Q_AELU 003-2018SES-FS-生物制剂-S003产品质量标准
产品标准 yy 0323-2018 不适用 说明

产品标准 yy 0323-2018 不适用说明产品标准 yy 0323-2018 不适用说明导言在医疗器械行业,产品标准是保障产品质量和安全性的重要依据。
然而,有时候我们会遇到一些情况,即产品标准 yy 0323-2018 不适用的情况。
这时候我们就需要对这种情况进行深入的了解和分析,以便更好地把握产品质量和安全性的要求。
本文将围绕产品标准 yy 0323-2018 不适用的情况展开讨论,探索其原因、影响以及应对之策。
1. 产品标准 yy 0323-2018 不适用的情况是什么?产品标准 yy 0323-2018 是一份涉及医疗器械行业的重要标准,它规定了相关产品的质量和安全性要求。
然而,在实际操作中,我们有时候会发现某些情况下这个标准并不适用。
一些新型医疗器械可能由于技术创新和尚未形成统一认知而不适用于该标准;又或者一些特殊用途的产品由于自身特点也不适用于该标准。
这些情况都需要我们深入思考和分析。
2. 产品标准 yy 0323-2018 不适用的原因是什么?产品标准 yy 0323-2018 的不适用可能是由于多种因素造成的。
技术创新和产品更新迭代可能会导致现有标准跟不上产品发展的步伐,从而出现不适用的情况。
一些特殊用途的医疗器械可能因为其独特的功能和风险特点而不适用于现有的产品标准。
国际标准的差异以及监管要求的不同也可能导致产品标准 yy 0323-2018 不适用。
3. 产品标准 yy 0323-2018 不适用的影响是什么?产品标准 yy 0323-2018 的不适用可能会对医疗器械行业产生一系列影响。
不适用的情况可能导致产品质量和安全性无法得到有效保障,从而增加了使用该产品的风险。
不适用情况可能加大了监管部门的审核和审批难度,增加了企业的研发成本和周期。
不适用还可能影响国际贸易和合作,给企业带来一定的市场壁垒。
4. 如何应对产品标准 yy 0323-2018 不适用的情况?针对产品标准 yy 0323-2018 的不适用情况,我们需要采取有效的措施应对。
生物制品制造业清洁生产评价指标体系

.生物制品制造业(生物制剂)清洁生产评价指标体系(征求意见稿)国家发展和改革委员会发布环境保护部工业和信息化部目录前言..............................................................................................................................................II 1适用X围 (1)2规X性引用文件 (1)3术语和定义 (1)4评价指标体系 (3)5评价方法 (6)6指标解释与数据来源 (7)前言为贯彻《中华人民XX国环境保护法》和《中华人民XX国清洁生产促进法》,指导和推动生物制品制造企业依法实施清洁生产,提高资源利用率,减少和避免污染物的产生,保护和改善环境,制定生物制品制造业(生物制剂)清洁生产评价指标体系(以下简称“指标体系”)。
本指标体系依据综合评价所得分值将清洁生产等级划分为三级,Ⅰ级为国际清洁生产领先水平;Ⅱ级为国内清洁生产先进水平;Ⅲ级为国内清洁生产基本水平。
随着技术的不断进步和发展,本评价指标体系将适时修订。
本清洁生产评价指标体系为首次发布。
本指标体系由国家发展改革委员会、环境保护部会同工业和信息化部组织制订。
本标准起草单位:正丰易科环保科技研究中心XX、中国环境科学研究院、中国生物技术股份XX、中国标准化研究院。
本指标体系由国家发展和改革委员会、环境保护部和工业和信息化部共同负责解释。
本指标体系自公布之日起试行。
1适用X围本指标体系规定了血液制品企业清洁生产的一般要求。
本指标体系将清洁生产指标分为六类,即生产工艺及设备要求、资源和能源消耗指标、资源综合利用指标、污染物产生指标、产品特征指标和清洁生产管理指标。
本指标体系适用于血液制品生产企业的清洁生产审核、清洁生产潜力与机会的判断以及清洁生产绩效评定和清洁生产绩效公告制度,也适用于新建或改扩建项目环境影响评价、环保核查、行业准入等管理制度。
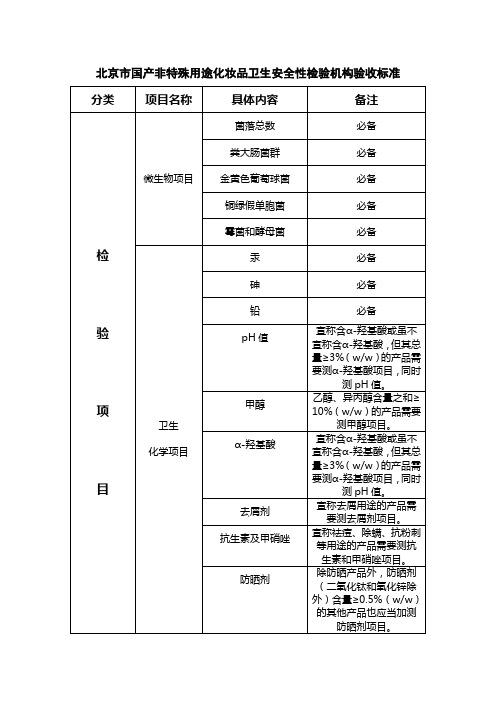
北京国产非特殊用途化妆品卫生安全性检验机构验收标准
毒理学项目
急性皮肤刺激性试验
(易触及眼睛的发用产品、指(趾)甲类、芳香类)
急性眼刺激性试验
次皮肤刺激性试验
(一般护肤产品、易触及眼睛的护肤产品、一般彩妆品、眼部彩妆品、护唇及唇部彩妆品)
pH计
超声波提取器
高速离心机
毒理学实验室
超净工作台
1/1000天平
1/10000天平
游标卡尺
台式离心机
4℃~-20℃低温冰箱
超净工作台
消毒设备
裂隙灯
分光光度计(可见、紫外)
电子天平(1/1000和1/10000)
台秤
检
验
人
员
具有中级以上技术职称的人数不少于实验室检验技术人员总人数的50%
技术负责人应当具有正高级技术职称,并具有十年以上相关工作经验
甲醇
乙醇、异丙醇含量之和≥10%(w/w)的产品需要测甲醇项目。
α-羟基酸
宣称含α-羟基酸或虽不宣称含α-羟基酸,但其总量≥3%(w/w)的产品需要测α-羟基酸项目,同时测pH值。
去屑剂
宣称去屑用途的产品需要测去屑剂项目。
抗生素及甲硝唑
宣称祛痘、除螨、抗粉刺等用途的产品需要测抗生素和甲硝唑项目。
防晒剂
北京市国产非特殊用途化妆品卫生安全性检验机构验收标准
分类
项目名称
具体内容
备注
检
验
项
目
微生物项目
菌落总数
必备
粪大肠菌群
必备
金黄色葡萄球菌
必备
铜绿假单胞菌
必备
霉菌和酵母菌
必备
Q_LAS 003-2019精油企业标准

漯河市艾生生物科技有限公司企业标准Q/LAS003—2019精油2019-11-12发布2019-11-20实施漯河市艾生生物科技有限公司发布前言本标准按照GB/T1.1—2009的规定进行编写。
本标准由漯河市艾生生物科技有限公司标准公司提出并起草。
本标准主要起草人:刘军辉。
精油1.范围本标准规定了精油的产品组成、规格、要求、检验规则、标志、包装、运输和贮存。
本标准适用于将老姜艾草玫瑰橄榄熏衣草当归人参藏红花等分别组成的精油。
2规范性引用文件下列文件对于本文件的应用是必不可少的。
凡是注日期的引用文件,仅所注日期的版本适用于本文件。
凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T191包装储运图示标志GB15979一次性使用卫生用品卫生标准QB/T1507日用香精中华人民共和国药典(2010版)二部中华人民共和国卫生部消毒技术规范(2002版)中华人民共和国卫生部化妆品卫生规范(2007版)中华人民共和国卫生部消毒产品标签说明书管理规范(2005年版)中华人民共和国卫生部消毒产品生产企业卫生规范(2009年版)JJF1070定量包装商品净含量计量检验规则国家质量监督检验检疫总局令(2005)第75号定量包装商品计量监督管理办法3要求3.1原料质量要求3.1.1产品配方中所用原料应符合国家有关法律法规,原料中醋酸氯己定、香精、水质量要求应符合表1规定。
3.1.2其他原料应符合相应供应商企业标准要求。
表1原料质量要求原料名称质量等级质量要求醋酸氯己定纯度≥98.0%《中华人民共和国药典》(二部)(2010版)香精化妆品级QB/T1507生产用水纯化水《中华人民共和国药典》(二部)(2010版)3.2感官要求感官要求应符合表2规定。
表2感官要求项目要求外观无色透明液体,不分层,无悬浮物或沉淀气味具有本品特型香味,无异味3.3理化指标理化指标应符合表3规定表3理化指标项目指标醋酸氯己定,%0.18~0.22pH值 5.5~8.5铅,mg/L<40砷,mg/L<10汞,mg/L<13.4微生物指标应符合表4的规定。
透明质酸钠医疗器械三类标准_概述及解释说明

透明质酸钠医疗器械三类标准概述及解释说明1. 引言1.1 概述透明质酸钠作为一种重要的医疗器械,被广泛应用于医疗和美容领域。
它具有保湿、润滑和填充作用,广泛应用于皮肤护理、关节润滑、眼部手术等多个医疗领域。
透明质酸钠在医疗器械三类标准中占据重要地位,其标准的制定和实施对确保产品质量和安全至关重要。
1.2 文章结构本文将围绕透明质酸钠医疗器械三类标准展开讨论。
首先介绍透明质酸钠医疗器械三类标准的概述,包括定义和背景、分类与特点以及应用领域和重要性。
接下来将详细解释这些标准的内容和要求,并探讨相关法规与具体规定。
此外,还会探讨这些标准的实施与管理机制。
最后,我们将探讨在实施透明质酸钠医疗器械三类标准过程中可能遇到的挑战,并提出相应的对策。
文章最后将总结重点观点及主要发现结果,同时展望透明质酸钠医疗器械三类标准在未来的发展前景。
1.3 目的本文的目的是为读者提供一个全面而清晰地了解透明质酸钠医疗器械三类标准的介绍和解释说明。
通过对标准内容和要求、相关法规与具体规定以及标准实施与管理机制等方面的深入探讨,我们希望能够增加对透明质酸钠医疗器械三类标准认识的理解,并为相关行业提供实施这些标准时可能遇到的挑战与对策。
同时,我们也将展望透明质酸钠医疗器械三类标准未来的发展前景,为该领域的从业者和研究人员提供参考和借鉴。
2. 透明质酸钠医疗器械三类标准概述2.1 定义和背景透明质酸钠医疗器械属于中国国家药品监督管理局(CFDA)所制定的三类医疗器械标准之一。
透明质酸钠是一种生物可降解的多糖,广泛应用于各个领域的医疗器械中。
它具有良好的生物相容性和生物降解性,被广泛用作填充剂、润滑剂以及局部治疗药物的载体。
2.2 分类和特点根据透明质酸钠医疗器械的特点和使用范围不同,可以将其分为不同的分类。
常见的分类包括注射用透明质酸钠、眼科用透明质酸钠、皮肤填充材料等。
不同分类的透明质酸钠医疗器械在成分、毒性评估、效果持久性等方面存在差异。
医疗器械的ech的残留量标准

医疗器械的ech的残留量标准医疗器械的残留量标准是为了确保医疗器械在使用后不存在过多的残留物,以保障患者的安全和医疗质量。
不同类型的医疗器械有不同的残留物限制标准,主要包括化学残留物、微生物污染和放射性残留物等方面。
以下是一些关于医疗器械残留量标准的相关参考内容。
1. 化学残留物标准:化学残留物是指在使用某种医疗器械后残留下来的化学物质,包括药物残留、有机溶剂、重金属、防腐剂等。
根据不同的医疗器械和使用场景,有重金属的残留标准,如重金属铅、汞的残留量标准。
一般来说,医疗器械的化学残留物限制标准参考ISO 10993-10《Biological evaluation of medical devices -- Part 10: Tests for irritation and skin sensitization》以及USP Chapter <1663>《Assessing Extractables in Non-Biological Matrices for Medical Devices》等国际标准。
2. 微生物污染标准:微生物污染是指在使用医疗器械过程中,细菌、真菌、病毒等微生物附着或残留在器械表面或内部,可能引起感染的一种情况。
根据不同的器械和使用场景,有不同的微生物限制标准,如空气中的细菌数量、器械表面的菌落形成单位等。
医疗器械的微生物污染标准参考ISO 11737-1《Sterilization of medical devices -- Microbiological methods -- Part 1: Determination of a population of microorganisms on products》等国际标准。
3. 放射性残留物标准:放射性残留物是指在使用放射性医疗器械后残留下来的放射性物质。
这类残留物应符合国家放射卫生安全标准,如《医疗机构放射卫生安全管理规定》、《医疗设备辐射防护规定》等法律法规。
医疗器械生物学评价标准目录
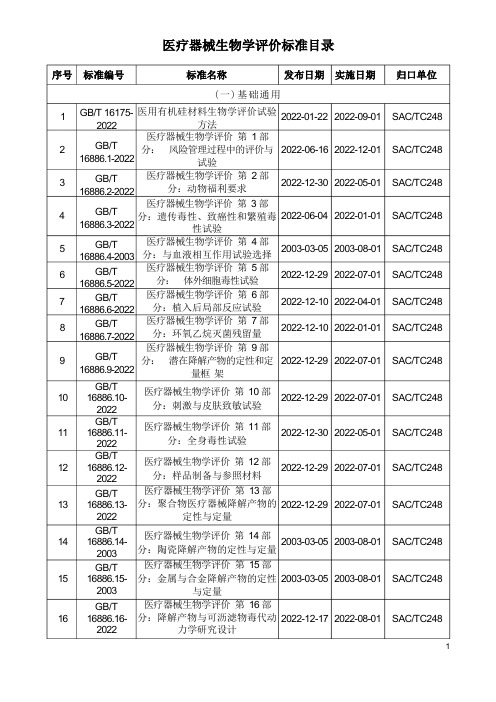
(一)基础通用1 2 3 4 5 6 7 8 910111213141516 GB/T 16175-2022GB/T16886.1-2022GB/T16886.2-2022GB/T16886.3-2022GB/T16886.4-2003GB/T16886.5-2022GB/T16886.6-2022GB/T16886.7-2022GB/T16886.9-2022GB/T16886.10-2022GB/T16886.11-2022GB/T16886.12-2022GB/T16886.13-2022GB/T16886.14-2003GB/T16886.15-2003GB/T16886.16-2022医用有机硅材料生物学评价试验方法医疗器械生物学评价第1 部分:风险管理过程中的评价与试验医疗器械生物学评价第2 部分:动物福利要求医疗器械生物学评价第3 部分:遗传毒性、致癌性和繁殖毒性试验医疗器械生物学评价第4 部分:与血液相互作用试验选择医疗器械生物学评价第5 部分:体外细胞毒性试验医疗器械生物学评价第6 部分:植入后局部反应试验医疗器械生物学评价第7 部分:环氧乙烷灭菌残留量医疗器械生物学评价第9 部分:潜在降解产物的定性和定量框架医疗器械生物学评价第10 部分:刺激与皮肤致敏试验医疗器械生物学评价第11 部分:全身毒性试验医疗器械生物学评价第12 部分:样品制备与参照材料医疗器械生物学评价第13 部分:聚合物医疗器械降解产物的定性与定量医疗器械生物学评价第14 部分:陶瓷降解产物的定性与定量医疗器械生物学评价第15 部分:金属与合金降解产物的定性与定量医疗器械生物学评价第16 部分:降解产物与可沥滤物毒代动力学研究设计2022-01-222022-06-162022-12-302022-06-042003-03-052022-12-292022-12-102022-12-102022-12-292022-12-292022-12-302022-12-292022-12-292003-03-052003-03-052022-12-172022-09-012022-12-012022-05-012022-01-012003-08-012022-07-012022-04-012022-01-012022-07-012022-07-012022-05-012022-07-012022-07-012003-08-012003-08-012022-08-01SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC2481718192021GB/T16886.17- 2005 GB/T16886.18- 2022 GB/T16886.19- 2022 GB/T16886.20- 2022 YY/T 1512-2022医疗器械生物学评价 第 17 部分:可沥滤物允许限量的建立 医疗器械生物学评价 第 18 部分:材料化学表征 医疗器械生物学评价 第 19 部 分:材料物理化学、形态学和表 面特性表征 医疗器械生物学评价 第 20 部 分:医疗器械免疫毒理学试验原 则和方法 医疗器械生物学评价风险管理过 程中生物学评价的实施指南2005-11-042022-12-302022-12-302022-12-102022-07-17 2022-04-01 2022-05-01 2022-05-01 2022-01-012022-07-01 SAC/TC248 SAC/TC248SAC/TC248SAC/TC248SAC/TC248YY/T 0870.1-医疗器械遗传毒性试验 第 1 部 2022 分:细菌回复突变试验 医疗器械遗传毒性试验 第 2 部YY/T 0870.2-变试验 医疗器械遗传毒性试验 第 3 部 24 分:用小鼠淋巴瘤细胞进行的 2022-07-24 TK 基因突变试验医疗器械遗传毒性试验 第 4 部25 分:哺乳动物骨髓红细胞微核试 2022-06-17 验医疗器械遗传毒性试验 第 5 部26 分:哺乳动物骨髓染色体畸变试 2022-06-17 验 YY/T 0870.6- 医疗器械遗传毒性试验 第 6 部27 2022-07-24 2022 分:体外哺乳动物细胞微核试验2022-10-012022-06-01 2022-08-012022-07-01 2022-07-01 2022-08-01SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248(三)繁殖和发育毒性28293031YY/T 1292.1- 医疗器械繁殖和发育毒性试验第2022 1 部份:筛选试验 YY/T 1292.2- 医疗器械繁殖和发育毒性试验第2022 2 部份:胚胎发育毒性试验 YY/T 1292.3- 医疗器械繁殖和发育毒性试验第2022 3 部份:一代繁殖毒性试验 YY/T 1292.4- 医疗器械繁殖和发育毒性试验第2022 4 部份:两代繁殖毒性试验 2022-03-02 2022-01-012022-03-02 2022-01-012022-01-26 2022-01-012022-02-28 2022-01-01SAC/TC248SAC/TC248SAC/TC248SAC/TC248(四)补体激活YY/T 0878. 1- 医疗器械补体激活试验 第 1 部32 2022- 10-21 2022- 10-01 SAC/TC2482022 分:血清全补体激活23 分:体外哺乳动物细胞染色体畸 2022-05-31 YY/T 0870.5-YY/T 0870.4-YY/T 0870.3-2022-10-21 2022 22 2022 2022 202233 34 3536 YY/T 0878.2- 2022YY/T 0878.3- 2022YY/T 0879.1- 2022YY/T 0879.2- 2022 医疗器械补体激活试验 第 2 部 分:血清旁路途径补体激活 医疗器械补体激活试验 第 3 部 分:补体激活产物(C3a 和 SC5b-9)的测定 (五)致敏医疗器械致敏反应试验 第 1 部分:小鼠局部淋巴结试验 (LLNA )放射性同位素掺入法 医疗器械致敏反应试验 第 2 部 分:小鼠局部淋巴结试验 (LLNA ) BrdU-ELISA 法2022-03-022022-07-24 2022-10-21 2022-03-022022-01-01 2022-08-01 2022-10-01 2022-01-01SAC/TC248SAC/TC248SAC/TC248SAC/TC248(六)免疫原性评价37 3839404142434445 YY/T 1465.1- 医疗器械免疫原性评价方法 第1 2022 部份:体外 T 淋巴细胞转化试验 医疗器械免疫原性评价方法 第2 部份:血清免疫球蛋白和补体成 分测定 ELISA 法 医疗器械免疫原性评价方法 第3 部份:空斑形成细胞测定 琼脂 固相法 医疗器械免疫原性评价方法 第4 部份:小鼠腹腔巨噬细胞吞噬鸡 红细胞试验半体内法 医疗器械免疫原性评价方法 第5 部份:用 M86 抗体测定动物源 性医疗器械中α-Gal 抗原清除率 医疗器械免疫原性评价方法 第6 部份:用流式细胞术测定动物脾 脏淋巴细胞亚群 医疗器械免疫原性评价方法 第7 部份:流式液相多重蛋白定量技 术 (七)降解YY/T 0511- 多孔生物陶瓷体内降解和成骨性 2022 能评价试验方法 YY/T 1775.1- 可吸收医疗器械生物学评价 第1 2022 部份:可吸收植入物指南 2022-01-262022-01-262022-07-292022-03-282022-07-292022-07-242022-03-092022-12-302022-03-09 2022-01-01 2022-01-01 2022-06-01 2022-04-01 2022-06-01 2022-08-01 2022-04-012022-06-01 2022-04-01SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248(八)临床前动物研究和临床研究YY/T 1754. 1- 医疗器械临床前动物研究 第 1部46 2022-09-27 2022-09-01 SAC/TC2482022 分:总则YY/T 1465.2-2022 YY/T 1465.3-2022 YY/T 1465.4-2022 YY/T 1465.5-2022 YY/T 1465.6-2022 YY/T 1465.7-2022医疗器械临床前动物研究 第 2部YY/T 1754.2-型48 医疗器械临床调查 1997-08-27 1998-01-01 SAC/TC248(九)微生物控制标示“无菌”医疗器械的要求49 第1 部份:最终灭菌医疗器械的 2022-07-02 2022-03-01要求标示“无菌”医疗器械的要求50 第2 部份:无菌加工医疗器械的 2022-07-02 2022-03-01要求YY/T 0618- 医疗器械细菌内毒素试验方法常51 2022-02-28 2022-01-012022 规监控与跳批检验山东中心山东中心SAC/TC248(十)动物源性医疗器械52 53 54 55 565758 596061 YY/T 0771.1- 动物源医疗器械 第 1 部份:风 2022 险管理应用YY/T 0771.2- 动物源医疗器械 第 2 部份:来 2022 源、采集与处置的控制 动物源医疗器械 第 3 部份:病YY/T 0771.3-2022YY 0970-2022 (十一)其他 YY/T 1500- 医疗器械热原试验 单核细胞激2022 活试验 人全血 ELISA 法 YY/T 1649.1- 医疗器械与血小板相互作用试验 2022 第 1 部份:体外血小板计数法 医疗器械与血小板相互作用试验 第 2 部份:体外血小板激活产物 (β -TG 、PF4 和 TxB2)的测定 YY/T 1651.1- 医疗器械溶血试验 第 1 部份:2022 材料介导的溶血试验 YY/T 1670.1- 医疗器械神经毒性评价 第 1 部 2022 分:评价潜在神经毒性的试验选 2022-03-31 2022-03-31 2022-12-302022-03-02 2022-10-212022-07-292022-05-312022-10-232022-05-31 2022-07-24 2022-04-01 2022-04-01 2022-06-01 2022-01-01 2022-10-012022-06-012022-06-012022-10-012022-06-012022-08-01SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248SAC/TC248毒和传播性海绵状脑病(TSE ) 因子去除与灭活的确认 动物源医疗器械 第 4 部份:传 YY/T 0771.4- 播性海绵状脑病(TSE )因子的 2022 去除和/或者灭活及其过程确认分 析的原则 含动物源材料的一次性使用医疗 器械的灭菌液体灭菌剂灭菌的确 认与常规控制 47 分:诱导糖尿病大鼠皮肤损伤模 2022-09-27 2022-09-01 SAC/TC248YY/T 1649.2-2022 YY/T 0615.2-YY/T 0615.1-YY/T 0297-2022202220221997择指南YY/T 1770.1- 医疗器械血栓形成试验第1 部2022 分:犬体内血栓形成试验2022-03-09 2022-04-01 SAC/TC24862。
生物制药标准修订发布稿 2010-5-24

国务院环境保护主管部门规定的执行国家污染物排放标准水污染物特别排放限值地域范围内的生物制药企业或生产设施向环境水体排放水污染物,适用相应的国家污染物排放标准水污染物特别排放限值。生物制药企业和生物医药研发机构排放恶臭污染物、环境噪声适用相应的国家污染物排放标准,产生固体废物的鉴别、处理和处置适用相应的国家固体废物污染控制标准。
本标准主要起草人:修光利,张萍,黄雪娟,詹天珍,陈少雄,张大年,侯丽敏,傅利英,徐利行
上海市生物制药行业污染物排放标准
1 适用范围
本标准规定了生物制药企业或生产设施、生物医药研发机构的水污染物和大气污染物排放限值。
本标准适用于现有生物制药企业或生产设施、生物医药研发机构水污染物和大气污染物排放管理,以及生物制药企业或生产设施、生物医药研发机构建设项目的环境影响评价、环境保护设施设计、竣工环境保护验收及其投产后的水污染物和大气污染物防治和管理。本标准也适用于制备动物用药物的生物制药企业或生产设施、生物医药研发机构。
3.9
指经国家或市人民政府批准的自然保护区范围内水域、饮用水水源准保护区。法律、法规禁止排放的水域除外。
3.10一般
指特殊保护水域以外的其放到企业法定边界外的废水量。包括与生产有直接或间接关系的各种外排废水(含厂区生活污水、冷却废水、厂区锅炉和电站废水等)。
3.12单位产品基准排水量
GB/T11901水质悬浮物的测定重量法
GB/T 11903水质色度的测定
GB/T11914水质化学需氧量的测定重铬酸盐法
GB/T 13197水质甲醛的测定乙酰丙酮分光光度法
GB/T13554-1992高效空气过滤器
GB/T 14204水质烷基汞的测定气相色谱法
上海市生物制药行业污染物排放标准

生物制药行业污染物排放标准The discharge standard of pollutants for bio-pharmaceutical industry(发布稿)目次前言 (II)1 范围 (1)2 规范性引用文件 (1)3 术语和定义 (1)4 技术内容 (2)4.1 水污染物排放标准 (2)4.2 大气污染物排放标准 (7)5 标准的实施与监督 (14)附录A(规范性附录) 污水混合排放时污染物最高允许排放浓度计算方法 (15)附录B(资料性附录) 生物制药企业的主要挥发性有机物 (16)附录C(资料性附录) 污染物及有关产品中英文名称对照表 (16)前言为贯彻《中华人民共和国环境保护法》、《中华人民共和国水污染防治法》、《中华人民共和国大气污染防治法》、《中华人民共和国海洋环境保护法》,加强生物制药行业污染物的排放控制,保障人体健康,维护生态平衡,结合上海市实际情况,制定本标准。
本标准主要有以下特点:1. 适用于生物制药企业水污染物、大气污染物排放管理;2.对不同生物制药生产过程规定了不同的排放标准;3.规定了现有生物制药企业达到更加严格的排放限值的时限。
生物制药企业的噪声控制、固体废物控制按国家和本市的有关规定执行。
本标准实施之日起,生物制药企业水污染物排放按本标准执行,不再执行DB31/199-1997《污水综合排放标准》;生物制药企业大气污染物排放按本标准执行,不再执行GB16297-1996《大气污染物综合排放标准》。
本标准为首次发布。
本标准的附录A为规范性附录,附录B、附录C为资料性附录。
本标准由上海市环境保护局提出并归口。
本标准由华东理工大学、上海市生物医药行业协会负责起草。
本标准由上海市人民政府2006年10月13日批准。
本标准由上海市环境保护局负责解释。
生物制药行业污染物排放标准1 范围本标准规定了生物制药企业的水污染物、大气污染物排放标准值。
本标准适用于生物制药企业的水污染物排放管理、大气污染物排放管理,以及生物制药建设项目的环境影响评价、建设项目环境保护设施设计、竣工验收及其投产后的污染控制和管理。
口腔微生态调节型产品质量要求-2023标准

口腔微生态调节型产品质量要求1 范围本文件规定了口腔微生态调节型产品的术语和定义、技术要求、检验方法、检验规则、标志、包装、运输和贮存。
本文件适用于宣称具有口腔微生态调节功效的产品,包括口腔喷雾剂、漱口水、牙膏等。
2 规范性引用文件下列文件中的内容通过文中的规范性引用而构成本文件必不可少的条款。
其中,注日期的引用文件,仅该日期对应的版本适用于本文件;不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T 601 化学试剂标准滴定溶液的制备GB/T 602 化学试剂杂质测定用标准溶液的制备GB/T 603 化学试剂试验方法中所用制剂及制品的制备GB/T 6682 分析实验室用水规格和试验方法GB/T 8372—2017 牙膏GB 15979 一次性使用卫生用品卫生标准QB/T 2945—2012 口腔清洁护理液3 术语和定义下列术语和定义适用于本文件。
口腔微生态oral microecology由口腔内微生物构成的动态生态平衡系统,各种口腔微生物之间的相互作用有助于人体抵御外界不良刺激的侵袭,对于维护人体的健康发挥重要的作用。
4 要求一般要求产品应具有口腔微生态调节功效,通过功效评价试验。
技术要求4.2.1 口腔喷雾剂、漱口水口腔喷雾剂、漱口水技术指标应符合表1的规定。
表1 技术指标4.2.2 牙膏牙膏应符合表2的规定。
表2 技术指标5 试验方法本文件所用试剂和水,除特殊规定外,均为分析纯试剂和符合GB/T 6682规定的三级水。
本文件中滴定分析用标准溶液、杂质测定用标准溶液、试验方法所用制剂和制品,除特殊规定外,均按GB/T 601、GB/T 602和GB/T 603之规定制备。
对于牙膏产品按称重量方法计算最后检测结果的指标采取先挤出20mm膏体,弃去,然后再挤样称量的取样方法。
功效评价试验见附录A。
口腔喷雾剂、漱口水5.2.1 香型按鼻嗅法进行检验。
5.2.2 澄清度在室温和自然光线条件下,将25mL样品倒入比色管内,距离30cm处目测观察。
三类体外诊断试剂《产品技术要求》

【下载本文档,可以自由复制内容或自由编辑修改内容,更多精彩文章,期待你的好评和关注,我将一如既往为您服务】医疗器械产品技术要求编号:*****检测试剂盒(胶体金法)1.产品型号/规格及划分说明试剂盒规格为10人份/盒,每个试剂盒内装10包检测试剂、1瓶抽提液A、1瓶抽提液B、10个采样管、10个小滴管和1份说明书。
2. 性能指标2.1物理检查2.1.1外观包装盒整洁,各组分齐全;抽提液瓶盖严紧,无漏液;盒签和瓶签清晰完整,标识无误;品名、批号和有效期清楚;说明书清晰完整。
2.1.2膜条宽度膜条宽度3.8±0.2mm。
2.1.3液体移行速度液体移行速度应不低于10mm/min。
2.1.4装量抽提液装量为4.0ml±0.2ml/瓶。
2.2阳性参考品符合率10份***国家阳性参考品检测结果应全部为阳性,阳性参考品符合率(+/+)应为10/10或3份企业阳性参考品检测结果应全部为阳性,阳性参考品符合率(+/+)应为3/3。
2.3阴性参考品符合率10份***国家阴性参考品检测结果应全部为阴性,阴性参考品符合率(-/-)应为10/10或10份企业阴性参考品检测结果应全部为阴性,阴性参考品符合率(-/-)应为10/10。
2.4重复性使用***国家重复性参考品或企业重复性参考品进行检测,结果应为阳性,且显色一致无明显差别。
2.5最低检出限使用***国家最低检出限参考品或企业最低检出限参考品进行检测,结果应为阳性,最低检出限应不高于1×105个菌/ml。
2.6批间差使用***国家重复性参考品或企业重复性参考品对三批试剂盒进行检测,结果应符合2.4的规定。
2.7稳定性试剂盒在规定的存储条件下存放至有效期末或37℃条件下放置14天,分别检测2.2~2.5项,结果应符合各项目的要求。
3. 检验方法3.1 物理检查3.1.1外观在自然光下目视检查,结果应符合2.1.1的要求。
3.1.2 膜条宽度随机抽取5支试剂条,用游标卡尺测量宽度,结果应符合2.1.2的要求。
解读《关于拟微球藻油等12种“三新食品”的公告》(2024年第5号)

解读《关于拟微球藻油等12种“三新食品”的公告》(2024年第5号)文章属性•【公布机关】国家卫生健康委员会•【公布日期】2024.10.10•【分类】法规、规章解读正文解读《关于拟微球藻油等12种“三新食品”的公告》(2024年第5号)一、新食品原料解读材料(一)拟微球藻油拟微球藻油是以拟微球藻(Nannochloropsis?gaditana)为原料,经乙醇提取、过滤、纯化、浓缩等工艺制成。
拟微球藻油主要成分为总脂肪(其中EPA含量≥20g/100g),且含有蛋白质、碳水化合物等营养成分。
国家卫生健康委员会2021年第5号公告已批准拟微球藻(Nannochloropsis?gaditana)为新食品原料。
本产品推荐食用量≤2克/天。
根据《中华人民共和国食品安全法》和《新食品原料安全性审查管理办法》规定,国家卫生健康委员会委托审评机构依照法定程序,组织专家对拟微球藻油的安全性评估材料审查并通过,认可其食用安全性和具有食品原料的属性。
新食品原料生产和使用应当符合公告内容以及食品安全相关法规要求。
鉴于拟微球藻油在婴幼儿、孕妇和哺乳期妇女人群中的食用安全性资料不足,从风险预防原则考虑,上述人群不宜食用,标签及说明书中应当标注不适宜人群和食用限量。
该原料的食品安全指标按照公告规定执行。
(二)前花青素前花青素是以松科松属植物海岸松(Pinuspinaster?Aiton)的树皮为原料,经研磨、乙醇提取、过滤、浓缩、干燥等工艺制成。
其主要成分为前花青素(≥50.0g/100g),且含有碳水化合物、膳食纤维、蛋白质、脂肪等营养成分。
欧盟批准海岸松的树皮可用于膳食补充剂。
本产品推荐食用量≤150毫克/天(以前花青素含量50g/100g计,超过该含量的按照实际含量折算)。
当摄入含有此类物质的其他食品时,应该注意每日摄入总量不超过每日推荐食用量。
根据《中华人民共和国食品安全法》和《新食品原料安全性审查管理办法》规定,国家卫生健康委员会委托审评机构依照法定程序,组织专家对前花青素的安全性评估材料审查并通过,认可其食用安全性和具有食品原料的属性。
上海市食品药品监督管理局2018年第1期医疗器械监督抽检质量通告-2018年第16号

上海市食品药品监督管理局2018年第1期医疗器械监督抽检质量通告正文:----------------------------------------------------------------------------------------------------------------------------------------------------上海市食品药品监督管理局2018年第1期医疗器械监督抽检质量通告2018年第16号为了加强对医疗器械的质量监督管理,规范市场秩序,保障医疗器械产品安全有效,2018年第一季度上海市食品药品监督管理局组织了对本市医疗器械生产、经营和使用单位的质量监督抽检,共完成检验382件。
现将质量抽检不符合标准的产品予以通告(见附表)。
相关抽样单位应依据《医疗器械监督管理条例》等有关法律法规对不符合标准规定的产品进行调查处置,同时加强本辖区内相关企业的日常监管,督促企业查清原因,召回相关产品,制定并落实整改措施,消除安全隐患。
对不符合标准规定的产品及相应企业,我局将进一步加强跟踪检查和抽验力度。
附表:2018年第1期医疗器械监督抽检不符合标准规定产品情况上海市食品药品监督管理局2018年4月19日附表2018年第1期医疗器械监督抽检不符合标准规定产品情况编号产品名称型号规格批号或编号标示生产单位被抽样单位不合格项目检验依据抽样单位检测单位1医用缝合针170123△1/2 11x24上海汉康医疗器械有限公司上海汉康医疗器械有限公司物理性能YY0043-2005杨浦区市场监督管理局上海市医疗器械检测所2医用缝合针170123○1/2 6x12上海汉康医疗器械有限公司上海汉康医疗器械有限公司物理性能YY0043-2005杨浦区市场监督管理局上海市医疗器械检测所3非吸收性外科缝线(蚕丝线)A88861600-31 4-0 15×24" 60cm16B0A苏州市嘉盛医疗用品有限公司上海荣嘉实业有限公司线径YZB/苏1603-2013产品注册标准徐汇区市场监督管理局上海市医疗器械检测所4黑色编织丝线M6001 60厘米*24条无纺布长方形耳挂式口罩5只/袋17.5cm*9.5cm-3层201609A04佳合医材(苏州)有限公司国药控股菱商医院管理服务(上海)有限公司线径GB9706.1-2007杨浦区市场监督管理局上海市医疗器械检测所5骨科外固定螺钉10138 皮质骨螺钉120/30mm 杆6mm 螺纹4.5/3.5mmB1039164Orthofix Srl上海中智恒康医疗器械有限公司耐腐蚀性国械注进20153462712产品技术要求徐汇区市场监督管理局上海市医疗器械检测所6一次性医用外科口罩"耳挂式17.8cmX8.9cm"5411000733美迪康医用材料(上海)有限公司美迪康医用材料(上海)有限公司口罩带沪械注准20162640540产品技术要求松江区市场监督管理局上海市医疗器械检测所2018年第1期医疗器械监督抽检(标识标签说明书等项目)不符合标准规定产品名单编号产品名称型号规格批号或编号标示生产单位被抽样单位不合格项目检验依据抽样单位检测单位1超声骨密度仪SONOST3000AC1UA 1604038Osteosys Co., Ltd.上海先德医疗系统有限公司设备或设备部件的外部标记GB9706.1-2007徐汇区市场监督管理局上海市医疗器械检测所2验光仪RMK-2001403200227上海雄博精密仪器股份有限公司上海雄博精密仪器股份有限公司设备或设备部件的外部标记;清洗、消毒或灭菌措施GB9706.1-2007;YY0673-2008宝山区市场监督管理局上海市医疗器械检测所3视力投影仪ACP-1001706110520上海雄博精密仪器股份有限公司上海雄博精密仪器股份有限公司设备或设备部件的外部标记GB9706.1-2007宝山区市场监督管理局上海市医疗器械检测所4视力投影仪ACP-8CP-ACP-080106078上海雄博精密仪器股份有限公司上海雄博精密仪器股份有限公司设备或设备部件的外部标记GB9706.1-2007宝山区市场监督管理局上海市医疗器械检测所5电动手术床DS-IIM537上海派可斯医疗器械有限公司上海派可斯医疗器械有限公司设备或设备部件的外部标记GB9706.1-2007宝山区市场监督管理局上海市医疗器械检测所——结束——。
无菌医疗器械质量检验相关标准

无菌医疗器械质量检验相关标准(一)医疗器械生物相容性试验GB/T 16886.3 医疗器械生物学评价第 3部分:遗传毒性、致癌性和生殖毒性试验GB/T 16886.4 医疗器械生物学评价第 4部分:与血液相互作用试验选择GB/T 16886。
5 医疗器械生物学评价第 5部分:体外细胞毒性试验GB/T 16886。
6 医疗器械生物学评价第 6部分:植入后局部反应试验GB/T 16886.7 医疗器械生物学评价第 7部分:环氧乙烷灭菌残留量GB/T 16886.8 医疗器械生物学评价第 8部分:生物学试验参照样品的选择和定性GB/T 16886。
10 医疗器械生物学评价第 10 部分:刺激与致敏试验GB/T 16886.11 医疗器械生物学评价第 11 部分:全身毒性试验GB/T 16886.12 医疗器械生物学评价第 12 部分:样品制备和参照样品GB/T 16886.13 医疗器械生物学评价第 13 部分:聚合物医疗器械降解产物的定性与定量GB/T 16886。
14 医疗器械生物学评价第 14 部分:陶瓷降解产物的定性与定量分析GB/T 16886。
15 医疗器械生物学评价第 15 部分:金属与合金降解产物的定性与定量GB/T 16886。
16 医疗器械生物学评价第 16 部分:降解产物与可溶出物的毒代动力学研究设计)GB/T 16886.17 医疗器械生物学评价第 17 部分:根据健康风险评价确定可溶出物允许极限的方法GB/T 16886.18 医疗器械生物学评价第 18 部分:材料化学定性GB/T 16886.19 医疗器械生物学评价第 19 部分:材料理化、机械和形态定性GB/T 16886.20 医疗器械生物学评价第 20 部分:医疗器械免疫毒理学试验原理和方法GB/T16175 医用有机硅材料生物学评价试验方法GB/T 16886。
9 医疗器械生物学评价第 9部分:潜在降解产物的定性与定量总则YY/T 0473 外科植入物聚交酯共聚物和共混物体外降解试验YY/T 0474 外科植入物用聚L-丙交酯树脂及制品体外降解试验YY/T 0567。
生物制品各剂型生产环境的洁净级别要求及认证要点

生物制品认证要点生物制品系指以天然或人工改造的微生物,寄生虫生物毒素或生物组织及代谢产物等为起始材料,采用生物学,分子生物学或生物化学等相应技术制成,并以相应分析技术控制中间产物和成品质量的生物活性制品,用于预防,治疗和诊断传染病或其他疾病.其硬件认证要点如下.不同生产工序操作必须有效隔离,不得相互妨碍.厂房洁净室(区)内表面(墙,地面,三棚等)应平整光滑,无裂缝,接口严密,无颗粒物脱落,并能耐受清洗和消毒,无积尘,不长霉.洁净室(区)的水,电,汽,建筑管线必须暗装.生产厂房必须按生产工艺和产品质量要求划分洁净级别,洁净室(区)内空气的尘埃粒子数和微生物数应符合规定,结果须予以记录.生物制品生产环境的空气洁净度级别要求:(1)100级:灌装前不经除菌过滤的制品其配制,合并,灌封,冻干,加塞,添加稳定剂,佐剂,灭活剂等;(2)10 000级:灌装前需经除菌过滤的制品其配制,合并,精制,添加稳定剂,佐剂,灭活剂,除菌过滤,超滤等;(3)100 000级:原料血浆的合并,非低温提取,分装前的巴氏消毒,轧兽及制品最终容器的精洗等;口服制剂其发酵培养密闭系统环境(暴露部分需无菌操作);酶联免疫吸附试剂的包装,配液,分装,干燥;胶体金试剂,聚合酶链反应试剂(PCR),纸片法试剂等体外免疫试剂;深部组织创伤用制品和大面积体表创面用制品的配制,灌装.洁净室(区)的窗户,天棚及进入室内的管道,风口,灯具与墙壁或天棚的连接部位均应密封.100级洁净室(区)和无菌制剂灌封室不得设水池或地漏.生物制品生产车间必须与青霉素类,头孢菌素类,激素类,抗肿瘤类化学药品,同位素药品生产车间严格分开.同位素类生物制剂的生产厂房须符合国家关于放射保护的要求,并应获得同位素使用许可证.10. 生物制品的生产用菌毒种与非生产用菌毒种,生产用细胞与非生产用细胞,强毒与弱毒,死毒与活毒,脱毒前与脱毒后,含牛血清与不含牛血清的制品,活疫苗与灭活疫苗,人血液制品,预防制品等加工或灌装不得同时在同一生产厂房内进行,其贮存要严格分开.不同种类的活疫苗的处理及灌装应彼此分开.强毒微生物及芽胞菌制品的区域与相邻区域应保持相对负压,并有独立的空气净化系统.卡介苗生产厂废话结核菌素生产厂房必须与其他制品生产厂房严格分开,其生产设备要专用.芽胞菌操作直至灭活过程完成之前必须使用专用设备.炭疽杆菌,肉毒梭状芽胞杆菌和破伤风梭状芽胞制品须在相应专用设施内生产.聚合酶链反应试剂(PCR)的生产和检定试剂,在使用阳性样品时,必须有符合相应规定的防护措施和设施.生产人免疫缺陷病毒(HIV)等检测试剂,在使用阳性样品时,必须有符合相应规定的防护措施和设施.以人血,人血浆或动物脏器,组织为原料生产制品必须使用专用设备,并与其他生物制品的生产严格分开.强毒微生物以及目前尚无有效预防治疗措施的有关病原微生物的加工处理,生产制品须在严格隔离的专用生产厂房内进行,操作致病性微生物须符合生物安全等级要求.不合格,回收或退回产品必须单独存放,并有明显标志.与制品直接接触的设备必须表面光洁,平整,易清洗,耐腐蚀,不与所加工的制品发生化学变化或吸附制品.纯化水,注射用水的生产设备必须保证水质质量.贮水罐,输入管道,管件阀门必须为无毒,耐腐蚀的材料制造.贮不罐密闭,通气口必须有不脱落纤维疏水性的除菌过滤装置,输水管道线能防止滞留,并易于拆洗,消毒.生物制品生产用菌毒种,细胞须按《中国生物制品规程》要求建立菌毒种,细胞原始种子库存和生产种子库.菌毒种,细胞来源必须历史清楚,能溯源,传代谱系清楚,并有记录.有菌毒种,细胞全面检定记录,出入库数量与登记数相符,在适宜条件下分库存放. 物料的贮存条件下不得使其受潮,变质,污染或发生差错.麻醉药品,剧毒药品,放射性药品的验收,贮存,保管,使用,销毁必须严格执行国家有关规定.粉针剂认证要点注射用无菌粉针末简称粉针.凡是在不溶液中不稳定的药物,如青霉素G,先锋霉素类及一些医用酶制剂(胰蛋白酶,辅酶A等)及血浆等生物制剂均需制成注射用无菌分末.根据生产工艺条件和药物性质不同,将冷冻干燥法制得的粉末,称为冻干针;而用其他方法如灭菌溶剂结晶法,喷雾干燥法制得的称为注射无菌分装产品.粉针剂的生产必须在无菌室内进行,其硬伯认证要点如下.洁净室应气密,分装(灌封)车间不得设水池和地漏,水电,工艺管线应暗装.粉针剂的分装,压塞,无菌内包装材料最终处理后勤的暴露环境为100级.生产青霉素类,头孢菌素类原料药的精制,干燥,包装厂房和其制剂生产车间与其他厂房严格分开,有独立的空调系统,室内保持相对负压.青霉素类,头孢菌素类原料的分装线为专用,不做其他抗生素或其他药品分装用.称量,精洗瓶工序,无菌衣准备,轧盖工序的环境洁净度要求最低为100 000级.配液,无菌更衣室,无菌缓冲走廊的空气洁净度级别为10 000级.灌装压塞和灭菌瓶贮存的洁净度级别为100级或10 000级背景下局部100级.洁净室(区)与非洁净室(区)之间必须设置缓冲设施.直接接触药品的包装材料最后一次精洗用水应符合注射用水质量标准.10 000级洁净(区)使用的传输设备不得穿越较低级别区域.10. 与药品直接接触的设备表面光洁,平整,易清洗,耐腐蚀,不与所加工的药品发生化学变化或吸附所加工的药品.11. 灭菌设备内部工作情况用仪表监测,监测仪表定期校正并有完整的记录.12. 纯水,注射朋水的生产设备要定期验证确保水的质量.13. 贮水灌,输水管道,管件,阀门等应为无毒,耐腐蚀的材质制造.14. 分装不得采用手工刮板分装工艺.大输液认证要点大输液又名可灭菌大容量注射剂,是指将配制好的药液灌入大于50 ml的输液瓶或袋内,加塞,加盖,密封后用蒸汽热压灭菌而制备的灭菌注射剂.其硬件认证要点主要如下.大输兴的稀想,过滤,内包装材料(如胶塞,绦纶膜,容器)的最终处理环境为10 000级. 灌装操作环境为100级.浓配或采用密闭系统的稀配,轧盖的环境为100 000级.洁净室要气密,水电,工艺管线应暗装,灌封间不得设水池和地漏.传输设备穿越不同区域要有净化措施.与药液直接接触的设备表面光洁,平整,易清洗,耐腐蚀,不与所加工的药品发生化学变化或吸附所或工的药品.灭菌设备具有自动监测,记录装置,其能力应与生产批量相适应.与药液直触的设备,容器具,管路,阀门,输送泵等应采用优质耐腐蚀材质,管路的安装尽量减少连(焊)接处.过滤器材不得吸附药液组份和释放异物.禁止使用含有石棉的过滤器材.原料,辅料及包装材料的贮存条件不得使其受潮,变质,污染或易于发生差错.清洗瓶用水必须使用注射用水,不得用旧回收瓶进行生产.小容量注射剂认证要点小容量注射剂指将配制好药淮灌入小于50ml安瓿内的注射剂.其硬件认证要点如下. 不同生产操作能有效隔离,不得互相防碍.厂房内的墙,地面天花板平整光洁,无裂隙,无脱落尘粒物质及起壳,不易积尘,不长霉. 洁净厂房级别要求:最终灭菌的小容量注射剂稀配,过滤,灌封,安瓿的干燥,冷却应在1万级环境下进行;浓配或采用密闭系统的稀配可以在10万级环境下进行.非最终灭菌的小容量注射剂,灌装前不需除菌滤过药液配制,注射剂的灌封,安瓿干燥灭菌后的冷却应为局部100级.灌溉装前需除菌滤过的药液配制应在1万级环境.其他洁净室(区)应为10万级.洁净厂房的水电,工艺管线应暗装.洁净室应气密.100级洁净车间和无菌制剂灌装室不得设水池和地漏.激素类,抗肿瘤类化学药品的生产使用专用设备,空调系统的排气经过净化.中药制剂的生产操作区应与中药材的前处理,提取,浓缩以及动物脏器,组织的洗涤或处理等生产操作区严格分开.不合格,回收或退回产品应单独存放.生产设备与生产要求相适应,便于生产操作和维修,保养.与药品直接接触的设备表面光洁,平整,易清洗,耐腐蚀,不与所加工的药品发生公演变化或吸附所加工的药品,不得有颗粒性物质脱落.1万级洁净室(区)使用的传输设备不得穿越较低洁净级别区域.灭菌柜应具有自动监测,记录装置,监测仪表定期校正并有完整的记录.纯化水,注射用水的生产设备能保证水质量标准.贮不罐,输水管道,管件阀门等应为无毒,耐腐蚀的材质制造.管路的安装应尽量减少连(焊)接处.过滤器材不得吸附药液组份和释放异物,禁止使用含有石棉的过滤器材.贮水罐密闭,通气口应安装不脱落纤维的疏水性除菌滤器.输水管线能防止滞留,并易于拆洗,消毒.更衣室,盥洗间,消毒设施不得对洁净区产生不记影响.10万级以上区域的洁净工作服应在洁净室(区)内洗涤,干燥,整理,必要时应按要求灭菌.洁净室(区)应使用无脱落物,易清洗,易消毒的卫生工具,卫生工具要存放于对产品不造成污染的指定地点,并应限定使用区域.原料,辅料及包装材料的贮存条件不得使其受潮,变质污染或易于发生差错.精洗瓶用水质量必须符合中国药典规定的注射用水质量标准.不得使用直颈安瓿生产.安瓿封口不得采用顶端熔封,安瓿封口后要有适当方法检学习漏.洁净室(区)在静态条件下检测的尘埃粒子数,浮游菌数或沉降菌数必须符合规定,应定期监控动态条件下的洁净状况.洁净室(区)的净化空气如可循环使用,应采到有效避免污染和交叉污染的措施.质量管理部门根据需要设置的检验,中药标本,留样观察及其他各类实验室应与药品生产区分开.生物检定,微生物限度检定和放射性同位素检定要分定进行.滴眼剂认证要点滴眼剂系指以药物配制蝗供眼用的澄明溶液或混悬液,用以诊断或治疗眼部疾患.其硬件认证要点如下.滴眼剂内包装物(瓶,塞,盖)的洗涤,干燥应在100 000级的洁净厂内进行.供角膜创伤或手术用滴眼剂的配制和灌装环境应为10 000级,其他眼用药品的暴露工序最低为100 000级洁净室内水电,工艺管线应暗装.供角膜创伤或手术用的滴眼剂,其内包装物的灭菌设备应定期验证,并不破坏内包物形态,无残留物.传输设备穿越不同区域要有净化措施.与药液直接接触的设备表面光洁,平整,易清洗,耐腐蚀,不与所加工的药品发生化学变化或吸附所加工的药品.供角膜创伤或手术用的滴眼剂,其无菌滤过后的贮淮灌应密闭,排气口应装置采用疏水性材质的灭菌滤器.原料,辅料包装村料的贮存条件不得使其受潮,变质,污染或易于发生差错.口服固体制剂认证要点口服固体制剂包括片剂,胶囊,颗粒剂等.其硬件认证要点如下.不同生产操作能有效隔离,不得互相妨碍.厂房内的墙,地面,天花板平整光洁,无裂隙,无脱落尘粒物质及起壳,不易积尘,不长霉. 洁净厂房级别要求:口服固体药品的暴露工序,其生产洁净室(区)空气洁净度等级不低于30万级.直接接触药品的包装材料最终处理的暴露工序洁净度级别应与其药品生产环境相同产尘量大的洁净室(区)经捕尘处理仍不能避免交叉污染时,其空气净化系统不得利用回风.空气洁净度级别相同的区域,产尘量大的操作室应保持相对负压.洁净室应气密.洁净厂房的水电,工艺管线应暗装.生产β-内酰胺结构类药品必须使用专用设备和独立的空气净化系统,并与其他药品生产区域严格分开.室内保持相对负压.避孕药品的生产厂房应与其他药品生产厂房分开,并装有独立的专用空气净人系统.生产性激素避孕药品的空气净化系统的气体排放应经净化处理.10. 激素类,抗肿瘤类化学药品的生产有专用设备及捕尘设施和空气净化系统,排入的废气应经净化处理.当不可避免与其他药品交替使用同一设备和空气净化系统时,应采用有效的防护,清洁措施和必要的验证.11. 干燥设备进风口应有过滤装置,出风口应有防止空气倒流装置.12. 中药制剂的生产振作区应与中药材的前处理,提取,浓缩以动物脏器,组织的洗涤或处理等生产操作区严格分开.13. 中药材炮制中的蒸,妙,炙,煅等炮制操作分别在其生产规模相适应的生产厂房(或车间)内进行,并有良好的通风,除烟,除尘,降温等设施.14. 不合格,回收或退回产品应单独存放.15.生产设备与生产要求相适应,便于生产操作和维修,保养.16.与药品直接接触的设备表面光洁,平整,易清洗,耐腐蚀,不与所加工的药品发生化学变化或吸附所加工的药品.17.药品生产采用的传送设备越不同洁净级别的生产车间,应有防止污染的措施.18.配料工艺用水及直接接触药品的设备,器具最后一次洗涤用水应符合纯化水质量标准.纯化水的生产设备能保证水质量.19.贮水罐密闭,通气应安装不脱落纤维的疏水性除菌滤器.输水管线能防止滞留,并易于拆洗,消毒.20.更衣室,盥洗室,消毒设施不得对洁净区产生不良影响.22.原料,辅料及包装材料的贮存条件不得使其爱潮,变质,污染或易于发生差错.23.称量,配料,粉碎,过筛,混合,压片,包衣生产设施或生产设备应有捕尘装备或防止交叉污染的隔离措施.24.硬胶囊剂充填和片剂,硬胶囊剂,颗粒剂分装应采用机械设备.口服液体制剂认证要点口服液体制剂是指药物分散在液体分散介质中的供内服的液态制剂.包括糖浆剂,口服液,酒剂,合剂,煎膏剂,汤剂等.其硬件认证要点如下.厂房洁净室内的墙,地面,三花板平整光洁,无裂隙,无脱落粒物质及起壳,不易积尘,不长霉.洁净室内水电工艺管线应暗装.非最终灭菌的口服液体制剂的配制,滤过,灌封空气洁净度级别最低为十万级;最终灭菌口服液体制剂的暴露工序空气洁净度级别最低为300 000级.直接接触药品的包装材料最终处理的暴露工序洁净度级别应与其药品生产环境相同. 中成药口服液体制剂其中药材炮制中的蒸,妙,炙,煅等炮制操作分别在与其生产规模相适应的生产厂房(或车间)内进行,并有良好的通风,除烟,除尘,降温等设施.生产设备与生产要求相适应,便于和产操作和维修,保养.与药品直接接触的设备表面光洁,平整,易清洗,耐腐蚀不与所加工的药品发生公演变化或吸附所加工的药品.灭菌设备内部工作情况用仪器监测,监测仪表定期校正并有完整的记录.贮水罐,输水管道,管件阀门等应为无毒,耐腐蚀的材质制造.纯水生产设备能保证水的质量.原料,辅料及包装材料的贮存条件不得使受潮,变质,污染或易于发生差错.原料药认证要点:原料药系指以化学合成,DNA重组技术,发酵,酶反应方法或从天然物质中提取制得,它是加工药物制剂的主要原料.为了确保制剂队主品的质量,原料药的精制,干燥,包装的操作应符合《药品生产质量管理规范》要求.其硬件认证要点如下.不同生产工序操作能有效隔离,不得相互妨碍.厂房洁净室(区)内表面(墙,地成,天棚等)应平整光滑,无裂缝,接口严密,无颗粒物脱落,并能耐受清洗和消毒,不易积尘,不长霉.洁净室(区)的水,电,汽,建筑管线必须暗装.生产厂房必须按生产工艺和产品质量要求划分洁净级别,洁净室(区)内空气的尘埃粒子数和微生物数应符合规定,结果须予记录.原料药精制,干燥,包装生产环境的空气洁净度级别要求:法定药品标准中列有无菌检查项目的原料药,其暴露环境应为10 000级背景下局部100级;其他原料药的生产暴露环境不低于300 000级.洁净室应气密.生产青霉素类等高致敏性原料药的精制,干燥,包装必须使用独立的厂房与设施,室内保持相对负压,并与其制市生产车间分开.生产β-内酰胺结构类的原料药的精制,干燥,包装必须使用专用的设备和独立的空气净化系统,并与其他药品生产区域严格分开.避孕药品生产的厂房应与其他药品生产厂房分开,使用独立的专用的空气净化系统. 生产激素类,抗肿瘤类化学药品应避免与其他药品使用同一设备和空气净化系统.不合格,回收或退回产品应单独存放,并有明显标志.易燃,易爆,有毒,有害物质的生产和贮存的厂房设施应符合国家的有关规定.生产设备与生产要求相适应,便于生产操作和维修,保养.与药品直接接触的设备,工具,容器表面光洁,平整,易清洗消毒.洁净室(区)内设备保温层应平整,光洁,不得有颗粒性物质脱落.10 000级洁净室(区)使用的传输设备不得穿越较低级别区域.灭菌柜应具有自动监测,记录装置,其能力应与生产批量相适应.纯化水,注射用水的生产设备能保证水质量.贮水罐,输水管道,管件阀门等应为无毒,耐腐蚀的材质制造.贮水罐密闭,排气口有无菌过滤装置,输不管线能防止滞留.并易于拆洗,消毒.物料应分开,并按规定条件贮存,不得使其受潮,变质,污染或易于发生差错.麻醉药品,精神药品,毒性药品(包括药材)的验收,贮存,保管,使用,销毁等应严格执行国家有关规定.中药制剂认证要点:中药制剂系指以中药材,中药饮片或中药提取物为原料,以加工制成的具有一定剂型和规格的制剂.其硬件认证要点如下.厂房内地面,墙壁,天棚等内表面应平整,易于清洁,不易脱落,无霉迹,应对加工生产不造成污染.洁净室(区)水电,工艺管线应暗装.中药无菌制剂生产环境的空气洁交度级别要求:最终灭菌药品10 000级:小容量注射剂的配液,过滤,灌封;直接接触药品的包装材料的最终处理;非最终灭菌药品100级或10 000级背景下局部100级:灌装前不需除菌滤过的药淮配制;注射剂的灌封,分装和压塞;直接接触药品的包装材料最终处理后的暴露环境;1 0 000级:灌装前需除菌滤过的药液配制;100 000级:轧盖,直接接触药品的包装材料最后一次精洗的最低要求;其他无菌药品10 000级:供角膜创伤或手术用滴眼剂的配制和灌装.中药非无菌制剂生产环境的空气洁净度级别要求:(1)100 000级:非最终灭菌口服淮体药品的暴露工序;深部组织创伤外用药品,眼用药品的暴露工序;除直肠用药外的腔道用药的暴露工序;(2)300 000级:最终灭菌口服液体药品的暴露工序;口服固体药品的暴露工序;表皮外用药品暴露工序;直肠用药的暴露工序.(3)直接接触药品的包装材料最终处理的暴露工序洁净度级别应与其药品生产环境相同.5. 非创伤面外用制剂及其他特殊的中药制剂生产厂房门窗应能密闭,必要时有良好的除湿,排风,除尘,降温等设施.用于直接入药的净药材和干膏的配料,粉碎,混合,过筛等厂房应能密闭,有良好的通风,除尘等设施,人员,物料进出及生产操作应参照洁净室(区)管理.6. 洁净室应气密.7. 100级洁净室(区)和无菌制剂灌装区不得设水池和地漏.8. 中药制剂的生产操作区应与中药材的前处理,提取,浓缩以及动物脏器,组织的洗涤或处理等生产操作区严格分开.9. 中药材炮制中的蒸,炒,炙,煅等厂房与其生产规模相适应,并有良好的通风,除尘,除烟,降温等设施.10.人合格,回收或退回产品应单独存放并有效隔离.11.生产设备与生产要求相适应,便于生产操作和维修,保养.与药品直接接触的设备,工具,容器应表面光洁,平整,易清洗消毒,耐腐蚀,不与的加工的药品发生化学变化或吸附加工的药品,是易产生脱落物,避免污染药品.10 000级洁净室(区)使用的传输设备不得穿越其他较低级别区域.灭菌柜应具有自动监测,记录装置,其能力应与生产批量相适应.纯化水,注射用水的生产设备能保证水质量.贮水罐,输水管道,管件阀门等应为无毒,耐腐蚀的材质制造.贮水罐密闭,排气口有无菌过滤装置,输水管线能防止滞留,并易于拆洗,消毒.物料应分区,并按规定条件贮存,不得使其受潮,变质,污染或易于发生差错.麻醉药品,毒性药材的验收,贮存,保管,使用,销毁等应严格执行国家有关规定.毒性药材应使用专门的设备,容器及辅助设施进行生产.外用药认证要点:外用药剂型通常包括:洗剂(溶液型,混悬型及乳浊液型),软膏剂(油脂性,乳剂性)搽剂,糊剂,酊剂,醑剂,膜剂,栓剂,硬膏剂,气雾剂,凝胶剂,滴耳剂,滴鼻剂,眼膏剂等,其硬件认证要点如下.不同生产操作能有效隔离,不得互相妨碍.厂房内的天花板平整光洁,无裂隙,无脱落尘粒物质及起壳,不易积尘,不长霉.洁净区内的水电,工艺管线应暗装.一般外用药的生产从配料到灌封洁净度要求为不低于300 000级,不能在最后容器中灭菌的油膏,霜膏,悬浮液,乳化液用药的制备和灌封,洁净要求为不低于100 000级. 中药制剂中的膏药,橡皮膏,外用酊剂,外用散剂等非创伤面外用药制剂,要求生产厂房门窗应能密闭,必要时有良好的除湿,排风,除尘,降温等设施,人员,物料进出及生产操作应参照洁净区(室)管理.洁净室应气密.不合格,加收或退回产品应单独存放.生产设备与生产要求相适应,便于生产操作和维修,保养.与药品直接接触的设备表面光滑,平整,易清洗,耐腐蚀,不与所加工的药品发生化学或吸附所或工的药品.软膏剂,眼膏剂,栓剂等配制和灌装的生产设备,管道应方便清洗和消毒.不同洁净级别区域之间的传送设备要有防止污染的措施.纯水(去离子水,蒸馏水)的生产设备能保证不质量.贮不罐,输水管道,管件阀门等应为无毒,耐腐蚀的材质制造.贮水罐密闭,排气口有无菌过滤装置,输水管道能防止滞留,并易于拆洗,消毒.更衣室,盥洗间,消毒设施不得对洁净区产生不影响.原料,辅料及包装材料的贮存条件不得使其受潮,变质,污染或易于发生差错。
【优质】药物的质量标准PPT

方法进行全面校正。 熔点判断:中国药典要求初熔和全熔两个读数。
外文名(英文名):按INN命名。
熔吸点收测 系定数法的溶-测--毛定剂细法管:检测定查法::测定供试品前,应先检查所用的溶剂,在测
E1cm1吸%收定度义。:溶解浓度定为1供%(试g/ml)品、光所路长用度为的1cm波的 长附近是否符合要求,不
4.晶型 检查晶型的意义:同一中药物,由于其晶
胞的大小和形状的不同,结晶的结构不同, 出现多晶现象。不同晶型的药物其生物利用 度不同。
检查方法:IR法、X-射线衍射法 示例:无味氯霉素中无效晶型的检查
5. 吸收系数的测定法: 纯度要求 安全性 药品的有效性:是以动物试验为基础,最终以临床疗效来评价。
得有干扰吸收峰。 Organic Chemistry》命名。
熔点判断:中国药典要求初熔和全熔两个读数。 吸收系数的测定法:
E1cm1%定义:溶解浓度为1%(g/ml)、光路长度为1cm的
吸收度。
(四)检查有效性试验
1.检查项包括:有效性 均一性
(三)鉴别
1.鉴别方法的特点: 化学鉴别法:操作简便、快速、价廉、应用广。 仪器鉴别法:UV、IR、TLC应用最广。
②不化同学 晶法型与的仪药(器物法其2相生)结物合利合,用每度理种不药同性品。一般新选用药2~ 质量标准的研究阶段,检查的项目
吸收系数的测定法:
E1cm1吸%收定度义应。:溶尽解浓可度为能1%(的g/m多l)、做光路,长度但为1c在m的制定该药质量标准的内容时应合
熔点测定法---毛细管测定法: 安全性:热原检查、毒性试验、刺激性试验、过敏性试验、升压或降压物质检查。
E 定义:溶解浓度为1%(g/ml)、光路长度为 参3.考文药I献U品P资A质C料量公1的标布c查准m的阅制《1及定%N整的am理基en础cla:ture of
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Q/AELU 赛恩斯环保股份有限公司企业标准
Q/AELU 003-2018 SES-FS-生物制剂-S003
2018-11-01发布2018-12-01实施
赛恩斯环保股份有限公司发布
前言
本标准按照GB/T1.1-2009给出的规则起草本标准由赛恩斯环保股份有限公司提出
本标准起草单位:赛恩斯环保股份有限公司本标准主要起草人:廖圆
SES-FS-生物制剂-S003
1 范围
本标准规定了重金属废水处理SES-FS-生物制剂-S003产品的技术要求、采样、试验方法、检验规则以及标志、包装、运输、贮存、安全。
本标准适用于赛恩斯环保股份有限公司生产的,用于含COD及重金属废水处理的SES-FS-生物制剂-S003。
2 规范性引用文件
下列文件中的条款通过本标准的引用而成为本标准的条款。
凡是注日期的引用文件,仅所注日期的版本适用于本标准。
凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本标准。
GB 191-2000包装储运图示标志
GB/T 601 化学试剂标准滴定溶液的制备
GB/T 602 化学试剂杂质测定用标准溶液的制备(GB/T602-2002)
GB/T 603 化学试剂试验方法中所用制剂及制品的制备(GB/T 603-2002)
GB/T 6678 化工产品采样总则
GB/T 6680 液体化工产品采样通则
GB 6682 分析实验室用水规格和试验方法
3 技术要求
3.1 外观
红棕色液体或青绿色固体。
3.2 技术指标
SES-FS-生物制剂-S003产品应符合表1要求。
表1 SES-FS-生物制剂-S003产品指标
指标项目指标
波美度/°Bé,20摄氏度≥24 模拟废水处理效果(以Zn计)/mg/L ≤ 1.0
模拟废水处理效果(以COD计)/mg/L ≤50
4 采样
4.1产品按批检验。
生产企业以成品槽、一天或一个班次生产的SES-FS-生物制剂-S003溶液为一批。
用户以每次收到的SES-FS-生物制剂-S003为一批。
4.2 SES-FS-生物制剂-S003用槽车、储槽装运时,建议用GB/T 6680中规定的适宜取样器,从深度不同的上、中、下三处采取等量的代表性样品混合。
4.3 SES-FS-生物制剂-S003用塑料桶(瓶)包装时,应按GB/T 6678中规定的采样单元数随机抽取样品。
4.4将采取的样品分装于两个清洁、干燥的带磨口塞的棕色广口瓶中,密封。
每瓶样品量不得少于200mL。
一瓶用于检验,一瓶用于备检。
样品瓶上应贴上标签,并注明:生产企业名称、产品名称、型号规格、批号或生产日期、采样量、采样日期及取样人姓名等。
5 试验方法
除非另有说明,在分析中仅使用分析纯试剂和GB/T 6682中规定的三级水或相应纯度的水。
试验中所需标准溶液、制剂及制品,在没有规定时,均按GB/T 601、GB/T 602、GB/T 603规定制备。
5.1 波美度的测定
5.1.1 测定原理
由波美计在被测液体中达到平衡状态时所浸没的深度读出该液体的波美度。
5.1.2 仪器设备
波美计:刻度值0-35°Bé
恒温水浴:可控温度20±1℃
温度计:分度值为1℃
量筒:250~500mL
5.1.3 测定步骤
将SES-FS-生物制剂-S003注入清洁、干燥的量筒内,不得有气泡。
将量筒置于20±1℃的恒温水浴中,待温度恒定后,将波美计缓缓地放入试样中,待波美计在试样中稳定后,读出波美计弯月面下缘的刻度(标有读弯月面上缘刻度的波美计除外),即为20℃试样的波美度。
样品温度与波美度有着一定的关系,当没有恒温水浴条件时需要同时检测样品温度,按照标准温度20℃折算成波美度,温度如高于20℃时,其读数应加上补正值;低于20℃时应减去补正值。
表2 SES-FS-生物制剂-S003波美度与温度补正表
温度(℃)补正度数温度(℃)补正度数温度(℃)补正度数
3 -0.58 13 -0.18 23 +0.18
4 -0.54 14 -0.14 24 +0.24
5 -0.5 15 -0.1 25 +0.3
6 -0.46 16 -0.08 26 +0.36
7 -0.42 17 -0.06 27 +0.42
8 -0.38 18 -0.04 28 +0.48
9 -0.34 19 -0.02 29 +0.54
10 -0.3 20 0 30 +0.6
11 -0.26 21 +0.06 31 +0.66
12 -0.22 22 +0.12 32 +0.72 5.1.4允许差
平行测定结果之差绝对值不大于0.5%。
取测定结果的算术平均值为报告结果。
5.2 模拟废水COD及重金属(以Zn计)去除效果
5.2.1实验方法
5.2.1.1 模拟废水
称取0.1g苯酚,0.440g硫酸锌(ZnSO4·7H2O)溶于水,移入1000mL的容量瓶中,稀释至刻度,配制出含COD及重金属的模拟废水。
此模拟废水中COD 约为250mg/L,Zn为100mg/L。
5.2.1.2 PAM溶液
准确称取1gPAM,在持续搅拌下溶于1L水中。
5.2.1.3 氧化剂
赛恩斯环保股份有限公司提供的,配合SES-FS-生物制剂-S003使用的药剂。
5.2.1.4试验流程
烧杯取500mL模拟废水置于磁力搅拌器进行搅拌,加入0.5mLSES-FS-生物制剂-S003,同时加入0.4mL氧化剂进行协同氧化反应30min,加入石灰乳调pH值至10.5进行水解反应15min,加入1mLPAM溶液搅拌2min,静置5min,取上清液经滤纸过滤待测。
5.2.2检测方法
5.2.2.1COD检测方法
水质化学需氧量的测定重铬酸盐法(HJ 828—2017)
5.2.2.2重金属检测方法(以Zn计)
水质铜、锌、铅、镉的测定原子吸收分光光度法(GB 7475-1987)
6 检验规则
6.1 SES-FS-生物制剂-S003应由生产企业按照本标准规定的试验方法和检验规则对产品质量进行检验,应保证所有出厂的产品都符合本标准的要求。
6.2每批次出厂的SES-FS-生物制剂-S003应附有质量证明书,内容包括:生产企业名称、产品名称、型号、批号或生产日期、执行标准号。
6.3用户有权按本标准规定对收到的SES-FS-生物制剂-S003进行检验,检验其质量是否符合本标准的要求。
6.4如果检验结果有一项指标不符合本标准要求时,应重新加倍在包装单元中采取有代表性的样品进行复检,复检结果中即使有一项指标不符合本标准的要求,则整批产品为不合格。
6.5当供需双方对产品质量发生异议时,应由有资质的检验机构仲裁检验。
7 标志、包装、运输和贮存
7.1 标志
出厂的SES-FS-生物制剂-S003的外包装上应有明显牢固的标志,内容包括:生产企业名称、地址、产品名称、型号规格、净质量、执行标准、批号或生产日期。
7.2 包装
SES-FS-生物制剂-S003宜用塑料桶(瓶)或槽车包装。
其中塑料桶包装采用双层桶盖,内盖扣严,外盖旋紧。
7.3 运输
运输过程中要求密封,避免雨淋;装运容器要求防腐并保持包装完整、标志清晰;运输时应避免撞击及其它化学物质污染。
7.4 贮存
贮存时不要暴晒,存放于阴凉、通风的库房内,贮存温度不低于0℃,有效贮存期为6个月。
应与氧化剂、酸类等分开存放,切忌混储。
储区应备有合适的材料收容泄漏物。
7.5 安全
SES-FS-生物制剂-S003产品具有一定的腐蚀性,操作人员在进行作业时应戴防护眼镜、胶手套等防护用具以避免身体直接接触。