solvents that are used or produced in the manufacture or purification of active substances,excipients, or medicinal product.Although manufacturers may choose to test the medicinal product,a cumulative method may be used to calculate the residual solvent levels in the medicinal product from the levels in the ingredients used to produce the medicinal product.If the calculation results in a level equal to or below that recommended in this guideline,no testing of the medicinal product for residual solvents need be considered.If however,the calculated level is above the recommended level,the medicinal product should be tested to ascertain whether the formulation process has reduced the relevant solvent level to within the acceptable amount. Medicinal product should also be tested if a solvent is used during its manufacture.This guideline does not apply to potential new active substances,excipients,or medicinal products used during the clinical research stages of development,nor does it apply to existing marketed medicinal products.The guideline applies to all dosage forms and routes of administration.Higher levels of residual solvents may be acceptable in certain cases such as short term(30days or less)or topical application.Justification for these levels should be made on a case by case basis.See Appendix2for additional background information related to residual solvents.3.GENERAL PRINCIPLES3.1.CLASSIFICATION OF RESIDUAL SOLVENTS BY RISK ASSESSMENTThe term“tolerable daily intake”(TDI)is used by the International Program on Chemical Safety(IPCS)to describe exposure limits of toxic chemicals and“acceptable daily intake”(ADI)is used by the World Health Organisation (WHO)and other national and international health authorities and institutes.The new term“permitted daily exposure”(PDE)is defined in the present guideline as a pharmaceutically acceptable intake of residual solvents to avoid confusion of differing values for ADI’s of the same substance.Residual solvents assessed in this guideline are listed in Appendix1by common names and structures.They were evaluated for their possible risk to human health and placed into one of three classes as follows:Class1solvents:Solvents to be avoidedKnown human carcinogens,strongly suspected human carcinogens,and environmental hazards.Class2solvents:Solvents to be limitedNon-genotoxic animal carcinogens or possible causative agents of other irreversible toxicity such as neurotoxicity or teratogenicity.Solvents suspected of other significant but reversibletoxicities.Class3solvents:Solvents with low toxic potential Solvents with low toxic potential to man;no health-based exposure limit is needed.Class3solvents have PDEs of 50mg or more per day.3.2.METHODS FOR ESTABLISHING EXPOSURE LIMITS The method used to establish permitted daily exposures for residual solvents is presented in Appendix3.Summariesof the toxicity data that were used to establish limits are published in Pharmeuropa,Vol.9,No.1,Supplement April 1997.3.3.OPTIONS FOR DESCRIBING LIMITS OF CLASS2 SOLVENTSTwo options are available when setting limits for Class2 solvents.Option1:The concentration limits in ppm stated in Table2 can be used.They were calculated using equation(1)below by assuming a product mass of10g administered daily.(1)Here,PDE is given in terms of mg/day and dose is givenin g/day.These limits are considered acceptable for all substances, excipients,or products.Therefore this option may be applied if the daily dose is not known or fixed.If all excipients and active substances in a formulation meet the limits givenin Option1,then these components may be used in any proportion.No further calculation is necessary provided the daily dose does not exceed10g.Products that are administered in doses greater than10g per day should be considered under Option2.Option2:It is not considered necessary for each component of the medicinal product to comply with the limits givenin Option1.The PDE in terms of mg/day as stated in Table2can be used with the known maximum daily dose and equation(1)above to determine the concentrationof residual solvent allowed in a medicinal product.Such limits are considered acceptable provided that is has been demonstrated that the residual solvent has been reducedto the practical minimum.The limits should be realistic in relation to analytical precision,manufacturing capability, reasonable variation in the manufacturing process,and the limits should reflect contemporary manufacturing standards. Option2may be applied by adding the amounts of a residual solvent present in each of the components of the medicinal product.The sum of the amounts of solvent per day should be less than that given by the PDE.Consider an example of the use of Option l and Option2 applied to acetonitrile in a medicinal product.The permitted daily exposure to acetonitrile is4.1mg per day;thus,the Option1limit is410ppm.The maximum administered daily mass of a medicinal product is5.0g,and the medicinal product contains two excipients.The composition of the medicinal product and the calculated maximum content of residual acetonitrile are given in the following table. Component Amount informulationAcetonitrilecontentDailyexposure Active substance0.3g800ppm0.24mg Excipient10.9g400ppm0.36mg Excipient2 3.8g800ppm 3.04mg Medicinal product 5.0g728ppm 3.64mg Excipient l meets the Option l limit,but the drug substance, excipient2,and medicinal product do not meet the Option l limit.Nevertheless,the product meets the Option2limit of 4.l mg per day and thus conforms to the recommendations in this guideline.Consider another example using acetonitrile as residual solvent.The maximum administered daily mass of a medicinal product is5.0g,and the medicinal product contains two excipients.The composition of the medicinal product and the calculated maximum content of residual acetonitrile is given in the following table.Component Amount informulation AcetonitrilecontentDailyexposureActive substance0.3g800ppm0.24mg Excipient10.9g2000ppm 1.80mg Excipient2 3.8g800ppm 3.04mg Medicinal product 5.0g1016ppm 5.08mgIn this example,the product meets neither the Option1 nor the Option2limit according to this summation.The manufacturer could test the medicinal product to determine if the formulation process reduced the level of acetonitrile.If the level of acetonitrile was not reduced during formulation to the allowed limit,then the manufacturer of the medicinal product should take other steps to reduce the amount of acetonitrile in the medicinal product.If all of these steps fail to reduce the level of residual solvent,in exceptional cases the manufacturer could provide a summary of efforts made to reduce the solvent level to meet the guideline value, and provide a risk-benefit analysis to support allowing the product to be utilised containing residual solvent at a higher level.3.4.ANALYTICAL PROCEDURESResidual solvents are typically determined using chromatographic techniques such as gas chromatography. Any harmonised procedures for determining levels of residual solvents as described in the pharmacopoeias should be used,if feasible.Otherwise,manufacturers would be free to select the most appropriate validated analytical procedure for a particular application.If only Class3solvents are present,a non-specific method such as loss on drying may be used.Validation of methods for residual solvents should conform to ICH guidelines“Text on Validation of Analytical Procedures”and“Extension of the ICH Text on Validation of Analytical Procedures”.3.5.REPORTING LEVELS OF RESIDUAL SOLVENTS Manufacturers of pharmaceutical products need certain information about the content of residual solvents in excipients or active substances in order to meet the criteria of this guideline.The following statements are given as acceptable examples of the information that could be provided from a supplier of excipients or active substances to a pharmaceutical manufacturer.The supplier might choose one of the following as appropriate:—Only Class3solvents are likely to be present.Loss on drying is less than0.5per cent.—Only Class2solvents X,Y,...are likely to be present.All are below the Option1limit(Here the supplier would name the Class2solventsrepresented by X,Y,...)—Only Class2solvents X,Y,...and Class3solvents are likely to be present.Residual Class2solvents are below the Option1limit and residual Class3solvents are below0.5per cent.If Class1solvents are likely to be present,they should be identified and quantified.“Likely to be present”refers to the solvent used in the final manufacturing step and to solvents that are used in earlier manufacturing steps and not removed consistently by a validated process.If solvents of Class2or Class3are present at greater than their Option1limits or0.5per cent,respectively,they should be identified and quantified.4.LIMITS OF RESIDUAL SOLVENTS4.1.SOLVENTS TO BE AVOIDEDSolvents in Class1should not be employed in the manufacture of active substances,excipients,and medicinal products because of their unacceptable toxicity or their deleterious environmental effect.However,if their use is unavoidable in order to produce a medicinal product with a significant therapeutic advance,then their levels should be restricted as shown in Table1,unless otherwise justified. 1,1,1-Trichloroethane is included in Table1because it is an environmental hazard.The stated limit of1500ppm is based on a review of the safety data.Table1.–Class1solvents in pharmaceutical products(solvents that should be avoided)Solvent Concentration limit(ppm)Concern Benzene2CarcinogenCarbon tetrachloride4Toxic and environmental hazard 1,2-Dichloroethane5Toxic1,1-Dichloroethene8Toxic1,1,1-Trichloroethane1500Environmental hazard 4.2.SOLVENTS TO BE LIMITEDSolvents in Table2should be limited in pharmaceutical products because of their inherent toxicity.PDEs are given to the nearest0.1mg/day,and concentrations are given to the nearest10ppm.The stated values do not reflect the necessary analytical precision of determination.Precision should be determined as part of the validation of the method. Table2.–Class2solvents in pharmaceutical products Solvent PDE(mg/day)Concentration limit(ppm) Acetonitrile 4.1410 Chlorobenzene 3.6360 Chloroform0.660 Cyclohexane38.838801,2-Dichloroethene18.71870 Dichloromethane 6.06001,2-Dimethoxyethane 1.0100N,N-Dimethylacetamide10.91090N,N-Dimethylformamide8.88801,4-Dioxane 3.83802-Ethoxyethanol 1.6160 Ethyleneglycol 6.2620 Formamide 2.2220 Hexane 2.9290 Methanol30.030002-Methoxyethanol0.550 Methylbutylketone0.550 Methylcyclohexane11.81180N-Methylpyrrolidone 5.3530 Nitromethane0.550 Pyridine 2.0200Solvent(mg/day)Concentration limit(ppm)Sulfolane 1.6160 Tetrahydrofuran7.2720Tetralin 1.0100 Toluene8.98901,1,2-Trichloroethene0.880Xylene*21.72170*usually60per cent m-xylene,14per cent p-xylene,9per cent o-xylene with17per cent ethyl benzene4.3.SOLVENTS WITH LOW TOXIC POTENTIAL Solvents in Class3(shown in Table3)may be regardedas less toxic and of lower risk to human health.Class3 includes no solvent known as a human health hazard at levels normally accepted in pharmaceuticals.However,there are no long-term toxicity or carcinogenicity studies for many of the solvents in Class3.Available data indicate that they are less toxic in acute or short-term studies and negative in genotoxicity studies.It is considered that amounts of these residual solvents of50mg per day or less(correspondingto5000ppm or0.5per cent under Option l)would be acceptable without justification.Higher amounts may also be acceptable provided they are realistic in relation to manufacturing capability and good manufacturing practice. Table3.–Class3solvents which should be limited by GMP or other quality-based requirementsAcetic acid HeptaneAcetone Isobutyl acetateAnisole Isopropyl acetate1-Butanol Methyl acetate2-Butanol3-Methyl-1-butanolButyl acetate Methylethylketonetert-Butylmethyl ether MethylisobutylketoneCumene2-Methyl-l-propanolDimethyl sulphoxide PentaneEthanol1-PentanolEthyl acetate1-PropanolEthyl ether2-PropanolEthyl formate Propyl acetateFormic acid4.4.SOLVENTS FOR WHICH NO ADEQUATE TOXICOLOGICAL DATA WAS FOUNDThe following solvents(Table4)may also be of interest to manufacturers of excipients,active substances,or medicinal products.However,no adequate toxicological data on which to base a PDE was found.Manufacturers should supply justification for residual levels of these solvents in pharmaceutical products.Table4.–Solvents for which no adequate toxicologicaldata was found1,1-Diethoxypropane Methylisopropylketone1,1-Dimethoxymethane Methyltetrahydrofuran2,2-Dimethoxypropane Petroleum etherIsooctane Trichloroacetic acidIsopropyl ether Trifluoroacetic acidGLOSSARYGenotoxic carcinogens:Carcinogens which produce cancer by affecting genes or chromosomes.LOEL:Abbreviation for lowest-observed effect level.Lowest-observed effect level:The lowest dose of substance in a study or group of studies that produces biologically significant increases in frequency or severity of any effects in the exposed humans or animals.Modifying factor:A factor determined by professional judgement of a toxicologist and applied to bioassay data to relate that data safely to humans.Neurotoxicity:The ability of a substance to cause adverse effects on the nervous system.NOEL:Abbreviation for no-observed-effect level.No-observed-effect level:The highest dose of substanceat which there are no biologically significant increases in frequency or severity of any effects in the exposed humans or animals.PDE:Abbreviation for permitted daily exposure.Permitted daily exposure:The maximum acceptable intake per day of residual solvent in pharmaceutical products.Reversible toxicity:The occurrence of harmful effectsthat are caused by a substance and which disappear after exposure to the substance ends.Strongly suspected human carcinogen:A substance for which there is no epidemiological evidence of carcinogenesis but there are positive genotoxicity data and clear evidence of carcinogenesis in rodents.Teratogenicity:The occurrence of structural malformations in a developing foetus when a substance is administered during pregnancy.APPENDIX 1.LIST OF SOLVENTS INCLUDED IN THE GUIDELINESolvent Other Names Structure Class Acetic acid Ethanoic acid CH 3COOH Class 3Acetone 2-Propanone Propan-2-oneCH 3COCH 3Class 3Acetonitrile CH 3CNClass 2AnisoleMethoxybenzene Class 3BenzeneBenzol Class 11-Butanol n -Butyl alcohol Butan-1-ol CH 3[CH 2]3OH Class 32-Butanol sec -Butyl alcohol Butan-2-olCH 3CH 2CH(OH)CH 3Class 3Butyl acetate Acetic acid butyl ester CH 3COO[CH 2]3CH 3Class 3tert -Butylmethyl ether 2-Methoxy-2-methylpropane (CH 3)3COCH 3Class 3Carbon tetrachloride TetrachloromethaneCCl 4Class 1ChlorobenzeneClass 2Chloroform Trichloromethane CHCl 3Class 2CumeneIsopropylbenzene(1-Methylethyl)benzeneClass 3CyclohexaneHexamethylene Class 21,2-Dichloroethanesym -Dichloroethane Ethylene dichloride Ethylene chloride CH 2ClCH 2ClClass 11,1-Dichloroethene 1,1-Dichloroethylene Vinylidene chloride H 2C=CCl 2Class 11,2-Dichloroethene 1,2-Dichloroethylene Acetylene dichloride ClHC=CHCl Class 2Dichloromethane Methylene chlorideCH 2Cl 2Class 21,2-DimethoxyethaneEthyleneglycol dimethyl ether MonoglymeDimethyl cellosolve H 3COCH 2CH 2OCH 3Class 2N,N -Dimethylacetamide DMA CH 3CON(CH 3)2Class 2N,N -Dimethylformamide DMFHCON(CH 3)2Class 2Dimethyl sulphoxideMethylsulphinylmethane Methyl sulphoxide DMSO (CH 3)2SOClass 31,4-Dioxane p -Dioxane[1,4]DioxaneClass 2Ethanol Ethyl alcohol CH 3CH 2OH Class 32-Ethoxyethanol CellosolveCH 3CH 2OCH 2CH 2OH Class 2Ethyl acetateAcetic acid ethyl esterCH 3COOCH 2CH 3Class 3Ethyleneglycol 1,2-Dihydroxyethane 1,2-Ethanediol HOCH 2CH 2OH Class 2Ethyl etherDiethyl ether Ethoxyethane 1,1′-Oxybisethane CH 3CH 2OCH 2CH 3Class 3Ethyl formate Formic acid ethyl ester HCOOCH 2CH 3Class 3Formamide MethanamideHCONH 2Class 2Formic acid HCOOHClass 3Heptane n -Heptane CH 3[CH 2]5CH 3Class 3Hexane n -HexaneCH 3[CH 2]4CH 3Class 2Isobutyl acetate Acetic acid isobutyl ester CH 3COOCH 2CH(CH 3)2Class 3Isopropyl acetate Acetic acid isopropyl ester CH 3COOCH(CH 3)2Class 3Methanol Methyl alcohol CH 3OH Class 22-Methoxyethanol Methyl cellosolve CH 3OCH 2CH 2OH Class 2Methyl acetate Acetic acid methyl ester CH 3COOCH 3Class 33-Methyl-1-butanolIsoamyl alcohol Isopentyl alcohol 3-Methylbutan-1-ol (CH 3)2CHCH 2CH 2OHClass 3Methylbutylketone 2-Hexanone Hexan-2-one CH 3[CH 2]3COCH 3Class 2MethylcyclohexaneCyclohexylmethaneClass 2Methylethylketone2-Butanone MEKButan-2-one CH 3CH 2COCH 3Class 3Methylisobutylketone4-Methylpentan-2-one 4-Methyl-2-pentanone MIBKCH 3COCH 2CH(CH 3)2Class 32-Methyl-1-propanol Isobutyl alcohol 2-Methylpropan-1-ol (CH 3)2CHCH 2OHClass 3N -Methylpyrrolidone1-Methylpyrrolidin-2-one1-Methyl-2-pyrrolidinoneClass 2Nitromethane CH 3NO 2Class 2Pentane n -Pentane CH 3[CH 2]3CH 3Class 31-PentanolAmyl alcohol Pentan-1-ol Pentyl alcohol CH 3[CH 2]3CH 2OHClass 31-Propanol Propan-1-ol Propyl alcohol CH 3CH 2CH 2OH Class 32-Propanol Propan-2-olIsopropyl alcohol (CH 3)2CHOH Class 3Propyl acetate Acetic acid propyl esterCH 3COOCH 2CH 2CH 3Class 3PyridineClass 2Sulfonane Tetrahydrothiophene1,1-dioxide Class 2TetrahydrofuranTetramethylene oxideOxacyclopentaneClass 2Tetralin1,2,3,4-Tetrahydronaphthalene Class 2TolueneMethylbenzene Class 21,1,1-Trichloroethane Methylchloroform CH 3CCl 3Class 11,1,2-Trichloroethene Trichloroethene HClC=CCl 2Class 2Xylene*Dimethybenzene XylolClass 2*usually 60per cent m -xylene,14per cent p -xylene,9per cent o -xylene with 17per cent ethyl benzene.APPENDIX 2.ADDITIONAL BACKGROUNDA2.1.ENVIRONMENTAL REGULATION OF ORGANIC VOLATILE SOLVENTSSeveral of the residual solvents frequently used in the production of pharmaceuticals are listed as toxic chemicals in Environmental Health Criteria (EHC)monographs and the Integrated Risk Information System (IRIS).The objectives of such groups as the International Programme on Chemical Safety (IPCS),the United States Environmental Protection Agency (USEPA)and the United States Food and Drug Administration (USFDA)include the determination of acceptable exposure levels.The goal is protection of human health and maintenance of environmental integrity against the possible deleterious effects of chemicals resulting from long-term environmental exposure.The methods involved in the estimation of maximum safe exposure limits are usually based on long-term studies.When long-term study data are unavailable,shorter term study data can be used with modification of the approach such as use of larger safety factors.The approach described therein relates primarily to long-term or life-time exposure of the general population in the ambient environment,i.e.ambient air,food,drinking water and other media.A2.2.RESIDUAL SOLVENTS IN PHARMACEUTICALS Exposure limits in this guideline are established by referring to methodologies and toxicity data described in EHC and IRIS monographs.However,some specific assumptions about residual solvents to be used in the synthesis and formulation of pharmaceutical products should be taken into account in establishing exposure limits.They are:1)Patients (not the general population)use pharmaceuticalsto treat their diseases or for prophylaxis to prevent infection or disease.2)The assumption of life-time patient exposure is not necessary for most pharmaceutical products but may be appropriate as a working hypothesis to reduce risk to human health.3)Residual solvents are unavoidable components inpharmaceutical production and will often be a part ofmedicinal products.4)Residual solvents should not exceed recommended levels except in exceptional circumstances.5)Data from toxicological studies that are used to determine acceptable levels for residual solvents should have been generated using appropriate protocols such as those described for example,by OECD and the FDA Red Book.APPENDIX 3.METHODS FOR ESTABLISHING EXPOSURE LIMITSThe Gaylor-Kodell method of risk assessment (Gaylor,D.W.and Kodell,R.L.Linear Interpolation algorithm for low dose assessment of toxic substance.J.Environ.Pathology,4,305,1980)is appropriate for Class 1carcinogenic solvents.Only in cases where reliable carcinogenicity data are available should extrapolation by the use of mathematical models be applied to setting exposure limits.Exposure limits for Class 1solvents could be determined with the use of a large safety factor (i.e.,10000to 100000)with respect to the no-observed-effect level (NOEL).Detection and quantification of these solvents should be by state-of-the-art analytical techniques.Acceptable exposure levels in this guideline for Class 2solvents were established by calculation of PDE values according to the procedures for setting exposure limits in pharmaceuticals (Pharmacopeial Forum ,Nov-Dec 1989),and the method adopted by IPCS for Assessing Human Health Risk of Chemicals (Environmental Health Criteria 170,WHO,1994).These methods are similar to those used by the USEPA (IRIS)and the USFDA (Red Book )and others.The method is outlined here to give a better understanding of the origin of the PDE values.It is not necessary to perform these calculations in order to use the PDE valuestabulated in Section 4of this document.PDE is derived from the no-observed-effect level (NOEL),orthe lowest-observed effect level (LOEL),in the most relevant animal study as follows:The PDE is derived preferably from a NOEL.If no NOEL is obtained,the LOEL may be used.Modifying factors proposed here,for relating the data to humans,are the same kind of “uncertainty factors”used in Environmental HealthCriteria (Environmental Health Criteria 170,World Health Organisation,Geneva,1994),and “modifying factors”or。
EU GMP 附录3:放射性药品的生产-中文

原则放射性药品的生产应当符合《药品生产质量管理规范》的第1 部分和第2 部分的原则,本附录涉及具体的实践,这可能是特定的放射性药品。
注i:在放射药品(医院或某些药店),使用上市许可或国家许可的发生器和工具制备放射性药品的情况,不包括在本指南内,除非由国家要求。
注ii:根据辐射防护规定,应确保任何医疗照射都在医生的临床责任之下。
在诊断和治疗核医学实践中应该有医学物理专家。
注iii:本附录也适用于在临床试验中所用的放射性药品。
注iv:放射性药品的运输是由国际原子能协会(IAEA)和辐射防护的要求规范。
注v :人们认识到,除本附录中描述的方法之外,有可接受的方法,能够实现质量保证的原则。
其他方法应进行验证,并提供至少具有等同于本附录所规定的质量保证水平。
前言1、放射性药品的生产和处理潜在的危险。
风险的程度取决于辐射的类型,辐射的能量和放射性同位素的半衰期。
必须特别注意防止交叉污染,对放射性核素污染物的残留以及废物处理。
2、由于其放射性药品的保质期较短,有些放射性药品可能在完成全部的质量控制测试前就已经被放行。
在这种情况下,对整个放行过程的准确和详细的描述,包括所涉及的人员的责任和质量保证系统的有效性的持续评估是必不可少的。
3、本指南适用于工业生产,核中心/研究所和PET中心进行的以下各类产品的生产和质量控制采用的生产过程:∙放射性药品∙正电子发射放射性药物(PET)∙放射性药品生产的放射性前体∙放射性核素发生器生产类型非GMP *GMP第2 部分与第1 部分(新增),包括相关附录放射性药品正电子发射放射性药物放射性前体反应器/回旋加速器生产化学合成纯化步骤加工,制备与分装无菌或最终灭菌放射性核素发生器反应器/回旋加速器生产加工*从回旋加速器到合成装置的目标与传送系统可以被认为是活性物质生产的第一步。
4、最后的放射性药品生产企业应描述和证明原料药和成品药品的生产步骤,以及《药品生产质量管理规范》(第1部分或第2部分)在具体的加工/生产步骤中的应用。
欧洲GMP正文(中文稿)

本资料由药智网收集整理欧洲药品生产质量管理规范欧洲药品生产质量管理规范目录第一章质量管理................................................................................................4原则4质量保证 (4)药品生产质量管理规范(GMP)...........................................................................4质量控制(QC).. (5)产品质量回顾..................................................................................................6第二章人员......................................................................................................8原则8通则8关键人员 (8)培训9人员卫生.......................................................................................................10第三章厂房和设备.. (11)原则11厂房11设备13第四章文件....................................................................................................14原则14通则14文件要求 (15)第五章生产......................................................................................................20原则20通则20生产过程中对交叉污染的预防 (21)验证21原料21生产操作:中间产品和待包装产品 (22)包装材料 (22)包装操作 (22)成品23不合格、回收料和退货物料 (24)第六章质量控制.............................................................................................25原则25通则25质量控制实验室规范 (25)销售产品的稳定性考察..................................................................................27第七章委托生产与委托检验 (29)原则29通则29委托方 (29)受托方 (29)目录本资料由药智网收集整理本资料由药智网收集整理 欧洲药品生产质量管理规范合同30第八章投诉与召回 (31)原则31投诉31召回31第九章自查 (33)原则33欧洲药品生产质量管理规范第一章质量管理原则生产许可证持有厂家只能生产医药产品,以确保药品符合其预期的使用目的,符合销售许可证的要求,并不因药品安全性、质量或药效方面的问题而给患者带来风险。
USP 35 1113中文版

自己翻译,非专业,有错误改正,勿拍砖〈1113〉细菌鉴定35(1)版167页,该章包括标题正在修订。
本章阐述微生物鉴定方法,方法的选用及验证。
〈1113〉微生物特性确定、鉴定及菌株分型前言在检测药品原辅料,生产用水,生产环境,中间体以及终端产品市的微生物时,需要对其特性进行确定。
这些包括适当的鉴定及菌株分型(标注,一些项目在章节末端提及)。
日常的微生物特性确定包括菌落形态、细胞形态(杆菌、球菌、细胞集合、产胞方式),革兰氏染色或者其他染色方法,和特征生化反应(氧化酶,过氧化氢酶,凝固酶反应)。
在非无菌药品生产过程和一些无菌制剂的生产环境中,微生物鉴定到这个水平足以达到风险评估的目的。
在某些情况下,可以用一些更确定的方法鉴定微生物种属。
除此之外,有些方法进行菌株鉴定对于确定微生物来源的研究很有用。
当微生物在以异常高频率或者数量超趋势出现在特殊种类的产品中时,微生物鉴定试验尤为频繁。
另外,微生物鉴定可用于无菌生产,当无菌实验中出现的阳性和从失败的无菌工艺模拟收回的污染样品如培养基灌装试验,都需要进行鉴定。
微生物鉴定系统是基于不同的分析方法,其限制可能源于固定的方法或者/和数据库的限制。
微生物鉴定是通过一个既定的标准菌株根据匹配特性(基因型和/或表型)确定其种属。
如果某种微生物在数据库中不存在,无法鉴定,制造商应当回顾所建立鉴定系统的数据库的宽度及适用性。
使用者应当考虑哪种系统是最符合实际使用要求的。
考虑到这些限制和鉴定所要达到的等级(属,种,株),使用者还要选择合适的方法用于日常药品的微生物鉴定。
在USP通用测试章特别提到的非无菌产品的微生物检验:指定微生物检查62,这一章表示用于鉴定实验确认的微生物需要通过选择培养基或者特征培养基培养并具有确定的形态特征。
同样,在USP通用测试章无菌试验71中规定:在从试验中分离出的微生物确定之后,此种(或这些种)微生物的生长可以毫不含糊地归咎于与物料和/或者进行无菌试验过程中所使用的方法,该实验无效。
欧洲药典适用性证书化学纯度和微生物质量文件的内容

欧洲药典适用性证书化学纯度和微生物质量文件的内容申请表—新COS证书请求连同相关附件都要完成(可从EDQM网站下载)。
文件应该按照CTD格式提供除非有正当理由。
参考插入指南以协助申请人。
它仍然是声请人的责任以保证所有立法和指南,修订或维持,适当时在申请中考虑。
参照的指南在每一部分提供这部分所期望的有用的信息。
然而,这个列别表不应该被当做综合万象。
药用物质(2034),发酵产品和TSE风险产品通则的要求应该被重视,适当时。
申请连同1或2个一次检测足够量(通常10g)的有代表性的商业批的样品提供给EDQM 证书秘书处。
适用时。
要求修订专论是杂质样品也要求提供,和/或如果用附加的方法控制相关物质附加到证书中,EDQM实验室可能检查。
1.4 专家信息专家的简历要显示出他在这个领域的经验。
2.3QOS按照QOS格式给一个文件内容的摘要。
期望QOS应该讨论欧洲药典专论控制活性物质质量的能力。
特别是申报的潜在杂质,或则选择方法的必然性。
尤其注意可能的杂质检测被忽略是要给出合理的理由,例如由于任意批号的实际值都是未检出或者使用特殊的方法生产不可能存在。
这个报告应该签名和日期。
3.2.S.1综合信息物质商业化历史:在欧洲被许可的药品许可历史摘要包含用指确定方法生产的物质,国家名称、产品名称和商业化日期。
要清晰说明产品是否用作兽用。
关于物质递交国家许可当局的活性主文件也要提供。
管理形式的相关部分应该给出这些信息。
声明:生产者要提供生产者按照递交文件和GMP指南履行的签字的声明,最好使用管理的形式。
3.2.S1.1命名欧洲药典专论名称,INN名,其他名称以及文件中使用的任何实验室代号。
3.2.S1.2一般属性对于物理特性,如果不止一个级别被生产,生产者不妨递交一个或更多的文件取决于是否单独申请,例如是:压缩的,特殊粒度,特定的多晶构形(专论不限制单晶的)。
在任何情况下不同质量都要符合专论指定的一般水平。
如果一个文件中有多于一个的级别(如仅要求一个证书),批分析结果,涉及的杂质档案,应该包括所有级别。
欧洲药典-凡例(中英文对照)

欧洲药典-凡例1.1. GENERAL STATEMENTSThe General Notices apply to all monographs and other texts of the European Pharmacopoeia.总论的内容适用于各论和欧洲药典中的其它章节。
The official texts of the European Pharmacopoeia are published in English and French. Translations in other languages may be prepared by the signatory States of the European Pharmacopoeia Convention. In case of doubt or dispute, the English and French versions are alone authoritative.欧洲药典以英语和法语形式发行,欧洲药典委员会的签署国可将药典内容译成其它语言,但若发生争议,应以英语和法语版为权威。
In the texts of the European Pharmacopoeia, the word "Pharmacopoeia" without qualification means the European Pharmacopoeia. The official abbreviation Ph. Eur. may be used to indicate the European Pharmacopoeia.在欧洲药典中,如无特殊规定,“药典”是指欧洲药典,缩写Ph. Eur.也指欧洲药典。
The use of the title or the subtitle of a monograph implies that the article complies with the requirements of the relevant monograph. Such references to monographs in the texts of the Pharmacopoeia are shown using the monograph title and reference number in italics.文章中如果引用了各论中的标题和副标题意味着文章内容符合相关各论的要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3.1.3. 聚烯烃定义聚烯烃是通过乙烯或丙烯的聚合而成,或是通过这些不超过25%的高同系物的物质或羧酸或酯的共聚作用获得。
某些材料可能是聚烯烃的混合物。
成品添加一定数量的添加剂到聚合物中是为了优化它们的化学性质,物理性质和机械性能,为了使它们适用于预定用途。
所有的这些添加剂都是选自附件列表,并指出了每一种产品中的最大允许含量。
产品中最多包含有三种抗氧化剂,一种或几种润滑剂或抗粘连剂以及当材料必须提供光照保护时,还要添加二氧化钛作为遮光剂。
– 二叔丁基对甲酚(增塑剂07):限量:0.125%–四钛季戊四醇松香酸酯[3-(3,5-二叔丁基-4-羟苯基)丙酸酯](增塑剂09):限量:0.3%–1,3,5-三羟甲基氨基甲烷(3,5-二叔丁基-4-邻羟苄基)- 三嗪-2,4,6(1H,3H,5H)-三酮, (增塑剂 13): 限量: 0.3%– 二乙烯[3,3-二[3-(1,1-dimethylethyl)-4-羟苯基]丁酸甲酯] (增塑剂08):限量:0.3%– 二(十八烷基)二硫化物(增塑剂15)限量:0.3%4,4′,4″-(2,4,6-三甲基苯-1,3,5-triyltrismethylene)–三羟甲基氨基甲烷[2,6-二(1,1-dimethylethyl)苯酚](增塑剂10)限量:0.3%2,2′-二(octadecyloxy)-5,5′-spirobi[1,3,2-dioxaphosphinane](增塑剂 14): 限量:0.3 %;– didodecyl 3,3′-硫代二丙酸(增塑剂16): 限量: 0.3 %;– dioctadecyl3,3′-硫代二丙酸(增塑剂 17): 限量:0.3 %;– 三羟甲基氨基甲烷[2,4-二(1,1-dimethylethyl)苯基] 亚磷酸盐 (增塑剂 12): 限量:0.3 %;– 增塑剂 18: 限量: 0.1%;–琥珀酸二甲酯和 (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)乙醇的共聚物 (增塑剂 22): 限量:0.3%上面列出的抗氧化添加剂总含量不超过0.3%。
–铝碳酸镁:限量:0.5%;–烷基酰胺:限量:0.5%;–烯烃酰胺:限量:0.5%;–硅铝酸钠:限量:0.5%;– 二氧化硅: 限量:0.5%;–苯甲酸钠: 限量:0.5%;– 脂肪酸酯及其盐类: 限量:0.5%;–磷酸三钠: 限量:0.5%;–液态石蜡: 限量:0.5%;– 氧化锌: 限量:0.5%;– 滑石: 限量:0.5%;–氧化镁: 限量:0.5%;– 硬脂酸钠或硬脂酸锌或两者的混合物:限量:0.5%;–二氧化钛: 限量:4%;材料提供者须能证明样品组成的数量和质量都符合要求。
特性外观:粉末,珠子,颗粒或在转换后,不同厚度的片材或容器。
溶解性:几乎不溶于水,溶于热的芳香烃,几乎不溶于乙醇,乙烷和甲醇。
它们的软化温度在 65 °C和 165 °C之间,它们燃烧时都会有蓝色火焰。
鉴定:如果有必要,将实验用的材料样品裁剪成边长最大尺寸不超过1CM的片状。
A.红外吸收分光光度法(2.2.24)准备:在0.25g样品中加入10ml的甲苯R并在回流冷凝器下加热约15分钟;放置几滴从氯化钠滑落获得的溶剂,然后在80°C的烘箱上将溶剂烘干。
最大吸收值:2920 cm− 1, 2850 cm− 1, 1475 cm− 1,1465 cm− 1, 1380 cm− 1, 1170 cm−1, 735 cm− 1 和 720 cm− 1.所得出的光谱和材料选出的标准样品的光谱是一样的。
如果材料以表格的形式进行检查,鉴定应该直接决定裁剪成合适大小的片状。
B.它符合相应的现场添加剂的补充测试。
C.在一个铂钳锅内,用1g硫酸氢钾R混合20mg,然后加热直到完全熔化。
允许对其进行冷却并加入20ml的稀硫酸。
轻轻地进行加热。
过滤掉产生的溶剂。
在滤液中添加1ml的磷酸和1ml的强过氧化氢溶液R。
如果该物质是不透明的钛白色,那么将会变成橙黄色。
试验:如果有必要,将实验用的材料样品裁剪成边长最大尺寸不超过1CM的片状。
S1试液.使用S1试液需要4小时的准备。
称取25g材料,置于带磨砂口的硼硅玻璃烧瓶中,加入500ml的注射用水R,然后加热回流约5个小时。
冷却并转移到烧杯中。
保留部分提取液用于测试S1试液的外观,然后用烧结玻璃过滤器过滤剩余溶液。
S2试液.精密称取2.0g材料,置于带磨砂口的圆底硼硅玻璃烧瓶中。
加入80ml的甲苯R然后加热搅拌回流约90分钟。
冷却至60°C然后加入120ml的甲醇R并持续搅拌。
用烧结玻璃过滤器将溶液进行过滤。
取25ml由40ml的甲苯R和60ml的甲醇R混合的溶液来冲洗烧瓶和过滤器,合并洗液和滤液,并用相同的混合溶剂稀释至250ml。
同法制备空白溶液。
S3试液.称取100g的材料,置于带磨砂口的圆底硼硅玻璃烧瓶中。
加入250ml的0.1M盐酸,加热搅拌回流1个小时。
冷却并收集溶液。
S1试液的外观。
清晰无色。
酸度或碱度。
量取100ml的S1试液,加入0.15ml的BRP指示剂R。
加入约1.5ml的0.01M氢氧化钠溶液,直到溶液颜色蓝色。
量取100ml的S1试液,加入0.2ml的甲基橙溶液R,加入约1ml的0.01M盐酸,直到溶液变为黄色再变为橙色。
吸光率:S1试液在波长范围220nm-340nm之间的吸光率最大值不超过0.2。
还原性物质。
量取20ml的S2试液,加入1ml的稀盐酸R和20ml的0.002M高锰酸钾。
加热回流3分钟然后直接冷却。
加入1g碘化钾R然后直接用0.01M硫代硫酸钠进行滴定测量法测量,用0.25ml的淀粉溶液R作为指示液。
同法空白滴定。
滴定体积的区别小于3.0ml。
可溶于己烷的物质。
称取10g材料,置于250ml带磨砂口的的圆锥形硼硅酸盐玻璃烧瓶中。
加入100ml的己烷然后加热搅拌回流4个小时。
在冰水中冷却然后用烧结玻璃过滤器迅速过滤(过滤时间必须少于5分钟;如有必要可将溶液加压来加速过滤)并让溶液保持在0°C左右。
量取20ml的滤液,放在恒重过的硼硅酸盐玻璃皿上,在水浴中蒸发。
将滤渣放在100-105°C 的烘箱上进行干燥一小时。
所得滤渣不得大于标准样品的10%,而且不超过5%。
可提取出的铝:限量:1ppm。
电感耦合等离子原子发射光谱法。
测试溶液:S3试液。
标准溶液。
用铝标准溶液(200ppm Al)R加0.1M的盐酸稀释成标准溶液。
波长:铝发射的波长为396.15nm,光谱采用396.25nm。
确定所用的盐酸中没有铝。
可提取出的钛:限量:1ppm。
电感耦合等离子原子发射光谱法。
测试溶液:S3试液。
标准溶液。
用钛标准溶液(100ppm Ti)R加0.1M的盐酸稀释成标准溶液。
波长:钛发射的波长为336.12nm,光谱采用336.12nm。
确定所用的盐酸中没有钛。
可提取出的锌:限量:1ppm。
原子吸收光谱测定法。
测试溶液:S3试液。
标准溶液。
用锌标准溶液(10ppm Zn)R加0.1M的盐酸稀释成标准溶液。
来源:锌空心阴极电子管。
波长:213.9nm雾化装置:空气乙炔火焰确定所用的盐酸中没有锌。
可提取出的重金属:限量:2.5ppm将50ml的溶液S3在水浴中蒸发至5ml然后加水R稀释成20.0ml。
取12ml溶液按照测试A进行。
用2.5ml的铅标准溶液(10ppm Pb)准备标准溶液。
硫酸灰分:限量:1.0%,称取5.0g试验。
此限制不适用于被二氧化钛处理过的材料。
补充测试只有在材料定期检查时,这些测试可全部或部分进行实施。
酚类抗氧化剂。
液相色谱法。
溶剂混合:氰化甲烷R,四氢呋喃R(50:50 V/V)S21试液。
精密量取50ml的S2试液,在45°C真空蒸干后,使残渣溶于5.0ml的混合溶剂中。
用S2试液的空白溶液同法制备空白样品。
S22试液。
精密量取50ml的S2试液,在45°C真空蒸干后,将残渣溶于5.0ml的二氯甲烷中。
用S2试液的空白溶液同法制备空白样品。
S23试液.精密量取50ml的S2试液,在45°C真空蒸干后,将残渣溶于5.0ml的相同量的氰化甲烷R和10g/l的四氢呋喃R里的叔丁基化过氧氢溶液的混合剂。
封上烧瓶静置1小时。
准备一个空白溶液。
仅在有必要对规定的所检查的物质组成进行酚类抗氧化剂分析时准备以下标准溶液。
标准溶液(a)。
溶解25.0mg的二叔丁基对甲酚CRS(增塑剂07)和60.0mg的增塑剂08CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的该溶液稀释成50.0ml。
标准溶液(b)。
溶解60.0mg的增塑剂09CRS和60.0mg的增塑剂10CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释成50.0ml。
标准溶液(c)。
溶解60.0mg的增塑剂11CRS和60.0mg的增塑剂12CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶液(d)。
溶解25.0mg的增塑剂07CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释到50.0ml。
标准溶液(e)。
溶解60.0mg的增塑剂08CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释到50.0ml。
标准溶液(f)。
溶解60.0mg的增塑剂13CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
标准溶液(g)。
溶解60.0mg的增塑剂09CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
标准溶液(h)。
溶解60.0mg的增塑剂10CRS于10.0ml的混合溶剂中。
用混合溶剂将2.0ml的此溶剂稀释至50.0ml。
标准溶液(i)。
溶解60.0mg的增塑剂11CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶剂(j)。
溶解60.0mg的增塑剂12CRS于10.0ml的二氯甲烷R中。
用二氯甲烷R将2.0ml的此溶液稀释成50.0ml。
标准溶液(k)。
溶解20.0mg的增塑剂18CRS于10.0ml的同等量的氰化甲烷R和10g/l的四氢呋喃R里的叔丁基化过氧氢溶液的混合剂。
允许放在密封容器内静置一个小时。
用混合溶剂将2.0ml的此溶液稀释至50.0ml。
A.如果该物质检查出含有增塑剂07和/或增塑剂08,请实施以下测试:-尺寸:l = 0.25 m, O = 4.6mm;-固定相:十八烷基甲硅烷基硅胶的气相色谱R (5 μm).移动相:水R,氰化甲烷R流量:2 mL/min检测方法:分光光度计,280nm注入:20μL的 S21试液,相应的空白溶液,标准溶液(a),和要么标准溶液(d)要么标准溶液(e)要么标准溶液(d)和(e)。