巯基检测
自由巯基的检测

Free Sulfhydryl in Recombinant Monoclonal AntibodiesWei Zhang*and Marta J.CzuprynWyeth BioPharma,Genetics Institute Campus,One Burtt Road,Andover,Massachusetts01810 Monoclonal antibody(mAb)therapy applications have been growing rapidly in recentyears.Like other recombinant protein drugs,therapeutic mAb’s need to be wellcharacterized to ensure their structural and functional integrity.IgG mAb’s arecomposed of two heavy and two light chains covalently linked by interchain disulfidebonds.Each domain of the heavy or light chain contains one additional disulfide bond.Native IgG mAb’s,with completely formed disulfide bonds,should not bear any freesulfhydryl.This report describes detection and quantification of free sulfhydryl inrecombinant mAb’s produced in Chinese hamster ovary(CHO)cells using a fluorescenttechnique.The method utilizes the fluorescent probe N-(1-pyrenyl)maleimide(NPM).The purified mAb’s appear to be homogeneous under native conditions with ap-proximately0.02mol of free sulfhydryl per mole of protein.Upon denaturation,minorspecies related to the mAb’s are observed on sodium dodecyl sulfate polyacrylamidegel electrophoresis(SDS-PAGE),and the free sulfhydryl level is determined to beapproximately0.1mol/mol of protein.These results suggest that a small portion ofthese recombinant mAb’s lack in intermolecular disulfide bonds but remain nonco-valently associated under native conditions.The formation of the free sulfhydrylcontaining mAb species is likely to occur during the culture process and/or proteinfolding process in the endoplasmic reticulum(ER).IntroductionThe rapid advancement of recombinant monoclonal antibody(mAb)therapy has necessitated detailed char-acterization and analysis of this protein family.Thera-peutic mAb’s derived by recombinant DNA technology, like other genetically engineered protein pharmaceuti-cals,require extensive and stringent characterization of purity,structural integrity,and stability,as molecular modifications may result in undesirable adverse biologi-cal effects,such as increased immunogenicity.Though the general structural features of antibodies have been well documented in literature(1,2),a number of studies have revealed structural variations of recombinant mAb therapeutics.These include isoaspartate formation re-sulting from asparagine deamidation and/or aspartate isomerization(3-5),processing of C-terminal lysine resides(6),methionine and cysteine(Cys)oxidation(7), variability in glycosylation(8-10),mutation introduced during transfection(11),and crossover event between variable genes of heavy and light chains during tran-scription(12).These modifications,due to host-dependent and/or in vitro process-dependent factors,are epigenetic and thus not predicable from the genetic structure of construct alone.They may alter the biological activities or pharmacokinetic properties of therapeutic mAb’s and consequently require substantive characterization to ensure product quality and to provide understanding of structure/function relationships.IgG antibodies have a four-chain structure composed of two heavy chains(HCs)and two light chains(LCs) covalently linked by interchain disulfide bridges(Figure 1).This four-chain structure is also maintained by strong,noncovalent interactions between the N-terminal half of HC-LC pairs and between the C-terminal regions of HC. In addition to interchain disulfide bonds,one intrachain disulfide bond is present and is shielded within each -barrel domain of HC and LC(1,2).While the disulfide linkage between HC and LC always involves the last Cys residue,Cys213according to the Kabat numbering system(13)of the LC,the Cys residue from the HC varies,depending on the IgG subtype(13).In IgG1mAb, the fifth Cys residue(Cys217)of HC forms an inter HC and LC disulfide bond.In IgG2and IgG4mAb’s,it is the third Cys amino acid of HC(Cys123)that links HC and LC.In the hinge region,different IgG subtypes have different numbers of disulfide bond connecting the HCs. In IgG1and IgG4,the two HCs are covalently linked by two disulfide bonds,while in IgG2,there are four disulfide bonds in its hinge region(1,2).In a correctly folded IgG antibody,regardless of the subtype,all Cys residues are involved in disulfide bond formation,there-fore,no free sulfhydryl should be present.During our work on recombinant mAb’s,we detected the presence of some free sulfhydryl as well as nonco-valently associated mAb fragments.We investigated the noncovalently associated species in mAb produced in Chinese hamster ovary(CHO)cells.To this end,we analyzed the structural integrity of these molecules under native and denaturing conditions.Free sulfhydryl content in the mAb’s was determined following deriva-tization with a fluorescent probe.Our results suggest that these noncovalently associated mAb fragments may result from incomplete formation of interchain disulfide bonds within the mAb molecules.*To whom correspondence should be addressed.Email: wxzhang@.Tel:(978)247-2997.Fax:(978)247-2834.509Biotechnol.Prog.2002,18,509−51310.1021/bp025511z CCC:$22.00©2002American Chemical Society and American Institute of Chemical EngineersPublished on Web04/11/2002Materials and MethodsMaterial.The recombinant mAb’s analyzed in the present work were produced in CHO cells and purified at Wyeth BioPharma,Genetics Institute Campus in Andover,MA.Samples included one IgG1,two IgG2(IgG2-I and IgG2-II herein),and one IgG4mAb.N -Acetyl-L -cysteine (Ac-Cys),N -(1-pyrenyl)maleimide (NPM),and N ,N -dimethylformamide (DMF)were obtained from Sigma.Size Exclusion Chromatography (SEC -HPLC).SEC -HPLC analysis of was performed on a Waters Alliance system with a Waters model 2487dual wave-length absorbance detector and a refrigerated autosam-pler controlled at 4°C.A TosoHaas G3000SW XL (7.8mm ×300mm,5µm,250Å)column was equilibrated with a mobile phase consisting of 10mM sodium phosphate,250mM NaCl,pH 7.2at a flow rate of 0.75mL/min.A 20-µl sample of mAb solution at a concentration of approxi-mately 1mg/mL was injected.The elution was monitored at 214nm.The analysis time was 20min.Sodium Dodecyl Sulfate Polyacrylamide Gel Elec-trophoresis (SDS -PAGE).The mAb samples were analyzed using precast Novex 4-20%Tris-Glycine gels.Nonreducing sample buffer consisted of 125mM Tris-HCl,pH 6.8,30%(v/v)glycerol,4%(w/v)SDS,and 0.05%(w/v)bromphenol blue.Reducing sample buffer was made by mixing 5vol of 1M dithiothreitol and 95vol of nonreducing buffer.Next,10µL of the mAb solutions at a concentration of 1mg/mL was mixed with equal volume of sample buffer.The mixture was heated at 100°C for 5min for nonreducing SDS -PAGE and 1min for the reducing gel.The gels were run at 150V (constant voltage)for 1.5h.After electrophoresis,the gels were stained in 0.05%(w/v)Coomassie blue and destained using 10%(v/v)acetic acid,15%(v/v)methanol.Measurement of Sulfhydryl.Sulfhydryl content of the mAb’s was measured by a fluorescent assay.Free sulfhydryl was derivatized by adding 10µL of 2mM NPM in DMF to the following three mAb sample prepara-tions:100µL of mAb solution diluted with 500µL of phosphate-buffered saline (PBS),pH 7.3;100µL of mAb solution diluted with 500µL of 6M guanidine hydro-chloride (GuHCl)in PBS buffer,pH 7.3;100µL of mAb solution diluted with 500µL of 6M GuHCl in PBS,pH 7.3,incubated under argon at 37°C for 2h.After theNPM solution was added,the mixtures were incubated at room temperature for 5min and quenched with 10µL of 50%acetic acid.A set of Ac-Cys standards in PBS,pH 7.3with the concentration ranging from 0.25to 25µM,and a PBS,pH 7.3blank were also prepared by the above procedure.Fluorescence measurement was performed on a Waters model 474scanning fluorescence detector equipped with a cuvette holder.This setting allows measurement of the fluorescence of a sample without using a chromatographic system and therefore simplifies sample handling.A quartz cuvette with 1.0-cm path length was used for fluorescence measurement.The excitation wavelength was set at 330nm,and the emission wavelength was set at 380nm.All samples were analyzed in triplicate.The measured sulfhydryl content was expressed as mole of sulfhydryl per mole of mAb.ResultsSEC -HPLC.Under the native chromatographic con-ditions in this work,all four mAb’s eluted between 10and 11min as a single peak (Figure 2),consistent with a molecular weight of ca.150kDa.The observed level of high molecular weight species (earlier eluting material)was below 1%for these mAb’s.No low molecular weight protein species were detected (the peak at ∼15min was related to salts in the buffer).These results confirmed the identities of these recombinant mAb’s,and they appeared to be homogeneous under the native SEC chromatographic conditions.SDS -PAGE.The four mAb’s had similar banding patterns in both reducing and nonreducing SDS -PAGE (Figure 3).Under reducing conditions,predominantly a single HC band migrating at approximately 50kDa and a LC band migrating at approximately 25kDa were observed (Figure 3A).The band below the LC of the IgG4mAb (Figure 3A,lane 6)was the result of deamidation-related peptide bond cleavage,as identified by electro-blotting and Edman sequencing (data not shown).Under nonreducing conditions,all four mAb’s migrated pre-dominantly as a single band of approximately 150kDa,corresponding to a full mAb molecule consisted of 2HC and 2LC (Figure 3B).However,an additional array of minor bands was also present with apparent molecular weights lower than 150kDa (Figure 3B).Peptidemap-Figure 1.Schematic drawing of IgG antibody.Each mAb consists of two HCs and two LCs covalently linked by interchain disulfide bonds.In IgG1and IgG4antibodies,two disulfide bonds are in the hinge region connecting the two HCs,whereas in IgG2antibody four disulfide bonds are in the hinge region.In addition,Cys213of the LC forms inter-HC and LC disulfide bond with Cys217in IgG1,or Cys123in IgG2and IgG4.In all IgG antibodies,each -barrel domain of the HC and LC contains one intrachain disulfide linkage.ping of these mAb’s detected very little peptide bond cleavage (data not shown).Electroblotting and subse-quent Edman sequencing of these lower bands suggested that they may be mAb missing one LC,HC dimer,a half molecule (one HC and one LC),single HC and LC (data not shown).The bands migrating around 25kDa were all identified to be LC-related (Figure 3B).It has been reported that proteins containing disulfide bonds bind less SDS than their reduced forms (14).Hence,the formation of the multiple LC bands may be due to incomplete reduction of the -barrel disulfide bonds in the LC (15).Under the denaturing SDS -PAGE condi-tions,mAb minor species were resolved,while under the native SEC -HPLC conditions,the mAb’s eluted as one peak.Therefore,these minor species are most likely present in solution as noncovalently associated mAb molecules,or partially linked via disulfide bond.Measurement of Sulfhydryl.To elucidate the origin of these mAb minor species under denaturing conditions,free sulfhydryl content was measured.Though Ellman’s assay has been used for quantification of protein sulfhy-dryl,it lacks adequate sensitivity for sulfhydryl content below the low micromolar range (16).Therefore,the sulfhydryl level in the mAb’s was assessed by a fluores-cent assay.NPM was used as the fluorescent probe because of its specificity,sensitivity,and ease of use (17).In particular,NPM is essentially nonfluorescent until sulfhydryl conjugated,which allows measurement of the fluorescence of the sample without prior reagent separa-tion (17).The fluorescence emission intensities of a series of Ac-Cys standards derivatized by NPM were used to establish a standard curve of fluorescence vs sulfhydryl concentration.The standard curve was linear from 0.25to 25µM with a R 2value of 0.9999(Figure 4)and was then used to determine the sulfhydryl content in the mAb samples.The results were summarized in Figure 5.When measured under nondenaturing conditions in PBS,less than 0.02mol of free sulfhydryl was detected per mole of the IgG2or IgG4mAb,while the IgG1mAb was found to contain approximately 0.03mol of sulfhydryl.To access the potentially buried sulfhydryl,the mAb’s were dena-tured with 5M GuHCl and then modified with NPM.In all cases,sulfhydryl level increased to nearly 0.08mol/mol of protein,3-4times higher than that in the native state.In a third set of experiments,the mAb samples in 5M GuHCl were incubated at 37°C for 2h.The two IgG2mAb’s were found to contain approximately 0.1mol of sulfhydryl per mol of protein,while the IgG1and IgG4mAb’s contained approximately 0.09sulfhydryl per pro-tein molecule.These results indicated that all mAb’s analyzed have similar levels of sulhydryl with the major-ity not accessible in the native state of protein.The increased sulfhydryl levels under denaturingconditionsFigure 2.SEC -HPLC profiles of recombinant mAb’s.(A)IgG1mAb.(B)IgG2-I mAb.(C)IgG2-II mAb.(D)IgG4mAb.Figure 3.SDS -PAGE profiles of recombinant mAb’s under (A)reducing and (B)nonreducing ne 1,molecular weight marker;lane 2,blank;lane 3,IgG1mAb;lane 4,IgG2-I mAb;lane 5,IgG2-II mAb;lane 6,IgG4mAb.also implied the presence of noncovalently associated mAb molecules in solution.DiscussionThe sulfhydryl group of Cys is potentially the most reactive nucleophile of all the functional side chains in proteins.It can exist in the reduced free state or the oxidized state to form disulfide bond.The state of sulfhydryl is often important to the structure and func-tion of a protein (18).Therefore,an assessment of the presence and state of sulfhydryl groups in recombinant protein pharmaceuticals allows an evaluation of their structural and functional integrity.In IgG antibodies,all Cys residues form inter-or intramolecular disulfide bonds,and therefore no free sulfhydryl should exist (Figure 1).However,in the present study,all four recombinant IgG mAb’s produced in CHO cells were found to contain free sulfhydryl groups,and the measured level was elevated upon denaturation.This implied that some Cys residues in these mAb’s stay in the reduced state and do not form either inter-or intrachain disulfide bond.The elevated accessibility of free sulfhydryl under denaturing condi-tions suggested that the majority of these sulfhydryls may reside within a solvent-inaccessible region.This result,combined with those from SEC -HPLC and SDS -PAGE,indicated the presence of noncovalently associated mAb molecules under native conditions.The noncovalent interchain interactions are known to be strong enough to maintain the four-chain structure of IgG antibodies,as reduced and alkylated IgG1did not measurably dissociate under native conditions (1,2).It has been shown that a chimeric IgG1mAb in which the hinge Cys residues were mutated to Ser was able to maintain thefour-chain structure in solution but was found to be predominantly of the half molecule form on nonreducing SDS -PAGE gels (19).Therefore,in the present study,the noncovalently associated species were undetectable under native SEC -HPLC conditions and were only revealed under denaturing conditions,such as SDS -PAGE and in GuHCl solution.The presence of free sulfhydryl groups in these mAb’s implied incomplete formation of disulfide bonds among the detected mAb fragments,which is required for covalent assembly of the four-chain folding of IgG anti-bodies.In CHO and other eukaryotic cells,disulfide bond formation and protein folding proceed within the lumen of the endoplasmic reticulum (ER),where protein oxida-tion takes place upon translocation of nascent peptides into the ER lumen (20).For antibodies,disulfide bond formation is additionally complicated with the balanced transcription of two different genes and the assembly of four polypeptide chains.It has been found that assembly of LC and HC to a complete antibody molecule is the primary rate-limiting step in antibody production and can be speeded up by a high expression rate of LC (21).Therefore,it is likely that the incomplete disulfide bond formation of these mAb’s occurs in vivo in the ER.It is worth pointing out that during the production of the mAb’s analyzed in the present study,low concentrations of both cystine and cysteine were used in the cell culture media,so it is also possible that formation of these hypodisulfide-bonded species occurs in vitro after the mAb’s are secreted into the culture media.However,during the production of recombinant murine interleukin 12in CHO cells using culture media containing cystine and cysteine,a glutathione adduct was found,indicating the incomplete disulfide bond formation in vivo (22).Though the origin of the incomplete disulfide bond formation of these mAb’s is unknown at this time,the presence of free sulfhydryl and noncovalently associated species may be undesirable.Also,accumulation of mis/unfolded proteins has been reported to cause ER stress (23,24)and,subsequently,apoptosis and loss of cell viability (25-27).Moreover,the long-term effect of these undisulfide-bonded mAb species on the stability and biological activity remains to be determined.ConclusionsWe have measured the free sulfhydryl content in recombinant mAb’s by a fluorescent assay.The detected sulfhydryl level was approximately 0.02and 0.1mol/mol of protein under native and denaturing conditions,respectively.The mAb’s appeared to be homogeneous as assessed by SEC -HPLC and reducing SDS -PAGE.However,in addition to the bands corresponding to the intact mAb’s,an array of minor binds was observed on nonreducing SDS -PAGE gels.These results suggest that noncovalently associated species are present in these mAb’s and that these mAb fragments may result from incomplete disulfide bond formation between the HC and LC.The formation of these undisulfide-bonded mAb fragments may occur during the protein folding process in the ER and/or the cell culture process.The long-term effects of these free sulfhydryl-containing minor species on the stability and potency of these mAb’s require further investigation.The fluorescent assay for assess-ment of free sulfhydryl is sensitive and quantitative and is applicable to production monitoring and structural analysis of other recombinant protein and mAb pharma-ceuticals.Figure 4.Ac-Cys fluorescence standard curve.The fluorescence was measured after derivatization with NPM.The excitation and emission wavelengths were set at 330and 380nm,respectively.The curve was linear in the range of 0.25-25µM.Figure 5.Free sulfhydryl content in recombinant mAb’s.Experimental conditions are described in Materials and Meth-ods.AcknowledgmentWe would like to thank Dr.Hubert Scoble for his support on this project and Drs.Mark Hardy,Yin Luo, and Lisa Marzilli for their critical review of the manu-script.We are grateful to Philip Boyle and Jeff Banas for their technical assistance.References and Notes(1)Dorrington,K.J.The structural basis for the functionalversativility of immunoglobulin G.Can.J.Biochem.1974, 56,1087-1101.(2)Burton,D.R.Immunoglobulin G:functional sites.Mol.Immunol.1985,22,161-206.(3)Cacia,J.;Keck,R.;Presta,L.G.;Frenz,J.Isomerization ofan aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE:identifica-tion and effect on binding affinity.Biochemistry1996,35, 1897-1903.(4)Perkins,M.;Theiler,R.;Lunte,S.;Jeschke,M.Determina-tion of the origin of charge heterogeneity in a murine monoclonal antibody.Pharm.Res.2000,17,1110-1117. (5)Harris,R.J.;Kabakoff,B.;Macchi,F.D.;Shen,F.J.;Kwong,M.J.;Andya,D.S.;Shire,J.;Bjork,N.;Totpal,K.;Chen,A.B.Identification of multiple sources of charge heterogeneityin a recombinant antibody.J.Chromatogr.B2001,752,233-245.(6)Harris,R.J.Processing of C-terminal lysine and arginineresidues of proteins isolated from mammalian cell culture.J.Chromatogr.A1995,705,233-245.(7)Kroon,D.J.;Baldwin-Ferro,A.;Lalan,P.Identification ofsites of degradation in a therapeutic monoclonal antibody by peptide mapping.Pharm.Res.1992,9,1386-1393.(8)Weitzhandler,M.;Hardy,M.;Co,M.S.;Avdalovic,N.Analysis of carbohydrates on IgG preparations.J.Pharm.Sci.1994,83,1670-1675.(9)Sheeley,D.M.;Merrill,B.M.;Taylor,L.C.Characterizationof monoclonal antibody glycosylation:comparison of expres-sion systems and identification of terminal R-linked galactose.Anal.Biochem.1997,247,102-110.(10)Bihoreau,N.;Ramon,C.;Lazard,M.;Schmitter,J.M.Combination of capillary electrophoresis and matrix-assisted laser desorption ionization mass spectrometry for glycosyl-ation analysis of a human monoclonal anti-Rhesus(D)anti-body.J.Chromatogr.B1997,697,123-33.(11)Harris,R.J.;Murnane,A.A.;Utter,S.L.;Wagner,K.L.;Cox,E.T.;Polastri,G.D.;Helder,J.C.;Sliwkowski,M.B.Assessing genetic heterogeneity in production cell lines: detection by peptide mapping of a low level Tyr to Gln sequence variant in a recombinant antibody.Bio/Technology 1993,11,1293-1297.(12)Wan,M.;Shiau,F.Y.;Gordon,W.;Wang,G.Y.Variantantibody identification by peptide mapping.Biotechnol.Bioeng.1999,62,485-488.(13)Kabat,D.J.;Wu,T.T.;Reid-Miller,M.;Perry,H.M.;Gottesman,K.S.Sequences of Proteins of ImmunologicalInterest,4th ed;U.S.Department of Health and Human Services:Washington,DC,1987.(14)Pitt-Rivers,R.;Impiombato,F.S.A.The binding of sodiumdodecyl sulphate to various proteins.Biochem.J.1968,109, 825-830.(15)Kikuchi,H.;Goto,Y.;Hamguchi,K.Reduction of the buriedintrachain disulfide bond of the constant fragment of the immunoglobulin light chain:global unfolding under physi-ological conditions.Biochemistry1986,25,2009-2013. (16)Wright,S.K.;Viola,R.E.Evaluation of methods for thequantitation of cysteines in proteins.Anal.Biochem.1998, 265,8-14.(17)Winters,R.A.;Zukowski,J.;Ercal,N.;Matthews,R.H.;Spitz,D.R.Analysis of glutathione,glutathione disulfide, cysteine,homocysteine,and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl)maleimide.Anal.Biochem.1995,227,14-21.(18)Liu,T.-Y.The role of sulfur in proteins.In The Proteins,3rd ed;Neuvath,H.,Hill,R.L.,Eds.;Academic Press: London,1977;pp329-402.(19)Gillies,S. D.;Wesolowski,J.S.Antigen binding andbiological activities of engineered mutant chimeric antibodies with human tumor specificities.Hum.Antibod.Hybridomas 1991,1,47-54.(20)Frand,A.R.;Cuozzo,J.W.;Kaiser,C.A.Pathways forprotein disulfide bond formation.Trends Cell Biol.2000,10, 203-210.(21)Strutzenberger,K.;Borth,N.;Kunert,R.;Steinfellner,W.;Katinger,H.Changes during subclone development and aging of human antibody-producing recombinant CHO cells.J.Biotechnol.1999,69,215-226.(22)Nickbarg,E.B.;Vath,J.;Pittman,D.D.;Leonard,J.E.;Waldburger,K.E.;Bond,M.Structural characterization of the recombinant P40heavy chain subunit monomer and homodimer of murine IL-12.Bioorg.Chem.1995,23,380-396.(23)Dorner,A.J.;Wasley,l.C.;Raney,P.;Haugejorden,S.;Green,M.;Kaufman,R.J.The stress response in Chinese hamster ovary cells.J.Biol.Chem.1990,265,22029-22034.(24)Kaufman,R.J.Stress singaling from the lumen of theendoplasmic reticulum:coordination of gene transscriptional and translational controls.Genes Dev.1999,13,1211-1233.(25)Al-Rubeai,M.;Singh,R.P.Apoptosis in cell culture.Curr.Opin.Biotechnol.1998,9,152-156.(26)Mastrangelo,A.J.;Betenbaugh,M.J.Overcoming apop-tosis:new methods for improving protein-expression systems.Trends Biotechnol.1998,16,88-95.(27)Murphy,T.C.;Woods,N.R.;Dickson,A.J.Expression ofthe transcription factor GADD153is an indicator of apoptosis for recombinant Chinese hamster ovary(CHO)cells.Biotech-nol.Bioeng.2001,75,621-629.Accepted for publication March5,2002.BP025511Z。
ELLMAN试剂法测定自由巯基和二硫键

E L L M A N试剂法测定自由巯基和二硫键This manuscript was revised by the office on December 22, 2012E L L M A N试剂测定自由巯基试验基于的原理:5,5’-dithiobis-2-nitrobenzoicacid(DTNB)5,5-二硫基-双(2-硝基苯甲酸)5,5-二硫基-双(2-硝基苯甲酸)(DTNB)在412nm没有吸收,与巯基反应后,生成2-硝基-5-巯基苯甲酸(TNB)[1]。
TNB2-在412nm有很强的吸收,可以用于对肽段的自由巯基进行定量分析[2]。
DTNBTNB2-根据文献记载,TNB的吸光系数在13.6*103M-cm-~14.25*103M-cm-之间[3,4]。
吸光度测量的最灵敏范围在0.2-0.7之间。
A=εbc,其中A为吸光度,ε是摩尔吸收光系数或消光系数,ε单位为升/(摩尔·厘米)[L/(mol·cm)]。
以吸光度下限0.2来计算(吸光系数取14.15*103M-cm-),需要TNB的浓度为c=0.2/(14.15*103LM-cm-*1cm)=1.4*10-5mol/L(1.4*10-8mol/ml),需要蛋白浓度为1.4*10-5mol/L*18790g/mol=0.2631g/L=0.2631mg/ml.我们的条件可以达到这个检测限度。
ELLMAN试剂法测定自由巯基主要的影响因素有:1.EDTA的适量加入有助于TNB显色的稳定和成梯度线性关系[5]。
2.在不同缓冲液中,TNB的最大吸收波长略微不同,所以它们在412nm的吸收也不3.同,要根据选择的缓冲液来确定[5]。
另外,TNB的分光光度法分析对SDS很敏感[6]。
4.DTNB随着pH的升高,降解速度加快。
在pH7.0,其降解速度为0.02%/h,在5.pH值8.0,其降解速度为0.2%/h,随着pH值得升高,降解速度加快,在pH12时,15min之内会完全降解[7,8]。
ELLM试剂法测定自由巯基和二硫键

E L L M试剂法测定自由巯基和二硫键Prepared on 22 November 2020ELLMAN试剂测定自由巯基试验基于的原理:5,5’-dithiobis-2-nitrobenzoicacid(DTNB)5,5-二硫基-双(2-硝基苯甲酸)5,5-二硫基-双(2-硝基苯甲酸)(DTNB)在412nm没有吸收,与巯基反应后,生成2-硝基-5-巯基苯甲酸(TNB)[1]。
TNB2-在412nm有很强的吸收,可以用于对肽段的自由巯基进行定量分析[2]。
DTNBTNB2-根据文献记载,TNB的吸光系数在*103M-cm-~*103M-cm-之间[3,4]。
吸光度测量的最灵敏范围在之间。
A=εbc,其中A为吸光度,ε是摩尔吸收光系数或消光系数,ε单位为升/(摩尔·厘米)[L/(mol·cm)]。
以吸光度下限来计算(吸光系数取*103M-cm-),需要TNB的浓度为c=(*103LM-cm-*1cm)=*10-5mol/L*10-8mol/ml),需要蛋白浓度为*10-5mol/L*18790g/mol=L=ml.我们的条件可以达到这个检测限度。
ELLMAN试剂法测定自由巯基主要的影响因素有:1.EDTA的适量加入有助于TNB显色的稳定和成梯度线性关系[5]。
2.在不同缓冲液中,TNB的最大吸收波长略微不同,所以它们在412nm的吸收也不3.同,要根据选择的缓冲液来确定[5]。
另外,TNB的分光光度法分析对SDS很敏感[6]。
4.DTNB随着pH的升高,降解速度加快。
在,其降解速度为%/h,在5.pH值,其降解速度为%/h,随着pH值得升高,降解速度加快,在pH12时,15min之内会完全降解[7,8]。
6.摩尔吸收光系数在不同的温度下不同,随温度的升高而下降[4].试验方案主要材料:1.材料PEG-G-CSF批号:080229浓度mlG-CSF批号:080126浓度ml10k超滤膜PALL2.试剂SequencingGradeModifiedTrypsin,Promega,lot#237826。
ELLMAN试剂法测定自由巯基和二硫键

ELLMAN试剂测定自由巯基试验基于的原理:5,5’-dithiobis-2-nitrobenzoic acid (DTNB) 5,5-二硫基-双(2-硝基苯甲酸) 5,5-二硫基-双(2-硝基苯甲酸)(DTNB)在412nm没有吸收,与巯基反应后,生成2-硝基-5-巯基苯甲酸(TNB)[1]。
TNB2-在412nm有很强的吸收,可以用于对肽段的自2-吸光度0.2来计算-*1cm)1.4*101.2.不3.同,要根据选择的缓冲液来确定[5]。
另外,TNB的分光光度法分析对SDS很敏感[6]。
4.DTNB随着pH的升高,降解速度加快。
在pH7.0,其降解速度为0.02%/h,在5.pH值8.0,其降解速度为0.2%/h,随着pH值得升高,降解速度加快,在pH 12时,15min之内会完全降解[7,8]。
6.摩尔吸收光系数在不同的温度下不同,随温度的升高而下降[4].试验方案主要材料:1.材料PEG-G-CSF 批号:080229 浓度4.32mg/mlG-CSF 批号:080126 浓度6.9mg/ml小瓶。
NOB液:0.1%TFA/90%乙腈/H2O3.仪器质谱仪:BRUKER DALTONICS MALTI-TOF-TOF autoflexⅢ(厂内编号KC2007-011)Beckman 22R台式离心机(厂内编号AM-039)Beckman DU-800 紫外分光光度计(厂内编号KC2007-005)恒温循环仪:JULABO F12-ED(厂内编号KC2008-003)反相柱:Symmetry C18 5um 300à高压液相仪器:,(UV/Visible Detector)试验过程:一、缓冲液替换PEG-G-CSF和G-CSF进行缓冲液替换,超滤替换缓冲液为50mM NH4HCO3,稀释中加入G-CSF+DTT相分离收样:SF+DTT相分离集到之间的交由崔文喜冻干四、ELLMAN试剂测定自由巯基由于收集到的PEG肽段已经是自由巯基,所以可以跳过还原二硫键这一步。
巯基检测方法

巯基检测方法
巯基是一种含有硫原子的官能团,常见于生物分子中的半胱氨酸和辅酶A等化合物中。
巯基的检测方法主要有以下几种:
1. 二硫苏糖法:该方法是利用巯基与二硫苏糖反应生成可见光吸收的化合物,从而检测巯基的存在。
该方法操作简单,灵敏度高,适用于生物样品中巯基的检测。
2. 五氯酚法:该方法是利用巯基与五氯酚反应生成可见光吸收的化合物,从而检测巯基的存在。
该方法操作简单,但灵敏度较低,适用于含有较高浓度巯基的样品。
3. 硫酸铜法:该方法是利用巯基与硫酸铜反应生成可见光吸收的化合物,从而检测巯基的存在。
该方法操作简单,但灵敏度较低,适用于含有较高浓度巯基的样品。
4. 氰化物法:该方法是利用巯基与氰化物反应生成可见光吸收的化合物,从而检测巯基的存在。
该方法操作简单,但灵敏度较低,适用于含有较高浓度巯基的样品。
总的来说,巯基的检测方法各有优缺点,需要根据具体实验要求选择合适的方法。
同时,为了保证实验结果的准确性,需要注意样品的处理和实验条件的控制。
化妆品中巯基乙酸等8种原料的检验方法2023年
附件18化妆品中巯基乙酸等8种原料的检验方法Thioglycollic acid and other7kinds of components in cosmetics1范围本方法规定了高效液相色谱法测定化妆品中巯基乙酸等8种原料的含量。
本方法适用于液态水基类、膏霜乳液类化妆品中巯基乙酸等8种原料含量的测定。
本方法所指的8种原料包括巯基乙酸、甘油巯基乙酸酯、巯基乙酸甲酯、二硫代二甘醇酸二铵、巯基乙酸乙酯、巯基乙酸异丙酯、巯基乙酸丁酯、巯基乙酸异辛酯。
2方法提要样品处理后,采用高效液相色谱仪分离,二极管阵列检测器检测,根据保留时间和紫外光谱定性,峰面积定量,以标准曲线法计算含量。
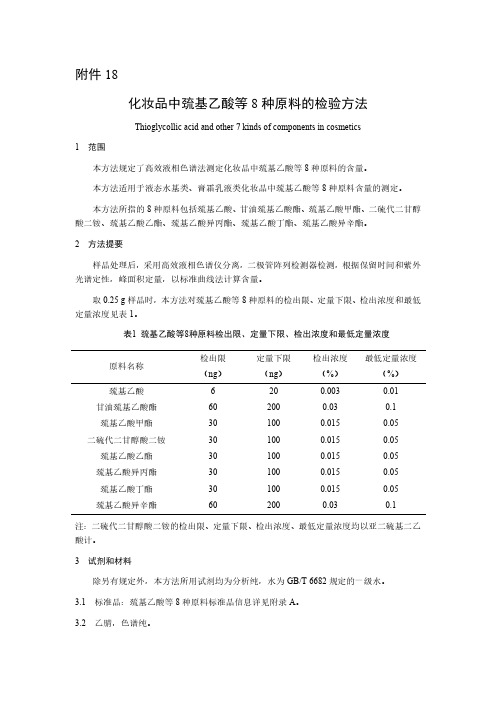
取0.25g样品时,本方法对巯基乙酸等8种原料的检出限、定量下限、检出浓度和最低定量浓度见表1。
表1巯基乙酸等8种原料检出限、定量下限、检出浓度和最低定量浓度原料名称检出限(ng)定量下限(ng)检出浓度(%)最低定量浓度(%)巯基乙酸6200.0030.01甘油巯基乙酸酯602000.030.1巯基乙酸甲酯301000.0150.05二硫代二甘醇酸二铵301000.0150.05巯基乙酸乙酯301000.0150.05巯基乙酸异丙酯301000.0150.05巯基乙酸丁酯301000.0150.05巯基乙酸异辛酯602000.030.1注:二硫代二甘醇酸二铵的检出限、定量下限、检出浓度、最低定量浓度均以亚二硫基二乙酸计。
3试剂和材料除另有规定外,本方法所用试剂均为分析纯,水为GB/T6682规定的一级水。
3.1标准品:巯基乙酸等8种原料标准品信息详见附录A。
3.2乙腈,色谱纯。
3.3磷酸,优级纯。
3.4磷酸溶液(0.1%):取磷酸(3.3)1mL,加水至1000mL,混匀。
3.5磷酸溶液(0.05%):取磷酸(3.3)0.5mL,加水至1000mL,混匀。
3.6含0.05%磷酸的乙腈溶液:取磷酸(3.3)0.5mL,加乙腈(3.2)至1000mL,混匀。
ELLMAN试剂法测定自由巯基和二硫键
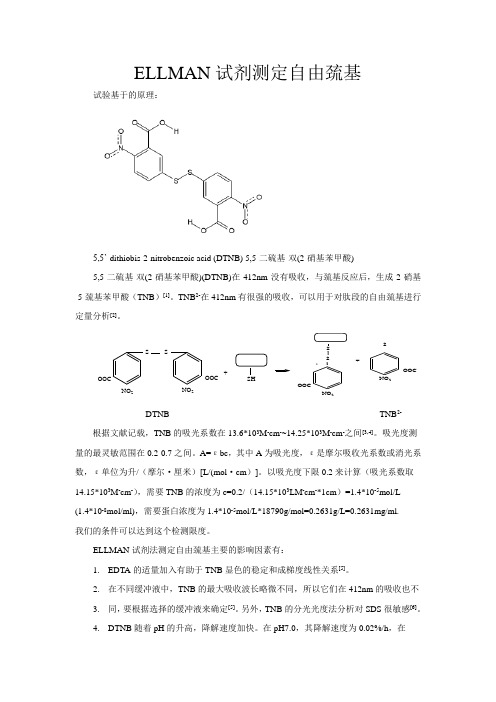
ELLMAN 试剂测定自由巯基试验基于的原理:5,5’-dithiobis-2-nitrobenzoic acid (DTNB) 5,5-二硫基-双(2-硝基苯甲酸)5,5-二硫基-双(2-硝基苯甲酸)(DTNB)在412nm 没有吸收,与巯基反应后,生成2-硝基-5-巯基苯甲酸(TNB )[1]。
TNB 2-在412nm 有很强的吸收,可以用于对肽段的自由巯基进行定量分析[2]。
SS NO 2NO 2-OOC-OOC+SH S -SNO 2NO 2-OOC-OOC+SDTNB TNB 2- 根据文献记载,TNB 的吸光系数在13.6*103M -cm -~14.25*103M -cm -之间[3,4]。
吸光度测量的最灵敏范围在0.2-0.7之间。
A=εbc ,其中A 为吸光度,ε是摩尔吸收光系数或消光系数,ε单位为升/(摩尔·厘米)[L/(mol ·cm )]。
以吸光度下限0.2来计算(吸光系数取14.15*103M -cm -),需要TNB 的浓度为c=0.2/(14.15*103LM -cm -*1cm )=1.4*10-5mol/L (1.4*10-8mol/ml),需要蛋白浓度为1.4*10-5mol/L*18790g/mol=0.2631g/L=0.2631mg/ml. 我们的条件可以达到这个检测限度。
ELLMAN 试剂法测定自由巯基主要的影响因素有:1. EDTA 的适量加入有助于TNB 显色的稳定和成梯度线性关系[5]。
2. 在不同缓冲液中,TNB 的最大吸收波长略微不同,所以它们在412nm 的吸收也不3. 同,要根据选择的缓冲液来确定[5]。
另外,TNB 的分光光度法分析对SDS 很敏感[6]。
4. DTNB 随着pH 的升高,降解速度加快。
在pH7.0,其降解速度为0.02%/h ,在5.pH值8.0,其降解速度为0.2%/h,随着pH值得升高,降解速度加快,在pH 12时,15min之内会完全降解[7,8]。
化妆品中巯基乙酸的检测方法

附件1化妆品中巯基乙酸的检测方法(离子色谱法)1 范围本方法规定了离子色谱法测定化妆品中巯基乙酸的含量。
本方法适用于化妆品中巯基乙酸及其盐类含量的测定。
2 方法提要样品中的巯基乙酸经水溶解提取后,用离子色谱仪分离巯基乙酸根与无机离子,电导检测器检测,以保留时间定性,峰面积定量。
本方法巯基乙酸的检出限5.8ng,定量下限20ng。
取样量为0.5g时,检出浓度为46μg/g,最低定量浓度0.15mg/g。
3 试剂和材料除另有规定外,本方法所用试剂均为分析纯或以上规格,水为GB/T 6682规定的一级水。
3.1 巯基乙酸,优级纯。
3.2 甲醇,优级纯。
3.3 硫酸溶液[φ(H2SO4)=10%]:取硫酸(ρ20=1.84g/ml)10mL,缓慢加入到90mL水中,混匀。
3.4 盐酸溶液[φ(HCl)=10%]:取盐酸(ρ20=1.19g/ml)10mL,加入90mL水中,混匀。
3.5 淀粉溶液(10g/L):称取可溶性淀粉1g,加水5mL调成溶液,再加入沸水95mL,煮沸,并加水杨酸0.1g或氯化锌0.4g 防腐。
3.6 氢氧化钠溶液(500g/L):称取氢氧化钠50g,加水适量使溶解并至100mL。
再量取一定量用经超声脱气的水稀释到淋洗液浓度。
3.7 重铬酸钾标准溶液[c(1/6K2Cr2O7)=0.1000mol/L]:准确称取已于120℃±2℃电烘箱中干燥至恒重的重铬酸钾基准物质4.9031g,溶于水并转移至1000mL量瓶中,定容至刻度,摇匀。
3.8 硫代硫酸钠标准溶液(0.1mol/L):称取硫代硫酸钠(Na2S2O3〃5H2O)26g(或无水硫代硫酸钠16g)溶于1000mL 新煮沸放冷的水中,加入氢氧化钠0.4g或无水碳酸钠0.2g,摇匀,贮存于棕色瓶内,放置两周后过滤,用重铬酸钾标准溶液标定其浓度,标定方法如下:准确吸取重铬酸钾标准溶液(3.7)25.00mL于500mL碘量瓶中,加碘化钾2.0g和硫酸溶液(3.3)20mL,立即密塞,摇匀,于暗处放置10min。
ELLMAN试剂法测定自由巯基和二硫键

ELLMAN试剂测定自由巯基试验基于的原理:5,5’-dithiobis-2-nitrobenzoicacid(DTNB)5,5-二硫基-双(2-硝基苯甲酸)5,5-二硫基-双(2-硝基苯甲酸)(DTNB)在412nm没有吸收,与巯基反应后,生成2-硝基-5-巯基苯甲酸(TNB)[1]。
TNB2-在412nm有很强的吸收,可以用于对肽段的自由巯基进行定量分析[2]。
DTNBTNB2-在/(摩TNB 1.4*10-51.2.3.4.5.之6.1.材料PEG-G-CSF批号:080229浓度4.32mg/mlG-CSF批号:080126浓度6.9mg/ml10k超滤膜PALL2.试剂SequencingGradeModifiedTrypsin,Promega,lot#237826。
20ug/小瓶。
加入20ul50mMNH4HCO3,pH7.8溶解,配成1ug/ul的酶液。
1MDTT刘春凤提供TFA(三氟乙酸):TEDIALot#705114乙腈:FisherScientificLot#055848StarterkitforMALDI-TOFMS:BRUKERDALTONICS,LotNO2007-208241-001(includingα-Cyano-4-hydroxycinnamicacid(HCCA),peptidecalibrationstandard,proteincalibrationstandardI,proteincalibrationstandardⅡ)PEG肽段反相分离流动相A液:0.1%TFA/H2OB液:1)取取2)按质量比1:40往PEG-G-CSF中加入Trypsin10ul,往G-CSF中加入Trypsin10ul,同时各加入1ul1MDTT使终浓度为5mM。
另做一空白对照,往300ul50mMNH4HCO3中加入Trypsin15ul。
37℃反应,开始时间为。
三、肽段反相分离收样:TT离喜冻干四、温度:①加入100ul反应缓冲液到样品管和对照管中,在412nm测定吸收值,吸收值调节到0.②加入100ul反应缓冲液到对照管中,加入100ulELLMAN试剂到样品管中,在412nm测定吸收值A DTNB。
一种检测巯基的近红外有机小分子探针、其制备方法及应用[发明专利]
![一种检测巯基的近红外有机小分子探针、其制备方法及应用[发明专利]](https://img.taocdn.com/s3/m/39bea574daef5ef7bb0d3c30.png)
专利名称:一种检测巯基的近红外有机小分子探针、其制备方法及应用
专利类型:发明专利
发明人:罗稳,李景华,丁旭,房茹,赵永梅,赵凯
申请号:CN202010315888.3
申请日:20200421
公开号:CN111349069A
公开日:
20200630
专利内容由知识产权出版社提供
摘要:本发明公开一种检测巯基的近红外有机小分子探针、其制备方法及应用,属于化学分析技术领域。
所述有机小分子探针的化学结构如式(I)表示:(I)其制备方法如下:将2‑氯‑1‑(2,4‑二羟基苯基)乙酮、6‑(二甲基氨基)‑2‑萘醛和有机溶剂混合,然后滴入碱溶液,室温反应后,浓缩有机相,用酸中和,得到中间体。
中间体与2,4‑二硝基苯磺酰氯在有机溶剂中反应后,分离提纯得到产物。
本发明制备方法简单易行,所得到的小分子探针可实现巯基的紫外和荧光双响应,检测细胞内的巯基物质。
具有灵敏度高、响应快速、特异性高等诸多优点,而且可以用于活体成像,在化工、环境、生物医药等领域具有广阔的应用前景。
申请人:河南大学
地址:475001 河南省开封市明伦街85号
国籍:CN
代理机构:郑州联科专利事务所(普通合伙)
代理人:张丽
更多信息请下载全文后查看。
ELLMAN试剂法测定自由巯基和二硫键

E L L M A N试剂测定自由巯基试验基于的原理:5,5’-dithiobis-2-nitrobenzoicacid(DTNB)5,5-二硫基-双(2-硝基苯甲酸)5,5-二硫基-双(2-硝基苯甲酸)(DTNB)在412nm没有吸收,与巯基反应后,生成2-硝基-5-巯基苯甲酸(TNB)[1]。
TNB2-在412nm有很强的吸收,可以用于对肽段的自由巯基进行定量分析[2]。
DTNBTNB2-根据文献记载,TNB的吸光系数在13.6*103M-cm-~14.25*103M-cm-之间[3,4]。
吸光度测量的最灵敏范围在0.2-0.7之间。
A=εbc,其中A为吸光度,ε是摩尔吸收光系数或消光系数,ε单位为升/(摩尔·厘米)[L/(mol·cm)]。
以吸光度下限0.2来计算(吸光系数取14.15*103M-cm-),需要TNB的浓度为c=0.2/(14.15*103LM-cm-*1cm)=1.4*10-5mol/L(1.4*10-8mol/ml),需要蛋白浓度为1.4*10-5mol/L*18790g/mol=0.2631g/L=0.2631mg/ml.我们的条件可以达到这个检测限度。
ELLMAN试剂法测定自由巯基主要的影响因素有:1.EDTA的适量加入有助于TNB显色的稳定和成梯度线性关系[5]。
2.在不同缓冲液中,TNB的最大吸收波长略微不同,所以它们在412nm的吸收也不3.同,要根据选择的缓冲液来确定[5]。
另外,TNB的分光光度法分析对SDS很敏感[6]。
4.DTNB随着pH的升高,降解速度加快。
在pH7.0,其降解速度为0.02%/h,在5.pH值8.0,其降解速度为0.2%/h,随着pH值得升高,降解速度加快,在pH12时,15min之内会完全降解[7,8]。
6.摩尔吸收光系数在不同的温度下不同,随温度的升高而下降[4].试验方案主要材料:1.材料PEG-G-CSF批号:080229浓度4.32mg/mlG-CSF批号:080126浓度6.9mg/ml10k超滤膜PALL2.试剂SequencingGradeModifiedTrypsin,Promega,lot#237826。
生物药游离巯基检测意义

生物药游离巯基检测意义生物药游离巯基是指生物药物中含有的具有游离巯基(-SH)的基团。
巯基是一种常见的生物活性基团,对于生物药物的活性、抗氧化性、储存和运输等方面具有重要影响。
因此,生物药游离巯基的检测对于药物质量和效果影响重大。
1.反应生物药游离巯基的含量对于药物质量和效果影响重大。
生物药游离巯基的含量直接影响药物的活性和效果。
一些生物药物需要经过细胞内巯基的氧化还原反应才能发挥药效,因此游离巯基的含量对于药物的疗效和安全性具有重要意义。
此外,生物药游离巯基的含量还会影响药物的稳定性,包括药物的储存和运输等环节。
2.生物药游离巯基的含量可以反映该生物药在体内的活性状态。
生物药游离巯基的含量可以反映该生物药在体内的活性状态,对于判断药物的作用机制和不良反应也具有一定的指导意义。
例如,一些药物在体内需要通过氧化还原反应才能发挥作用,如果生物药游离巯基含量不足,则会影响药物的活性;相反,如果生物药游离巯基含量过高,则可能产生不良反应甚至毒性。
3.检测生物药游离巯基的存在可以帮助判断该药物是否具有抗氧化性。
生物药游离巯基具有抗氧化性,可以保护细胞免受氧化应激损伤。
一些药物需要经过氧化还原反应才能发挥作用,如果生物药游离巯基含量过低,则会影响药物的抗氧化效果;相反,如果生物药游离巯基含量过高,则可能对细胞产生损伤甚至引发炎症反应。
因此,检测生物药游离巯基的存在可以帮助判断该药物是否具有抗氧化性以及抗氧化能力的强弱。
4.生物药游离巯基的含量对于药物的储存和运输也有重要影响。
生物药游离巯基的含量会影响药物的稳定性,对于药物的储存和运输也具有重要影响。
一些生物药物需要在低温、干燥、避光的条件下储存,以保持其稳定性和有效性。
如果生物药游离巯基含量过高,可能会导致药物的不稳定性和易变性,从而影响药物的质量和效果。
因此,检测生物药游离巯基的含量可以帮助评估药物的储存和运输条件,以保证药物的质量和效果。
5.检测生物药游离巯基的含量有助于研究该药物的作用机制和不良反应。
化妆品中巯基乙酸的检测方法

化妆品中巯基乙酸的检测方法(征求意见稿)1.使用范围本方法规定了采用液相色谱法测定化妆品中巯基乙酸(CAS:68-11-1)的方法。
本方法适用于头发烫卷剂或烫直剂、脱毛膏类化妆品中巯基乙酸的测定。
2.方法提要样品在经过提取后,经高效液相色谱仪分离,紫外检测器检测,根据保留时间定性,峰面积定量,以标准曲线法计算含量。
本方法对巯基乙酸的检出限为0.004μg,定量下限为0.0132μg。
若取0.25g样品,本方法对巯基乙酸的检出浓度为35.6μg /g,最低定量浓度为118.7μg/g。
3.试剂和材料除另有规定外,所用试剂均为分析纯,水为一级实验用水。
3.1巯基乙酸,纯度≥99%。
乙腈,色谱纯。
乙腈水溶液,乙腈+水(10+90)。
磷酸二氢钾(KH2PO4),色谱纯。
磷酸,优级纯。
巯基乙酸标准储备液:取巯基乙酸0.05g,精确到0.0001g,置50 mL棕色容量瓶中,用乙腈水溶液()溶解并定容至刻度,摇匀,配成质量浓度为1.0 g/L的标准储备溶液。
巯基乙酸标准工作溶液:精密配制浓度分别为5.0μg/mL、20.0μg/mL、50.0μg/mL、80.0μg/mL、110.0μg/mL和150.0 μg/mL的系列巯基乙酸标准工作溶液。
4.仪器高效液相色谱仪,配紫外检测器。
涡旋混合仪。
超声波清洗器。
分析天平:感量0.0001g。
5.测定步骤样品前处理准确称取试样0.25g,精确至0.001g,置于25mL具塞刻度管中,加入20 mL 乙腈水溶液(),涡旋1 min,振摇,超声(功率:400W)提取30 min,取出,冷却至室温后定容,混匀,取上层液经0.45 μm滤膜过滤后稀释10倍,稀释液作为待测样液,备用。
测定5.2.1色谱条件色谱柱:C18柱,250 mm×4.6mm,5 μm(耐酸柱);流动相:乙腈+LKH2PO4(磷酸调pH=)(10+90);流速:mL/min;检测波长:215nm;柱温:30℃;进样量:20μL。
巯基酯成品硫含量的测定

巯基酯成品硫含量检验规程一、仪器和试剂1、试剂无水乙醇 1+1盐酸溶液 淀粉指示剂5g/l2、仪器250ml 锥形瓶 电子分析天平3、0.05mol/L 碘酸钾标准溶液的配制及标定配制:称取18g 碘酸钾水中,90.15g 碘化钾,3.23g 氢氧化钠溶解于400ml 水中,稀释至10L ,储存于广口瓶中,摇匀标定:称取0.1g 标准锡(含量为99.99%),加50ml 盐酸溶解,再加50ml 无二氧化碳的水和1.3g 铝丝于锥形瓶中加热至反应完成后,冷却至常温,然后加2ml 淀粉指示剂,用配制好的碘酸钾溶液滴定至淡蓝色为终点,并保持30S 不褪色,同时做空白计算:)0V -V (0.05935m99.99C ⨯⨯=式中:C-碘酸钾标准滴定溶液浓度的准确数值,mol/LV-滴定消耗碘酸钾标准溶液体积,mlV 0-空白滴定消耗碘酸钾标准溶液体积,mlm-标准锡质量准确数值,ml二、检测步骤准确称取0.2000g 左右的样品,加50ml 无水乙醇、1ml( 1+1)盐酸溶液摇匀后用0.05mol/L 碘酸钾标准溶液滴定至浅黄色30s 不褪色为终点,记录体积V 。
计算:%100.32C 1m V W ⨯=式中:m----样品的质量,gC----碘酸钾标液的浓度,mol/LV----碘酸钾标液的滴定体积,ml巯基酯成品硫含量检验规程一、仪器和试剂1、试剂无水乙醇2、仪器250ml 锥形瓶 电子分析天平3、0.05mol/L 硫代硫酸钠标准溶液配制:称取26g 硫代硫酸钠(Na2S2O3.5H2O )(或16g 无水硫代硫酸钠),加0.2g 无水碳酸钠,溶于2000mL 水中,缓缓煮沸10min ,冷却。
放置两周后过滤储存于广口瓶中标定:称0.09g 干燥至恒重的重铬酸钾,置于碘量瓶中,加25g 水,2g 碘化钾及20ml 硫酸溶液(20%),摇匀,于暗处放置10分钟,加150ml 水,用硫代硫酸钠标准滴定溶液滴定,近终点加2ml 淀粉指示剂,继续滴定至溶液由蓝色变亮绿色,同时做空白计算:M V -V m 1000C 0⨯⨯=)(式中:m-重铬酸钾质量准确数值,gV-滴定消硫代硫酸钠标准溶液体积,mlV 0-空白滴定消耗硫代硫酸钠标准溶液体积,mlM-重铬酸钾的摩尔质量数,g/mol[M ( K 2Cr 2O 7)=49.031]4、0.05mol/L 碘标准溶液的配制及标定配制:称取13g 碘及35g 碘化钾,溶于100mL 水中,稀释至2000ml ,摇匀储存于棕色广口瓶中标定:量取35-40 ml 配制好的碘溶液,置于碘量瓶中,加150ml 水,用硫代硫酸钠标准滴定溶液滴定,近终点加2ml 淀粉指示剂,滴定至溶液蓝色消失,同时做水消耗碘的空白:量取250ml 水,加0.15-0.2ml 配制好的碘液及2ml 淀粉指示剂用硫代硫酸钠标准滴定溶液滴定至溶液蓝色消失计算:43121V -V C V -V C ⨯=)(式中: V 1-硫代硫酸钠标准滴定溶液的体积数值,mlV 2-空白试验消耗硫代硫酸钠标准溶液体积,mlC 1-硫代硫酸钠标准滴定溶液浓度的准确数值,mol/LV3-碘溶液体积准确数值,mlV3-空白试验加入碘溶液体积准确数值,ml二、检测步骤准确称取0.2000g 左右的样品,加50ml 无水乙醇、摇匀后用0.05mol/L 碘标准溶液滴定至浅黄色30s 不褪色为终点,记录体积V 。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
巯基检测方法
1. RP-HPLC法测定巯基含量
采用色谱柱Kromasil-C18 (250×4.6mm, 5μm),流动相A(0.1%TFA)和流动相B(甲醇)梯度洗脱:流动相B 40%~80%,0~10min,然后80% B保持5min,流速0.8mL/min,检测波长327nm,得到NTB标准曲线y=3.67059x+0.14123,回收率101.9%,RSD=l.17%,从而建立了一种高灵敏度巯基检测方法。
2. 采用分子荧光光谱法作为反应条件,用反相高效液相色谱梯度洗脱法测定巯基
用OPA、丹酰氯、茚三酮与半胱氨酸反应,测其可见紫外吸收光谱及荧光光谱;在不同PH、温度、反应时间条件下,用OPA与半胱氨酸反应测其荧光度;分别吸收0.1mmol/L半胱氨酸溶液0、20、40、60、80、100 μl,各加入10 μ
lH
2O
2
,室温下反应30min,然后加热蒸干,残渣用200μl OPA衍生液,定容至5 ml,
4 min时测其荧光光谱。
取pH8.4的硼酸缓冲溶液 5μl,混合10次;加入OPA 衍生液2μl,混合进样走HPLC。
梯度条件:洗脱液B所占比例0min为0,17min 线性增加至60%,17.5min线性增加至100%,20min洗脱结束。
激发波长为340nm,荧光检测波长为450nm。
3.柱前衍生高效液相色谱-紫外检测法
以tris(2-carboxylethyl) –phosphine (TCEP)为还原剂,7–fluorbenzo–2–oxa –1,3–diazole–4-sulfonate(SBD-F)为衍生剂,N-乙酰半胱氨酸为内标,C8色谱柱分离,流动相为甲醇 -磷酸盐缓冲液(pH =3. 0),梯度洗脱 ,385 nm处检测。
线性范围为8. 3~1042. 6 μmol/L,最低检测限为 0. 42μmol/L,日内精密度为 1. 67%~1. 86%,日间精密度为 2. 08%~3. 06 %,平均回收率为 98. 1%~103. 2 %。
4. 电化学脱附与荧光技术联用
将样品固定在烷基硫醇自组装膜修饰的金电极表面 ,通过荧光试剂马来酰亚胺与游离巯基反应原位标记 GSH,恒电位条件下脱附电极表面吸附物 ,检测脱附物在 0.1 mol·L-1 .KOH溶液中的荧光强度。
2008.8.28。