氯吡格雷与普拉格雷的代谢机制
氯吡格雷片机制

氯吡格雷片机制氯吡格雷片机制是一种药物作用机制,主要涉及抑制血小板的聚集,从而预防和治疗因血小板高聚集状态引起的心、脑及其他动脉循环障碍疾病。
以下是关于氯吡格雷片机制的详细解释。
首先,氯吡格雷是一种血小板抑制剂,其主要作用机制是通过选择性地抑制ADP(二磷酸腺苷)与血小板受体的结合,从而抑制血小板的聚集。
ADP是一种在血小板活化过程中起重要作用的物质,它能够促进血小板的聚集和活化。
氯吡格雷通过阻断ADP与血小板受体的结合,能够有效地抑制血小板的聚集,从而减少血栓的形成。
其次,氯吡格雷的作用不仅仅局限于抑制ADP诱导的血小板聚集。
它还能够通过阻断其他激动剂诱导的血小板聚集,进一步减少血栓的形成。
这种广泛的抑制作用使得氯吡格雷在预防和治疗动脉粥样硬化血栓形成方面具有显著的效果。
在临床上,氯吡格雷主要用于预防和治疗因血小板高聚集状态引起的心、脑及其他动脉循环障碍疾病,如心肌梗死、缺血性脑卒中、外周动脉疾病等。
通过抑制血小板的聚集,氯吡格雷能够有效地减少这些疾病的发生和发展,提高患者的生活质量。
然而,需要注意的是,氯吡格雷在使用过程中可能会引起一些不良反应,如出血等。
因此,在使用氯吡格雷时,需要根据患者的具体情况和疾病类型进行个体化的剂量调整,并密切监测可能出现的不良反应。
此外,氯吡格雷的吸收和代谢主要发生在肝脏,因此肝功能异常的患者在使用氯吡格雷时需要注意调整剂量或避免使用。
同时,氯吡格雷与其他药物的相互作用也需要关注,以避免可能的药物相互作用导致的不良反应。
总之,氯吡格雷片机制是一种通过抑制血小板聚集来预防和治疗动脉粥样硬化血栓形成的药物作用机制。
它在临床上具有广泛的应用价值,但需要注意可能的不良反应和药物相互作用。
通过合理的使用和管理,氯吡格雷能够为患者带来更好的治疗效果和生活质量。
常用抗血小板药物的作用机制,这篇讲清楚了

常用抗血小板药物的作用机制,这篇讲清楚了*仅供医学专业人士阅读参考干货来了!动脉粥样硬化血栓形成是影响心、脑血管和外周动脉的全身系统性疾病,而其中血小板的活化和聚集在动脉粥样硬化血栓形成和发展中起着重要作用。
那么血小板在血栓形成过程中都发生了哪些变化呢?血小板的变化在血管损伤部位,血小板暴露于细胞外基质(ECM)的成分中。
血小板表面的糖蛋白(GP)受体与这些成分相互作用,特别是胶原蛋白和血管性血友病因子(VWF),导致血小板附着于损伤的血管内皮。
激活的血小板开始发生形状变化,导致内源性二磷酸腺苷(ADP)和血栓素A2(TXA2)的释放,并刺激血小板和其他细胞表面凝血酶的形成。
这些刺激进一步作用于周围的血小板,导致更多的血小板活化、聚集。
同时血小板表面的整合蛋白GP Ⅱb/Ⅲa发生构象变化,活化的GP Ⅱb/Ⅲa结合粘合蛋白,特别是纤维蛋白原和VWF;这一过程会导致血小板聚集。
血小板聚集物与纤维蛋白和凝血酶的相互作用最终导致血栓形成(见下图)。
复杂的细胞内信号传导过程的激活会导致各种刺激因子的产生和释放,包括TXA2、凝血酶和ADP,它们通过与各自的G蛋白耦合受体结合来发挥作用(见下图)。
抗血小板药物的分类根据药物作用于血小板活化聚集过程中的不同环节,常用抗血小板药物可以分为以下几类。
1.TXA2抑制剂阿司匹林是临床上广泛应用的血栓素抑制剂,是目前各类指南推荐的抗血小板治疗的基本药物。
阿司匹林通过对环氧酶(COX-1)的作用直接抑制TXA2合成,抑制血小板黏附聚集活性。
阿司匹林其它作用包括介导血小板抑制的嗜中性一氧化氮/环磷酸鸟苷以及参与各种凝血级联反应和纤溶过程。
2.ADP P2Y12受体拮抗剂ADP存在于血小板内的高密度颗粒中,与止血及血栓形成有关。
血小板ADP受体调控ADP浓度,人类血小板有3种不同ADP受体:P2Y1、P2Y12和P2X1受体。
其中P2Y12受体在血小板活化中最重要。
P2Y12受体拮抗剂通过抑制P2Y12受体,干扰ADP介导的血小板活化。
氯吡格雷

氯吡格雷(Clopidogrel),属于噻吩吡啶类抗血小板药物,第二代ADP受体拮抗剂。
氯吡格雷为无活性的药物前体,需经肝细胞内细胞色素P450酶系活化,其中约85%被酯化为无活性的代谢产物经肠道代谢,仅有约15%氯吡格雷被活化生成具有活性的代谢产物发挥其抗血小板药理作用。
主要机制:为选择性的、不可逆的抑制二磷酸腺苷(ADP)与血小板受体P2Y12的结合及继发ADP介导的糖蛋白GPIIIb/IIIa复合物的活化从而抑制血小板聚集。
火化后的氯吡格雷主要是与血小板P2Y12受体结合,阻断其与ADP结合位点,从而持久的抑制继发的腺苷酸环化酶的激活,抑制血小板的活性。
氯吡格雷反应多样性的定义:氯吡格雷在临床上作为抗血小板制剂其疗效使大多数患者明显受益,然而,仍有一部分患者不可避免的出现并发症,研究发现,不同患者对氯吡格雷的反应呈现明显的个体差异,这种对氯吡格雷呈现低应答(Low responder)或无应答(Clopidogrel nonresponse)的现象称之为氯吡格雷抵抗(Clopidogrel resistance,CR)。
目前学者们将临床上患者对氯吡格雷呈现不同应答状态的现象称之为氯吡格雷反应多样性(Clopidogrel Response Diversity,CRD)。
氯吡格雷反应多样性的定义:?CRD的相关因素:CYP2C19酶基因多态性、糖尿病、体重指数等因素有关。
脂溶性他汀类药物包括阿托伐他汀、辛伐他汀等和除泮托拉唑外的质子泵抑制剂可通过竞争性抑制影响氯吡格雷活化、增加氯吡格雷应答和无应答几率。
CYP2C19酶作为细胞色素P450药物代谢酶家族中的重要成员,在不同种族和人群中具有显著差异。
有研究指出,CYP2C19酶基因多态性与该酶活性密切相关。
不同研究对氯吡格雷翻一个多样性产生的机制看法不同,目前大多数学者认为导致氯吡格雷反应多样性的原因有以下几个方面:1、C YP2C19基因多态性与氯吡格雷反应多样性所谓基因多态性(polymorphism ),是指在一个生物群体编码的基因序列中,存在由一个或多个不连续等位基因(allele)发生突变。
P2Y12受体拮抗剂

P2Y12受体拮抗剂是一类作用于血小板P2Y12受体, 对二磷酸腺苷引起的血小板聚集起抑制作用的药物, 临床上主要用于预防和治疗心血管疾病的血栓事件.P2Y12受体拮抗剂与阿司匹林联用的双重抗血小板治疗方案, 是各种指南推荐、临床上经常使用的心血管病抗栓治疗方案.目前, 临床上可供选用的P2Y12受体拮抗剂有氯吡格雷、普拉格雷和替格瑞洛, 这些药物各自有哪些作用特点, 疗效和平安性又有何不同?氯吡格雷氯吡格雷是第二代P2Y12受体拮抗剂(注:第一代P2Y12受体拮抗剂为1979年上市的噻氯匹定, 其副作用较多, 在临床应用中逐渐被氯吡格雷所取代), 其在化学结构上属噻吩并吡啶类化合物, 是前体药物, 需要在肝脏中通过细胞色素 P450(CYP 450)酶代谢成为活性代谢物后, 才会不成逆地抑制P2Y12受体, 抑制血小板的聚集反应.因此, 氯吡格雷抗血小板活性的发挥存在延迟现象, 即起效时间比力长.氯吡格雷在临床应用中存在一些缺陷, 包括:消除半衰期较长, 个体不同较年夜, 部份患者服用该药后未发生抗血小板效果即“氯吡格雷抵当”, 与质子泵抑制剂(PPI)合用时可能会升高不良反应的发生率.目前, 已经明确CYP 2C19是与氯吡格雷抵当相关的代谢酶之一, 美国食品与药物管理局(FDA )已增加了氯吡格雷的黑框警告, 建议临床医生选用氯吡格雷前对患者进行基因检测, 对弱代谢患者应增加剂量.普拉格雷普拉格雷是第三代P2Y12受体拮抗剂, 同氯吡格雷相同, 此药也是噻吩并吡啶类化合物和前体药物, 需要在体内转化为其活性代谢产物后, 才会不成逆地抑制P2Y12受体从而发挥作用.研究显示, 与氯吡格雷的标准剂量或更年夜剂量相比, 普拉格雷对血小板的抑制作用更快、更继续、更强.同时, 由于普拉格雷的强抑制血小板聚集的作用, 也增加了其出血风险.替格瑞洛与噻吩并吡啶类药物(氯吡格雷和普拉格雷)的化学结构分类分歧, 替格瑞洛是一种环戊烷三唑并吡啶类的新型抗血小板药物, 故之前的中文名“替卡格雷”现已更换为“替格瑞洛”.与氯吡格雷和普拉格雷相比, 替格瑞洛是第一个可以口服却不需要生物转化就可直接发挥药效且可与P2Y12受体可逆结合的抗血小板药物.因其与受体的结合是可逆性的, 故一天须服药2 次.替格瑞洛具有快速起效、非前体药物可直接作用、不受个体基因不同影响等优势, 且与血小板可逆结合, 停药后血小板功能迅速恢复.与普拉格雷类似, 替格瑞洛对P2Y12受体的抑制效果也要强于氯吡格雷.替格瑞洛的疗效已经PLATO研究证实, 被国内外多部指南列于一线推荐位置, 欧洲指南更是在2011年将替格瑞洛的推荐级别列于氯吡格雷之前, 在替格瑞洛或普拉格雷不能使用的患者中才推荐使用氯吡格雷.上述三种药物主要药理学性质和药代动力学参数比力见表.小结氯吡格雷临床适应证较广泛, 禁忌证相对较少, 是目前临床上双重抗血小板治疗的首选药物, 但其疗效受到基因多态性的限制, 存在起效缓慢、可能与PPI 发生药物间相互作用等缺陷, 限制了其在临床中的应用.与氯吡格雷相比, 普拉格雷和替格瑞洛起效更迅速、个体不同小、对血小板抑制活性更高, 但出血风险亦增加, 也在一定水平上限制了其临床应用.分歧的P2Y12受体拮抗剂具有分歧的作用特点, 在疗效和平安性方面各具优势, 孰优孰劣不能一概而论.临床医生在选用P2Y12受体拮抗剂前, 应首先对患者进行评估, 可根据分歧的患者情况选择分歧的药物.临床医生在考虑药物作用特点和个体差此外同时, 亦应权衡药物的疗效和平安风险, 从而提高药物的临床治疗效果、减少不良事件的发生, 使患者最年夜水平受益.氯吡格雷的局限性氯吡格雷上市至今已有二十年时间, 期间各国学者进行了年夜量的临床研究, 极年夜的推动了ACS患者抗栓的进展, 增进了以氯吡格雷为基础的抗栓战略不竭优化及演变.尽管氯吡格雷非常优秀, 但自身仍存在一定局限性, 因此有学者检验考试研发能够克服氯吡格雷缺陷的新型药物.首先是氯吡格雷拮抗血小板的力度偏弱, 平均IPA在55%左右;其次氯吡格雷的前体药无效, 需经过肝脏代谢(两个步伐)后才华发挥作用, 这一过程中氯吡格雷代谢产物易与其他药物发生相互作用, 招致药效的个体间不同较年夜;再者是氯吡格雷起效较慢, 75 mg/天需要4~5天才华到达满意效果, 300 mg/天需4~6小时起效, 600 mg/天需要2小时, 但仍无法满足急诊PCI需求;最后是氯吡格雷作用不成逆, 患者停药后5~7天才华恢复血小板活性, 难以应对年夜出血事件或需紧急手术的情况.新型P2Y12受体抑制剂目前已经做到二期、三期临床或上市使用的四种新型药物包括普拉格雷、替格瑞洛、坎格瑞洛和依诺格雷.普拉格雷是最早呈现的有望取代氯吡格雷的药物, 与氯吡格雷相比, 普拉格雷有两个优点:一是起效时间稍快, 1个多小时即可发挥作用;虽然普拉格雷也需要经过肝脏代谢(一步代谢),但减少了个体不同;二是比氯吡格雷更有效的抑制血小板活性,对血小板活性的压制可达60%~70%.基础研究显示普拉格雷较氯吡格雷略有进步, TRITON-TIMI 38研究就验证了这一观点, 但其结果其实不理想, 因为普拉格雷在减少患者事件的同时也增加了年夜出血风险, 尤其是在≥75岁及有卒中史患者中, 该药在急诊PCI患者中有一定用武之地.替格瑞洛与氯吡格雷相比, 替格瑞洛是非前体药, 不需要经肝脏代谢, 进入人体后快速起效, 半小时对血小板的抑制作用即可达40%, 2小时可达80%, 而且个体不同比普拉格雷更小;第二是效果可逆, 当患者发生年夜出血或需紧急手术时可及时处置.PLATO研究在43个国家的862家中心纳入了18624例患者, 结果发现受试者终点事件下降, 且出血事件无显著性增加, 因此近期的年夜量指南都予以了优势推荐.坎格瑞洛坎格瑞洛是一种静脉用ADP受体拮抗剂, 注射5分钟内即可到达满意的血小板抑制效果, 防止了口服药经吸收、代谢才华发挥作用的过程, 可用于急诊患者的快速抗血小板治疗.早期研究包括CHAMPION-PCI与CHAMPION-PLATFORM, 各入选5000余例冠心病患者进行治疗, 结果其实不理想, 但给出了一定希望;另一项是CHAMPION PHOENIX研究, 其设计方案较以往有所改进, 更贴合临床实践, 结果坎格瑞洛组患者死亡、心梗及血栓事件明显减少, 可是部份年夜出血事件增加.针对坎格瑞洛的荟萃分析显示, 坎格瑞洛治疗对总体死亡事件无明显影响, 但支架内血栓明显减少, 心梗事件有下降趋势, 但无统计学不同;小出血及中等水平出血有所增加.因此该药的临床应用仍存在很多困惑, 静脉用药与口服用药的切换过程也存在问题.。
氯吡格雷是二磷酸腺苷 (ADP) 受体抑制剂
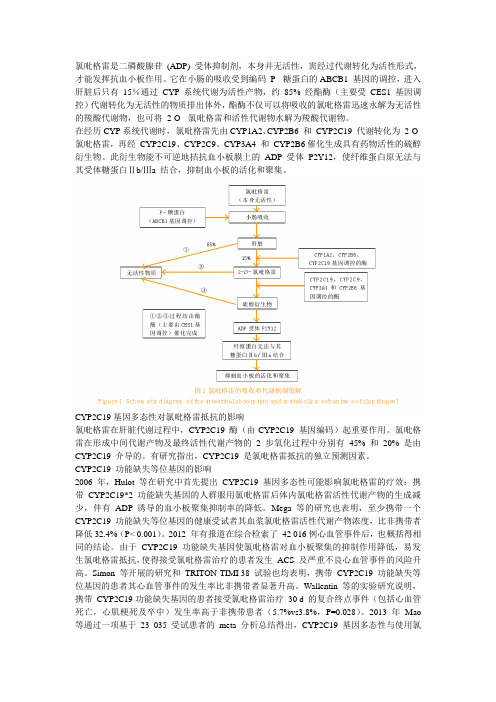
氯吡格雷是二磷酸腺苷(ADP) 受体抑制剂,本身并无活性,需经过代谢转化为活性形式,才能发挥抗血小板作用。
它在小肠的吸收受到编码P- 糖蛋白的ABCB1 基因的调控,进入肝脏后只有15%通过CYP系统代谢为活性产物,约85% 经酯酶(主要受CES1基因调控)代谢转化为无活性的物质排出体外,酯酶不仅可以将吸收的氯吡格雷迅速水解为无活性的羧酸代谢物,也可将2-O- 氯吡格雷和活性代谢物水解为羧酸代谢物。
在经历CYP系统代谢时,氯吡格雷先由CYP1A2、CYP2B6 和CYP2C19 代谢转化为2-O- 氯吡格雷,再经CYP2C19、CYP2C9、CYP3A4 和CYP2B6催化生成具有药物活性的硫醇衍生物。
此衍生物能不可逆地拮抗血小板膜上的ADP 受体P2Y12,使纤维蛋白原无法与其受体糖蛋白Ⅱb/Ⅲa 结合,抑制血小板的活化和聚集。
CYP2C19基因多态性对氯吡格雷抵抗的影响氯吡格雷在肝脏代谢过程中,CYP2C19 酶(由CYP2C19 基因编码)起重要作用。
氯吡格雷在形成中间代谢产物及最终活性代谢产物的 2 步氧化过程中分别有45% 和20% 是由CYP2C19 介导的。
有研究指出,CYP2C19 是氯吡格雷抵抗的独立预测因素。
CYP2C19 功能缺失等位基因的影响2006 年,Hulot 等在研究中首先提出CYP2C19基因多态性可能影响氯吡格雷的疗效:携带CYP2C19*2 功能缺失基因的人群服用氯吡格雷后体内氯吡格雷活性代谢产物的生成减少,伴有ADP 诱导的血小板聚集抑制率的降低。
Mega 等的研究也表明,至少携带一个CYP2C19 功能缺失等位基因的健康受试者其血浆氯吡格雷活性代谢产物浓度,比非携带者降低32.4%(P< 0.001)。
2012 年有报道在综合检索了42 016例心血管事件后,也概括得相同的结论。
由于CYP2C19 功能缺失基因使氯吡格雷对血小板聚集的抑制作用降低,易发生氯吡格雷抵抗,使得接受氯吡格雷治疗的患者发生ACS 及严重不良心血管事件的风险升高。
氯吡格雷机制

氯吡格雷机制氯吡格雷(Clopidogrel)是一种选择性、不可逆性的ADP受体拮抗剂,是一种抑制血小板聚集的抗血小板药物。
它通过阻断ADP受体P2Y12的功能,抑制血小板激活和聚集,从而预防血管内血小板凝块的形成,降低了心血管疾病的风险。
氯吡格雷机制主要是通过与ADP受体P2Y12结合,阻断ADP 对血小板的激活作用。
血小板是血液中的细胞片段,主要功能是在血管受损时形成血栓,帮助止血。
然而,当血小板过度激活时,会导致血小板聚集,形成血栓,堵塞血管导致心脑血管疾病。
ADP是一种在血小板活化和聚集过程中发挥重要作用的物质。
当血管受损时,血小板会释放ADP,并结合于ADP受体P2Y12上,从而触发一系列化学反应,使更多的血小板活化和聚集,形成血栓。
氯吡格雷作为一种抗血小板药物,可以与ADP受体P2Y12结合,阻断ADP对P2Y12的结合,从而抑制血小板的活化和聚集。
具体来说,氯吡格雷在肝脏内经细胞酯化酶产生活性代谢物(氯吡格雷酯化物),该代谢物通过血液循环直接到达血小板,与P2Y12结合,阻断ADP对P2Y12的结合位点,从而抑制血小板的激活。
ADP受体P2Y12的激活是血小板凝集的关键步骤,所以氯吡格雷的抑制作用可以减少血小板的聚集,从而预防血栓的形成。
一旦血管受损,血小板受到刺激,释放出ADP,与P2Y12结合,导致血小板聚集,形成血栓。
而氯吡格雷的作用就是阻断ADP与P2Y12的结合,从而防止血小板聚集和血栓的形成。
氯吡格雷还可以通过其他机制发挥作用。
例如,氯吡格雷与血小板膜上的其他受体结合,如糖蛋白IIb/IIIa受体,从而阻止血小板的黏附和聚集。
同时,氯吡格雷还可以降低细胞内钙离子浓度,减少血小板的收缩能力,进一步抑制血小板的活化和聚集。
总之,氯吡格雷机制主要是通过与ADP受体P2Y12结合,阻断ADP对血小板的激活作用,从而抑制血小板的聚集和血栓的形成。
此外,氯吡格雷还可以通过其他机制发挥作用,如阻止血小板黏附和聚集,降低血小板收缩能力。
关于氯吡格雷体内的药动学过程的描述

文章大纲:一、概述- 引出氯吡格雷的背景和重要性- 概述本文要探讨的内容二、氯吡格雷的药代动力学1. 药物在体内的吸收过程2. 药物在体内的分布过程3. 药物在体内的代谢过程4. 药物在体内的排泄过程三、影响氯吡格雷药代动力学的因素1. 芳龄因素2. 性莂因素3. 肝功能和肾功能4. 药物相互作用四、氯吡格雷的临床应用和剂量调整1. 不同疾病状态下的药代动力学变化2. 如何根据药代动力学参数进行剂量调整五、总结和展望- 总结氯吡格雷的药代动力学特点- 展望未来在氯吡格雷研究方面的发展方向文章内容(篇幅约3000字,仅供参考):氯吡格雷是一种抗血小板药物,被广泛应用于心血管疾病的预防和治疗。
了解氯吡格雷在体内的药代动力学过程对于正确使用和调整药物剂量至关重要。
1. 氯吡格雷的药代动力学氯吡格雷在体内的药代动力学过程包括吸收、分布、代谢和排泄四个方面。
药物经口服给药后,进入胃肠道吸收到血液循环中,随后通过肝脏进行首过效应,一部分药物被代谢,剩余的药物进入全身循环。
药物在体内主要分布在血液和组织器官中,尤其是在肝脏、肾脏和心脏等靶器官中富集。
药物在体内主要通过肝脏的细胞色素P450酶系统进行代谢,再经过肝脏或肾脏的排泄从体内排出。
2. 影响氯吡格雷药代动力学的因素药代动力学参数受多种因素影响,包括芳龄、性莂、肝功能、肾功能以及与其他药物的相互作用等。
随着芳龄的增长,药物的代谢和排泄速度会下降,因此老年患者在使用氯吡格雷时需要谨慎剂量调整。
性莂因素也会影响药物的药代动力学过程,女性患者在经期和妊娠期对药物的代谢能力可能会发生变化。
肝功能和肾功能对药物的代谢和排泄起着重要作用,患者如果存在肝肾功能不全的情况,需要根据具体情况进行剂量调整。
氯吡格雷与其他药物的相互作用也需要引起重视,某些药物可能影响氯吡格雷的代谢和排泄过程,从而影响其疗效和安全性。
3. 氯吡格雷的临床应用和剂量调整在临床实践中,根据患者的芳龄、性莂、肝肾功能以及与其他药物的联合应用情况,医生需要综合考虑氯吡格雷的药代动力学参数,合理调整药物的剂量和给药方案,以确保药物的疗效和安全性。
氯吡格雷的使用需要注意4点
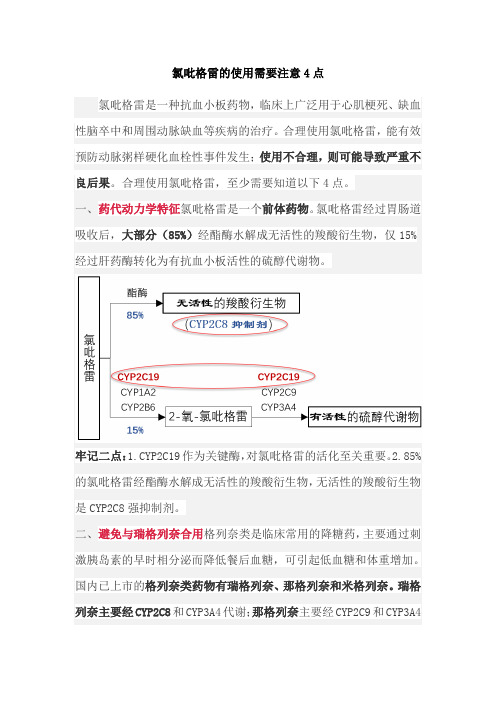
氯吡格雷的使用需要注意4点氯吡格雷是一种抗血小板药物,临床上广泛用于心肌梗死、缺血性脑卒中和周围动脉缺血等疾病的治疗。
合理使用氯吡格雷,能有效预防动脉粥样硬化血栓性事件发生;使用不合理,则可能导致严重不良后果。
合理使用氯吡格雷,至少需要知道以下4点。
一、药代动力学特征氯吡格雷是一个前体药物。
氯吡格雷经过胃肠道吸收后,大部分(85%)经酯酶水解成无活性的羧酸衍生物,仅15%经过肝药酶转化为有抗血小板活性的硫醇代谢物。
牢记二点:1.CYP2C19作为关键酶,对氯吡格雷的活化至关重要。
2.85%的氯吡格雷经酯酶水解成无活性的羧酸衍生物,无活性的羧酸衍生物是CYP2C8强抑制剂。
二、避免与瑞格列奈合用格列奈类是临床常用的降糖药,主要通过刺激胰岛素的早时相分泌而降低餐后血糖,可引起低血糖和体重增加。
国内已上市的格列奈类药物有瑞格列奈、那格列奈和米格列奈。
瑞格列奈主要经CYP2C8和CYP3A4代谢;那格列奈主要经CYP2C9和CYP3A4代谢;米格列奈主要通过与葡萄糖醛酸结合排泄,极少量经CYP2C9代谢。
因为,85%的氯吡格雷经酯酶水解成无活性的羧酸衍生物,无活性的羧酸衍生物可显著抑制CYP2C8。
若氯吡格雷与瑞格列奈合用,可抑制瑞格列奈的体内代谢,使瑞格列奈血药浓度升高3.9~5.1倍,增加低血糖风险,因此氯吡格雷应避免与瑞格列奈合用。
三、能与哪些质子泵抑制剂合用?氯吡格雷可增加胃肠道出血风险。
为了预防氯吡格雷引起的胃肠道损害事件,临床广泛合用质子泵抑制剂。
氯吡格雷是前体药物,在体内经CYP2C19转化后,才能成为能抑制血小板聚集的活性物质。
奥美拉唑、埃索美拉唑能和氯吡格雷竞争CYP2C19,导致氯吡格雷活性过程受阻,可能影响其抗血小板活性。
如果必须合用质子泵抑制剂,可选择兰索拉唑、泮托拉唑、雷贝拉唑和艾普拉唑等。
氯吡格雷(原研药)药品说明书:质子泵抑制剂药品说明书:四、餐前服还是餐后服?氯吡格雷的常规用法用量为:每次75mg,每日一次,与或不与食物同服。
氯吡格雷机制

氯吡格雷机制
氯吡格雷是一种抗血小板药物,常用于预防和治疗心脑血管疾病,特别是冠心病和脑血管疾病。
它的药理机制主要涉及抑制血小板聚集和血栓形成。
具体来说,氯吡格雷通过抑制血小板表面的ADP(腺苷二磷酸)受体P2Y12的激活,干扰ADP与其受体之间的结合,从而抑制ADP诱导的血小板聚集和凝血过程。
这种抑制作用可持续数天,因为氯吡格雷与P2Y12受体形成不可逆结合。
此外,氯吡格雷还可通过其他机制发挥作用。
例如,它可以抑制腺苷转化酶,增加腺苷水平,从而对血管内皮细胞和血小板产生保护作用。
此外,氯吡格雷还可抑制血小板释放的血管收缩物质,如血栓素A2和血小板活化因子4。
总的来说,氯吡格雷通过多种机制抑制血小板聚集和血栓形成,从而减少心脑血管疾病的风险。
然而,使用氯吡格雷时需要注意其可能引起的出血风险,并遵循医生的建议和监测。
氯吡格雷的说法、解释法

一)抗血小板药1、氯吡格雷的个体化治疗氯吡格雷是目前应用最广泛的抗血小板凝集药物,临床患者存在较大的药效差异,美国食品药品监督管理局(FDA)于2010年3月12 日发布氯吡格雷与其相关的基因多态性对该药疗效的警示。
氯吡格雷为前体药,氯吡格雷经肠道转运体ABCB1转运入血,通过CYP2C19代谢转化为初步活性代谢物2-氧代-氯吡格雷,初步活性代谢物2-氧代-氯吡格雷主要经PON 1转化为活性巯基衍生物,进而发挥抗血小板作用。
随着大量研究表明,氯吡格雷的疗效与PON 1、CYP2C19*2、CYP2C19*3、ABCB1基因多态性有密切关系,氯吡格雷的出血风险与CYP2C19*17有着密切关系。
(1) 基于CYP2C19代谢型的个体化治疗CPIC指南建议:1)对于CYP2C19超快代谢型(UM)患者,可按照氯吡格雷说明书的推荐剂量和服用方法,注意出血风险;2)对于CYP2C19中间代谢型(IM)患者,增加氯吡格雷低反应性的风险。
考虑换药,普拉格雷不或更小程度上由CYP2C19代谢,但与氯吡格雷相比出血风险增加。
或换用其他替代治疗。
3)对于CYP2C19慢代谢型(PM)患者,增加氯吡格雷低反应性的风险。
考虑换药,普拉格雷不或更小程度上由CYP2C19代谢,但与氯吡格雷相比出血风险增加。
或换用其他替代治疗。
(2) 基于ABCB1代谢型的个体化治疗转运体ABCB1基因的多态性,明显影响氯吡格雷的代谢。
若其编码区第3435位从碱基C变为碱基T,则转运效率下降,生物利用度下降。
带有ABCB1 3435TT者,心血管事件发生率为15.5%,比CC者10.7%高,HR为1.72。
因此,对于rs1045642位点TT基因型的患者,初始给药剂量和维持剂量均为双倍。
(3) 基于PON1代谢型的个体化治疗若PON1基因的第576位碱基G突变为A时,其水解2-oxo-clopidogrel生成活性巯基衍生物的能力降低,氯吡格雷抑制血小板的活性下降。
氯吡格雷 代谢基因

氯吡格雷代谢基因引言氯吡格雷(Clopidogrel)是一种常用的抗血小板药物,广泛用于预防心脑血管疾病的发生。
然而,由于个体间药物代谢能力存在差异,导致氯吡格雷在不同个体中的药效和不良反应表现存在差异。
这种差异主要与患者体内的代谢基因型有关。
本文将重点讨论氯吡格雷代谢基因,探讨其对药物疗效和安全性的影响。
氯吡格雷代谢途径氯吡格雷是一种伞根类抑制剂(ADP受体拮抗剂),通过抑制血小板聚集来预防血栓形成。
它需要在体内经过两步酶促反应才能转化为活性代谢产物。
首先,氯吡格雷需要被肝细胞中的细胞色素P450酶系统(CYP450)催化转化为活性中间产物2-氯苄基硫脲(2-oxo-clopidogrel)。
然后,2-oxo-clopidogrel再被酶羧酸酯酶-2(CES2)催化转化为最终的活性代谢产物。
氯吡格雷代谢基因氯吡格雷的代谢主要受到多个基因的影响,其中最为重要的是CYP2C19和CES2基因。
CYP2C19基因CYP2C19基因编码一种肝脏中的细胞色素P450酶,对氯吡格雷的代谢起关键作用。
根据人群遗传多态性,CYP2C19基因型可分为正常代谢型(EM)、缓慢代谢型(PM)和超快代谢型(UM)。
•EM型:具有正常的酶活性,能够有效地将氯吡格雷转化为活性代谢产物。
•PM型:由于突变等原因导致酶活性降低,使得氯吡格雷转化能力减弱。
携带PM型基因的患者需要较高剂量才能达到相同的疗效。
•UM型:突变引起酶活性增强,导致药物转化速度加快,可能增加药物毒副作用风险。
CES2基因CES2基因编码羧酸酯酶-2,也是氯吡格雷代谢的重要酶。
CES2基因多态性会导致酶活性的变化,进而影响氯吡格雷的代谢。
据研究表明,某些CES2基因型可能与氯吡格雷的疗效和不良反应有关。
例如,携带某些突变型CES2基因的患者可能在服用氯吡格雷后出现更严重的出血风险。
个体化用药考虑到CYP2C19和CES2基因对氯吡格雷代谢的影响,个体化用药策略已经被提出。
常用抗血小板药物总结

常用抗血小板药物总结
抗血小板药物是一类常用的药物,用于预防和治疗血小板功能异常所导致的血
栓形成。
在临床上,抗血小板药物常用于预防心脑血管疾病。
以下是几种常用的抗血小板药物:
1. 阿司匹林(aspirin):阿司匹林是最常用的抗血小板药物之一。
它通过抑制
血小板中的Cyclooxygenase(COX)酶,从而减少血小板生成的血小板聚集因子,防止血小板凝聚形成血栓。
2. 氯吡格雷(clopidogrel):氯吡格雷是另一种常用的抗血小板药物。
它通过
抑制ADP受体,阻断ADP的作用,从而防止血小板聚集。
3. 依达拉奉(prasugrel):依达拉奉是一种新一代的抗血小板药物。
它与氯吡
格雷具有类似的作用机制,但相比之下,依达拉奉具有更快、更强的抗血小板作用。
4. 替格瑞洛(ticagrelor):替格瑞洛也是一种新一代的抗血小板药物。
与氯吡
格雷和依达拉奉不同的是,替格瑞洛不需要经过肝脏代谢,因此作用更加可靠并且具有预测性。
5. 普拉格雷(prasugrel):普拉格雷是一种比氯吡格雷更强的新一代抗血小板
药物,可以减少心血管疾病患者的心肌梗死、中风和死亡风险。
这些抗血小板药物在医生的指导下使用,可有效预防和治疗血小板相关的疾病。
然而,每种药物都有自身的适应症和禁忌症,患者在使用前应咨询专业医生,并遵循医生的建议和用药指导。
氯吡格雷的使用,患者必须清楚这10点用药事项

氯吡格雷的使用,患者必须清楚这10点用药事项氯吡格雷是前体药物,必须通过CPY2C19酶代谢,才可以生成抑制血小板聚集的活性代谢物,用于预防和治疗因血小板高聚集引起的心、脑及其他动脉循环障碍疾病。
它适用于近期发作的脑卒中、心肌梗死和确诊的外周动脉疾病,以及急性冠脉综合征的患者。
但是,在用药的时候,有10个要点患者需要特别注意。
1. 禁忌症对氯吡格雷过敏、严重的肝损伤、活动性出血如消化道溃疡出血或颅内出血等情况要禁用。
2. 常规剂型有25mg和75mg两种,给药时要看清楚规格。
3. 对于不同症状的患者用药剂量不同a.对于急性冠脉综合征的患者,均应该以单次300mg为负荷剂量,75mg每日一次服用,服用时间视患者具体情况而定。
合用阿司匹林不超过100mg/日。
对于年龄超过75岁的ST段抬高性急性心肌梗死患者,不使用氯吡格雷负荷剂量。
在症状出现后应尽早开始联合治疗,并至少用药4周。
b.近期心肌梗死患者(从几天到35天内),近期缺血性卒中患者(从7天到6个月内)或确诊外周动脉性疾病的患者推荐剂量为每天75mg。
不能耐受者可以选择饭后服用,一般没有规定具体吃药时间,但是要每天定时吃药。
4. 搭配禁忌•氯吡格雷由CPY2C19酶代谢,奥美拉唑与其会产生竞争性抑制,导致氯吡格雷的效用降低,增加心肌梗死不良事件。
故在选择胃黏膜保护剂合用时应选择CPY2C19抑制作用轻的泮托拉唑。
•抑制CYP2C19的药物包括奥美拉唑、埃索美拉唑、氟伏沙明、氟西汀、吗氯贝胺、伏立康唑、氟康唑、氯苄匹啶、环丙沙星、西咪替丁、卡马西平、奥卡西平、氯霉素等,均不建议和氯吡格雷合用。
•接受阿司匹林、非甾体抗炎药、肝素、血小板糖蛋白IIb/IIIa (GP IIb/IIIa)拮抗剂、选择性5-羟色胺再摄取抑制剂(SSRI)和5-羟色胺去甲肾上腺素再摄取抑制剂(SNRIs)或溶栓药物治疗病人合用氯吡格雷,会增加出血风险,不建议合用。
•因可能使出血加重,不推荐氯吡格雷与口服抗凝药如华法林合用。
普拉格雷的简介(精制研究)

普拉格雷FNOS O O简介:普拉格雷(Prasugrel)化学名称为2-乙酰氧基-5-(环丙基羰基-2-氟苯基)-4,5,6,7-四氢噻吩(3,2-c)吡啶。
结构式见图,它是日本Daiichi Sankyo Co.公司开发的,日本Ube公司生产。
现在在美国市场与Eli Lilly公司合作的一种口服抗血小板药物,该药物于2009年2月23日获欧盟批准使用,同年7月10日经FDA批准在美国上市(商品名Effient),应用于治疗动脉粥样硬化和急性冠状动脉综合征。
P2Y12受体阻滞剂为噻吩并吡啶类化合物,包括噻氯吡啶、氯吡格雷和普拉格雷。
噻氯吡啶第一个用于临床的噻吩并吡啶类衍生物,由法国Sanofi公司开发,1978年首次在法国上市。
1991年FDA批准在美国上市。
我国于1988年批准进口,商品名为“抵克利得”(Ticlid)。
2003年5月专利到期。
由于其存在骨髓抑制、白细胞减少、再生障碍性贫血、血小板减少症等严重的不良反应,使其逐渐退出抗血栓市场。
氯吡格雷是赛诺菲-安万特公司研究开发的抗血小板聚集药物,商品名为“Plavix”(波立维)。
1997年11月FDA批准了Plavix作为抗凝药,用于治疗急性冠状动脉综合征(ACS)。
经过10年的临床使用,其目前己是抗血小板聚集临床上的标准治疗药物。
2008年该药物全球销售额为70亿美元。
氯吡格雷存在着一个较大的弱点,那就是它的治疗效力较低,这也为后来者介入这一市场打开了通道。
普拉格雷是继噻氯吡啶(ticlopidine) 和氯吡格雷( clopidogrel)之后的第3代噻吩吡啶类的抗血小板药。
是目前是目前抗血小板聚集临床上标准治疗药物氯毗格雷的有力竞争者,可夺取氯吡格雷的市场份额,具有“重磅炸弹级”产品的潜力,预计到20巧年其销售将达到巧亿美元。
与氯吡格雷相似,普拉格雷的作用靶点也是PZY12受体,也是在体内代谢后产生活性。
不同之处主要在它抑制血小板聚集的能力更强、更有效。
氯吡格雷抵抗相关机制及对策

药代动力学差异却非常显著,因此认为这种个体问的变异性 可能与药物吸收及转化有关。、 3.药物的相互作用:凡由(:YP3A4代谢或能抑制该酶的
药物均有可能干扰cPG的代谢过程,削弱抗血小板效应。研
究结果显示,需要cYP3A4代谢的亲脂性他汀类药物可能损
伤cPG的抗血小板效应,导致cPG抵抗。然而,这些数据具 有争议性,因为也有研究证实亲脂性他汀类药物与cPG抗 血小板效应并不相关州。与cPG竞争cYP3A4活性的药物(如
・71・
・综述与讲座・ 氯吡格雷抵抗相关机制及对策
洪晓明
李兴德
氯吡格雷(clopido殍el,cPG)是临床应用最为广泛的新型 噻吩吡啶类抗血小板聚集药物之一,其作用明显优于阿司匹
地与血小板表面二磷酸腺苷(ADP)P2Y12受体结合,减少ADP
结合位点,阻断ADP对腺苷酸环化酶的抑制作用,促进环单 磷酸腺苷(cAMP)依赖和前列腺素E.(PGE。)刺激的舒血管物
ADP诱导下,服药前基线值与服药后血小板最大聚
集率的差值≤10%的患者被判定为cPG抵抗。另外,流式细 胞仪检测ADP刺激后GPⅡb/Ⅲa受体和P.选择素的表达, 血栓弹性描记法测定ADP诱导下血小板.纤维蛋白凝块长 度,以及检测P2Y12受体含量和vAsP磷酸化指数等均可作 为评估cPG抵抗的方法。然而,检测方法的不同导致cPG抵 抗发生率的报道差异较大,为5%一44%。
阿司匹林、氯吡格雷、替罗非班等抗血小板药物机制、作用特点、药代动力学及出血风险处理等并发症处理

阿司匹林、氯吡格雷、替罗非班等抗血小板药物机制、作用特点、药代动力学及出血风险处理等并发症处理血小板聚集药物是脑梗死急性期、二级预防的主要治疗药物,它们通过作用于血栓形成的不同阶段,达到抗血小板聚集的作用。
血小板聚集形成血栓的过程在细胞外基质,血小板表面的糖蛋白受体及GPIb-IX-V 与血管内皮细胞释放的血管性血友病因子等相互作用,导致血小板附着在胶原蛋白上形成活化后的血小板。
在整合素αIIbβ3介导下,纤维蛋白原通过桥联的方式,两端与不同的αIIbβ3 结合,最终导致血小板的聚集。
激活的血小板还会释放内源性二磷酸腺苷和血栓素A2,最终导致更多血小板的活化与聚集。
不同抗血小板聚集药物作用机制抗血小板聚集药物作用特点阿司匹林1)机制:作为最经典的抗血小板药物,阿司匹林可以不可逆的抑制血小板环氧化酶(COX-1),导致TXA2 生成减少,从而抑制血小板的聚集。
2)起效时间:阿司匹林的达峰时间约为 0.3~2 小时,清除半衰期与剂量相关;肠溶片较普通片吸收时间可延长3~6 小时。
因此,快速起效时可选择嚼服。
3)功能恢复时间:由于阿司匹林不可逆的抑制环氧化酶活性,因此,血小板功能的恢复需要等待血小板的再生,即完全停药后 7~10 天。
4)代谢途径:阿司匹林经由肾脏代谢,因此,使用时需考虑肾功能情况。
禁用于合并氨甲蝶呤时,可能会减少其肾清除。
5)合并用药:同样作用于水杨酸的NSAID,如布洛芬等,合并时也需要谨慎。
此外,促进尿酸排泄的药物,如苯磺唑酮等,也可能需要谨慎。
6)注意事项:阿司匹林还可能导致支气管痉挛并引起哮喘发作,因此,也需要考虑患者是否合并哮喘。
7)服用时间:普通剂型的阿司匹林通常用于退热止痛,肠溶型的阿司匹林不会在酸性的胃肠道环境下溶解,而会进入碱性的环境,以尽量减少对胃肠道的刺激,建议餐前服用。
同时由于阿司匹林可以直接破坏消化道黏膜,应用时需注意其消化道出血并发症。
氯吡格雷1)机制:氯吡格雷经过CYP450 酶代谢后,生成的活性代谢产物可以不可逆抑制ADP 与血小板P2Y12 受体的结合,从而抑制血小板的聚集。
关于氯吡格雷体内的药动学过程的描述

关于氯吡格雷体内的药动学过程的描述【关于氯吡格雷体内的药动学过程的描述】1. 介绍氯吡格雷(Clopidogrel)是一种用于预防心脑血管疾病的抗血小板药物,被广泛应用于临床治疗中。
它通过抑制血小板聚集,减少血栓形成,从而降低心血管事件的发生率。
本文将围绕氯吡格雷在体内的药物动力学过程展开详细描述,帮助读者更深入地了解这一药物的作用机制以及在临床治疗中的应用。
2. 肝素的代谢氯吡格雷首先需要在体内被代谢为其活性代谢产物,这个代谢过程主要发生在肝脏中。
肝脏中的细胞色素P450酶系统起着至关重要的作用,它能将氯吡格雷代谢为其活性的二氢代谢产物。
这个代谢过程对于氯吡格雷的药效具有重要影响,也为我们解释了为什么一些患者对氯吡格雷的反应会有差异。
3. 药物的吸收和分布氯吡格雷口服后能够在消化道被有效吸收,但由于其受到肝脏的首过效应影响,生物利用度相对较低。
在体内的分布方面,氯吡格雷主要分布在血浆中,并且能够结合大约98%的血浆蛋白,这意味着在体内的很长一段时间内,只有少量的氯吡格雷能够以自由态存在,这对于维持其血浆浓度具有影响。
4. 药物的代谢和排泄经过肝脏的代谢后,氯吡格雷的代谢产物会进一步被排泄出体外。
其中70%的代谢产物通过尿液排泄,30%则通过粪便排泄。
了解氯吡格雷在体内的代谢和排泄过程,对我们合理使用该药物,避免不良反应产生具有重要意义。
5. 个人观点和理解我个人对氯吡格雷在体内药动力学过程的深入了解,不仅可以帮助医生更好地指导患者用药,还能够指导临床研究更加有效地开展。
在未来,相信随着对药物代谢、作用机制等方面认识的不断深入,将更有助于我们合理使用该药物,提高其治疗效果,减少不良反应的发生。
总结回顾通过对氯吡格雷在体内的药动力学过程的描述,我们了解到其在人体内的吸收、分布、代谢和排泄等过程。
深入了解药物在体内的过程,有助于我们更加全面、深刻地认识该药物,并指导其在临床中的应用。
以上就是对“关于氯吡格雷体内的药动力学过程的描述”的文章撰写,希望能够帮助您更好地理解这一主题。
氯吡格雷片机制

氯吡格雷片机制全文共四篇示例,供读者参考第一篇示例:氯吡格雷片是一种用于预防和治疗心血管疾病的药物,具有抑制血小板聚集和减少心血管事件风险的作用。
其主要成分是氯吡格雷,通过干扰血小板聚集的生物化学途径来发挥药效。
氯吡格雷是一种P2Y12受体拮抗剂,能够抑制ADP介导的血小板激活和聚集。
在炎性环境中,血小板会通过释放ADP、TXA2和5-羟色胺等介质,促进血栓形成和动脉粥样硬化的进展。
而氯吡格雷可以选择性地结合于血小板P2Y12受体上,阻断ADP的结合,从而抑制血小板的激活和聚集,减少血栓的形成,降低心血管事件的风险。
氯吡格雷片的机制主要可以分为以下几个方面:2. 保护心血管内皮功能:氯吡格雷通过减少血小板的活化和聚集,可以降低血管内皮细胞的受损程度,保护血管内皮功能。
这对于预防动脉粥样硬化等心血管疾病的发生具有积极意义。
3. 减少炎症介质的释放:ADP的释放不仅可以促进血小板激活和聚集,还会引起炎症反应,促进动脉粥样硬化的进展。
氯吡格雷的抑制作用可以减少炎症介质的释放,降低炎症反应,降低心血管事件的风险。
4. 提高血流动力学:血小板的激活和聚集会导致血栓形成,阻塞血管,影响血流动力学,增加心血管疾病的风险。
氯吡格雷通过抑制血小板的激活和聚集,可以改善血流动力学,保护心血管健康。
氯吡格雷片通过抑制血小板的激活和聚集,减少血栓形成,保护血管内皮功能,降低炎症反应,改善血流动力学等多种机制,来预防和治疗心血管疾病,降低心血管事件的风险。
患者在服用氯吡格雷片的过程中应密切监测血小板功能和不良反应,以确保药物的有效性和安全性。
【本段字数:443】值得注意的是,虽然氯吡格雷片在预防和治疗心血管疾病中具有重要作用,但在使用过程中也需要注意一些潜在的风险和注意事项。
氯吡格雷片属于抗血小板药物,对血小板功能的影响比较强,因此患者在使用过程中应密切监测血小板计数和功能,避免出现出血等不良反应。
患者在使用氯吡格雷片的同时应遵循医生的建议,严格按照用药剂量和时间进行服用,不可自行停药或更改剂量,以免影响药效和安全性。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1
问题的提出
分析1991~2000年18个药物ADR报告,发现发生 率最高的27个药物中(包括Carbamazepine, fluoxetine, Ibuprofen, Imipramine, Isoniazid, Naproxen, Rifampin, Teophylline, Phenytoin, Verapamil , Warfarin等),有16个(57%)药物的ADR发生至少与一 种药酶基因多态性(主要与poor metabolism)有关;反 之,随机抽查药物中,也有7~12%的药物与基因相关,提 示我们,如依据患者个体的基因多态性给药即可大大 减少ADR的发生!
7
FDA1945~2005要求121个药品标明 药物基因组生物标记物相关信息
8
Pharmacotherapy 2008;28(8):992–998
药物基因学的临床应用
Enzyme/Disease
Gene
• Glucose-6-PhosphateDehydrogenase
G6PD
Deficiency (G6PD Def)
• N-Acetylation & Tuberculosis
NAT2
•Cytochrome P450 Enzyme ━Drug Metabolism
CYP2D6
• Warfarin & Coagulation
CYP2C9
VKORC1
•Thiopurine S-Methyltransferase
TPMT
4
20世纪:千人一药一量!
One drug fits all ! One dose fits all !
5
21世纪:一人一药一量
“量体裁衣”给药
6
努力做到个体化给药
What is Personalized Medicine?
━“5R”
To the Right patient Give the Right drug At the Right time In the Right dose By the Right route
18
CYP2C19抑制剂
♣抗溃疡药:奥美拉唑>兰索拉唑>埃索拉唑>潘托拉唑>
雷贝拉唑(最弱);
♣抗抑郁药:氟西汀、氟伏沙明、帕罗西汀(弱)、舍曲林
(弱); ♣抗癫痫药:丙戊酸、奥卡西平、非氨酯、托吡酯(弱); ♣调脂药:氟伐他汀、洛伐他汀 ♣其他:氟康唑、酮康唑、胺碘酮、吲哚美辛、口服避孕药、
西咪替丁、氯雷他定。
❖ 甲基化(Methylation) methyltransferase
❖ 磺化Sulphation Glutathione S-transferases Sulfotransferases
❖ 乙酰化Acetylation N-acetyltransferases(NAT) Amino acid N-acyl transferase
❖ Cytochrome P450 monooxygenase system ❖ Flavin-containing monooxygenase system ❖ Alcohol dehydrogenae and aldehyde dehydrogenase ❖ Monoamine oxidase ❖ Co-oxidation by peroxidases
❖ 葡萄糖醛酸化Glucuronidation UDP-glucuronosyltransferases(UGT) Mercapturic acid biosynthesis
13
CYP450的命名
14
CYP450主要家族与其同工酶
15
主要被CYP2C19代谢的药物-1
❖ 抗惊厥药
பைடு நூலகம்
美芬妥英 (4氧化) 苯妥英 (4氧化,微量) 乙苯妥因 (4氧化) 地西泮 (N去甲基) 苯巴比妥 (3氧化) 海索比妥 (3氧化) 甲苯比妥 (3氧化)
JAMA 2001; 286: 2270-2279 2
个体化安全用药的钥匙
—药物基因学与药物基因组学
3
药物基因组学与药物基因学的区别
※ Pharmacogenetics:研究单个基因与个体药物
反应差异的相关性
※Pharmacogenomics:研究多个基因或整个基因
组与个体药物反应差异的相关性 目前已将二者统一称为PGx.
17
其他被CYP2C19代谢的药物(底物)
地西潘 艾斯西酞普兰 氟硝西潘 氟西汀 吗氯贝 胺 舍曲林 曲米帕明 美芬妥英 卡立普多 环磷酰胺 异环磷酰胺 奈非那韦 氯 胍 R-华法林 普萘洛尔 甲 苯磺丁脲 伏立康唑 伊曲韦林 苯妥英 地西潘 多 虑平 美沙酮 奋乃静 雷尼替汀 他莫昔芬 氯吡格雷
& Cancer
• ACE Inhibitors, Antidepressants, Diabetes, Asthma, etc
9
参与药物代谢的主要酶系
10
药物代谢途径
Phase Ⅰ
Phase Ⅱ
Phase Ⅱ
11
I 相反应(Phase I reaction)
主要使原形药物降解或前体药物活化
氧化(Oxidation)
19
CYP450的基因组学
20
为什么药效不一样?
21
Pharmacogenomics
Drug targets
Drug transporters
Drug metabolizing enzymes
Pharmacodynamics
Pharmacokinetics
Variability in efficacy or toxicity
还原(Reduction)
❖ NADPH-cytochrome P450 reductase ❖ Reduced cytochrome P450
水解(Hydrolysis)
❖ Esterases ❖ Epoxide hydrolase
12
Ⅱ相反应 (Phase II reaction)
主要是结合反应,增加极性,易于排出
16
主要被CYP2C19代谢的药物-2
❖ 抗溃疡药 奥美拉唑(5氧化,80%) 兰索拉唑(5氧化,>50%) 泮托拉唑 (5氧化,>50%) 埃索拉唑(5氧化,>50%) 雷贝拉唑 (5氧化,<10~20%)
❖ 抗抑郁药 阿米替林(N-去甲基) 氯米帕明(N去甲基
丙咪嗪 (N去甲基) 西酞普兰(Citalopram,N去甲基)