9342eec MDD 中文介绍
MDD指令93_42_EEC(中英文对照版最终版)

第 1 页 共 72页 Edited By Stephen Ling. Any question, please contact with me Via Email: ibat@
Whereas certain medical devices are intended to administer medicinal products within the meaning of Council Directive 65/65/EEC of 26 January 1965 on the approximation of provisions laid down by law, regulation or administrative action relating to proprietary medicinal products; whereas, in such cases, the placing on the market of the medical device as a general rule is governed by the present Directive and the placing on the market of the medicinal product is governed by Directive 65/65/EEC; whereas if, however, such a device is placed on the market in such a way that the device and the medicinal product form a single integral unit which is intended exclusively for use in the given combination and which is not reusable, that single-unit product shall be governed by Directive 65/65/EEC; whereas a distinction must be drawn between the abovementioned devices and medical devices incorporating, inter alia, substances which, if used separately, may be considered to be a medicinal substance within the meaning of Directive 65/65/EEC; whereas in such cases, if the substances incorporated in the medical devices are liable to act upon the body with action ancillary to that of the device, the placing of the devices on the market is governed by this Directive; whereas, in this context, the safety, quality and usefulness of the substances must be verified by analogy with the appropriate methods specified in Council Directive 75/318/EEC of 20 May 1975 on the approximation of the laws of the Member States relating to analytical, pharmaco-toxicological and clinical standards and protocols in respect of the testing of proprietary medicinal products; 鉴于部分医疗器械是符合 1965 年 1 月 26 日理事会第 65/65/EEC 号指令, 与专卖医药产品有关的法律, 法规或管理行 为所订的实施规定, 鉴于医疗器械的上市基本上由本指令规范, 但医疗产品的上市则受 65/65/EEC 号指令规范; 鉴于若 有某种器械须与其他医疗产品组成一完整的产品而上市销售, 使用, 且无法二次使用时, 则该组合产品应受 65/65/EEC 号指令规范; 鉴于前述的医疗器械和包含医疗物质且该物质单独使用时符合 65/65/EEC 号指令规定的医疗器械应加以 区别; 鉴于前述包含于医疗器械的医疗物质若对人体产生作用以辅助医疗器械的作用时, 则该医疗器械的上市应由本指 令规范; 鉴于 1975 年 5 月 20 日 75/318/EEC 号理事会指令[制定各会员国在测试专利医疗产品方面有关分析药物毒性 和临床的标准及调查书的法律调和], 医疗物质的安全, 品质及效用在前述情况下则须依该指令明定的适当方法加以证 实;
MDD_93-42-EEC医疗器械指令_全部(中文)

欧洲共同体理事会关于医疗器械的93/42/EEC指令1993年6月14日欧洲共同体理事会考虑到建立欧洲经济共同体的条约,特别是其第100a条;考虑到欧洲共同体委员会提交的议案;考虑到与欧洲议会合作;考虑到经济与社会委员会的意见;鉴于应在欧洲共同体内部市场范围内正式通过必要的措施;鉴于欧洲共同体内部市场是一个确保商品、人员、服务和资本自由流通的无内部边界的区域;鉴于各成员国有关医疗器械的安全、健康保护和工作特性的法律、法规和行政条款的内容和范围是不同的;鉴于各成员国之间对这类器械的认证和检验程序存在差异;鉴于这种差异在欧洲共同体内形成贸易壁垒;鉴于各国针对医疗器械的使用所制定的有关患者、使用者及其他人员的安全和健康保护的条款应予以协调,以保证此类器械在欧洲共同体内部市场自由流通;鉴于协调条款必须与成员国为管理直接或间接与这类器械有关的公共健康和医疗保险计划的资金筹措而采取的措施相区别;鉴于因而只要遵守欧洲共同体法律,这些条款并不影响各成员国实施上述措施的能力;鉴于医疗器械应向患者、使用者及第三方提供高水平的保护并达到制造商赋予其的性能水平;鉴于因此保持和提高各成员国已达到的保护水平是本指令的基本目的之一;鉴于在1965年1月26日欧洲共同体理事会关于使有关特许专卖药品的法律、法规或行政措施趋于一致的65/65/EEC指令中,某些医疗器械是用于施药的;鉴于在这种情况下,医疗器械投放市场通常由本指令管理,而药品投放市场由65/65/EEC指令管理;鉴于如果这种器械投放市场的方式使器械与药品构成一种规定只供组合使用且不能再次使用的整体,则这个单一整体产品应由65/65/EEC指令管理;鉴于必须将上述器械与包含某种物质,特别是当其单独使用时,按65/65/EEC指令可视为药物的医疗器械相区别;鉴于在这种情况下,若这种物质能配合医疗器械对人体产生辅助作用,则这类器械投放市场由本指令管理;鉴于这类物质的安全性、质量和有效性必须比照欧洲共同体理事会1975年5月20日关于使成员国有关分析标准、药物毒理学标准和临床标准及特许专卖药品试验协议的法律趋于一致的75/318/EEC指令规定的适当方法加以验证;鉴于本指令附录中的基本要求和其他要求,包括对“减小”或“降低”危险的任何引用,在解释和实施时必须考虑设计时的技术现状和实际做法及高水平的健康和安全相适应的技术与经济条件;鉴于按照1985年5月7日欧洲共同体理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计和生产的规定必须限于满足基本要求所必须的条款;鉴于因为这些要求是基本的,因此它们应取代各国相应的条款;鉴于实施基本要求应审慎考虑设计时的技术水平和与高水平的健康及安全保护相适应的技术、经济条件;鉴于1990年6月20日欧洲共同体理事会关于使成员国有关有源植入式医疗器械的法律趋于一致的90/385/EEC指令是新方法指令在医疗器械领域中的首次应用案例;鉴于为了使欧洲共同体的统一规定适用于所有医疗器械,本指令基本上是以90/385/EEC指令的条款为依据的;鉴于为此必须修订90/385/EEC 指令,以便放入本指令规定的一般性条款;鉴于电磁兼容性问题是医疗器械安全的一个组成部分;鉴于就1989年5月3日欧洲共同体理事会关于使成员国有关电磁兼容的法律趋于一致的89/336/EEC指令而言,应包含这方面的专门规定;鉴于本指令应包括有关发射电离辐射的器械在设计和制造方面的要求;鉴于本指令既不影响1980年7月15日欧洲共同体理事会80/836/Euratom指令对有关保护公众和工人免受电离辐射危险的基本安全标准的指令进行修订所要求的授权,也不影响欧洲共同体理事会1984年9月3日对接受医疗检查和治疗的人员的辐射防护规定了基本措施的84/466/Euratom指令的实施;鉴于欧洲共同体理事会1989年6月12日关于采取措施鼓励改善工人工作中的安全和健康的89/391/EEC指令以及有关这一问题的专门指令应继续予以实施;鉴于为了证实符合基本要求并使这种符合得到验证,需要制定欧洲协调标准来防止与医疗器械的设计、制造和包装有关的危险;鉴于这类欧洲协调标准是由非官方机构制定的,应保持其非强制性的地位;鉴于为此欧洲标准化委员会(CEN)和欧洲电工标准化委员会(CENELEC)按照1984年11月13日欧洲共同体委员会与这两个机构之间签署的合作总指导原则,被认可为批准协调标准的主管机构;鉴于在本指令中,协调标准是受欧洲共同体委员会委托,由上述两机构之一,或两个机构共同根据欧洲共同体理事会1983年3月18日关于在技术标准和法规领域提供信息程序的83/189/EEC指令,依照上述总指导原则而批准的技术规范(欧洲标准或协调文件);鉴于对协调标准进行修订,欧洲共同体委员会应得到根据83/189/EEC指令建立的常设委员会的帮助;鉴于应采取的措施必须按欧洲共同体理事会87/373/EEC决定中规定的程序Ⅰ而规定;鉴于在特定领域,欧洲药典专著这类现有的形式应包括在本指令的范围内;鉴于因此有几部欧洲药典专著可视为等同于上述协调标准。
医疗器械200747EC指令(9342EEC指令的修订案)

DIRECTIVE 2007/47/EC OF THE EUROPEAN PARLIAMENT AND OF THECOUNCILof 5 September 2007amending Council Directive 90/385/EEC on the approximation of the laws ofthe Member Statesrelating to active implantable medical devices, Council Directive 93/42/EECconcerning medicaldevices and Directive 98/8/EC concerning the placing of biocidal products onthe market医疗器械指令更改的分析(针对2007/47/EC)September 5, 2007, the European Union issued a new Directive (2007/47/EC) that will affect all manufacturers selling medical devices in Europe.2007年9月5日,欧盟发布了一个将影响所有在欧洲出售医疗器械的制造商的新指令(2007/47的/EC)。
Essentially, this new Directive is the first significant modification to the Medical Device Directive since 1993 and there are several changes you may need to prepare for WELL BEFORE it becomes mandatory in March 2010. Here is a brief overview of major changes本质上讲,这份新指令是自1993年起,对医疗器械指令的第一个重大的修改,在2010年3月强制执行之前,你们需要好好准备它相应的改变。
欧盟医疗器械指令9342EEC指令

►M3 Directive 2001/104/EC of the European Parliament and of the Council of 7 December 2001
►M4 Regulation (EC) No 1882/2003 of the European Parliament and of the Council of 29 September 2003 ►M5 Directive 2007/47/EC of the European Parliament and of the Council of 5 September 2007
MDD 93/42/EEC指令
1
Module 2 – MDD指令
医疗器械指令 (MDD) 93/42/EEC 1993-06-14 COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993 concerning medical devices
►M1 Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 ►M2 Directive 2000/70/EC of the European Parliament and of the Council of 16 November 2000
十八:不正确地使用C E 标示 十九:决定不予批准或予以限制 二十:保密规定 二十一:相关法令修改和废止 二十二:附则 二十三:本指令向各成员国发送
6
Event, Date, Location, Author
第1章 定义和范围 医疗器械:
是指任何仪器、设备、器具、材料或者其它物品, 不论是单独使用还是 组合使用的, 包括使用所需软件在内, 由制造者为下列预期用途(目的)用于 人类的,这些目的是: - 疾病的诊断、预防、监护、治疗或者 缓解; - 损伤或残疾的诊断、监护、治疗、缓解或者补偿; - 解剖或生理过程的研究、替代或者补偿; - 妊辰控制; 其作用于人体体表及体内的主要设计作用不是用药理学、免疫学 或代谢的手 段获得,但可能有些手段参与并起一定辅助作用。
手持式频谱分析仪 (N9342C, N9343C, N9344C)

是德N9342C/43C/44C 手持式频谱分析仪请注意:安捷伦电子测量仪器部已经转为是德科技有限公司。
关于此方面详细信息,请访问用户手册Notices© Keysight Technologies, Inc. 2012-2017No part of this manual may be reproduced in any form or by any means (including electronic storage and retrieval or translation into a foreign language) without prior agreement and written consent from Keysight Technologies, Inc. as governed by United States and international copyright laws. Trademark AcknowledgmentsManual Part NumberN9342-90003EditionEdition 5, October2017Printed in ChinaPublished by:Keysight TechnologiesNo 116 Tianfu 4th street Chiengdu, 610041 C hina WarrantyTHE MATERIAL CONTAINED IN THIS DOCUMENT IS PROVIDED “AS IS,” AND IS SUBJECT TO BEING CHANGED, WITHOUT NOTICE, IN FUTURE EDITIONS. FURTHER, TO THE MAXIMUM EXTENT PERMITTED BY APPLICABLE LAW, KEYSIGHT DISCLAIMS ALL WARRANTIES, EITHER EXPRESS OR IMPLIED WITH REGARD TO THIS MANUAL AND ANY INFORMATION CONTAINED HEREIN, INCLUDING BUT NOT LIMITED TO THE IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS FOR A PARTICULAR PURPOSE. KEYSIGHT SHALL NOT BE LIABLE FOR ERRORS OR FOR INCIDENTAL OR CONSEQUENTIAL DAMAGES IN CONNECTION WITH THE FURNISHING, USE, OR PERFORMANCE OF THIS DOCUMENT OR ANY INFORMATION CONTAINED HEREIN. SHOULD KEYSIGHT AND THE USER HAVE A SEPARATE WRITTEN AGREEMENT WITH WARRANTY TERMS COVERING THE MATERIAL IN THISDOCUMENT THAT CONFLICT WITHTHESE TERMS, THE WARRANTYTERMS IN THE SEPARATEAGREEMENT WILL CONTROL.Technology LicensesThe hardware and/or softwaredescribed in this document arefurnished under a license and may beused or copied only in accordancewith the terms of such license.U.S. Government RightsThe Software is “commercialcomputer software,” as definedby Federal Acquisition Regulation(“FAR”) 2.101. Pursuant to FAR12.212 and 27.405-3 andDepartment of Defense FARSupplement (“DFARS”) 227.7202,the U.S. government acquirescommercial computer softwareunder the same terms by whichthe software is customarilyprovided to the public.Accordingly, Keysight providesthe Software to U.S. governmentcustomers under its standardcommercial license, which isembodied in its End User LicenseAgreement (EULA), a copy ofwhich can be found at/find/sweulaThe license set forth in the EULArepresents the exclusive authorityby which the U.S. governmentmay use, modify, distribute, ordisclose the Software. The EULAand the license set forth therein,does not require or permit,among other things, thatKeysight: (1) Furnish technicalinformation related tocommercial computer softwareor commercial computersoftware documentation that isnot customarily provided to thepublic; or (2) Relinquish to, orotherwise provide, thegovernment rights in excess ofthese rights customarily providedto the public to use, modify,reproduce, release, perform,display, or disclose commercialcomputer software orcommercial computer softwaredocumentation. No additionalgovernment requirementsbeyond those set forth in theEULA shall apply, except to theextent that those terms, rights, orlicenses are explicitly requiredfrom all providers of commercialcomputer software pursuant tothe FAR and the DFARS and areset forth specifically in writingelsewhere in the EULA. Keysightshall be under no obligation toupdate, revise or otherwisemodify the Software. Withrespect to any technical data asdefined by FAR 2.101, pursuantto FAR 12.211 and 27.404.2 andDFARS 227.7102, the U.S.government acquires no greaterthan Limited Rights as defined inFAR 27.401 or DFAR 227.7103-5(c), as applicable in any technicaldata.Safety NoticesA CAUTION notice denotes a hazard. Itcalls attention to an operatingprocedure, practice, or the like that,if not correctly performed or adheredto, could result in damage to theproduct or loss of important data. Donot proceed beyond a CAUTIONnotice until the indicated conditionsare fully understood and met.A WARNING notice denotes a hazard.It calls attention to an operatingprocedure, practice, or the like that,if not correctly performed or adheredto, could result in personal injury ordeath. Do not proceed beyond aWARNING notice until the indicatedconditions are fully understood andmet.目录1简介介绍 2主要功能 2前端面板概述 5顶部面板概述 6显示屏标注 72使用指南检查货品包装和装箱清单 12电源要求 13交流电源线规格 14安全须知 15供电要求 17静电防护 17安装电池 18查看电池信息 18电池充电 18首次开机 20仪器开机 20准备工作 21设置界面 21检查设备信息 21设置开关机/预设置 21显示和音频调整 22通用系统设置 22定时开关机 23IP设置 23外部输入 23系统信息 24添加选件 24显示错误信息 25进行校准 25数据安全 26固件升级 26探头供电输出 26HSA PC 软件 27默认出厂设置 28基本测量 29联系是德科技 313开始测量测量多个信号 34同一屏上比较信号 34测量低电平信号 39减小输入损耗 39改善频率分辨率和精确度 44测量信号失真 45识别由频谱仪产生的失真 45三阶交调失真 48功率测量 50占用带宽 50邻道功率泄漏比 50信道功率 51脉冲响应传输测量 53测量低通滤波器阻带衰减 55反射测量 57使用反射测量方法测试回波损耗 59使用USB功率探头作功率测量 60使用USB功率探头作平均功率测量 62使用USB功率探头作峰值功率测量 64功率计设置 66频谱监测 69解调FM调频信号 71解调分析 73AM/FM解调分析 73ASK/FSK解调分析 75通道扫描仪 79扫描最强/最弱的N个信道 79序列扫描 80通道扫描设置 82电缆和天线测试 84测试前的准备 84反射测量 85故障定位测量 85文件 86查看文件列表 86保存文件 87保存用户状态文件 88删除文件 89加载文件 894按键说明Amptd 92参考电平 92衰减 92预放 92刻度 92刻度类型 93Y 轴单位 93高灵敏度 93参考偏移量 93幅度修正 94电阻 95PSD功能 95Display 96刻度线 96Y轴刻度 96显示线 96BW 97分辨率带宽 97视频带宽 97VBW/RBW 98平均类型 98Sweep 100扫描时间 100扫描 101单次扫描 101触发 101时间选通 102扫描设置 103Enter 105退出/清除 106频率 107自动调谐 107中心频率 107起始频率 107终止频率 107中心频率步进 108信道标准 108频率偏移 108低频通道(选件BB1) 108标记 109标记 109标记的轨迹 109模式 109标记移到 111功能 112标记列表 113读出 113放大/缩小 113差量参考 114全部关闭 114标记记录 114Peak 115搜索峰值 115左次峰值 115右次峰值 115峰峰值搜索 115连续峰值 115峰值表 116峰值标准 116测量 117占用带宽 117邻道功率比 117信道功率 119频谱监测 120频谱模板(SEM) 123模式 126频谱分析 126跟踪发生器 126功率计 129测量管理器 134解调分析 134天馈线测试 135通道扫描仪 138 Span 144扫宽 144全扫宽 144零扫宽 144上次扫宽 144Trace 145轨迹 145刷新 145最大值保持 145最小值保持 146静止 146空白 146检波 146平均次数 147平均时间 148重新平均 148Limit 149极限线 149极限模板 149设置极限模板 149极限类型 149蜂鸣器 150保存模板 150调用模板 1505错误信息错误信息表 1526按键结构图BW 158Sweep 158FREQ 159LIMIT 159MARKER 160Peak 161File/Mode - 任务序列 162Mode - 跟踪发生器 163Mode - 调制分析(AM/FM) 164Mode - 调制分析(ASK/FSK) 165 Mode - 天馈线测试 166Mode - 功率计 167Meas(1) 168Meas(2) 169Span 169System 170Trace 171简介1简介1简介介绍2介绍是德N934xC 是手持式射频频谱分析仪,其频率范围从100 kHz 覆盖到20 GHz。
9342eec中文

20 鉴于按照一般规则,医疗器械应标示 CE 标志,表明它们符合本指令的条款,使其能在欧共体内自由流 通,并按其预定用途投入使用;
23 鉴于证实符合基本要求可能意味着必须由制造商负责完成临床试验;鉴于为了完成临床试验,必须确定 保护公众健康和公共秩序的适当方式;
24 鉴于在欧共体级别一体化的医疗器械警戒系统可以更有效地完成健康保护和相关的控制; 25 鉴于本指令覆盖了 1976 年 7 月 27 日理事会第 76/764/EEC 号关于使成员国有关临床用汞柱式温度计最高
15 鉴于为了证实符合基本要求并使该符合性得以验证,有必要协调欧洲标准以避免与医疗器械的设计、制 造和包装有关的危险;鉴于这类欧洲协调标准是由非官方立法机构制定的,应保持其非强制性的性质; 鉴于到目前为止,欧洲标准化委员会(CEN)和欧洲电工标准化委员会(Cenelec),根据 1984 年 11 月 13 日执委会与这两个机构之间签署的合作通则,已被公认为是制定协调标准的职能机构;
Created by lisong Page 1 of 35
11 鉴于按照 1985 年 5 月 7 日理事会关于技术协调与标准化新方法的决议所规定的原则,有关医疗器械设计 和制造的规则必须限制在满足基本要求所必需的条款内;鉴于因为这些要求是最基本的,因此它们应取 代相应的国家规定;鉴于基本要求的实施应谨慎考虑设计当时的技术水平,并在符合健康和安全高度保 护的原则下考虑技术和经济因素;
17 鉴于在 1990 年 12 月 13 号决议关于用于技术协调指令不同合格评定程序的各阶段模式中,理事会制定了 协调合格评定程序;鉴于将这些模式用于医疗器械可使制造商和公告机构在合格评定中的责任根据有关 的器械型式予以确定;鉴于对这些模式的细节所作的补充,根据医疗器械必需验证的性质,证明是合理 的;
CE认证适用标准清单2012

EN 12470-4:2000
(21/03/2010)
2000A, 01.07.09
EN ISO 13485:2003/AC:2009
Medical devices - Quality management systems - Requirements for regulatory purposes
疗器械的生物学评价第1部分:评价和试验
EN ISO 10993-1:2003
(21/03/2010)
—
EN ISO 10993-1:2009/AC:2010(new)
First publication
EN ISO 10993-3:2009
Biological evaluation of medical devices - Part 3: Tests for genotoxicity, carcinogenicity and reproductive toxicity (ISO 10993-3:2003)
非侵入式血压表.第3部分:电机血压表的补充要求
EN 1060-3:1997
(31/05/2010)
2005A, 1.07.09
EN ISO 9919:2009
Medical electrical equipment - Particular requirements for the basic safety and essential performance of pulse oximeter equipment for medical use (ISO 9919:2005)
(IEC 60601-1:2005)
医疗电气设备.基本安全和主要性能的一般要求
MEDDEV2.7.1rev42016附中文

MEDDEV2.7.1rev42016附中文__.71 revision 4 June 2022年Guidelines on Medical Devices Clinical Evaluation:A Guide for Manufacturers And Notified Bodies Under Directives 9342EEC and __EEC__N __IONDG Internal Market, Industry, Entrepreneurship and SMEsConsumer, Environmental and Health TechnologiesHealth technology and Cosmetics备注:中文翻译中的临床调查=临床研究,评估=评价、设备=器械、数据=资料NoteThe present Guidelines are part of a set of Guidelines relating to questions of application of EC-Directives on medical Devices. They are legally not binding. The Guidelines have been carefully drafted through a process of intensive consultation of the various interested parties (competent authorities, Commission services, industries, other interested parties) during which intermediate drafts where circulated and comments were taken up in the document. Therefore, this document reflects positions taken by representatives of interest parties in the medical devices sector.These guidelines incorporate changes introduced by Directive 2022年/47/EC amending Council Directive 90/385/EEC and Council Directive 93/42/EEC.本指南为一系列与CE―医疗器械指令应用问题相关的指南中的一部分。
西门子FC9342 联动控制盘产品说明书
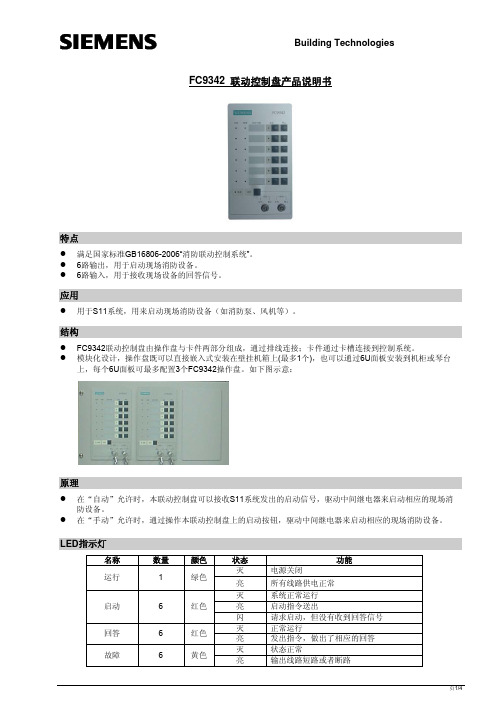
PIN.19 PIN.20
DC 24V 1# 联动请求信号 2# 联动请求信号 3# 联动请求信号 4# 联动请求信号 5# 联动请求信号 6# 联动请求信号 空
复位信号
状态反馈数据端 机壳地
DC24V电源正极
自动联动请求信号,由FS1120控制器输出的信号(如由E3L020 联动卡输出所提供的),低电平有效,在接收到持续有效的请求 信号且“自动”锁处于“允许”状态下,FC9342的相应通道则 进入启动状态,并通过卡件上的CZ201端子的相应端输出持续的 启动信号,用于控制现场设备。
DC 24V电源负极
页3/4
FC9342与E3L020联动卡接线示意图举例
z FC9342与E3L020共用DC24V电源时:
1 输出1 2
3 4 5 6 输出6 7
Building Technologies
2 3 4 5 6 7 JP100
FC9342卡件
E3L020
20
备注:E3L020的输出线路可根据实际使用选择(Input/Output 1# ~16#)
复位请求信号,由E3L020提供,低电平有效,当接收到有效的 复位脉冲后,FC9342复位。 常规情况下,必须与PIN.12短接来使用 机壳地
状态反馈数据
常规可不用(PIN.12必须与PIN.10短接)
自动锁允许状态
空 DC 0V
“自动”状态信号反馈,可向FS1120控制器反馈的状态信号(如与 E3L020连接,在“自动”处于“允许”的状态下,向E3L020发出持 续的低电平信号)。常规可不用
上,每个6U面板可最多配置3个FC9342操作盘。如下图示意:
原理
z 在“自动”允许时,本联动控制盘可以接收S11系统发出的启动信号,驱动中间继电器来启动相应的现场消 防设备。
欧盟医疗器械9342EEC

欧盟医疗器械9342EEC
凡是要出⼝到欧盟的医疗器械都需要通过医疗器械的认证。
欧盟的医疗器械指令为93/42/EEC。
93/42/EEC有23个Article和12个Annex。
Article主要介绍定义,范围和介绍。
Annex主要介绍认证模式,基本要求和分类。
基本要求:
Annex I是基本要求,所有的医疗器械必须符合基本要求,基本要求中可能有部分不适⽤,但适⽤部分必须符合基本要求。
分类:
Annex IX是分类的要求。
CE-MDD中分类可分为:
Class I;
Class IIa;
Class IIb;
Class III;
危险等级由低到⾼。
认证模式:
Annex II~VII都是认证模式,不同的分类选择不同的模式,⼀个分类可能有多个认证模式,具体认证模式选择由制造商决定,如果制造商不清楚,可以授权公告机构来决定。
公告机构:
公告机构是由欧盟授权,机构所在国的相关部门批准,批准后有⼀个公告号,可在欧盟⽹站上查询公告范围,起始⽇期和有效期。
N9342C手持频谱仪

MDD 93-42-EEC简介

2014/07/01,欧盟网站共计公布25类需进行CE
Marking的商品大类
1. 有源植入医疗器械; 2. 燃气炉具;
3. 载人的索道装置;
4. 建筑产品;
5. 能源相关产品生态设计;
6. 电磁兼容;
7. 使用于具有爆炸性环境中的设备和保护系统;
8. 民用爆破器材;
9. 热水锅炉;
10. 体外诊断试剂;
② ‘CE’,法语“Communate Europpene”的缩写,意思即“欧洲 共同体”;
③ 具有’CE’标识的商品,意味着该商品满足欧盟相关法规及协 调标准的要求,制造商负有相应责任。
CE Marking
目的 消除商品流通壁垒,保护公众利益.
CE Marking
适用范围
Member states of EU 欧盟成员国(28个,24种官方语言)
基本要求 一般要求 有关设计及架构的要求
EC符合性声明(完全品质保证系统)
内容 Definition,Scope Placing on the market and putting into service Essential Requirements Free movement, devices intended for special purposes Reference to standards Committee on standards and Technical regulations Committee on Medical device Safeguard clause Classfication Information on incidents occuring following placing of devices on the market Conformity assessment procedures Particular procedure for systems and procedure packs Decisions with regards to classfication, derogation clause Registration of persons responsible for placing devices on the market Clinical investigation Notified bodies CE marking Wrongly affixed CE Marking Decision in respect of refusal or restriction Confidentiality Repeal and amendment of Directives Implementation,transitional provisions This directive is addressed to the member states
ISO13485-2016与QSR820 MDD9342EEC 条款对照

ISO13485:2016版标准条款MDD93QSR820 MDD93/42/EEC(1993年6月14日)`a 文件的批准和发布b文件更改A评价供应商B采购文件(包括质量要求、当产品发生更改时及时通知组织,组织评估对最终产品的影响)A文件化的指导书B监视和控制过程参数C符合相关的标准或法规D过程及过程设备的确认E标准的工艺生产和过程更改(验证-确认-批准)建立设备管理程序校准(当精确度不符合要求时应采取补救措施和重新建立限制,并评价是否已对产品质量产生不利影响)1校准检查A特殊过程B过程参数(合格的人员;操作者或使用的主要设备要记录)A检验包括检验、试验进货检验不合格品的控制不合格品的评审和处置1建立程序并保存记录2返工程序和记录建立纠正和预防措施1分析过程、原因并用适当的统计技术发现重复出现的问题2调查产生的原因3识别需要采取的纠正预防措施4对纠正预防措施进行验证5实施和记录为纠正预防进行的更改6人员7提交管理评审标签的完整性标签的检验包装和搬运箱经过设计和构造,起到保护作用建立程序,防止损坏、污染及对产品质量的不利影响建立程序,防止出现混淆、损坏、质量下降、污染,确保废品、拒收产品或质量不好的产品被使用或交付建立出入库规范建立并保持产品交付记录投诉处理程序建立程序,抽样计划C 各产品应建立DMR(设备主文档)产品规范:如图纸、作业指导书D保存期限、生产过程规范,生产设备操作规程,质量保证程序,检验标准环境控制人员污染控制C若发生变化或过程的偏离,应评审和评价过程并重新确认并记录过程检验最终检验检验记录保存标签的贮存标签的操作控制号建筑物设备1保养计划2检查3校准原材料自动过程清线记录。
mdd9342eec简介

② “Directive 指令”是指设定所有欧盟成员国必须实现的目标的立法行为,但是 该设定目标的实现途径由各成员国自己决定。
注:http://europa.eu/eu-law/decision-making/legal-acts/index_en.htm
Turkey 土耳其
Kosovo科索沃
CE Marking
适用对象
面向欧盟成员国进行销售的规定商品。
① 对商品原产地无要求 ② 规定商品*
注:* 见下述网址 http://ec.europa.eu/enterprise/policies/single-marketgoods/cemarking/professionals/manufacturers/index_en.htm
2019/07/01,欧盟网站共计公布25类需进行CE Marking 的商品大类
1. 有源植入医疗器械; 2. 燃气炉具; 3. 载人的索道装置; 4. 建筑产品; 5. 能源相关产品生态设计; 6. 电磁兼容; 7. 使用于具有爆炸性环境中的设备和保护系统; 8. 民用爆破器材; 9. 热水锅炉; 10. 体外诊断试剂; 11. 升降机; 12. 低压器材; 13. 机械; 14. 测量仪器; 15. 医疗器械; 16. 环境噪声辐射; 17. 非自动称重仪器; 18. 个人防护设备; 19. 压力设备; 20. 烟火; 21. 无线电和电信终端设备; 22. 休闲用船只; 23. 电子电器设备中有害物质的限制; 24. 玩具安全; 25. 简单压力容器。
23项条款
条款号 Article 1 Article 2 Article 3 Article 4 Article 5 Article 6 Article 7 Article 8 Article 9 Article 10 Article 11 Article 12 Article 13 Article 14 Article 15 Article 16 Article 17 Article 18 Article 19 Article 20 Article 21 Article 22 Article 23
MDD9342EEC欧盟医疗器械分类总则介绍说明

醫療器械分類原則醫療器械之分類原則主要依據其特性,如非侵入式器材、侵入式器材、主動式器材,以及其它特殊原則。
而分類別的界定,則可根據MDD的guidelines、93/42/EEC Annex IX 中之敘述、或使用英國衛生署的disk來判斷,並由廠商自行決定。
依據MDD93/42/EEC附錄九中詳定18條規則,考慮醫療器械之設計及製造對人體可能帶來的危險程度,可將醫療器械分為以下4類: •Class I 低風險 (Low risk)•Class IIa 低到中風險 (Low to medium risk)•Class IIb 中風險 (Medium risk)•Class III 高風險 (High risk)產品分類規則:1、規則應用由器械的預期使用目的決定;2、如果器械是和其它器械配合使用,分類規則分別適用於每種器械;3、附件可以和其它一起使用的器械分開單獨分類;4、啟動或影響某種器械的軟件與器械屬於同一類型。
分類準則:時間:暫時(<60分鐘)、短期(<30天)、長期(>30天)創傷性:非創傷、通過孔徑創傷,外科創傷、植入。
適用位置:中央循環、中樞神經系統,其它地方。
能量供應:無源,有源。
MDD 93/42/EEC附錄IX 18條規則規則1?4、所有非創傷性器械均屬於I類,除非他們:用於儲存體液(血袋例外) II a類於Ila類或更高類型的有源醫療器械類 II a類改變體液成分II a∕II b類一些傷口敷料II a∕II b類規則5、侵入人體孔徑的醫療器械暫時使用(牙科壓縮材料、檢查手套) I類短期使用(導管、隱形眼鏡) II a類長期使用(正常牙線) II b類規則6-8、外科創傷性器械再使用的外科器械(鉗子,斧子) I類暫時或短期使用(縫合針。
外科手套) 11a類長期使用(假關節,眼內晶體) II b類與中央循環系統(CCS)或中樞神經系統接觸的器械 III類規則9、給予或交換能量的治療器械 II a類(肌肉刺激器、電鑽、皮膚光療機、助聽器)一種潛在危險方式工作的 II b類(嬰兒培養箱、高頻電刀、超聲碎石機、X光機)規則10、診斷器械提供能量(核磁共振,超聲診斷儀) II a類診斷∕監視體內放射藥物分佈 II a類(r照相機、正電子發射成像儀)診斷∕監視生理功能(心電圖、腦電圖) II a類危險情況下監視生理功能 II b類(手術中的血氣分析儀)發出電離輻射(X射線診斷議) II b類規則11控制藥物或其它物質進出人體的有源器械 II a類(吸引設備、供給泵)如以一種潛在危險方式工作 II b類(麻醉機、呼吸機、透析機、高壓氧艙)規則12.所有其它有源醫療器械屬於I類(觀察燈、牙科椅、輪椅、牙科用治療燈、記錄處理觀察診斷圖像用的有源器械)規則13、與醫用物質結合的器械(含殺精子的避孕套、含抗生素的牙髓材料) III類規則14、避孕用具(避孕套、子宮帽 II b類) II b/III類 (子宮內避孕器 III類)規則15、清洗或消毒的器械醫療器械(內窺鏡消毒) II a類接觸鏡(消毒液、護理液) II a類規則16、用於記錄X射線圖像的器械(X光片) II a類規則17、利用動物組織的器械(生物)心臟瓣膜、腸線、膠原)III類規則18、血袋 II b類。
MDD 9342EEC简介

2014/07/01,欧盟网站共计公布25类需进行CE Marking的商品大类
1. 有源植入医疗器械; 2. 燃气炉具; 3. 载人的索道装置; 4. 建筑产品; 5. 能源相关产品生态设计; 6. 电磁兼容; 7. 使用于具有爆炸性环境中的设备和保护系统; 8. 民用爆破器材; 9. 热水锅炉; 10. 体外诊断试剂; 11. 升降机; 12. 低压器材; 13. 机械; 14. 测量仪器; 15. 医疗器械; 16. 环境噪声辐射; 17. 非自动称重仪器; 18. 个人防护设备; 19. 压力设备; 20. 烟火; 21. 无线电和电信终端设备; 22. 休闲用船只; 23. 电子电器设备中有害物质的限制; 24. 玩具安全; 25. 简单压力容器。
Malta 马耳他
Czech Republic 捷克
Hungary 匈牙利
Netherlands 荷兰
Denmark 丹麦
Ireland 爱尔兰
Poland 波兰
Candidate countries 候选国家 (5个)
Iceland 冰岛
Montenegro 黑山
Serbia 塞尔维亚
The former Yugoslav Republic of Macedonia 马其顿
Turkey 土耳其
Kosovo科索沃
CE Marking
适用对象
面向欧盟成员国进行销售的规定商品。
① 对商品原产地无要求 ② 规定商品*
注:* 见下述网址 http://ec.europa.eu/enterprise/policies/single-marketgoods/cemarking/professionals/manufacturers/index_en.htm
2019年N9342C手持式频谱分析仪教程

一,设备:分为主机,充电器和天线三部分,(如下图是天线和主机)设备链接开机键与开机界面第1 system个界面图Tisp:这个界面我们扫频一般是用不到的,不要动这些默认设置第2 file个界面图Tisp:这个界面我们扫频一般是用不到的,不要动这些默认设置第3 limit个界面图Tisp:这个界面我们会用到F3保持最大值,在扫频界面波峰会保持最大值方便观察,分析。
F4保持最小值,在扫频界面波峰会保持最小值方便观察,分析。
F5静止,在扫频界面波峰会保持静止方便观察,拍照。
(下面省略F建的功能解释,他的功能,和他保持平行)Tisp:这个界面我们会用到F2,手动调到用下图的圆建,左右转圈即可调节大小Tisp:这个界面我们会用到F2,用圆键手动衰减调到零dbF3,把预放关掉,每按一次F3键表示开,关字符下面会有下滑线,下划线在那个字符下面,表示该功能被执行Tisp:这个界面我们扫频一般是用不到的,不要动这些默认设置因为这些设置会根据你后面扫频的频段设置而改变Tisp:这个界面是我们主要了解的界面和用到的界面因为设置频段是再这个界面F3起始频段设置F4终止频段设置设置方式就是先按下该功能键,这时是没变化的,要在输入数值时才会显示如下图输入完后,一般是按下F2MHz的比如输入1940:第8 shift+ save 是截屏组合键(先按shift,会后一个黄色S的图标出现再按save,而不是同时按)可以编辑截的图片的名字第9 peak键,可以标记波峰,或者一个频点,(最多可以标记6个位置)F1是序列号,没按一次,会到下一个数字,然后,按下peak就会出现标记数字,大圆键可以调整其位置也就是标记的频点。
二理论1定位是根据三线点:在干扰源周围的3个方向扫,其3个方向的信号最强方向会聚集一点A(A 就是干扰源的位置。
然后缩小范围,重复以上,最终可以初步判断,干扰物,最终确认需要把干扰源的信号屏蔽点,看先基站是否恢复正常。
MDD 93-42-EEC 基本要求检查表

基本要求检查表MDD 93/42/EEC Essential Requirements Checklist基本要求是MDD的最重要部分,它包括所有的医疗器械通用要求:一、基本要求(总要求)a)安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析);b)风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统);c)性能符合性(产品的基本要求);d)器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。
);e)器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。
二、基本要求的具体内容包括如下14条:1、器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。
2、生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应:首先应尽可能降低甚至避免危险其次,对无法避免的危险采取适当的防护措施,包括安装报警装置;最后,告知用户所提供防护措施的弱点及其可能带来的危险。
3、器械必须取得生产者期望获得的功能。
器械设计制造和包装应有利于第一条(2)(A)D 多规定的各项功能的发挥。
4、在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。
5、器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。
6、副作用的大小同器械的使用性能相比较可以为人们所接受。
7、化学、物理和生物性能8、感染和微生物污染。
9、组装和环境因素10、检测器械11、辐射防护12、带有能源或与其他能源相连接的器械13、生产者提供的操作信息14、如果需要根据医疗数据确定器械是否满足基本要求,如第六款的情形,有关数据必须按照附录Ⅹ的规定取得。
医疗器械的9342EEC定义适用范围

医疗器械的93/42/EEC定义适用范围93/42/EEC定义适用范围主要包括以下八个方面:1.本指令适用于医疗器械及其附件。
在本指令中,附件本身应被视为医疗器械。
医疗器械和附件以下均称为器械。
2.奥咨达医疗器械咨询在本指令中,适用以下定义:(1) “医疗器械”是指制造商预定用于人体以下目的的任何仪器、装置、器具、材料或其他物品,无论它们是单独使用还是组合使用,包括为其正常使用所需的软件:(奥咨达医疗器械咨询)——疾病的诊断、预防、监视、治疗或减轻;——损伤或残障的诊断、监视、治疗、减轻或修补;——解剖学或生理过程的探查,替换或变更;——妊娠的控制。
(只专注于医疗器械领域)医疗器械不是通过药理学、免疫学或代谢作用等方式在人体内或人体上达到其预定的主要作用,但这些方式有助于其功能的实现。
(2) “附件”是指本身虽然不是器械,但由其制造商专门指定与器械一起使用,使其能按照器械制造商预定的器械用途来使用的物品。
(3) “体外诊断用器械”是指制造商预定用于体外检查人体样品,目的在于提供人体生理状况、健康或疾病状况,或先天性异常等信息的任何器械,包括单独使用或组合使用的试剂,试剂产品、成套器材、仪器、设备或系统。
(4) “定制器械”是指按照执业医师开具的处方而专门制作的器械,由医师负责提供专门的设计特性,指定只适用于特定患者。
上述处方也可由具有执业资格的其他人开具。
为满足执业医生或其他任何专业人员特殊需要而成批生产的器械,不认为是定制器械。
(5) “临床检查用器械”是指预定供执业医生在适当的人类临床环境下进行附录Ⅹ中第2.1点所述的检查所使用的器械。
在临床检查中,任何具备专业资格、被准予从事此项检查的人员均可等同地被认可为具有正式资格的执业医生。
(6) “制造商”是指在以其名义将器械投放市场前负责器械的设计、制造、包装和标签的自然人或法人,无论这些工作是他自己完成的,还是由第三方代表他完成的。
本指令规定制造商必须履行的义务也适用于负责对一件或几件制成品进行装配,包装、加工、全面整修和/或加贴标志和/或对其作为一件器械规定其预定用途,以便其以自己的名义投放市场的自然人或法人。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
CE认证之MDD 介绍
目录
一:MDD作用及适用范围
二:医疗器械产品分类及规则
三:产品基本要求
四:制造企业需要提交的技术文件(TCF文件)五:申请认证公司需做的准备
六:认证流程
注:CE标志使用注意事项
附录:一:TCF文件包含的部分内容
一:MDD作用及适用范围
1 绝大部分医疗器械产品进入欧盟市场需要申办此项CE认证,欧盟借此进行医疗器械产品的上市审查及市场监管,对产品制造商来说也是一种贸易壁垒形式。
2 适用产品范围:
医疗仪器、器械、器具、材料或其它物品,无论它是单独使用还是联合使用,包括制造商预定其恰当应用所必需的软件。
3 不适用产品范围:
–90/385/EEC(AIMDD)指令覆盖的有源植入式医疗器械:任何借助外科手术,器械全部或者部分进入人体或自然腔道中;在手术过程结束后长期留在体内,或者这些器械部分留在体内至少30天以上,这些器械被认为是植入器械。
常用的有骨与关节替代物、心血管植入物、人工心脏瓣膜、眼内晶体植入物、血管支架等。
--98/79/EEC(IVD)指令覆盖下的体外诊断医疗器械
--以及药品,化妆品,个人防护用品和动物用医疗器械
4 指令适用国家:欧盟全体27个成员国
二:医疗器械产品分类规则
医疗器械指令 MDD 93/42/eec 按照危险程度将产品分为Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类,附录九中详定18条规则用来确定产品类别
1、规则应用由器械的预期使用目的决定;
2、如果器械是和其它器械配合使用,分类规则分别适用于每种器械;
3、附件可以和其它一起使用的器械分开单独分类;
4、启动或影响某种器械的软件与器械属于同一类型。
分类准则:
时间:暂时(<60分钟)、短期(<30天)、长期(>30天)
创伤性:非创伤、通过孔径创伤,外科创伤、植入。
适用位置:中央循环、中枢神经系统,其它地方。
能量供应:无源,有源。
规则1~4、所有非创伤性器械均属于I类,除非他们:
用于储存体液(血袋例外) II a类
于Ila类或更高类型的有源医疗器械类 II a类
改变体液成分 II a/II b类
一些伤口敷料 II a/II b类
规则5、侵入人体孔径的医疗器械
暂时使用(牙科压缩材料、检查手套) I类
短期使用(导管、隐形眼镜) II a类
长期使用(正常牙线) II b类
规则6-8、外科创伤性器械
再使用的外科器械(钳子,斧子) I类
暂时或短期使用(缝合针。
外科手套) IIa类
长期使用(假关节,眼内晶体) II b类
与中央循环系统(CCS)或中枢神经系统接触的器械 III类
规则9、给予或交换能量的治疗器械 II a类
(肌肉刺激器、电钻、皮肤光疗机、助听器)
以一种潜在危险方式工作的 II b类
(婴儿培养箱、高频电刀、超声碎石机、X光机)
规则10、诊断器械
提供能量(核磁共振,超声诊断仪) II a类
诊断/监视体内放射药物分布 II a类
(r照相机、正电子发射成像仪)
诊断/监视生理功能(心电图、脑电图) II a类
危险情况下监视生理功能 II b类
(手术中的血气分析仪)
发出电离辐射(X射线诊断议) II b类
规则11 控制药物或其他物质进出人体的有源器械 II a类
(吸引设备、供给泵)
如以一种潜在危险方式工作 II b类
(麻醉机、呼吸机、透析机、高压氧舱)
规则12.所有其他有源医疗器械属于I类
(观察灯、牙科椅、轮椅、牙科用治疗灯、记录处理观察诊断图象用的有源器械)
规则13、与医用物质结合的器械(含杀精子的避孕套、含抗生素的牙髓材料) III类
规则14、避孕用具(避孕套、子宫帽 II b类) II b/III类 (子宫内避孕器 III类)
规则15、清洗或消毒的器械
医疗器械(内窥镜消毒) II a类
接触镜(消毒液、护理液) II a类
规则16、用于记录X射线图象的器械(X光片) II a类
规则17、利用动物组织的器械(生物)心脏瓣膜、肠线、胶原
III类
规则18、血袋 II b类
三:申请CE认证的医疗器械基本要求
医疗器械必须满足指令93/42/EEC附录Ⅰ规定的基本要求
—必须是安全的;
—必须根据目前认可的工艺技术设计和制造;
—必须达到预期的性能;
—在规定的寿命期内必须保证产品的安全和性能。
—必须规定适当的运输和储存要求;
—副作用必须在可接受的范围内;
四:制造企业需要提交的技术文件(TCF文件)
1.作用:此技术文件用以说明产品符合本指令附录I中的基本要求,并有助于公告机构与国家检测机构的审核。
2.技术文件需要根据该产品对应的附录要求来制作。
产品与附录对应关系如下:III类医疗器械
-附录II,包括第四部分
-附录III+附录IV
-附录III+附录V
IIb类医疗器械
-附录II,不包括第四部分
-附录III+附录IV
-附录III+附录V
-附录III+附录VI
IIa类医疗器械
-附录II,不包括第四部分
-附录II+附录IV
-附录II+附录V
-附录II+附录VI
I类医疗器械,含无菌和/或测量功能
-附录II+附录IV
-附录II+附录V(无菌仪器必须采用这一途径)
-附录II+附录VI
I类医疗器械,无无菌和/或测量功能
-附录VII
3.技术文档需要的信息包括:
(1)证明产品符合基本要求的必要的技术性信息
(2)质量管理体系和产品质量流程的简要描述
(3)出具一份符合MDD要求的声明。
五:申请认证公司需做的准备
1 收集与认证产品有关的欧盟技术法规和欧盟(EN)标准,通过消化、吸收、纳入企业产品标准
2 企业严格按照以上产品标准组织生产,也就是把上述技术法规和EN标准的要求,贯彻到企业产品的设计开发和生产制造的全过程
3 企业必须按ISO9000+ISO13485标准建和维护质量体系,并取得ISO9000+ISO13485认证,质量体系审核前,企业应有至少三个月的质量体系运行记录,并完成1-2次内部质量体系审核
4 欧盟境外医疗器械制造商必须指定一个欧盟授权代表,并印在包装上,技术文件必须保存在欧盟授权代表处,并且需建立一套有效的“事故防范监督系统”,通过其欧盟授权代表对产品的事故报告、通告、召回等提供协助
六:认证流程
流程及要求如下:
1由企业提出认证申请,并提供相关产品资料
2协助企业进行医疗器械分类及确认认证模式
3与企业确认认证产品,报价签约
4企业向认证机构提交ISO9001+ISO13485质量体系文件
5认证机构将产品送交合格机构检测并获得检测报告
6企业进行产品技术文件制作(简称TCF文件)
7认证机构进行质量管理体系与TCF文件初审,指出问题,企业完善
8认证机构对企业的ISO9000+ISO13485质量体系和TCF文件进行正
式现场审核
9正式审核通过后,认证机构与企业签订框架协议,明确取得CE
证书后各方遵循原则和产品使用CE标志的范围,用户投诉
处理办法。
颁发CE标志证书
10每年进行定期复核
注:CE标志使用注意事项
欧盟内的产品属于欧盟指令范围内时,应在产品上贴附CE标志
不属于欧盟指令范围内的产品不能贴符CE标志
CE标志不代表产品的质量保证,而是表示该产品符合欧盟指令及相关标准的基本要求事项
CE标志可以在产品或产品的名牌,使用说明书,保证书上贴符
CE标志的贴符应便于看到,容易阅读,不易抹掉
CE标志不能与具有混淆可能性的其它任何标志使用
CE标志的大小应大于5 mm以上
CE标志已规定了标准图形
附录:
TCF文件包含的部分内容
1、产品名称、分类及引用标准条款的简要描述
2、产品概述(包括类型和预期用途)
a) 产品的历史沿革
b) 技术性能参数
c) 产品配合使用的附件、配合件和其它设备清单
d) 产品的图示与样品
e) 产品所用原材料及供应商
3、使用该产品的调和标准/或其它标准
4、风险分析评估结论和预防措施(EN1441 产品服务危险分析报告)
5、生产质量控制
a) 产品资料和控制文档(包括产品生产工艺流程图)
b) 产品的灭菌方法和确认的描述
c) 灭菌验证
d) 产品质量控制措施
e) 产品稳定性和效期的描述
6、包装和标识
a) 包装材料说明
b) 标签
c) 使用说明书
7、技术评价
a) 产品检验报告及相关文献
b) 技术概要及权威观点
8、潜在风险评价
a) 产品潜在风险测试报告及相关文献
b) 潜在风险的概要及权威观点
9、临床评价
a) 产品临床测试报告及相关文献
b) 临床使用概述及权威观点。