CODEHOP使用说明
在线商城系统说明书

在线商城系统说明书一、引言在线商城系统是一种基于互联网的电子商务系统,旨在提供一个便捷、安全、高效的购物平台。
本说明书旨在向用户介绍在线商城系统的特点、功能和使用方法,使用户能够更好地了解和使用该系统。
二、系统概述在线商城系统提供了以下功能:1. 商品浏览和搜索:用户可以通过系统浏览和搜索各类商品,查看商品详细信息和图片。
2. 购物车管理:用户可以将感兴趣的商品加入购物车,并随时查看购物车中的商品数量和总金额。
3. 订单管理:用户可以提交订单,选择支付方式并填写配送地址,系统会自动生成订单号并跟踪订单状态。
4. 会员中心:用户可以注册成为系统会员,享受会员特权,如积分累积、优惠券领取等。
5. 支付与配送:用户可以选择合适的支付方式,并填写收货地址,系统会安排商品配送并提供快递跟踪服务。
三、系统安装与设置1. 硬件要求:在线商城系统可以在各类主流计算机硬件上运行,建议使用稳定的网络环境以确保系统的流畅运行。
2. 软件要求:系统运行需要安装相应的服务器软件,如Apache、MySQL等,并兼容常用的浏览器。
3. 数据库配置:在数据库中创建相应的表格和字段,确保系统能够正确存储和读取相关数据。
4. 网站域名与主机配置:将系统部署到互联网上的特定域名下,配置合适的主机环境。
四、系统登录与权限管理1. 用户注册与登录:用户需要注册账号才能登录系统,并提供必要的个人信息,如用户名、密码、手机号码等。
2. 密码保护:系统应使用加密算法对用户密码进行保护,确保用户信息的安全性。
3. 权限管理:系统应对用户权限进行管理,根据用户身份赋予不同的操作权限,以确保系统的安全性和合法性。
五、商品管理1. 商品分类:系统应提供商品分类功能,方便用户查找具体商品。
2. 商品发布:商家可以通过系统发布新的商品,包括商品名称、价格、库存、详细描述、产品图片等信息。
3. 商品推荐:系统应根据用户的购买记录和浏览行为,智能地推荐相关商品,提高用户购买的几率。
CODEHOP使用流程

1.输入以下网址:/codehop.html
2.找到该行:
The input should be a set of local multiple alignments (blocks) of a group of related protein sequences. The alignments must be in Blocks Database format, such as in Block Maker output.
点击“block maker”进入格式制作。
3.向该页的以下框内输入保守区域的多个序列
输入格式为:
>名字1
……(同源蛋白保守区域序列1)
>名字2
……(同源蛋白保守区域序列2)
>名字3
……(同源蛋白保守区域序列3)
然后点击方框下发的“Make Blocks”
4.新的对话框页面上显示如下:
Making your blocks, please wait...
Your results will be available for approximately 24 hours here.
点击“here”.
5.在新的页面的底部点击[BLOCKS format]。
6.新的对话框中显示CODEHOP的格式,将该页面内的全部内容复制,粘贴至CODEHOP主页的方框(如下所示)中。
Paste your block(s) below:
7.调整方框下方的各个参数,点击方框右上方的“look for primers”即可。
科脉商业管理软件说明书

科脉商业管理系统使用说明一、系统安装二、基本设置1(后台)9001cx第一次进入2如人名或0001)3三、完善基本档案1),区域),4四、业务工作(一)库存管理12345、查实时库存输入编码(或直接在后侧选择相应类别)。
6。
78即自动计算出现差错(二)销售相关1734查找,(三)前台POS端设置1、前台零售操作步骤1POS,按的数字,完成一次交易。
(2,再按入条码,按,再按。
)会员卡收款:先输入所购买商品,按,输入卡号,按,完成退货。
(7)退出POS在当前收款状态下,按号即为小票号。
销售状态,按,条码,店内码作废,退货结算,人民币,前台日结,会员卡上面提到的设置,都是指第一次进入时需要设置,再次进入时是指在键盘上对应的按键。
在收银界面按某键时,提示XXX键没有定义,只要按一下,就好了。
仅供个人参考仅供个人用于学习、研究;不得用于商业用途。
For personal use only in study and research; not for commercial use.Nur für den persönlichen für Studien, Forsc hung, zu kommerziellen Zwecken verwendet werden.Pour l 'étude et la recherche uniquement à des fins personnelles; pas à des fins commerciales.толькодля людей, которые используются для обучения, исследований и не должны использоваться в коммерческих целях.以下无正文。
CODEHOP使用说明

CODEHOP使用说明CODEHOP是一种用于设计引子扩展PCR引物的方法,用于在没有已知序列信息的情况下扩增目标DNA片段。
它是一种基于保守区域的引物设计方法,可以在短序列的基础上快速设计出高度特异性的引物。
CODEHOP是由Michael A. Innis等人在1990年首次提出的,是一种被广泛应用于分子生物学和基因工程研究中的技术。
2.识别保守区域:在比对结果中,可以识别出具有高度保守性的区域。
这些保守区域即为CODEHOP引物设计的基础,因为这些区域相对稳定,可以用于设计特异性高的引物。
3.设计CODEHOP引物:通过使用一系列特殊的引物序列,在保守区域周围设计具有高度特异性的CODEHOP引物。
这些引物设计需要考虑以下几个因素:引物长度应为20-30个碱基,GC含量应在40-60%范围内,引物的末端应为CPG三个氨基酸,引物的内部序列应具有高度特异性。
此外,引物序列之间应有一定的相似性,以确保其在PCR反应中协同工作。
4.测试引物特异性:设计完成后,使用引物进行PCR反应,使用目标物种的基因组DNA作为模板,进行反应。
通过观察PCR产物的大小和特异性,评估CODEHOP引物的特异性。
CODEHOP引物设计方法的优点是快速、简单且高效。
它不需要已知的序列信息,仅通过保守区域的比对就能够设计出高度特异性的引物。
此外,CODEHOP引物可以广泛应用于各种分子生物学研究中,如基因克隆、基因表达和突变分析等。
然而,CODEHOP方法也存在一些限制。
首先,引物设计仅仅依赖于保守区域,可能导致引物的特异性不够高。
其次,CODEHOP方法需要在实验过程中进行引物的优化和测试,以确保其特异性和扩增效率。
总之,CODEHOP是一种快速、简单且高效的引物设计方法,可以用于扩增目标DNA片段。
通过比对已知序列,识别保守区域,并设计具有高度特异性的CODEHOP引物,研究人员能够在没有已知序列信息的情况下成功扩增目标DNA,为基因工程和分子生物学研究提供了强有力的工具。
网上购物商城使用说明书

网上购物商城使用说明书主要功能网上购物商城主要由前台会员模块和后台管理模块两部分组成。
❑前台功能模块前台模块主要包括会员注册、登录、修改个人信息、购物、查询购物情况和查看各种服务条款等功能。
❑后台管理模块后台模块主要包括后台管理员对会员、商品、仓库、订单和管理员的管理等功能。
会员在登录进入该网上购物商城后,不仅可以查看其各种服务条款,还可以选择查看各种商品的详细信息并购买。
管理员登录后,可以查看商品销售情况,及管理会员、商品、仓库和其自身的信息。
管理员还可以根据实际情况添加其他管理员以维护该网上购物商城的购物环境和安全。
操作注意事项用户在使用《网上购物商城》之前,应注意以下事项:(1)本系统后台管理员用户名为:51aspx,密码为:51aspx。
(2)单击前台首页的“后台入口”按钮,弹出登录页面,添加正确的用户名和密码进入后台。
(3)本系统后台的“仓库管理”模块虽然实现了添加、修改、删除等功能,但与前台并没有建立连接,前台的仓库类型是固定的。
(4)本系统后台的“订单管理”模块是按订货人统计而不是订单号。
业务流程在使用本系统时,请按照以下流程进行操作:1.前台前台中所有的功能模块只需用户单击相关超链接,便可进入信息展示页面。
(1)通过【会员管理】页面可以进行会员注册。
(2)通过【首页】页面可以查看商品信息及购买商品。
(3)通过【购物信息查询】页面可以查看购物记录。
注意:在“购物车”和“购物信息查询”模块中,用户需先通过首页进行“注册”,成为本站的会员后才能进行购物及查看购物记录。
下面给出商品的购买过程。
(1)单击导航区上的【首页】菜单按钮,进入如图1.1所示的界面。
– 1 –图1.1 首页页面(2)在此页面中单击“详细信息”按钮,进入商品详细信息页面,如图1.2所示。
图1.2 商品详细信息(3)如果您已经注册为会员,可以直接单击“购买”按钮进入购物车页面如图1.3所示,否则提示“您还没有登录,请登录后再购买,谢谢合作!”。
网上商城系统操作手册

网上商城系统操作手册目录1 系统描述 (1)1.1总体介绍 (1)1.2软件需求 (1)2系统后台管理介绍和操作说明 (1)2.1后台登陆 (1)2.2管理员信息管理 (1)2.2.1添加管理员 (1)2.2.2管理员列表 (1)2.2.3修改密码 (2)2.3 用户信息管理 (3)2.4 销售量查询 (3)2.5 商品信息管理 (3)2.5.1显示、修改、删除商品信息 (3)2.5.2添加商品信息 (4)2.5.3添加、删除商品类别 (4)3系统前台管理介绍和操作说明 (5)3.1商品浏览 (5)3.2用户管理 (5)3.2.1用户登录 (5)3.2.2修改密码 (5)3.2.3修改个人资料 (6)3.3购物记录查询 (6)3.4购物车管理 (6)1 系统描述1.1总体介绍整个项目分前台和后台两个项目开发。
前台用户可以在网上商城按类别浏览商品的信息,也可以搜索自己所需要的商品的内容,放入购物车以便购买,下订单,也可以修改自己的注册信息,察看自己的订单。
同时可以注册和编辑个人信息。
后台包括用户管理和商品管理和用户权限管理。
前台功能描述:➢商品浏览:热门商品浏览(首页显示点击率最高的商品列表)、新到商品浏览(首页显示最新添加的商品列表)、商品分类浏览,按商品名称搜索、商品详细信息;注册账户才可以购买商品,用户登录对用户身份进行验证,未注册用户只能浏览商品,不能完成结账;➢用户管理:注册新用户、登录、用户修改密码、用户个人资料管理;➢购物车管理:用户可以将感兴趣的商品加入购物车,便于商品购买,如果用户未登录则提示先登录后购买;➢订单管理:用户下订单后,可以对订单删除和更改商品,以及修改联系方式,系统判断客户账户里有没有足够的资金,如果没有足够的资金则给出提示,如有则可完成订单。
后台管理功能描述:➢管理员信息管理:登录;添加新管理员、删除管理员;修改密码;管理员日志(记录管理员的每个操作,由超级管理员进行查询);➢商品信息管理:添加、删除商品类别;添加、修改、删除商品信息;➢用户信息管理:查询用户信息、修改账户金额;➢销售量查询:查询某月的销售情况(包括每种商品的售出数量、相关订单数、销售收入)。
秘奥商务管理软件超市版的前台使用说明书1

系统简介为满足众多的中小型零售企业对销售管理的需要,秘奥软件有限公司开发出了一套产品简洁直观、易学易用的特点,将店面销售业务与整个系统无缝地链接起来,使店面的销售情况,随时可以传递、汇总到后台,为您解决了每日处理大量销售单据的烦恼,帮助您在最短的时间内准确无误地计算出各种商品的销售数量、销售金额和利润等数据,使您可以及时掌握仓库库存信息,安排采购计划。
系统由后台进销存系统与前台的POS销售组成。
后台可以录入收银员、货品、班次等基础资料等信息,前台主要作为零售的操作并把数据传到后台。
这些零售数据提交到服务器后,后台可查看这些单据,并通过零售日结功能来实现冲减库存、计算利润的过程,此外还可以通过各种报表查看商品销售情况,收银员收款、交款情况。
●可在局域网和互联网中应用,由总部(配送中心)系统和门店系统组成,即前台POS+后台MIS;●连网/断网自由切换的工作方式使您在任何网络环境下可工作;●集团化统一采购和门店独立采购相结合,统一配送、统一结算、统一定价、统一促销;●门店可以分多个柜组、多个导购员销售管理;●支持各种POS硬件设备:小票打印、顾显、钱箱、条码枪等;●全面消费卡支持:会员、积分、储值、折扣;●灵活多样的促销支持:按限时、按限量、按限时限量;●智能化错误提醒功能(错误条码、商品价格、数量等)●严格权限控制,对各个收银员可设置不同权限;●前台交接班日志,详细记录收银员的所有操作,方便核对;●强大报表系统,与灵活的自定义报表工具——强大的报表系统,保证了总部、门店之间信息的通畅;前台基本业务流程前台零售业务流程入下图:登录模式选择软件启动后将出现如下图登陆模式选择的对话框:连网模式:当选此模式时,在系统后台实时查看到前台的销售情况,前台并可实时取得后台更新的商品、柜组、导购等基本资料。
无需做上传与下载操作。
此工作模式缺点是前台一直与服务器保持连接,在服务器或网络较忙时会影响前台销售速度。
断网模式:当选此模式时,前台首先需要下载基础资料,然后就可以与服务器断开连接,需要上传销售单据时,再进行连接,后台才可以查看到有关的数据。
codesoft 6.0中文使用手册2

CodeSoft 中文说明简明手册·软件的安装 2 ·基本界面和操作步骤 3 ·设置打印机 4 ·标签的设置 5 ·条码的输入 6 ·数据库的输入 8 ·计数器的的设置 14·将数据库和计数器等变量导入条形码 15软件的安装步骤安装步骤:1.开机后启动 Windows95/98/2000.2.点击“开始”按钮,选择“程序”下的资源管理器,或在“开始”按钮处单击右键并选择资源管理器单击后,进入资源管理器。
3.将系统安装光盘安装盘插入对应的驱动器。
4.·运行光盘根目录下的“setup.exe”文件,·选择语言为“间体中文” ,·选择安装目录, 建议选择默认安装路径即可, 第一次安装会提示重新启动计算机 , 按确定即可, ·然后按“下一步”直到“结束”完成安装;5. 安装完毕请拷贝软件目录下的 CsUtil.dll,将安装路径目录下的此文件覆盖粘贴,如果第一步安装过程选择默认 C 盘安装路径,则只需找到 C:\program files\cs6下覆盖粘贴即可;如果上步的安装选择了新路径,则需找到和第一步相同的路径;6.安装完成以上两步,即可运行执行文件 Cs6.exe,运行 CODESOFT6.0条码软件,她可为您带来丰富的而强大的中文条码编辑打印功能。
7.严禁翻录或者拷贝用作其它商业用途,违者追究法律责任。
安装结果(默认路径 C:\Program Files\CS6 ,在安装目录下执行以下文件:Cs.exe 主执行文件基本界面和操作步骤基本界面:操作步骤:选择打印机:选择或添加打印机,设置打印机参数标签设置:设置标签的大小,边距插入对象:条码:条码种类,高度等信息数据库的导入计数器的设置选择打印机按 F5键或在工具栏上点击如图按钮,弹出对话框:如果打印列表中没有您对应的打印机,点击在右边的“添加”按钮,弹出对话框:在左边“安装”列表中选择对应打印机类型, (下面以 Zebra Z4M打印机为例接口选择 LPT1(即为串口接口 :如图所示:选择完毕后,点击“完成”按钮。
CODEHOP使用说明

CODEHOP : COnsensus-DEgenerate HybridOligonucleotide PrimersCODEHOP helpOn this page:Other pages: •General information • Getting started •Obtaining input alignments • CODEHOP program algorithm • Terms and parameters • CODEHOP manuscript • Strictness parameter details • Basic tips• PublicationGeneral informationThe CODEHOP program designs PCR (Polymerase Chain Reaction) primers from protein multiple-sequence alignments. The program is intended for cases where the protein sequences are distant from each other and degenerate primers are needed.The multiple-sequence alignments should be of amino acid sequences of the proteins and be in the Blocks Database format Proper alignments can be obtained by different methods .The result of the CODEHOP program are suggested degenerate sequences of DNA primers that you can use for PCR. You have to choose appropriate primer pairs, get them synthesized and perform the PCR.A CODEHOP primer is degenerate at the 3' core region, with a length of 11-12 bp across four codons of highly conserved amino acids, and is non-degenerate at the 5' consensus clamp region, with a length which depends on its desired annealing temperature, typically between 20 and 30bp:5' 3'--------------------===========non-degenerate degenerateconsensus clamp core#bases from temp 11-12 basesThe hybrid structure (5' consensus and 3' degenerate) of CODEHOP primers allow the PCR amplification to be specific during the early cycles from the original source DNA and selective during the late cycles from the PCR synthesized products:Schematic comparison of standard degenerate PCR (left) with the CODEHOP (right), illustrating regions of mismatch in primer-to-template annealing during early PCR cycles and in primer-to-product annealing during subsequent cycles. Vertical lines indicate nucleotide matches between primer (arrow) and template or synthesized product. The overall degeneracy is the product of degeneracies at each nucleotide position, so that the fraction of precisely hybridizing primers is 1/degeneracy.Obtaining input alignmentsInput alignments for the CODEHOP program must be in the Blocks Database format. Block multiple-sequence alignments are ungapped and usually local alignments. Local alignments cover only parts of the protein sequences. The regions between the blocks are the ones with no sequence similarity or where gaps must be inserted to align the sequences.You can get a multiple alignment from a group of related protein sequences using the Block Maker or other automated methods(such as ClustalW). The alignments can also be manually made or modified according to your knowledge of the proteins (position of the active sites orpost-translational modifications etc.). In any case the alignments passed to the CODEHOP program must be in the Blocks format. BlockMaker blocks need no reformatting. Appropriate parts of Clustal- or FASTA-formatted multiple alignments can be automatically made into blocks by the Blocks multiple alignment processor. Other types of multiple alignment can be semi-manually reformatted with the Blocks formatter. All of the above Blocks programs have links to send the resulting blocks directly to theCODEHOP program and also provide other information for evaluating the blocks (logos, trees, searches).More information on multiple alignments can be found in the notes for the ISMB97 tutorial on Introduction to making and using protein multiple alignments.Terms and parameters•Degeneracy measures how many different sequences are specified by the primer. The degeneracy of each position depends on the sum of the nucleotides appearing in it (1 to 4). The degeneracy of the primer is the product of the degeneracies of all of its positions.The degenerate core region is scored by its degeneracy, with lower degeneracy scored higher. A standard degenerate alphabet is used in the core region.•Degeneracy strictness specifies how to count nucleotide(s) with low occurrences. It is a parameter between 0 and 1. Strictness of 0 means that all nucleotides that actually appear in the position arecounted to calculate the degeneracy. Strictness of 1 means that only the nucleotides with the highest value in the position are counted.Intermediate strictness values give behavior in between. You can look at a detailed explanation for more information on how thestrictness is calculated.•Temperature is computed for the clamp region plus any adjacent non-degenerate positions in the core region. Differenttemperatures affect only the length of the clamp; highertemperatures result in longer clamps. Temperature is computed using the nearest-neighbor method of Rychlik, et al ( NAR 1990 18:21, 6409-6412). This method takes into consideration the primer and K+ (potassium ions, taken as 50 mM) concentrations.•Nucleotide runs in the clamp region can be limited (the default is5 in a row). If longer runs are encountered, they are broken up bytaking the next highest- scoring nucleotide in the position with the lowest-scoring run nucleotide.•The clamp score is a measure of how well the non-degenerate 5' clamp matches the block given a codon usage table. The score is a weighted average of the maximum scores in each column of the DNA PSSM. A perfect score of 100 is obtained if the clamp consists of invariant methionines and tryptophans, and a minimum score of 25 is obtained if A, C, G and T are equally likely at all positions. In computing the clamp score, positions are increasingly down-weighted asdistance from the degenerate 3' core increases. The clamp score is intended to estimate how well the clamp will stabilize the core inannealing to the target templates. All else being equal, the higher the clamp score the better. However, the clamp score is not relevant to stability of the clamp/product hybrid, which is estimated by the annealing temperature using the nearest neighbor method.•The core/clamp boundary can be restricted to always be on a codon boundary.•Genetic code tables are used to back-translate from protein to DNA.•Codon usage tables are derived from the Codon Usage Database.Tables currently available: request tables for other organisms here.The codon usage table is used to convert the amino acid PSSMconstructed from the input blocks into the DNA PSSM. It can also be used to determine the content of the clamp region.•Clamp residues can be selected as the most common codons of the consensus amino acids are used in the clamp. Else the clamp residues are the ones with maximum weight in the DNA PSSM, which may result in 'artificial' codons. If the clamp residues are taken from the DNA PSSM and more than one residue in a position have maximum weight, one of them is selected at random to keep the clamp non-degenerate.•The program output can show all possible primers or only the single most degenerate primer in each region.Basic tipsOnce you have a block(s) in the input window of the CODEHOP page you can "Look for primers" using the default parameters. You can adjust the setting according to your intended use of the primers and the results you got with the defaults. If you don't get predictions, or you don't like what you get we recommend to first raise the degeneracy to 256 or higher (if you dare ...) and retry. Next, you might try raising the strictness of the core region, for example to 0.1 or 0.25.Your target sequence(s) might be expected to be more similar to some specific sequence(s) in the input blocks. In this case you can bias the primers towards these sequences. Rather than raising the degeneracy or strictness, increase the weights of the specific sequences. The weights are the values following each sequence segment of the block. Usually the highest weight is 100. In the CODEHOP input window increase the weights of your specific sequences (say to 3 or 4 times the original weight or to 200 or 400). You can also remove individual sequences from the input blocks by down-weighing them to 0 (the minimal weight) if they are too divergent and prevent finding primers.For amplification, we recommend using AmpliTaq Gold with a 9' preheat (this provides an automatic hotstart - a hotstart of some kind isimportant). We have had success using the time-release feature with the addition of 15-20 extra cycles. The primer finding strategy of the CODEHOP program (Rose, et al, manuscript accepted by NAR) is different from the usual degenerate PCR strategies. It is desirable to keep annealing temperatures high - 60o C is OK if you have a 60o C clamp. We recommend trying the highest temperature that yields a clean PCR product. We have used "touchdown" PCR down to Tm-3o C or lower, say from 63o C down to a good clamp annealing temperature in -0.5 to -1o C increments, and the remaining cycles are carried out at the 53-57o C clamp annealing temperature for a 60o C clamp. The intent of the touchdown is to give the correct product a head start, because it is likely to anneal at a higher temperature than any failure product. Once the clamp 'takes over', then all primed products, whether correct or not, will be on an even footing, so we try to keep the stringency high in all cycles. With luck, it should not be necessary to gel-purify product, but may rather try cloning directly from the reaction mix if a single band of the expected size is obtained.We and a few other users were already successful in using the CODEHOP strategy and program to amplify various sequences from complicated and diverged genomes. Please let us know if you have any tips to pass on based on your experience using CODEHOP-predicted primers.PublicationResults obtained by this method should cite:"Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly-related sequences" by T.M. Rose, E.R. Schultz, J.G. Henikoff, S. Pietrokovski, C.M. McCallum and S. Henikoff, Nucleic Acids Research, 26(7):1628-1635.Genes identified using CODEHOP.CODEHOP programThe CODEHOP program designs a pool of primers containing all possible 11- or 12-mers for the 3' degenerate core region and having the most probable nucleotide predicted for each position in the 5' non-degenerate clamp region.The program consists of the following steps: (note the scheme on the right)1) A set of blocks is input, where a block is an aligned array of amino acidsequence segments without gaps that represents a highly conserved region ofhomologous proteins. A weight is provided for each sequence segment, which can beincreased to favor the contribution of selected sequences in designing the primer.A codon usage table is chosen for the target genome.2) An amino acid position-specific scoring matrix (PSSM) is computed for each blockusing the odds ratio method.3) A consensus amino acid residue is selected for each position of the block as thehighest scoring amino acid in the matrix.4) For each position of the block, the most common codon corresponding to the aminoacid chosen in step 3 is selected utilizing the user-selected codon usage table.This selection is used for the default 5' consensus clamp in step 8.5) A DNA PSSM is calculated from the amino acid matrix (step 2), genetic codetable and codon usage table. The DNA matrix has three positions for each position of the amino acid matrix. The score for each amino acid is divided amongits codons in proportion to their relative weights from the codon usage table, andthe scores for each of the four different nucleotides are combined in each DNAmatrix position. Nucleotide positions are treated independently when the scores arecombined. As an option, the highest scoring nucleotide residue from each positioncan replace the most common codons from step 4 that are used in the consensus clamp.6) The degeneracy is determined at each position of the DNA matrix based on thenumber of bases found there. As an option, a weight threshold can be specified suchthat bases that contribute less than a minumum weight are ignored in determiningdegeneracy.7) Possible degenerate core regions are identified by scanning the DNA matrix inthe 3' to 5' direction. A core region must start on an invariant 3' nucleotideposition, have length of 11 or 12 positions ending on a codon boundary, and have amaximum degeneracy of 128 (current default). The degeneracy of a region is theproduct of the number of possible bases in each position.8) Candidate degenerate core regions are extended by addition of a 5' consensus clampfrom step 4 or 5. The length of the clamp is controlled by a melting point temperaturecalculation (current default = 60o) and is usually ~20 nucleotides.9) Steps 7 and 8 are repeated on the reverse complement of the DNA matrix from step5 for primers corresponding to the opposite DNA strand.CODEHOP program scheme1) input - - - - - - - - - - - - - - - seq 1 Protein sequence block - - - - - - - - - - - - - - - seq 2- - - - - - - - - - - - - - - seq 3- - - - - - - - - - - - - - - seq 4- - - - - - - - - - - - - - - seq 5- - - - - - - - - - - - - - - etc.|| 2) transformation to AA PSSMV| | | | | | | | | | | | | | | Ala AA PSSM| | | | | | | | | | | | | | | Cys| | | | | | | | | | | | | | | Asp| | | | | | | | | | | | | | | Glu| | | | | | | | | | | | | | | Phe| | | | | | | | | | | | | | | Gly| | | | | | | | | | | | | | | His| | | | | | | | | | | | | | | Ile| | | | | | | | | | | | | | | Lys| | | | | | | | | | | | | | | etc.| || | 3) calculation of AA consensus sequence| V| - - - - - - - - - - - - - - - AA consensus sequence || || | 4) transformation to DNA consensus sequence| V| ------------------------------------------- DNA consensus sequence | || | 5) back-translation to DNA PSSM| V| ||||||||||||||||||||||||||||||||||||||||||| A DNA PSSM| ||||||||||||||||||||||||||||||||||||||||||| C| ||||||||||||||||||||||||||||||||||||||||||| G| ||||||||||||||||||||||||||||||||||||||||||| T| | || | | 6) calculation of degeneracies| | V| | ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ position degeneracy values | || | 7) identify degenerate regions ("===")| || 8) identify consensus regions for degenerate regions ("---")| |V V5' -------==== 3' CODEHOP primers output 3' ====--------- 5'。
实验2 引物设计与测序结果分析

学院:______ 班级:_______ 学号:_________ 姓名:__________ 成绩:______ 实验二引物设计及测序结果分析目的:1、掌握常规引物设计的原则及操作流程。
2、熟悉简并引物设计的原理及操作方法。
3、熟悉引物设计软件及在线引物设计工具的操作方法。
4、掌握使用相关软件及在线工具分析测序结果的方法。
内容:1、使用Primer Premier、Oligo、BLAST等软件及在线工具进行常规引物设计,并对引物扩增效率、特异性进行评价。
2、使用DNAMAN软件进行常规引物快速设计。
3、使用NCBI中的在线引物设计工具Primer-BLAST快速设计引物。
4、使用在线工具CODEHOP设计简并引物。
5、使用Chromas、BioEdit软件查阅测序结果峰图文件。
6、使用DNAMAN软件对测序序列进行编辑,进行序列拼接。
软硬件要求:联网计算机,预装Windows 7操作系统,预装IE或Chrome浏览器、英汉电子词典(有道词典或金山词霸),预装DNAMAN7、Primer Premier5、Oligo7、Chromas、BioEdit等生物信息学分析软件。
操作及问题:一、Primer Premier5、Oligo7、BLAST常规引物设计本部分操作将使用Primer Premier5、Oligo7、BLAST等软件及工具设计拟南芥AtBADH基因编码区全长特异引物。
(参考“第四章引物设计及测序结果分析”课件)(一)使用Primer Premier5搜索引物1、在NCBI数据中查找登录号为NM_001198470的序列记录,查阅相关信息,并下载序列将其保存为fasta格式文件。
问题1:该序列是什么类型的序列?该序列编码区在什么位置?2、打开Primer premier5软件,点击键ctrl+V将上一步中下载的序列粘贴入弹出的GeneTank窗口中(或者点击。
3、点击GeneTank窗口中左上角的Primer premier窗口中点击Search Criteria窗口中根据要求选择合适选项及参数,选定后,点击Search Progress窗口中有Search Results窗口;如没有出现数重新搜索引物。
ecshop操作流程
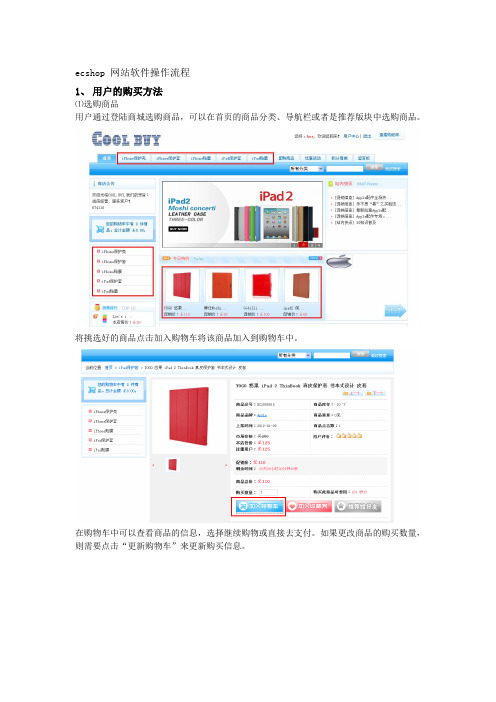
ecshop 网站软件操作流程1、用户的购买方法⑴选购商品用户通过登陆商城选购商品,可以在首页的商品分类、导航栏或者是推荐版块中选购商品。
将挑选好的商品点击加入购物车将该商品加入到购物车中。
在购物车中可以查看商品的信息,选择继续购物或直接去支付。
如果更改商品的购买数量,则需要点击“更新购物车”来更新购买信息。
点击去结算会出现让你输入用户名和密码的界面如果已经注册过该商城的会员,只需要在用户登录界面输入用户名和密码直接登录。
如果不是该商城的会员,需要填写用户名、电子邮件地址、密码、确认密码字段就可以注册会员了。
还可以选择“不打算登录,直接购买”选项,进行商品的购买,但是这样非会员无法享受购物积分、无法查询订单等,所以我们建议您花一分钟时间注册成为会员,这样就能享受整个网站强大的会员功能和多种优惠措施了。
注册会员成功后会出现填写收货人信息的页面,将页面中的信息填写完整,点击“配送至这个地址”会出现以下选择区域:①商品列表:核对商品列表,如有错误可以点击“修改”来改购物车中的商品信息②收货人信息:核对收货人信息,如有错误可以点击“修改”来改收货人地址③配送方式:选择适合自己的配送方式,可以到达收货人所在地区范围的④支付方式:会员自行选择商家提供的支付方式⑤商品包装:根据自己需要来选择⑥其他信息:若有红包,输入红包序列号,点击验证红包就可获取红包信息。
如有什么要求可填写订单付言核实完整个订单信息无误后,点击提交订单出现这个界面可以直接点“立即使用快钱支付”进行线上付款。
订单提交成功,可以在首页的“用户中心”查看点击我的订单查看订单信息会员也可在首页的订单查询中输入订单号查询订单状态2、产生订单处理方法:点击订单列表就可以查看订单详情可以看到该订单的订单号、下单时间、收货人、总金额、应付金额以及订单状态将鼠标放在订单号处,即可看到该订单的商品信息点击查看可以看到该订单的详细信息,可以对该订单的基本信息、其他信息、收货人信息、商品信息、费用信息作出修改核实信息无误后可以选择“确认”来确认该订单也可对该订单做出付款、取消、无效、售后的选择。
数字门店系统用户操作手册说明书

[User Manual]数字门店系统用户操作手册<Version 1.0>目录1引言 (3)2运行环境 (3)2.1主机及硬件设备 (3)2.2操作系统及配置依赖 (3)3系统初始化 (3)3.1系统安装及初始化 (3)4.系统功能操作说明 (4)4.1店端应用模块/功能操作说明 (4)4.1.1业务介绍 (4)4.1.2功能介绍 (4)4.1.3功能操作界面图示 (4)4.1.4操作指南 (4)4.2后台模块/功能操作说明 (10)4.2.1业务介绍 (10)4.2.2功能操作界面图示 (10)4.2.3操作指南 (11)4.3报表管理模块/功能操作说明 (16)4.3.1功能操作界面图示 (16)4.3.2操作指南 (16)1引言本文档为数字门店系统用户使用手册2运行环境2.1主机及硬件设备B/S架构,对使用端无硬件要求。
2.2操作系统及配置依赖3系统初始化3.1系统安装及初始化3.1.发布,网络环境已提供,包括:对应Wifi、相关域名、发布所使用的服务器、发布所涉及的探针、摄像头等硬件设备;3.2.服务器软件环境已安装,包括:操作系统MS Windows 2012 R2(IIS 8.5)以上,.net framework4.5.2以上,SQL Server 2016,Mongo DB 2.8.X(可选);本地硬件相关服务器,需部署内网,以便调用相关硬件;3.3.Sitecore已完成安装,所使用的版本 8.2 (rev. 171121);本文不涉及Sitecore 本身的安装,仅包括项目涉及的更新内容;3.4.Sitecore安装域名与端口,如:1234,本文之后的叙述中将以[http://Sitecore:port]代替,结尾不含路径反斜杠。
3.5.Sitecore网站数据源均设置于安装目录\App_Config\ConnectionStrings.config中,请勿在Web.config中单独设置。
引物设计几种引物设计软件的介绍及使用

8
精选ppt课件
9
引物选择
③ ②
④
①
精选ppt课件
10
选择标准
1.无二级结构(所有按钮都不是红色显示) 2.产物所处位置是否满足要求。 3. 两条primer 的Tm值相差不超过3℃,最好在
1℃内。引物的Tm值最好不低于60 ℃,以保证产 物的特异性。 4.引物GC含量是不是在40%~60%间。
Primer3 Very popular primer design tool for
designing primers for PCR, hybridization.
Primerfinder Easy to use, free, online primer
design program
MethPrimer A free online program for designing
probability of random primering. Batch mode
allows designing for multiple sequences. Users
can also calculate the melting temperature using
the nearest neighbor model.
primers for methylation specific PCR (MSP) and
bisulfite sequencing PCR.
Primo Primo online is a friendly PCR primer design
tool. It reduces PCR noise by lowering the
精选ppt课件
19
某超市-电脑使用手册

某超市-电脑使用手册某超市-电脑使用手册第一章:入门与登录1.1 电脑开机与连接超市提供的电脑已经连接好了电源和网络线,只需按下电源按钮即可开机。
请确保电脑后面的网线与网络连接正常。
1.2 用户登录在电脑启动后,屏幕会显示登录界面。
输入您的用户名和密码,点击“登录”按钮即可进入系统。
如果您没有账号,请联系超市管理员进行注册。
第二章:主界面与功能模块介绍2.1 主界面登录成功后,您将看到超市管理系统的主界面。
主界面的左侧是主菜单,展示了电子商务、库存管理、销售管理、会员管理、系统设置等主要功能模块的快捷入口。
2.2 功能模块2.2.1 电子商务在这个模块中,您可以查看最新的促销活动、商品展示、在线购物、订单管理等功能,方便您进行线上购物。
2.2.2 库存管理该模块涵盖了库存商品的添加、修改、删除、查询等功能,以及库存报表和库存预警等功能,支持您对超市商品的全面管理。
2.2.3 销售管理在这个模块中,您可以进行商品销售、退货、查询、报表查看等操作,方便您对超市销售数据的分析和管理。
2.2.4 会员管理该模块提供了会员信息的添加、修改、删除、查询等功能,以及会员等级设置、积分管理、会员报表等功能,方便您对会员数据的管理和分析。
2.2.5 系统设置在这个模块中,您可以对超市管理系统进行基本设置,包括用户管理、权限管理、系统参数设置等功能。
请注意不要随意更改系统设置,以免造成错误操作。
第三章:具体操作指南3.1 商品管理3.1.1 商品的添加在库存管理模块中,点击“商品管理”,然后选择“添加商品”。
按照提示填写商品的相关信息,如名称、价格、库存量等。
完成后,点击“确认”按钮保存商品信息。
3.1.2 商品的修改和删除在库存管理模块中,点击“商品管理”,然后选择“修改商品”或“删除商品”。
根据需要修改或删除相应的商品信息。
3.2 销售管理3.2.1 商品的销售在销售管理模块中,点击“新增销售”按钮。
选择要售出的商品,并输入销售数量和销售价格,点击“确认”按钮完成销售操作。
立通达 COD 电商 ERP 系统操作说明书

立通达COD电商ERP系统操作说明书2020年6月5日修订记录建议使用谷歌,Firefox浏览器操作后台,兼容性会比较的好!! 请务必爱动手、爱思考, 仔细操作,不要固执己见;遇到不明白的地方大胆尝试,不要慌;一、首次拿到后台的基础设置1.1 安全策略登陆后台,修改密码以及验证码,。
密码用于登录后台,验证码用于查看具体内容 , 二次密码提升安全性 ,为了系统安全,不要使用弱密码和常用密码,并且密码长度应该保证在12位及以上。
验证码用于查看数据时使用1.2如何开启登录IP访问限制1.3 网站基础设置,开管理员账号,设置登录用户名,密码以及真实姓名, 配置权限1.4 设置部门数据,二、网站基础设置首先解析域名,添加宝塔,这里就不再细说了, 具体看https以及添加宝塔方法操作文档2.1 添加分类,分类数据显示在网站首页导航, 添加网站时可设置对应的分类2.2 隐私协议等文章进入文章菜单下,编辑或者新增文章内容。
文章内容我们应该根据自己的实际情况进行编写,一篇文章可以支持多语言,在对应的语言添加就行了,文章内容包括但不限于:自己公司简介、联系方式、售后服务、隐私条款等相关信息,设置商城的paypal收款账号,目前仅支持paypal , 信用卡不支持2.3 图片域名设置这里设置本地图片域名,前期系统搭建都会配置好,如用CDN,则可以不设置,需联系技术配置好CDN, 设置以后所有的图片都使用这个域名, 图片为绝对路径地址如:/data/upload/123456.jpg2.4 超商店铺系统默认提供自动化抓取超商店铺数据,如遇到有些店铺更新不及时,也可以手动更新,状态为启用中的会在网站下单出给顾客使用,,关闭了的不能使用三、网站建站3.1 添加网站点击网站列表进入, 然后添加网站红色标题的内容为必填项,请务必按要求录入, 网站模板提供4套,154,155,156为类Shopify 模板, 147模板为类阿噗风格模板, 网站模板为必选项,因为要开企业广告账户,必须使用综合站模板, 具体的产品页面模板在建产品的时候可以控制.注: 本系统区分主域名跟二级域名,务必理解好, 如跟是两个独立的域名,域名设置不当,会造成出单后订单同步失败部门选择以后无法修改, 务必保存前确认好,选择地区: 决定是哪个国家的站,以及对应的货币符号,网站前端也有一些国家不同,要求也不同选择语言: 用于控制页面显示那种语言,目前支持国家语言香港(中国)/台湾(中国)/日本/泰国/柬埔寨/老挝/菲律宾/越南/马来西亚/新加坡/印度尼西亚/迪拜/伊朗/卡塔尔/阿联酋/沙特/韩国FB像素ID:当产品使用的是商城模板, 像素追踪用就是这里的像素ID ,如果是单页面模板,用的是具体填写的,后续再做说明FB客服:商城模板右侧按钮点击跳转后的FB客服付款方式: 用于控制完整显示哪些付款方式,其中711超商,全家超商,只对台湾有用导航品类: 显示的是一级分类数据,用于显示在网站顶部导航底部政策: 控制网站首页底部帮助文章, 在添加文章那的内容显示市场价: 用户控制商城网站使用显示市场价, (FB对过于夸张的市场价不友好)購物車按鈕:用于控制商城是否显示购物车按钮以及加入购物车, 用于推商城产品提高转化率3.1 添加产品进入网站产品, 点击添加产品,,进入添加产品页面3.1.1添加产品基本信息添加产品基本信息, 红色字体标注的,为必填项,必须要录入或选择,不然产品会下单异常接着往下来,细致讲解每一项的用处销售方式: 若要做套餐,则产品类型必须为套餐, 单品比较适合商城销售所属网站: 产品要挂在哪个网站下, 可以支持一个产品绑定多个产品,至少选择一个产品模板: 这是重点, 决定产品用什么模板销售,商城模板主要用的是网站选的模板详情页,如要单页面模板,一定要选择单页面模板.商城模板: 选择了以后使用的就是网站绑定的商城模板详情页,做商城销售必须选商城模板单页面模板: 需录入单页面使用的域名, 域名可以跟上面选择的网站一致,也可以不一样,后缀自动生成了,对应的地区, 模板使用的语言, 以及单独的FB像素ID ,选择模板,优先推荐style134, style133 ,转化率比较高,显示底部帮助: 控制单页面模板底部是否显示商城的文章显示返回主页: 控制单页面是否显示返回主页的按钮付款方式: 控制单页面使用的付款方式, 目前不支持paypay ,后台订单显示的标题:是只顾客下单了以后在订单,采购,仓库,财务,转寄这里显示的标题, 主要给公司内部人员看,填中文顾客看到的标题: 是指显示在模板上的标题,给顾客看,填对应国家的语言其他的细腻按照字面意思填就行了FB视频ID: 主要用于FB拍摄的视频或者FB直播的ID ,,主要针对商城,单页面不支持采购链接: 给公司采购看的,保存了以后无法修改, 第一次要录入好抖音像素: Tik Tok的追踪代码,直接复制Tik Tok给的那段头部追踪代码: 如要加客服插件,或Google追踪这些,FB像素代码不要加在这里,系统默认集成好了的,主图,轮播图上传,支持jpg,png,,不支持视频上传,轮播图上传了以后显示轮播,不显示主图轮播图的优先级高于主图如何添加属性,系统必须要至少有一个属性,否则无法生成SKU,点击添加属性按钮,左边属性是给自己看的,填中文就行了,,邮编是给客户看的,需要填推广国家对应的网站注意属性跟属性值的区别,比如属性, 通常是颜色这类,属性值是具体的颜色名称,如红色, 黑色,白色.如何添加详细描述产品基本信息就添加完了,3.2 做套餐在产品列表,点击做套餐按钮,如做套餐,编辑产品类型为套餐, 重新刷新即可出现按提示输入套餐,套餐显示的名称, 价格,排序,运费,以及允许购买数量, 套餐图片目前大部分模板是不支持的,请不要添加套餐产品,如一个套餐捆绑多个相同的产品,,产品需要选择属性,可以添加多次产品就行,如只有一个属性不需要选择多个属性,则可以直接添加数量就行,3.3 添加评论入口,默认所有产品都能添加评论,3.4 控制产品首页不显示设置首页不显示以后的效果是不在,在网站首页的Banner下产品列表中显示,,但是分类列表还是可以看到3.5 用于查看填好了属性以后生成的SKU ,可以修改SKU名称如添加了属性以后要修改,但是系统没同步,,则手动来这里修改对应的SKU名称,另外可以打印SKU条码3.6 复制产品注意看复制产品的页面是否生成新产品, 会生成一个新的产品同步到ERP系统,生成新的SKU ,如同一个产品只是复制不同的链接去推广,则默认选否, 这样可以保证多个站下单以后同步到订单,采购,仓库这些流程都是同一个产品,3.6 产品删除删除产品为软删除,删除后可以在回收站找回来,进行恢复建站的基本操作结算完毕.四、订单管理3.1 审核订单订单生成以后,系统会根据设定的规则自动化审核一遍,给出对应的参考以及重复数量重复数统计规则为重复数统计按下单电话,姓名完全相同则标红,数量包含本身订单数 , 另同一IP下单数超过1单也会标红已审核订单,可以一键直接通过,4.2 所有订单主要是提供综合查看订单信息功能, 订单信息可以控制账号权限,是否查看收货人信息,4.3 广告费导入广告费到系统,可以为ROI统计提供广告费数据4.3 ROI4.4 更新订单状态主要用于修改订单状态,标记各种环节的订单,比已采购了的,已签收,拒收之类的,企业版可以直接按系统的流程来走,无需通过这个功能来标记。
codesoft6.0使用手册
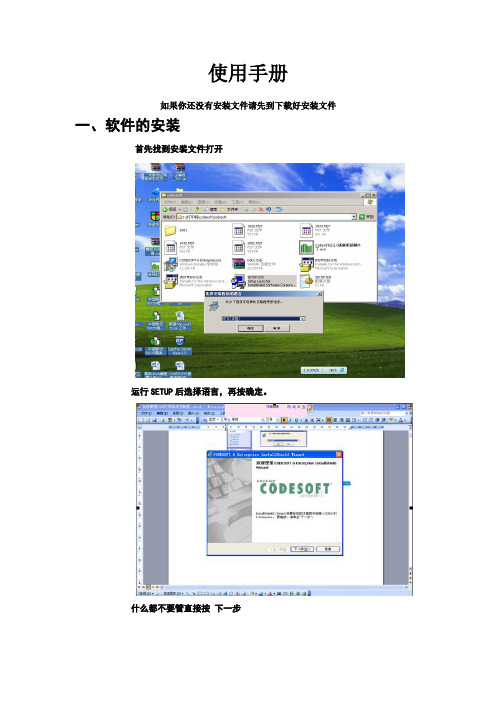
使用手册如果你还没有安装文件请先到下载好安装文件一、软件的安装首先找到安装文件打开运行SETUP后选择语言,再按确定。
什么都不要管直接按下一步选择我接受点击下一步填写用户名和单位,随便填的,填好后点击下一步更改安装目录,默认就好,直接点击下一步选择完全安装点击下一步点击安装出现安装进度稍等一会安装进度消失后,出现以上画面,点击完成 ,完成后桌面出现一个CS6 快捷方式,在這里值得注意,最好先不要運行CODESOFT.,因为此时运行的CS6是试用版,接下来我们要將其破解。
怎么破解呢首先我们找到安装盘中有一个Codesoft6[1].0破解版破解补丁.exe 的文件夹,直接运行安装这个破解程序文件,如下图所示:安装随即完成。
二、软件的使用常用的功能介绍,如下图所示打开CS6后,首先要设置页面大小,如下图所示页代表整个标签也就是说它的宽度是底纸的宽度;高度则是面纸的高度;页设置好了以后,我们就要设置标签大小,如下图:标签的宽度就是面纸的宽度,而高度和面纸的一样其中下面行.列的选择是根据标签的规格来确定的象上图的则是1行1列接下来,设置页面边距,设好后,标签大小算是完成了,如下图;图中所说的左側是指底纸边缘到面纸边缘的距离注意:设定标签大小的时候,填写里面的参数要非常准缺,要不能打印出来的内容偏位或标签跳张,一定要学会用以下公式:(标签)宽度 + (左边空)宽度 = (页)宽度(标签)高度 = (页)高度在标签页面上如何插入条码内容呢插入条码,如下图:把鼠标点击一下创建条码图标,然后出现一个插入条码的图标,再把图标移到标签页面上来,再点击一下,会出现条码的一些设定,如下图注意:校验字符:指的是条码的一个校验码,是电脑自动生成,一般不去理会,旋转:0度,90度,180度,270度指条码出打出来的方向。
注意:左边排出来的条码可以看出条码数字的变动,这里大家可以去试着改改就清楚了。
注意:这里只针对设定条码下方数据字体的大小默认 TRUE TYPE,也就是选择字体类型默认默认设置字体设置字体的宽度设置字体的大小,最后一项数据源是设定条码变量,连接条码,套用数据库用得到的,在在标签页面上如何插入文字内容呢如插入文字,如下图:把鼠标点击一下生成文本,然后出现一个大的十字架的图标,再把图标移到标签页面上来,再点击一下,会出现字本的一些设定,这里跟插入条码的动作是一样的,这里可直接输入内容。
奥派电子商务操作手册节选

在“电子支付实践”模块中选择“支付通”。
选择支付通模块的“服务商平台”进入,为服务商绑定一个银行账号。这样,支付通的用户才能使用支付通。
在“银行账户管理”中【新增账户】。
填写账户信息,银行商户编号在登录企业网上银行时可以看到。填写完成后点击【添加银行账户】,这样,服务商就成功绑定银行账户了。
【注意】:在该页面的下方,同样有操作帮助,学生也可按照该帮助进行实验。
单位注册信息。单位注册信息是在实验中有需要填写单位信息时系统默认的内容。在学生管理界面点击“我的实验”下的【单位注册信息】,填写部门的相关信息,点击【提交】,系统提示操作成功。
我的邮件。我的邮件是一个虚拟的邮件系统,软件内所有邮件均可通过该系统查收。点击“我的实验”下的【我的邮件】,可以查看到实验过程中收到的所有邮件信息。
点击想要查看的邮件主题,即可看到邮件的详细内容。
我的短消息。我的短消息是一个虚拟的手机短消息系统,实验过程中的所有短消息都可以通过该系统查收。点击“我的实验”下的【我的短消息】,可以看到实验过程中的收到的手机短消息。
个人信息。点击“我的实验”下的【个人信息】,可以对学生个人信息做一些修改。
修改完后点击【提交】即可。
按照以上方法,在工商银行申请王军的个人账户,在招商银行申请张玲的个人账户。申请的时候要注意,系统默认的是李明的资料,需要把相关信息改过来。另,在招商银行申请账户时,需要“角色选择”后选择相应的银行。
2.
点击【企业账户申请】。
按照申请要求,填写申请表,如图所示。填写完成后,点击表格下方的【申请】,等待银行柜台审核。
进行审批,点击【审批通过】。
三、
【实践涉及步骤】
服务商增加银行账户
支付通账户手机注册
小型网上购物系统使用说明书

小型网上购物系统使用说明书1.系统介绍随着互联网的深入应用,络购物的交易额占社会消费品零售总额的比例节节攀升,方便快捷的网络购物从一个新生事物变成了越来越多人选择的购物方式。
用户通过互联网,能足不出户,轻松闲逸地实现自己订购餐饮和食品,随着食天下网上订餐平台的兴起,网上订餐已经逐渐成为了一种潮流。
基于这种环境,网上购物系统受到广大商家的青睐。
网上购物系统分为前台登录和后台登录两种方式,其中,用户登录前台方式可以根据展示的商品网上购物商城主要由前台会员模块和后台管理模块两部分组成。
前台模块主要包括会员注册、登录、修改个人信息、购物、查询购物情况和查看收藏等功能。
后台模块主要包括后台管理员用户在后台登录页面可以对会员、商品、订单进行管理,同时也可以进行销售统计、修改密码等操作。
管理员还可以根据实际情况添加其他管理员以维护该网上购物商城的购物环境和安全。
2.系统文件1.在App_Code文件夹中有连接数据库的公共类(和)。
2.在App_Data文件中有被连接的数据库文件()。
3.在images文件夹中用于存放系统所需的图片。
4.在js文件夹中有控制系统界面的Javascript文件。
3.系统结构网上购物系统流程图如下:4.使用说明部署运行环境计算机操作系统:Windows 7 旗舰版 64位;所需安装的开发软件:Microsoft Visual Studio 2012和Microsoft SQL Server 2008;软件功能介绍界面介绍运行程序,出现网上购物系统登录界面,如图1所示:图1 用户登录界面用户注册在用户登录界面,用户可以选择直接登录或者注册,新用户注册个人信息时单击【注册账号】按钮书出现注册页面,如图2所示。
图2 用户注册页面填写此页面时需注意,文本框内全部为文本格式。
为方便用户在登陆后界面核对信息,个人信息页面还之后用户还可以上传个人照片,点击【选择文件】按钮,根据图片路径,选择图片,如图3所示。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
CODEHOP : COnsensus-DEgenerate HybridOligonucleotide PrimersCODEHOP helpOn this page:Other pages: •General information • Getting started •Obtaining input alignments • CODEHOP program algorithm • Terms and parameters • CODEHOP manuscript • Strictness parameter details • Basic tips• PublicationGeneral informationThe CODEHOP program designs PCR (Polymerase Chain Reaction) primers from protein multiple-sequence alignments. The program is intended for cases where the protein sequences are distant from each other and degenerate primers are needed.The multiple-sequence alignments should be of amino acid sequences of the proteins and be in the Blocks Database format Proper alignments can be obtained by different methods .The result of the CODEHOP program are suggested degenerate sequences of DNA primers that you can use for PCR. You have to choose appropriate primer pairs, get them synthesized and perform the PCR.A CODEHOP primer is degenerate at the 3' core region, with a length of 11-12 bp across four codons of highly conserved amino acids, and is non-degenerate at the 5' consensus clamp region, with a length which depends on its desired annealing temperature, typically between 20 and 30bp:5' 3'--------------------===========non-degenerate degenerateconsensus clamp core#bases from temp 11-12 basesThe hybrid structure (5' consensus and 3' degenerate) of CODEHOP primers allow the PCR amplification to be specific during the early cycles from the original source DNA and selective during the late cycles from the PCR synthesized products:Schematic comparison of standard degenerate PCR (left) with the CODEHOP (right), illustrating regions of mismatch in primer-to-template annealing during early PCR cycles and in primer-to-product annealing during subsequent cycles. Vertical lines indicate nucleotide matches between primer (arrow) and template or synthesized product. The overall degeneracy is the product of degeneracies at each nucleotide position, so that the fraction of precisely hybridizing primers is 1/degeneracy.Obtaining input alignmentsInput alignments for the CODEHOP program must be in the Blocks Database format. Block multiple-sequence alignments are ungapped and usually local alignments. Local alignments cover only parts of the protein sequences. The regions between the blocks are the ones with no sequence similarity or where gaps must be inserted to align the sequences.You can get a multiple alignment from a group of related protein sequences using the Block Maker or other automated methods(such as ClustalW). The alignments can also be manually made or modified according to your knowledge of the proteins (position of the active sites orpost-translational modifications etc.). In any case the alignments passed to the CODEHOP program must be in the Blocks format. BlockMaker blocks need no reformatting. Appropriate parts of Clustal- or FASTA-formatted multiple alignments can be automatically made into blocks by the Blocks multiple alignment processor. Other types of multiple alignment can be semi-manually reformatted with the Blocks formatter. All of the above Blocks programs have links to send the resulting blocks directly to theCODEHOP program and also provide other information for evaluating the blocks (logos, trees, searches).More information on multiple alignments can be found in the notes for the ISMB97 tutorial on Introduction to making and using protein multiple alignments.Terms and parameters•Degeneracy measures how many different sequences are specified by the primer. The degeneracy of each position depends on the sum of the nucleotides appearing in it (1 to 4). The degeneracy of the primer is the product of the degeneracies of all of its positions.The degenerate core region is scored by its degeneracy, with lower degeneracy scored higher. A standard degenerate alphabet is used in the core region.•Degeneracy strictness specifies how to count nucleotide(s) with low occurrences. It is a parameter between 0 and 1. Strictness of 0 means that all nucleotides that actually appear in the position arecounted to calculate the degeneracy. Strictness of 1 means that only the nucleotides with the highest value in the position are counted.Intermediate strictness values give behavior in between. You can look at a detailed explanation for more information on how thestrictness is calculated.•Temperature is computed for the clamp region plus any adjacent non-degenerate positions in the core region. Differenttemperatures affect only the length of the clamp; highertemperatures result in longer clamps. Temperature is computed using the nearest-neighbor method of Rychlik, et al ( NAR 1990 18:21, 6409-6412). This method takes into consideration the primer and K+ (potassium ions, taken as 50 mM) concentrations.•Nucleotide runs in the clamp region can be limited (the default is5 in a row). If longer runs are encountered, they are broken up bytaking the next highest- scoring nucleotide in the position with the lowest-scoring run nucleotide.•The clamp score is a measure of how well the non-degenerate 5' clamp matches the block given a codon usage table. The score is a weighted average of the maximum scores in each column of the DNA PSSM. A perfect score of 100 is obtained if the clamp consists of invariant methionines and tryptophans, and a minimum score of 25 is obtained if A, C, G and T are equally likely at all positions. In computing the clamp score, positions are increasingly down-weighted asdistance from the degenerate 3' core increases. The clamp score is intended to estimate how well the clamp will stabilize the core inannealing to the target templates. All else being equal, the higher the clamp score the better. However, the clamp score is not relevant to stability of the clamp/product hybrid, which is estimated by the annealing temperature using the nearest neighbor method.•The core/clamp boundary can be restricted to always be on a codon boundary.•Genetic code tables are used to back-translate from protein to DNA.•Codon usage tables are derived from the Codon Usage Database.Tables currently available: request tables for other organisms here.The codon usage table is used to convert the amino acid PSSMconstructed from the input blocks into the DNA PSSM. It can also be used to determine the content of the clamp region.•Clamp residues can be selected as the most common codons of the consensus amino acids are used in the clamp. Else the clamp residues are the ones with maximum weight in the DNA PSSM, which may result in 'artificial' codons. If the clamp residues are taken from the DNA PSSM and more than one residue in a position have maximum weight, one of them is selected at random to keep the clamp non-degenerate.•The program output can show all possible primers or only the single most degenerate primer in each region.Basic tipsOnce you have a block(s) in the input window of the CODEHOP page you can "Look for primers" using the default parameters. You can adjust the setting according to your intended use of the primers and the results you got with the defaults. If you don't get predictions, or you don't like what you get we recommend to first raise the degeneracy to 256 or higher (if you dare ...) and retry. Next, you might try raising the strictness of the core region, for example to 0.1 or 0.25.Your target sequence(s) might be expected to be more similar to some specific sequence(s) in the input blocks. In this case you can bias the primers towards these sequences. Rather than raising the degeneracy or strictness, increase the weights of the specific sequences. The weights are the values following each sequence segment of the block. Usually the highest weight is 100. In the CODEHOP input window increase the weights of your specific sequences (say to 3 or 4 times the original weight or to 200 or 400). You can also remove individual sequences from the input blocks by down-weighing them to 0 (the minimal weight) if they are too divergent and prevent finding primers.For amplification, we recommend using AmpliTaq Gold with a 9' preheat (this provides an automatic hotstart - a hotstart of some kind isimportant). We have had success using the time-release feature with the addition of 15-20 extra cycles. The primer finding strategy of the CODEHOP program (Rose, et al, manuscript accepted by NAR) is different from the usual degenerate PCR strategies. It is desirable to keep annealing temperatures high - 60o C is OK if you have a 60o C clamp. We recommend trying the highest temperature that yields a clean PCR product. We have used "touchdown" PCR down to Tm-3o C or lower, say from 63o C down to a good clamp annealing temperature in -0.5 to -1o C increments, and the remaining cycles are carried out at the 53-57o C clamp annealing temperature for a 60o C clamp. The intent of the touchdown is to give the correct product a head start, because it is likely to anneal at a higher temperature than any failure product. Once the clamp 'takes over', then all primed products, whether correct or not, will be on an even footing, so we try to keep the stringency high in all cycles. With luck, it should not be necessary to gel-purify product, but may rather try cloning directly from the reaction mix if a single band of the expected size is obtained.We and a few other users were already successful in using the CODEHOP strategy and program to amplify various sequences from complicated and diverged genomes. Please let us know if you have any tips to pass on based on your experience using CODEHOP-predicted primers.PublicationResults obtained by this method should cite:"Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly-related sequences" by T.M. Rose, E.R. Schultz, J.G. Henikoff, S. Pietrokovski, C.M. McCallum and S. Henikoff, Nucleic Acids Research, 26(7):1628-1635.Genes identified using CODEHOP.CODEHOP programThe CODEHOP program designs a pool of primers containing all possible 11- or 12-mers for the 3' degenerate core region and having the most probable nucleotide predicted for each position in the 5' non-degenerate clamp region.The program consists of the following steps: (note the scheme on the right)1) A set of blocks is input, where a block is an aligned array of amino acidsequence segments without gaps that represents a highly conserved region ofhomologous proteins. A weight is provided for each sequence segment, which can beincreased to favor the contribution of selected sequences in designing the primer.A codon usage table is chosen for the target genome.2) An amino acid position-specific scoring matrix (PSSM) is computed for each blockusing the odds ratio method.3) A consensus amino acid residue is selected for each position of the block as thehighest scoring amino acid in the matrix.4) For each position of the block, the most common codon corresponding to the aminoacid chosen in step 3 is selected utilizing the user-selected codon usage table.This selection is used for the default 5' consensus clamp in step 8.5) A DNA PSSM is calculated from the amino acid matrix (step 2), genetic codetable and codon usage table. The DNA matrix has three positions for each position of the amino acid matrix. The score for each amino acid is divided amongits codons in proportion to their relative weights from the codon usage table, andthe scores for each of the four different nucleotides are combined in each DNAmatrix position. Nucleotide positions are treated independently when the scores arecombined. As an option, the highest scoring nucleotide residue from each positioncan replace the most common codons from step 4 that are used in the consensus clamp.6) The degeneracy is determined at each position of the DNA matrix based on thenumber of bases found there. As an option, a weight threshold can be specified suchthat bases that contribute less than a minumum weight are ignored in determiningdegeneracy.7) Possible degenerate core regions are identified by scanning the DNA matrix inthe 3' to 5' direction. A core region must start on an invariant 3' nucleotideposition, have length of 11 or 12 positions ending on a codon boundary, and have amaximum degeneracy of 128 (current default). The degeneracy of a region is theproduct of the number of possible bases in each position.8) Candidate degenerate core regions are extended by addition of a 5' consensus clampfrom step 4 or 5. The length of the clamp is controlled by a melting point temperaturecalculation (current default = 60o) and is usually ~20 nucleotides.9) Steps 7 and 8 are repeated on the reverse complement of the DNA matrix from step5 for primers corresponding to the opposite DNA strand.CODEHOP program scheme1) input - - - - - - - - - - - - - - - seq 1 Protein sequence block - - - - - - - - - - - - - - - seq 2- - - - - - - - - - - - - - - seq 3- - - - - - - - - - - - - - - seq 4- - - - - - - - - - - - - - - seq 5- - - - - - - - - - - - - - - etc.|| 2) transformation to AA PSSMV| | | | | | | | | | | | | | | Ala AA PSSM| | | | | | | | | | | | | | | Cys| | | | | | | | | | | | | | | Asp| | | | | | | | | | | | | | | Glu| | | | | | | | | | | | | | | Phe| | | | | | | | | | | | | | | Gly| | | | | | | | | | | | | | | His| | | | | | | | | | | | | | | Ile| | | | | | | | | | | | | | | Lys| | | | | | | | | | | | | | | etc.| || | 3) calculation of AA consensus sequence| V| - - - - - - - - - - - - - - - AA consensus sequence || || | 4) transformation to DNA consensus sequence| V| ------------------------------------------- DNA consensus sequence | || | 5) back-translation to DNA PSSM| V| ||||||||||||||||||||||||||||||||||||||||||| A DNA PSSM| ||||||||||||||||||||||||||||||||||||||||||| C| ||||||||||||||||||||||||||||||||||||||||||| G| ||||||||||||||||||||||||||||||||||||||||||| T| | || | | 6) calculation of degeneracies| | V| | ~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ position degeneracy values | || | 7) identify degenerate regions ("===")| || 8) identify consensus regions for degenerate regions ("---")| |V V5' -------==== 3' CODEHOP primers output 3' ====--------- 5'。