计算化学9-常用计算化学应用软件及资源
物理化学实验中常用的数据处理软件及在化学中常见应用方法

物理化学实验中常用的数据处理软件及在化学中常见应用方法随着计算机技术的不断发展,数据处理软件在物理化学实验中的应用越来越普遍。
这些软件可以帮助实验人员处理实验数据,提高实验效率和准确度。
本文将介绍几种常用的数据处理软件及在化学中常见的应用方法。
一、常用的数据处理软件1. ExcelExcel是微软公司开发的一款电子表格软件,广泛应用于各个领域。
在物理化学实验中,Excel可以用来制作数据图表、计算平均值和标准偏差、进行线性回归等。
对于数据量较小的实验,Excel是一个简单易用的数据处理工具。
2. OriginOrigin是一款专业的科学数据分析和绘图软件,主要应用于科学研究、工程设计和教学等领域。
在物理化学实验中,Origin可以用来制作各种类型的图表、进行数据拟合和统计分析等。
Origin的功能非常强大,但学习起来也比较复杂。
3. MatlabMatlab是一款用于数学计算、数据分析和可视化的软件,被广泛应用于科学研究、工程设计和金融分析等领域。
在物理化学实验中,Matlab可以用来进行数据处理、信号处理和图像处理等。
Matlab的功能非常强大,但学习起来也比较困难。
二、在化学中的应用方法1. 数据图表的制作在物理化学实验中,数据图表是非常重要的,可以帮助实验人员更直观地了解实验结果。
在Excel中,可以选择不同的图表类型,如折线图、柱状图、散点图等,来展示实验数据。
在Origin中,可以制作更复杂的图表,如等高线图、三维图等,以展示更多的信息。
在Matlab中,可以利用其强大的绘图功能,制作各种复杂的图表。
2. 数据拟合和统计分析在物理化学实验中,常常需要对实验数据进行拟合和统计分析。
在Excel中,可以使用函数进行线性回归、非线性拟合和数据统计等。
在Origin中,可以使用各种拟合和统计分析工具,如最小二乘法拟合、方差分析等。
在Matlab中,可以使用其强大的数学计算和统计分析功能,进行各种数据拟合和统计分析。
常用化学软件介绍

mmCIF
*.cif
MSI ChemNote *.msm
MOPAC Input *.mop
Protein DB
*.pdb
ROSDAL
*.rdl
SMDFile
*.smd
SYBYL
*.sm1
SYBYL2
*.sm2
Tinker MM2 Input *.xyz
Tinker MM3
*.xyz
Tinker MM3
CH
127.9
C
138.2
CH
127.9
C
138.2
CH
127.9
C
138.2
CH3 CH3 CH3
24.9 24.9 24.9
128.5 0.7
-3.0 0.7 1.0
128.5 9.2
-0.1 -0.1
0.7 128.5
0.7 0.7 -3.0 1.0 128.5 -0.1 9.2 -0.1 0.7 128.5 -3.0 0.7 0.7 1.0 128.5 -0.1 -0.1 9.2 0.7 -2.3 24.3 0.6 2.3 -2.3 24.3 0.6 2.3 -2.3 24.3 0.6 2.3
5 Structure5
6 Structure6
C6H6 C6H5Br C4H4O C4H4S C4H5N C5H10
MolWeight 78.11184
Molname Benzene
LogP
MR
1.866 26.058
157.0079
Bromobenzene
2.754 33.6808
68.07396
TIFF
*.tif
Windows AVI Movie *.avi
化学常用软件应用简介(ChemOffice)

➢ 撤销操作,单击 【Undo】
旋转控制点
缩放控制点
• 3.1.3.4 检查结构错误和整理结构式
•3.1.1 主界面
状态栏
• ChemDraw主界面自上而下分为菜单栏、工具 栏和绘图窗口。绘图窗口左侧是垂直工具栏,其 中的工具和模板是化学专用的。有些模板按钮下 面带有 箭头,单击该按钮不松开,会在其右侧 弹出子工具栏。
【询问工具】 【箭头】 【轨道】 【绘图元素】 【括弧】【化学符号】
• 3.1.2 模板
Window】,弹出分析窗口,包含该化合物的分子简式、 摩尔质量、同位素分布图,元素分析组成比例等数据。 【View】/【Show Chemical Properties Window】, 弹出化学性质窗口,包含该化合物的沸点、熔点、临界 温度、临界压力、临街体积、Gibbs自由能、LogP、 MR、Herry’s Law、生成热、ClogP、CMR等。
• 3.1.3.12 快捷菜单和 快捷键
(1)快捷菜单
ChemDraw快捷菜单包 含了多种选项,使用快捷菜 单能完成常用编辑、属性设 置、模板选择等功能。
在选中的结构上单击鼠标 右键,会弹出快捷菜单。
• 3.1.3.6 根据结构得出化合物命名
如果确定了化合物的结构,想知道其系统命名,可 以借助ChemDraw。 ➢ 绘制化合物结构式 ➢ 选中此结构,【Structure】/【Convert Structure to Name】,即可在结构式下面出现系统命名。 ➢ 并非所有的结构式都能给出化合物名称,如果无法由 结构产生系统命名,ChemDraw会弹出窗口给出提示 信息。
计算化学实验_分子结构模型的构建及优化计算
计算化学实验_分⼦结构模型的构建及优化计算实验9 分⼦结构模型的构建及优化计算⼀、⽬的要求1.掌握Gaussian 和GaussView程序的使⽤。
2.掌握构建分⼦模型的⽅法,为⽬标分⼦设定计算坐标。
3.能够正确解读计算结果,采集有⽤的结果数据。
⼆、实验原理量⼦化学是运⽤量⼦⼒学原理研究原⼦、分⼦和晶体的电⼦结构、化学键理论、分⼦间作⽤⼒、化学反应理论、各种光谱、波谱和电⼦能谱的理论,以及⽆机、有机化合物、⽣物⼤分⼦和各种功能材料的结构和性能关系的科学。
Gaussian程序是⽬前最普及的量⼦化学计算程序,它可以计算得到分⼦和化学反应的许多性质,如分⼦的结构和能量、电荷密度分布、热⼒学性质、光谱性质、过渡态的能量和结构等等。
GaussView是⼀个专门设计的与Gaussian配套使⽤的软件,其主要⽤途有两个:构建Gaussian的输⼊⽂件;以图的形式显⽰Gaussian计算的结果。
本实验主要是借助于GaussView程序构建Gaussian的输⼊⽂件,利⽤Gaussian程序对分⼦的稳定结构和性质进⾏计算和分析。
三、软件与仪器1.软件:Gaussian03、GaussView计算软件,UltraEdit编辑软件。
2.仪器:计算机1台。
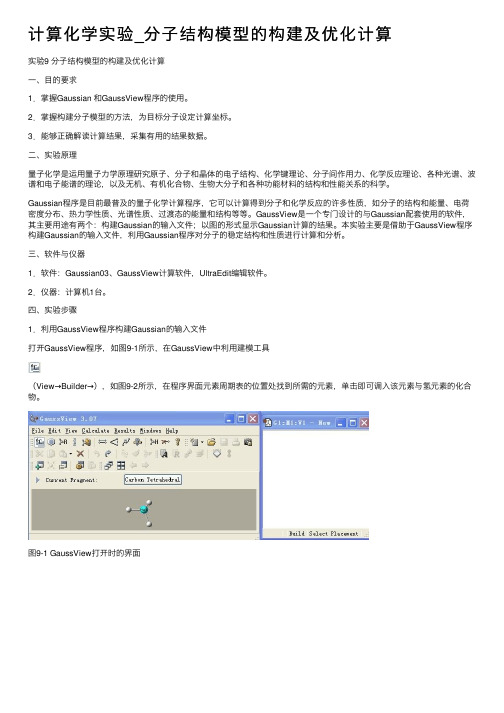
四、实验步骤1.利⽤GaussView程序构建Gaussian的输⼊⽂件打开GaussView程序,如图9-1所⽰,在GaussView中利⽤建模⼯具(View→Builder→),如图9-2所⽰,在程序界⾯元素周期表的位置处找到所需的元素,单击即可调⼊该元素与氢元素的化合物。
图9-1 GaussView打开时的界⾯图9-2点击Builder及双击图标后出现的元素周期表窗⼝图若要构建像⼄烷这样的链状分⼦,需要先点击⼯具栏中的按钮,常见的链状分⼦就显⽰在新打开的窗⼝中,如图9-3所⽰。
图9-3 常见链状官能团窗⼝图若要构建像苯、萘等环状结构的分⼦结构,需要双击⼯具栏中的按钮,常见的环状有机分⼦就显⽰在新打开的窗⼝中,如图9-4所⽰。
chemdoffice-软件使用简介

用于多类应用程序间互换信息的通用格式
一、Chemdraw简介
5 DARC-F1 (*.fld)——Questel DARC系统中存储结构的原本文件 格式 6 DARC-F1 Query(*.flq)——Questel DARC系统中存储查询的原本
文件格式
7 ISIS Reaction(*.rxn)——MDL开发的格式,用于存储元素反应信息 8 ISIS Sketch(*.skc)——在Windows或Macintosh环境下,存储并传输 到另外的ISIS应用程序中
一、Chemdraw简介
9 MDL MolFile (*.mol)——MDL(分子设计有限公司) MolFile文件 格式用于其它一些在Windows、Macintosh和Unix环境下的化学 数据库和绘画应用软件 10 Galactic Spectra(*.spc)——银河图谱文件格式
11 Jcamp Spectra(*.jdx,*.dx)——图谱文件格式,可读入紫外、
一、Chemdraw简介
菜单栏
工具栏
主工具图标板
编辑区
滚动栏 状态/信息栏
一、Chemdraw简介 主工具图标板
套索 实键 双键 虚键 切割键 切割楔键 黑体键 黑体楔键 空心楔键 波浪键 表格 长链 环丙烷环 环戊烷环 环庚烷环 环己烷椅式 环戊二烯环 蓬罩(选取框) 橡皮 文本 笔 箭头 轨道 绘图元素 基元 化学符号 弧形 质询工具 模板 环丁烷环 环己烷环 环辛烷环 环己烷椅式 苯环
一、Chemdraw简介
模板工具 官能团模板工具
一、Chemdraw简介
模板工具 己糖模板工具
一、Chemdraw简介
模板工具
苯环模板工具
一、Chemdraw简介
计算化学在化学化工中的应用综述

计算化学在化学化工中的应用综述摘要:计算化学在最近十年中是发展最快的化学研究领域之一,通过对具体的分子系统进行理论分析和计算,能比较准确地回答有关稳定性、反应机理等基本化学问题。
如今计算化学已被广泛用于材料、催化和生物化学等研究领域。
本文主要就计算化学的背景、计算化学常用的方法及其在化学化工中的应用等几个方面作一简单介绍。
关键词:计算化学原理材料催化应用引言计算化学是根据基本的物理化学理论(通常指量子化学、统计热力学及经典力学)及大量的数值运算方式,应用计算机技术,通过理论计算研究化学反应的机制和速率,总结和预见化学物质结构和性能关系的规律的学科。
计算化学是化学、计算机科学、物理学、生命科学、材料科学以及药学等多学科交叉融合的产物,而化学则是其中的核心学科[1]。
可以用来解释实验中各种化学现象,了解、分析实验结果,预测化学反应方向,还可以用来验证、测试、修正或发展较高层次的化学理论。
准确高效的理论计算方法也是计算化学领域中非常重要的一部分。
近二十年来,计算机技术的飞速发展和理论计算方法的进步使理论与计算化学逐渐成为一门新兴的学科[2]。
今天,理论化学计算和实验研究的紧密结合大大改变了化学作为纯实验科学的传统印象,有力地推动了化学各个分支学科的发展。
随着人们对“化学不再是纯实验科学”论断认识的不断提高,计算化学将在各个化学研究领域和交叉学科领域发挥作用。
特别是随着当前世界学科前沿的发展趋势,材料、生命、医药、环境等学科越来越被政府和科学家们重视,计算化学也将在这几个方面发挥重大作用[3]。
1 计算化学常用的方法及其介绍下面对计算化学中常用的几种理论计算方法作一个简单的介绍:1.1 从头算方法从头算方法仅使用一些最基本的物理常数(如光速、普朗克常数等)作为已知参数,完全利用数学工具来求解薛定锷方程,而不引入任何经验性质的化学参数。
由于绝大多数化学体系的薛定锷方程没有严格的解析解,只能在求解的过程中引入各种数学近似,使用数值解法得到结果。
计算化学的软件工具和数据资源

计算化学的软件工具和数据资源计算化学是一种基于计算机科学的化学研究方法,它利用计算机模拟来预测化学物质的物理化学性质和反应机理。
随着计算机科学和化学研究的迅速发展,计算化学的应用已经得到广泛的推广和应用。
为了更加有效地使用计算化学方法,研究人员需要掌握一些计算化学的软件工具和数据资源,让我们一起来看看这些有用的工具和资源。
1. 分子模拟软件分子模拟软件是计算化学研究的核心工具。
它可以用来模拟分子的运动和反应,预测分子的结构、能量和性质。
常用的分子模拟软件有Gromacs、Amber、LAMMPS、CHARMM等。
这些软件通常需要一定的计算机编程技能,但是它们提供了很强的自由度和控制力,可以满足不同研究需求的要求,同时也是训练计算化学研究人员的基本技能。
2. 密度泛函理论软件密度泛函理论(DFT)是一种计算电子结构的方法,它可以用来预测分子的几何构型、电子能级和电荷分布等。
常用的DFT软件有Gaussian、VASP、Quantum ESPRESSO等。
这些软件通常需要一定的物理、数学和计算机科学知识,但是它们提供了很强的准确性和可靠性,可以用来研究很多重要的化学问题。
3. 虚拟筛选软件虚拟筛选软件是用来寻找化学药物和分子杂交物的软件工具。
它利用基于计算机的分子模拟和化学信息检索技术,可以从大规模化学库中筛选出具有特定生物活性的化合物。
常用的虚拟筛选软件有Autodock、Vina、Glide等。
这些软件允许研究人员进行高通量筛选和分子设计,可以大大提高化学药物和分子杂交物的研发速度和成功率。
除了这些软件工具之外,计算化学研究人员还需要掌握一些数据资源,这些数据资源可以用来支持计算化学研究的可靠性和准确性。
4. 化合物数据库和手册化合物数据库和手册是计算化学研究人员必备的资源之一。
它们收集了大量的化学结构、性质和反应信息,包括分子式、结构式、物理化学性质、毒性信息、化学反应机理等。
常用的化合物数据库和手册有Beilstein、PubChem、ChemSpider、EPA等。
计算化学相关的免费的在线数据库及工具

计算化学相关的免费的在线数据库及工具文/Sobereva Last Update:2011-MAY-7这些是我平时收集的和计算化学/分子模拟有关的免费的在线的库和工具,既在线又免费的实用的网站是很有价值的。
其中有些对计算有直接帮助,有些则是提供计算所涉及的素材。
由于笔者的研究和生物分子结构问题有关,所以列表中包含不少偏生物的内容。
下列网址在写入本文时均可访问,若无法访问可尝试代理,若确实网址已改变请告诉我。
前面有√的代表比较重要。
很多在线工具需要Java运行环境。
如果有其它好的免费的化学相关的在线的库或工具欢迎回帖补充,我也会在日后逐渐补充。
1 在线信息数据库部分√ ChemSpider小分子信息整合数据库:简介:是当前众多的在线分子数据库的信息整合,便于用户搜索,数据来自200种数据库。
根据分子俗名、系统命名、Smile/InChI字符串、注册号、分子式等方式搜索,会列出分子平面结构、实验测定的和由ACD/Labs、EPISuite、ChemAxon软件预测的理化性质(LogP、LogD(PH=5.5和7.4时)、水溶性、分子体积、密度、沸点、闪点、蒸汽压、气化焓、折光率、可极化率、表面张力、SASA等),以及毒性、分子简介、Smile/InChI/InChIKey字符串、在其它分子数据库中的编号和链接、相关文章及专利、同义词、相关蛋白质、NMR/IR 光谱图等诸多信息,某些分子还可以链入web CSD获得三维结构。
点击左上角的分子图形窗口上方的3D标签,再点击下方的SAVE,可以获得由Marvin预测的分子的三维结构。
√ SDBS光谱数据库:http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi简介:很好的有机化合物光谱数据库,包含六类光谱:EI-MS、FT-IR、H-NMR、C13-NMR、ESR、Raman。
含3万余个化合物,其中以商业化学试剂为主,约2/3的数据是6碳至16碳的化合物。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
以上有不当之处,请大家给与批评指正, 谢谢大家!
19
原子电荷分布(布居分析)和自旋密度分布
分子的多极矩(永久偶极矩和四极至十六极矩)
NMR 屏蔽常数、化学位移及分子的磁化率、振动 园二色强度
电子亲和性与电离势、极化率与超极化率、静电 势与电子密度分布等
第一步工作准备输入文件。Gaussian程序对输入文件 有一定的格式要求,如图显示了一个完整的Gaussian 输入文件
常用计算化学应用软件及资源
Gaussian 软件
Gaussian的功能很强大 可以做从头算 半经验 DFT
Gaussian 软件
可用来研究分子的能量与几何结构 化学反应过渡态的能量与几何结构 振动频率分析、红外与拉曼光谱; 分子的热化学性质、键能 化学反应能 化学反应途径
研究分子轨道的能量与性质
ADF软件在材料科学和生命科学均有应用,但更侧 重于前者,尤其在重元素化学、无机化学、催化领 域非常流行。最新版的ADF加入了QM/MM方法, 可用于生物大分子体系的研究。
量子化学资源
International Journal of Quantum chemistry Journal of molecular modeling Journal of physical chemistry Journal of chemical theory and computation (美 国化学会2005年推出) Journal of molecular structure (Theochem) Reviews in Computational Chemistry (丛书) Journal of theoretical and computational chemistry Theoretical chemical accounts Journal of computational chemistry
Gaussian公司的官方网站
国际上著名的计算化学列表网站,开通较早,内有大量关于 计算化学的邮件列表。
北京大学化学系开设的量化计算论坛
厦门大学化学系开设的量化计算论坛
国内著名的量子化学论坛
国内著名的量子化学论坛
化学数据库(Data base)
分子结构库 晶体库 热力学数据库 药物库 高分子库 分子光谱、波谱图谱库 生物数据库(蛋白质、核酸、多糖库) 化学文献库
此外,它还常用作定量结构-性质(或活性)关系 研究中结构参数的计算。
ADF软件
ADF可以进行单点计算、几何优化、寻找过渡态、 计算力常数和热化学性质、跟踪反应路径、研究电 子结构、通过比较离子的激发态和基态而获得激发 能。
新版本的ADF包括了含时密度泛函理论,基组库中 包含了1~118号所有元素,而且对常见元素有不同 大小的基组,从最小的到高质量的。
准备好输入文件后,便可在Gaussian程序中读入 此输入文件进行计算
程序运算完后,Gaussian会给出正常结束的信息。此 时,可打开文本格式的输出文件,提取所需信息。
MOPAC 软件
MOPAC 软件是一款大众化、功能强大的半经验量化 程序
可以计算分子、自由基、离子和聚合物的电子结构、 光谱,热化学性质、同位素取代效应和力常数等。 其中的一个过渡态定位程序和两个过渡态优化程序 用于研究化学反应。