_药典临床用药须知_中药卷_2005年版_中含反药药对成方制剂收载情况与分析
079中国药典2005年版的变化对药品研发的影响分析——固体制剂相关改变带来的影响

发布日期20051015栏目化药药物评价>>化药质量控制标题中国药典2005年版的变化对药品研发的影响分析——固体制剂相关改变带来的影响作者马磊部门正文内容审评三部马磊摘要:以中国药典2005年版二部片剂通则的变化为例,探讨新版药典制剂通则的变化对口服固体制剂研发可能带来的影响。
关键词:中国药典2005年版制剂通则口服固体制剂2005年版二部附录的制剂通则中收载的口服固体制剂包括以下几类:片剂、胶囊剂、颗粒剂、散剂和丸剂,与2000版药典二部制剂通则相比,各制剂项下均有不同程度的变化。
其中以片剂和胶囊剂的修改内容最多,下面以片剂通则的变化为例,讨论这些变化可能对药品审评和研发带来的影响。
与上版中国药典(2000年版)相比,新版药典片剂通则的主要变化见下表:可溶片为2005年版中国药典的新增制剂,系指能溶解于水的非包衣片或薄膜包衣片。
可溶片在用前须溶于水中,以供口服、涂抹或含漱用。
可溶片应进行崩解时限检查,介质为水,温度15~25℃,除另有规定外,各片均应在30分钟内完全崩解并溶化。
如有1片不能完全溶解,应另取6片复试,均应符合规定。
所以可溶片的主药及辅料应具有较好的水溶性,对于水溶性差的药物,则不宜制成该类剂型;另外,还应综合考虑药品的适应症、给药途径及临床用药的需求情况,避免盲目开发。
以往药典中均未对薄膜衣片的包衣液残留问题提出相关的要求,新版药典对此进行了原则性规定:“必要时,薄膜包衣剂应检查残留溶剂”。
所以研发者今后应对包衣材料及溶剂的安全性给予充分关注,尽可能避免使用有毒有害的有机溶剂作为包衣液的溶剂。
从整体上讲,新版药典对各种剂型的定义更加清晰,要求更为明确。
如规定缓释片(控释片)系指在水中或规定的释放介质中缓慢地(恒速或接近恒速地)释放药物的片剂。
缓(控)片应进行释放度检查,缓释片的释放度曲线应不少于3个点,控释片的释放度曲线应不少于5个点。
药品研发者在考虑开发该类制剂时应当注意:与其相应的普通制剂比较,缓、控释制剂的给药频率应至少减少一半,或给药频率比普通制剂有所减少,但能显著增加患者的顺应性或疗效。
中国药典2005年版的变化对药品研发的影响分析

中国药典2005年版的变化对药品研发的影响分析★注射制剂相关改变带来的影响N中国药典2005年版已经于2005年7月1日正式执行,从整体上看,经过近年的发展,我国药典已经大大缩短了与医药发达国家的差距,并充分体现了我国的特色,上述特点在新版药典中得到了充分的体现,以注射制剂为例,与中国药典2000年版相比,新版药典在原基础上,根据是否可以直接使用和药物在制剂中的存在形式对注射剂型进行了细分,上述调整有利于药物研发设置质量控制指标时进一步模块化和规范化,充分体现了现代工程设计的优秀思路。
另一方面,在检查项目中,也参考国外医药研发发达国家经验,并结合我国实际,增加了可见异物检查项、调整了液颜色检查法和澄明度检查法,并删减了异常毒性和降压物质检查项。
进一步体现了目前注射制剂的制备和质控特点。
($Gv%x 以下,本文主要谈谈新版药典在注射制剂及其质控项目等方面所发生的变化。
一、制剂通则的变化F-8.Q与中国药典2000年版相比,新版药典在该部分进行了比较大的调整。
下表是中国药典1995、2000和2005年版在注射制剂方面的变化情况:Ihn从上表可以看出,新版药典仍然保留了注射液,静脉输液、注射用无菌粉针等内容,增加了供静脉滴注用大体积(除另有规定外,一般不小于100ml)注射液,也称为输液。
将直接使用的注射剂按容量大小进行了分类,并按药物在制剂中的存在形式划分为溶液型、混悬型和乳剂型,较旧版药典更为明晰。
由于上述变化,在质量控制方法,增加了对静脉用乳状液型注射液分散相的粒度控制-“90%应在1um以下,不得有大于5um的球粒”的描述。
并在原中国药典2000年版附加剂基础上增加了乳化剂、助悬剂。
1o\另外,间接使用的注射剂中在原无菌粉末基础上增加了浓溶液(Concentrates),体现了现代制药工业的需要。
并将前者的制备工艺进行了划分,分为喷雾干燥、溶媒结晶和冷冻干燥型。
i浓溶液在美国药典和日本药典没有收载,但欧洲药典和英国药典纳入了该制剂类型。
_中国药典_2005年版中药成方制剂显微鉴别项存在问题的探讨

玄参
五味清浊散 、十六味冬青丸 、脑得生 丸 、八味檀香散 、七厘散 、五虎散 、金 蒲胶囊 、跌打丸 、痛经丸
红花
再造丸
制何首乌
栀子金花丸 、青果丸
金银花
复方皂矾丸 、青娥丸
核桃仁
龙胆泻肝丸 (水丸 、大蜜丸 ) 、补中 益气丸 、得生丸 、舒肝和胃丸 、逍遥 丸 (水丸 、大蜜丸 )
柴胡 (北柴胡 、南柴胡 )
人参养荣丸 、桂附理中丸
含甘草的成方制剂
当归养血丸 明目地黄丸
羚羊清肺丸
处方中药材 丁香 丁香 丁香 大黄 川芎 川楝子 马钱子 甘草 炙甘草
甘草
白芍
玄参
荷叶丸
玄参
龙胆泻肝丸 (水丸 、大蜜丸 )
地黄
首乌丸
地黄
艾附暖宫丸 、化症回生片 、再造丸 、 济生肾气丸 、女金丸 、纯阳正气丸
化症回生片
肉桂 延胡索
种皮表皮细胞淡黄色 ,多角形 ,壁薄 ,下方叠 合有网纹细胞 。 (同肥儿丸 )
纤维束棕红色
纤维束棕红色 ,壁甚厚 ,有的周围细胞含草酸 钙方晶 ,形成晶纤维 。
不规则碎块无色或淡绿色 ,半透明 ,有光泽 , 有时可见细密波状纹理 。
种皮石细胞黄色或淡棕色 ,多破碎 ,完整者长 多角形 、长方形或不规则形 ,壁厚 ,有大的圆 形纹孔 ,胞腔棕红色 。
制剂名称 梅花点舌丸 、红灵散 麻仁丸
处方中药材 人工麝香 火麻仁
跌打活血散
没药
清肺消炎丸 、牛黄清心丸 (局方 ) 苦杏仁 (炒 )
标准规定 无定形团块淡黄棕色 ,埋有细小方形结晶 。 果皮石细胞淡黄色至红棕色 ,表面观多角形 ,壁厚 。 不规则碎块淡黄色 ,半透明 ,渗出油滴 ,加热后油滴溶化 , 现正方形草酸钙结晶 。
执行《中国药典》2005版一部附录中遇到的问题

执行《中国药典》2005版一部附录中遇到的问题《中国药典》2005版一部附录在丸剂重量差异检查项、水分测定法、栓剂融变时限检查、中药分散片检查、中药片剂脆碎度检查方面有不完善的地方,本文旨在对这些问题进行探讨,为《中国药典》2010年版的完善提供参考。
[Abstract] Pill weight variation check, moisture test, suppository melt time limit test, dispersible tablet of Chinese medicine test, tablets of Chinese medicine broke brittle test in Chinese Pharmacopoeia edition 2005 appendix 1 had imperfect places, and the problems were discussed in this paper, hoping to provide basis for the improvement of Chinese Pharmacopoeia edition 2010.[Key words] Chinese Pharmacopoeia edition 2005; Appendix; Problem《中国药典》2005版在凡例、品种的标准要求、附录的制剂通则和检验方法等方面均有较大的变化和进步。
《中国药典》2005版一部共收载品种1 146种,其中,新增154种,修订453种。
本版药典附录较2000版进行了较大的增修订,如农药残留测定法中增订了对12种有机磷和3种拟虫菊酯类农药的测定方法,不溶性微粒检查法中增订了小容量注射剂的检查,薄层色谱法中增订了系统适用性试验,微生物限度检查修订为按给药途径要求,并增加了方法验证试验。
《中国药典》2005版自2005年7月1日开始执行至今已3年多,作为基层药检所,在执行一部附录过程中有些不明确的地方,本文参考相关文献对此进行探讨,希望2010版药典能够明确。
中华人民共和国药典临床用药须知-《临床用药须知》是《中国药典》的配套用书

中华人民共和国药典临床用药须知,2010版临床用药须知,中国药典,中华人民共和国兽药典《中华人民共和国药典临床用药须知》作者: 国家药典委员会出版社: 中国医药科技出版社加入时间: 2011年7月13日总定价:1980元按批发价:1200元(五套团购价格可再议)中华人民共和国药典临床用药须知:2010年版中药饮片卷定价592元中华人民共和国药典临床用药须知: 2010年版化学药和生物制品卷定价:798元中华人民共和国药典临床用药须知:2010年版中药成方制剂卷定价:590元《中华人民共和国药典临床用药须知》(以下简称《临床用药须知》)是《中华人民共和国药典》(以下简称《中国药典》)配套丛书之一。
本版《临床用药须知》由国家药典委员会组织国内200多位一流的临床各科专家及相关药学专家编写,分三卷出版:①中药饮片卷,正文1268页,共介绍了656种药物,其中包括正品药物547种,附药109种。
正品药物按中文名称、汉语拼音名、药材来源、炮制、性味归经、功能主治、效用分析、配伍应用、鉴别应用、方剂举隅、成药例证、用法与用量、不良反应、使用注意、化学成分、药理毒理、本摘要、参考文献等分别撰写;②中药成方制剂卷,正文1068页,收载品种约1565个。
针对每类药物的方解、临床应用、药理毒理、不良反应、注意事项、用法与用量、参考文献等方面进行系统介绍。
为了便于临床使用,附录中还介绍了病证索引;③化学药和生物制品卷,正文1577页,收载品种1440余种。
本版的最大特点是对儿童用药剂量进行了全面的审查和修订。
同时,对每种药物的出处均作了标注,即是否是《中国药典》收录的药品,是否是国家基本药物,是否是医保药物,等等。
《临床用药须知》2010年版在继承前版的基础上,做了大量发展和创新性的工作,具有以下鲜明特色:①首次将“中药饮片”独立成卷,为中药材、饮片的规范使用提供了标准和依据。
②中药成方制剂卷、中药材与饮片卷对收载品种的药理毒理进行了独立介绍。
评《中国药典》2005年版(一部)

评《中国药典》2005年版(一部)
周富荣
【期刊名称】《中国药品标准》
【年(卷),期】2005(6)4
【摘要】《中华人民共和国药典》2005年版(一部)(以下简称“新版《药典》”),是按第八届药典委员会确定的设计方案和要求编制而成的,经国家食品药品监督管理局批准颁布实施。
于2005年7月1日起正式执行。
是建国以来第
八版药典,也是21世纪的第一版。
【总页数】5页(P26-30)
【作者】周富荣
【作者单位】北京市药品检验所,100035
【正文语种】中文
【中图分类】R9
【相关文献】
1.对2005年版《中国药典(一部)》的几点建议 [J], 萧汉文
2.对《中国药典》(2005年版一部)薄层色谱法中部分含苯、甲苯展开剂的改进 [J], 陈蔼;梁洁莹;李书渊
3.对《中国药典》2005年版一部成方制剂含量测定标准存在问题的探讨 [J], 刘德军
4.对《中国药典》2005年版一部存在问题的浅见 [J], 王幼鹏
5.试论按《中国药典》2005年版一部检验中药饮片的局限性 [J], 郭全兴
因版权原因,仅展示原文概要,查看原文内容请购买。
国家食品药品监督管理局关于执行《中国药典》2005年版有关问题的补充通知

国家食品药品监督管理局关于执行《中国药典》2005年版有
关问题的补充通知
【法规类别】药品管理
【发文字号】国食药监注[2006]59号
【发布部门】国家食品药品监督管理局(原国家药品监督管理局)(已撤销)
【发布日期】2006.02.27
【实施日期】2006.02.27
【时效性】现行有效
【效力级别】部门规范性文件
国家食品药品监督管理局关于执行《中国药典》2005年版有关问题的补充通知
(国食药监注[2006]59号)
各省、自治区、直辖市食品药品监督管理局(药品监督管理局):
根据《关于颁布和执行中国药典2005年版有关事宜的通知》(国食药监注﹝2005〕234号),《中国药典》2005年版已于2005年7月1日执行,现就执行《中国药典》2005年版的有关事宜补充通知如下:
一、《中国药典》2005年版(一部)对《中国药典》2000年版(一部)收载的葛根、黄柏、金银花三个多来源药材品种,按植物不同来源分列为两个药材品种,在中成药处方中亦将按两味药材管理。
生产含分列药材的中成药生产企业,应根据《中国药典》2005年版(一部)的药材来源及名称,重新确认处方中使用的药材名称。
凡处方中药味名称
需变更的中成药品种,应于2006年8月1日前向国家局提出修订国家药品标准的药品注册补充申请,其技术审核工作由国家药典委员会统一负责。
目前需中成药企业确认的分列药材名单如下:。
中国药典2005年版药典

(2)化工原料作为药用的条款删去; (3)强调同一原料,根据临床用药要求不同,须分别制 定相应的质量要求
? 第 条:包装、标签、说明书—分别按 SFDA要求
普通片(内涵)
片
2000 年版 未明确
片剂分类
10 种
缓、控释片定义 未明确
微生物限度检查 卫生部文件
光散射法用于有色容器及液 体(不用灯检法确认)
缓、控释胶囊定义 肠溶胶囊定义
微生物限度检查 残留溶剂 【水分】
胶囊剂
2000 年版 未明确 肠溶空心胶囊
卫生部文件 未明确
2005 年版 明确—同片剂 肠溶空心胶囊 肠溶包衣颗粒、小丸
(肠溶微丸胶囊问题)
(肠溶)空心胶囊 - 撤 原则性要求(执委会) 原则性要求
眼用制剂
2000 年版
2005 年版
滴眼剂、眼膏剂
眼用液体:滴眼、洗眼、眼 内注射
眼用半固体:眼膏、乳膏、 凝胶
眼用固体:眼丸、眼膜、眼 内插入
无
混悬型滴眼液要求
【重(装)量差异】 无
单剂量各种眼用制剂要求
糖浆剂
定义 【微生物限度】
2000 年版
2005 年版
含蔗糖60% (一部) 45% (一、二部)
65% (二部)
卫生部文件
按微生物限度检查法作为补 充
颗粒剂
分类与定义 【溶化性】
2000 年版 可溶、泡腾、肠溶、缓(控) 释
未明确
2005 年版
同2000 年版并给出定 义 另增混悬颗粒
明确混悬颗粒不要求做 溶化性检查(一部要求 混匀)
口服溶液剂、混悬剂、乳剂
分类与定义
中国药典2005版解读

为防止药物在胃内分解失效、对胃的 剌激或控制药物在肠道内定位释放,可对片 剂包肠溶衣;为治疗结肠部位疾病等,可对 片剂包结肠定位肠溶衣。 肠溶片除另有规定外,应进行释放度 检查。 片剂在生产与贮藏期间应符合下列规 定。 一、原料药与辅料混合均匀。含药量 小或含毒、剧药物的片剂,应采用适宜方法 使药物分散均匀。
三、 配制注射剂时,可根据药物的性质加入适 宜的附加剂。如渗透压调节剂、pH值调节剂、增溶剂、 助溶剂、抗氧剂、抑菌剂、乳化剂、助悬剂等。所用 附加剂应不影响药物疗效,避免对检验产生干扰,使 用浓度不得引起毒性或过度的刺激。常用的抗氧剂有 亚硫酸钠、亚硫酸氢钠、焦亚硫酸钠,一般浓度为 0.1%~0.2%;常用抑菌剂为0.5%苯酚、0.3%甲酚、 0.5%三氯叔丁醇等。多剂量包装的注射液可加适宜的 抑菌剂,抑菌剂的用量应能抑制注射液中微生物的生 长,加有抑菌剂的注射液,仍应用适宜的方法灭菌, 静脉输液与脑池内、硬膜外、椎管内用的注射液均不 得加抑菌剂。除另有规定外,一次注射量超过15ml的 注射液,不得加抑菌剂。
4.在不溶性微粒检查法中增 订小容量注射剂的检查;薄层色谱 法中增加了系统适用性试验;微生 物限度检查修订为按给药途径要求, 并增加了方法验证试验;无菌检查 法由培养7天修订为培养14天。
在指导原则中,修订了原料药与药物 制剂稳定性试验指导原则;缓释、控 释和迟释制剂指导原则,使之与实际 研究和生产情况更趋一致;并增加药 物引湿性试验指导原则和近红外分光 光度法指导原则等,对考察药品质量, 规范质量要求和统一药品标准起到指 导作用。
江苏省药品检验所 周帼雄
剂
型
Ⅰ A 片剂 Ⅰ B注射剂 Ⅰ C酊剂 Ⅰ D栓剂 Ⅰ E胶囊剂 Ⅰ F软膏剂 乳膏剂 糊剂(软膏剂) Ⅰ G眼用制剂(眼膏剂) Ⅰ H丸剂 Ⅰ J植入剂(新增) Ⅰ K糖浆剂 Ⅰ L气雾剂 粉雾剂 喷雾剂 Ⅰ M 膜剂 Ⅰ N颗粒剂Ⅰ O 口服溶液剂 口服混悬剂 口服乳剂(口服溶液剂、混 悬剂、乳剂) Ⅰ P 散剂 Ⅰ Q 耳用制剂(滴耳剂) Ⅰ R鼻用制剂(滴鼻剂) Ⅰ S 洗剂 冲洗剂 灌肠剂(洗剂) Ⅰ T 搽剂 涂剂 涂膜剂(搽剂) Ⅰ U 凝胶剂 Ⅰ V贴剂(透皮贴剂)
中国药典2005年版药材与饮片妊娠禁用_忌用和慎用药的分析

行,而探讨其相似度变化原因时,可进行镜像分析,由于药材11号样本的指纹图谱与标准指纹图谱在色谱峰比例上存在差异,造成相似度相对较低。
REFERENCES[1] MA C Y, DAI B C, LIN R C. HPLC quantitative analysis andfingerprint study of four flavonoids in Scutellaria baicalensis[J].Chin J Pharm Anal(药物分析杂志), 2003, 23(2): 83-86. [2] XIAO L H, WANG H Y, LI F M, et al. Comparative study onHPLC-FPS of Radix Scutellatiae of various sources [J]. J Shenyang Pharm Univ(沈阳药科大学学报), 2004, 21(1): 28.收稿日期:2008-09-05中国药典2005年版药材与饮片妊娠禁用、忌用和慎用药的分析白晓菊(国家药典委员会,北京 100061)摘要:本文在中国药典1995年版、2000年版和2005年版对妊娠禁用、忌用和慎用药材与饮片收载情况比较的基础上,探讨了妊娠禁用、忌用和慎用药与毒性和功能的关系,并对新版中国药典收载妊娠禁用、忌用和慎用药的表述提出了几点建议。
关键词:中国药典;药材与饮片;妊娠禁用;妊娠忌用;妊娠慎用;分析中图分类号:R921.2 文献标志码:A 文章编号:1007-7693(2009)09-0724-04Analysis of Medicinal Substances and Cut Crude Drugs with Forbidding, Contraindication or Caution for Pregnancy in Chinese Pharmacopoeia (2005)BAI Xiaoju(State Pharmacopoeia Commission, Beijing 100061, China)ABSTRACT: In this paper, Chinese medicinal substances and cut crude drugs with forbidding, contraindication or caution for pregnancy were analyzed, basing on comparison of Chinese Pharmacopoeia(CP) 1995, 2000 and 2005. Furthermore, relation between toxicity and function were discussed, and several suggestions on the description of drugs that were forbidden, contraindicated or cautious in pregnancy, which would record in CP2010 were pointed out.KEY WORDS: Chinese Pharmacopoeia; medicinal substances and cut crude drugs; be forbidden in Pregnancy; be contraindicated in pregnancy; be cautious in pregnancy; analysis中国药典2005年版收载药材与饮片551种,其中在【注意】项下收载孕妇慎用、忌服(用)和禁用的药材与饮片总计70种。
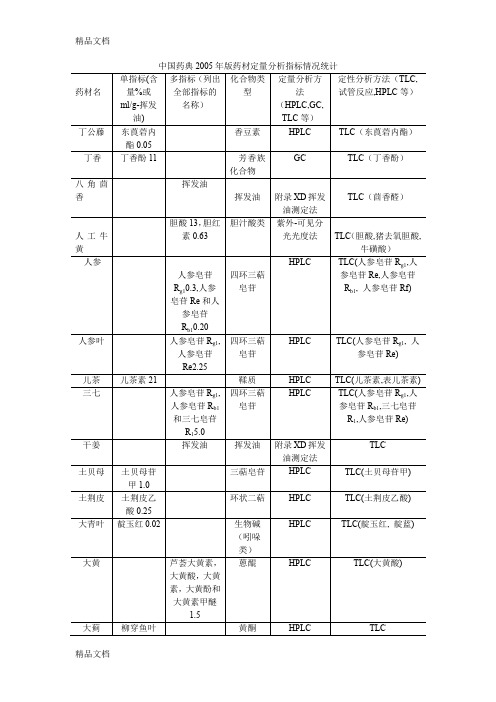
(整理)中国药典2005年版药材定量分析指标统计.
仙茅苷0.1
苷类
HPLC
TLC(仙茅苷)
白芍
芍药苷1.6
苷类(酚苷)
HPLC
TLC(芍药苷)
白芷
欧前胡素0.08
香豆素
HPLC
TLC()
白矾
含水硫酸铝钾KAl(SO4)2·12H2O(99.0)
无机物
滴定法
试管反应
白鲜皮
梣酮0.03
黄酮类
HPLC
TLC(梣酮)
炒瓜篓子
3,29-二苯甲酰基栝楼仁三醇0.06
胆酸6.0
胆红素35.0
胆汁酸类
胆汁色素类
薄层色谱扫描法,紫外-可见分光光度法
试管反应,TLC(胆酸,去氧胆酸)
何首乌
2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷(1.0)
二苯乙烯苷类
HPLC
TLC
制何首乌
2,3,5,4′-四羟基二苯乙烯-2-O-β-D-葡萄糖苷(0.7)
二苯乙烯苷类
生物碱
HPLC
TLC(盐酸小檗碱)
灯盏细辛
野黄芩苷0.3
黄酮类
HPLC
HPLC(野黄芩苷)
安息香
总香脂酸30.0
树脂
重量法
试管反应
防己
粉防己碱+
防己诺林碱1.6
生物碱
HPLC
TLC(粉防己碱,
防己诺林碱)
防风
升麻素苷+
5-O-甲基维斯阿米醇苷0.24
色原酮
HPLC
TLC(升麻素苷,
5-O-甲基维斯阿米醇苷)
无机物
滴定法
试管反应
西红花
西红花苷-I+
西红花苷-Ⅱ(10.0)
中国药典2005年版二部主要增修订情况

重视残留溶剂的控制,本版药典(二部)对残留 溶剂的检查推荐采用顶空毛细管气相色谱法,限 度与ICH一致。
收载化学合成原料药约100种,本版药典(二部) 24个品种增订残留溶剂检查。
本版药典(二部)在确保安全性的前提下,将热 原修改为细菌内毒素的品种有73个品种,增订细 菌内毒素检查的品种有112个,细菌内毒素判定标 准由“不得过…”修订为“应小于…”,避免肉眼观 察的不确定性。此外,42个品种删除异常毒性; 30个品种删除降压物质;
辅料设立专栏。
22/ 23
胶囊剂 微生物限度检查(卫生学?) 无菌检查 热原检查 细菌内毒素检查 吸光度(吸收度)
23/ 23
鼻用制剂与耳用制剂亦做了类似的调整。
14/ 23
乳膏剂 – 不同的修订。
糖浆剂 – 糖浆剂含糖量则根据实际并参照EP由65%修 改为45%,同时对微生物进行控制。
外用液体制剂 – 根据其剂型特点与用途增订了冲洗剂、灌肠剂、 涂剂及涂膜剂并要求进行微生物限度检查,必 要时,进行细菌内毒素及无菌检查如冲洗剂。
18/ 23
3.扩大HPLC在多组分原料及制剂中的 应用,重点加强品种要求
本版药典(二部)有223个品种采用HPLC方法 取代传统的容量法或紫外法或生物检定法。 – 例如盐酸小檗碱片与胶囊含量测定原采用滴 定法,多种生物碱均可参与滴定反应,本版 药典修订为HPLC法,可准确测定小檗碱的 含量;
18个小剂量药品增订含量均匀度检查。 70个原料药增订专属性红外鉴别。
4/ 23
目前,由于TOC测定仪器尚不普及,本版药典 (二部)在品种正文中仍沿用中国药典2000年 版的标准,但为与我国药品生产管理要求相适 应,纯化水与注射用水增订微生物限度检查, 限度要求与USP一致,纯化水限度为每1ml中 含细菌、霉菌和酵母菌总数不得过100个,注 射用水限度为每1ml中含细菌、霉菌和酵母菌 总数不得过10个。
对2005年版《中国药典》中药品种变化的探讨

对2005年版《中国药典》中药品种变化的探讨
范世明;黄泽豪;蔡珍华
【期刊名称】《福建中医药大学学报》
【年(卷),期】2006(016)002
【摘要】中国药典2005年版已于2005年7月1日正式执行了,本版药典较2000年版相比作了较大改动,质量标准也有了大幅度提高。
其中中药材品种及质量是整个中药产业链的首要一环,要确保中药饮片和中成药质量,首先必须抓好中药材这个最根本的源头。
在以往的《中国药典》中,中药材部分问题众多:一药多基原现象极为普遍;中药材的质量标准低下,与国际天然药物质量标准有些差距等。
鉴于此,2005年版《中国药典》以大量的调查研究为基础,对收载的中药材品种进行一系列修订和增订。
为了更好地学习并执行新版药典,笔者对其中有关“中药材品种变动”的内容做了认真的学习,并结合自身从事的药用植物学的工作特点进行了探讨。
【总页数】2页(P34-35)
【作者】范世明;黄泽豪;蔡珍华
【作者单位】福建中医学院药学系,福建,福州,350108;福建中医学院药学系,福建,福州,350108;福建中医学院药学系,福建,福州,350108
【正文语种】中文
【中图分类】R282
【相关文献】
1.2005年版《中国药典》中药标准的变化 [J], 钱忠直
2.关于2005年版《中国药典》附录部分的几点探讨 [J], 王红梅;陈启荣
3.中国药典2005年版(二部)主要变化 [J], 张培培;王平;王国荣
4.《中国药典》2005年版(二部)外用制剂相关变化及其意义 [J], 张星一;王平;左舒
5.对《中国药典》2010年版毒性中药品种的探讨 [J], 李红念;梅全喜
因版权原因,仅展示原文概要,查看原文内容请购买。
《中国药典》2005年版二部残留溶剂检查法介绍

复方阿米三嗪片(萝巴新)(二甲硫酸阿米三嗪)33≯410≯215萝巴新3≯3≯115盐酸马普替林3≯2≯110盐酸米托蒽醌3≯3≯710盐酸异丙嗪片4 ≯215盐酸异丙嗪注射液4 ≯215盐酸雷尼替丁4 ≯215盐酸雷尼替丁片4 ≯410盐酸雷尼替丁胶囊4 ≯410盐酸雷尼替啶注射液4 ≯610盐酸萘甲唑林3 ≯210柳氮磺吡啶4 ≯410 配制对照品溶液检查已知杂质的品种有盐酸乙胺丁醇、盐酸小檗碱、盐酸左旋咪唑、盐酸吗啡、癸氟奋乃静和氯硝西洋等40余个。
此外配制自身稀释对照溶液和配制对照品溶液,用以检查未知杂质及已知杂质并控制杂质斑点数的品种有盐酸氯米帕明(杂质斑点≯4个)和贝诺酯(杂质斑点≯4个)。
本版药典在薄层色谱法中增加了系统适用性试验和测定法,上述列举的国内外药典的实例可供采用薄层色谱法作为鉴别与有关物质测定的品种正文,在修订或方法研究时参考,可根据品种自身的特点与具体情况,选择系统适用性试验项下的有关要求订入标准中,以使检测方法严谨和完善,确保方法的准确性和重现性,以利于药品质量控制。
《中国药典》2005年版(二部)残留溶剂检查法介绍胡昌勤 刘颖(中国药品生物制品检定所,北京100050)I n troduction of Residua l Solven ts Test i n Ch i nese Pharmacopoe i a2005Ed ition(Volu m e ) H u Changqin and L iu Y ing(N ational Institu te f or the Control of P har m aceu tical and B iolog ical P rod ucts,B eij ing100050) 药品中的残留溶剂系指在原料药、辅料以及制剂生产中使用的,但在工艺过程中未能完全去除的有机挥发性化合物。
I CH(人用药品注册技术要求国际协调会)对残留溶剂的这一定义,明确了药品中残留溶剂的最基本特征,同时也包含了药品残留溶剂的测定具有如下特点:(1)残留溶剂的种类相对固定(I CH规定了69种);(2)在具体样品中具有不确定性;(3)残留量相对较低,一般在痕量或微量范围;(4)同一样品中不同溶剂的残留量相差较大。
《中华人民共和国药典》2005年版设计方案

委 员:丁建弥 于德泉 马大龙 仇士林 尹红章 王 平 王 姮(女) 王 健 王 祥 王小宁 王升启 王宁生 王宁利 王永炎 王永铭 王白露 王军志 王庆国 王佑春 王志清 王依婷(女) 王国治 王国荣 王英华(女) 王峥涛 王春龙 王晓平(女) 王晓良 王鹏富 王慕邹 王憬惺 车庆明 邓丙戌 冯 丽(女) 冯帆生 冯国平 冯国康 卢爱英(女) 叶飞云 尼玛顿珠 甘绍伯 田颂九 田瑞华 白慧良
郑筱萸 金于兰(女) 俞永新 俞如英(女) 姚乃礼 姚守拙 姚达木 赵 明(女) 赵 铠 赵进东 赵维良 钟大放 饶春明 倪道明 凌大奎 唐秋瑾(女) 唐家琪 徐连连(女) 徐建国 徐惠南(女) 徐愚聪 秦少容(女) 翁维良 袁勤生 诸骏仁 贾天柱 钱忠直 钱维清(女) 高学敏 高润霖 屠鹏飞 曹采方(女) 曹雪涛
《中国药典》2005年版整体设计
1.指导思想 坚持与时俱进的精神,广泛吸纳和应用现 代科学技术与研究成果;坚持继承与发展、 理论与实践相结合的方针;坚持科学、实 用、规范的原则;进一步完善我国的药品 标准体系;不断扩展《中国药典》在国际 社会的影响。
《中国药典》2005年版整体设计
2.总体目标 2.1.形成以《中国药典》为主体的国家药品标准体 系,实现《中国药典》一、二、三部的统一与规 范; 2.2.按照中药、化学药、生物制品的特点和实际, 采取扩大收载和更新淘汰并举的措施,积极开展 药品标准检验方法的研究工作,使本版药典从收 载品种到标准水平等方面力争全面提高; 2.3.努力保持《中国药典》的科学性、严肃性和先 进性,使我国药典成为在亚洲地区乃至国际上有 影响的药典之一。
《中国药典》2005年版科研项目 安排落实情况
(二)化学药部分:1088项,其中新增439 个品种,增修订项目涉及220个品种,增订 静脉注射剂不溶性微粒检查151个品种,增 修订细菌内毒素检查179个品种,另有3个 与附录有关的增修订项目。
2005版《中国药典》收载的中成药中石膏制法及用量的统计分析及探讨

云 南 中 医 中 药 杂 志
9 1
2 0 版 《 国药 典 》 载 的 中成 药 中石 膏 制法 及 用量 的 统 计 分 析 及 探 讨 05 中 收
黄 龙 ,范德胜
( 南三金 制 药有 限责任 公 司,湖 南 常德 4 5 0 ) 湖 1 0 1
2 蛤蚧定喘丸 7 2 8 2 9 橘红丸 鹭鸶咯丸
注: 以上统计序号按成方制剂在药典中的顺序排列。
表 2 不 同 剂 型 的 制 法 统 计 表
序号 成方制剂 品种数
石膏制剂方法
9 2
云 南 中 医 中 药 杂 志
21 年第 3 卷第7 00 1 期
昆明 地 区地 理 气 象 因素 与 感 染后 咳 嗽 ( 燥 伤 肺 型 ) 关 性 机 理 探讨 风 相
备 。统计 结 果 见 表 1 表 3 ~ 。
表 1 石膏在处方 中用量及制法统计表
2 清 解 服 5 热 毒日 液 l l 6 4 6 5 7 4 3 膏 , 0 o . 浸 煎煮。 分 和 次, 别为
Um l n
2 6
清眩丸
70 10 1.9 0 0 42 石膏粉碎成细粉, 泛丸即得 6l Z 40 石膏粉碎成细粉, 1 5 .9 泛丸即得 65 5 74 石膏 成细粉, 7 O .1 粉碎 直接人药 57 6 1.8 5 0 07 石膏粉碎成细粉, 直接人药
孔 毖 船 丛
石 膏为纤 维状 的集合 体 , 长 块状 、 呈 板块 状 或 不 规则 块 状 。白色 、 白色或淡黄色 , 的半 透明 。体重 , 灰 有 质软 , 断面 纵
牛 解 片 黄 毒
7 2 2 6 石 加 煎 次, 次2 。 8 0 0 5 4 膏 水 煮2 每 0 . h
对2005年版《临床用药须知》中药卷若干问题的商榷

对2005年版《临床用药须知》中药卷若干问题的商榷
孙桂明;孙世成
【期刊名称】《中国药品标准》
【年(卷),期】2010(011)004
【摘要】@@ 为了推动中成药的临床应用,促进我国中医药学科的发展,国家药典
委员会2005年编写的<临床用药须知>中药卷(以下简称<中药卷>)为第一版指导
临床中、西医合理使用中成药的专业书籍.该书是<中国药典>配套丛书之一.全书共收载1423种药品,每种药品的功能主治、方解、临床应用、药理毒理、不良反应、注意事项、用法用量都比较详细,这对指导我国卫生工作人员准确了解和全面使用<中药卷>中的各类药品,保证临床用药安全有效必将起到重大作用.因初次编写,时间紧、任务重,出现问题和错误也在所难免.
【总页数】3页(P243-245)
【作者】孙桂明;孙世成
【作者单位】威海现代妇科医院,威海,264200;威海市中医院,威海,264200
【正文语种】中文
【中图分类】R921.2
【相关文献】
1.关于《中国药典》2005年版中药材杂质检查项的商榷 [J], 王红;韦云川;韦继雯;李伟
2.对2005年版《临床用药须知》中有关问题的商榷 [J], 曾志海;李小龙
3.对《中国药典》2000年版二部临床用药须知的商榷 [J], 段利生;虎安定
4.2005年版《中国药典》(一部)中有关中药颗粒剂收录现状及存在问题商榷 [J], 周志祥
5.《2005年版中国药典临床用药须知》必须做皮试的药物 [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
2005年版《中国药典》(一部)中有关中药颗粒剂收录现状及存在问题商榷

2005年版《中国药典》(一部)中有关中药颗粒剂收录现状及
存在问题商榷
周志祥
【期刊名称】《中国药房》
【年(卷),期】2006(17)13
【摘要】目的:评价 2005年版<中国药典>(一部)中颗粒剂的收录现状与规范性问题.方法:对 2005年版<中国药典>(一部)中全部颗粒剂品种从制备方法、质量控制等方面进行分析.结果:制备工艺兼顾传统工艺与现代制备技术,现代分析技术在中药颗粒剂中得到了进一步的扩大应用;浓缩清膏的温度与比重没有统一规范,不同品种相同成分含量测定要求及限度指标等差异较大.结论: 2005版<中国药典>(一部)颗粒剂质量控制存在不合理的情况,需进一步完善.
【总页数】2页(P1033-1034)
【作者】周志祥
【作者单位】湖北红安县中医院,红安县,438400
【正文语种】中文
【中图分类】R921.2;R944.2+7
【相关文献】
1.《中国药典》2005年版一部表格形式商榷 [J], 杨丽平;朱嘉;卢海生
2.对2005年版《中国药典》一部阿胶鉴别方法的商榷 [J], 窦琦云;丁泽明
3.《中国药典》2005年版(一部)中含雄黄、朱砂制剂儿童用量商榷 [J], 邵家德;
邵乾
4.《中国药典》2005年版(一部)中含雄黄、朱砂制剂儿童用量商榷 [J], 邵家德; 邵乾
5.关于《中国药典》2005年版一部中槐花质量标准〔性状〕项商榷 [J], 王先教因版权原因,仅展示原文概要,查看原文内容请购买。
关于《中国药典》2005年版二部阿昔洛韦含量测定项下系统适用性试验溶液配制的商榷

关于《中国药典》2005年版二部阿昔洛韦含量测定项下系统
适用性试验溶液配制的商榷
杨兴明;黄慧芬
【期刊名称】《中国药品标准》
【年(卷),期】2008(9)4
【摘要】阿昔洛韦原料是《中国药典》2005年版二部收载的一种抗病毒药,鸟嘌呤是它的一种降解杂质。
其标准中含量测定项下系统适用性试验规定:取阿昔洛韦对照品溶液5mL,加入鸟嘌呤对照品储备溶液(取鸟嘌呤对照品10mg,置
100mL量瓶中,加0.4%的氢氧化钠溶液适量使溶解,并稀释至刻度,摇匀,即得)1mL,摇匀,取20“L注入液相色谱仪,记录色谱图,阿昔洛韦峰与鸟嘌呤峰分离度应符合规定。
实验中发现,按药典方法系统适用性溶液在不同的色谱柱得到的结果不同,专属性不强,所以笔者觉得系统适用性溶液的配制值得商榷。
【总页数】2页(P243-244)
【作者】杨兴明;黄慧芬
【作者单位】武汉市药品检验所,武汉430012;武汉市药品检验所,武汉430012【正文语种】中文
【中图分类】R921.2
【相关文献】
1.《中国药典》2000年版二部中有关颗粒剂项下值得商榷的问题 [J], 邵建强
2.关于《中国药典》2010年版盐酸组氨酸及谷氨酸中其他氨基酸项下系统适用性
要求的商榷 [J], 程速远;张慧;梁成罡
3.对《中国药典》2005年版二部地塞米松磷酸钠注射液含量测定方法的商榷 [J], 郑琰;刘淑华;李荣华
4.《中国药典》2005年版(二部)薄层色谱法系统适用性试验介绍 [J], 王祥
5.关于《中华人民共和国药典》2010年版二部氯霉素含量测定项下色谱条件的商榷 [J], 苗爱东;阚秀燕;杨静;李妍
因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药典临床用药须知 中药卷 ( 2005年版 ) 共 收载中成药品种 1 423个, 含十八反、十九畏药对的 成方制剂共 18个, 约占收载品种总数的 1. 26% , 其 中含十八反药对的成方 9个, 含十九畏药对的成方 9个。含 1组以上十八反药对 的成方有 3个, 均为 乌头类反药药对配伍使用。结果见表 1、表 2。
序号 N o.
6 7 8 9
成方制剂 Formu las
阳和解凝膏 ! ! 少林风湿跌打膏 消核片 ! ! ! ! 内消瘰疠丸 ! !
所含反药药对 Coup let m edicinal of incom patib lem edicinal 生川乌、生草乌、生附子、白蔹、白及 生川乌、生草乌、白蔹、白及 海藻、甘草 海藻、甘草
北京中医药大学学报 ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! 第 34卷
K ey words: Instructions of China Pharm acopoeia A pp lication; form ulas of Chinese m ed icina;l e ighteen incom patible m ed icina;l nineteen m ed icina l o f mu tua l antagon ism
表 1! 含十八反 药对的成方制剂收载情况 T ab le 1! Records of formu las contain in g coup let m edic inal of eigh teen incompat ib le m edic inal
序号 N o.
1 2 3 4 5
成方制剂 Form ulas
中华人民共和国药典 ( 一部 ) 中收载的中药配伍 禁忌 品种, 确定 十 八反、十九 畏 药对 配伍 的 查询 范围。
其中乌头包括川乌、草乌、附子及其炮制品; 半 夏包括法半夏、清半夏、姜半夏、竹沥半夏; 瓜蒌包括 瓜蒌皮、瓜蒌仁、全瓜蒌、天花粉; 贝母不分川贝母、 浙贝母、平贝母、湖北贝母、伊贝母; 白及包括不同写 法, 如白芨; 大戟包括京大戟、红大戟; 甘遂包括生甘 遂、制甘遂; 甘草包括生甘草、炙甘草; 诸参指人参 ( 包括山参、生晒参、生晒山参、园参、红参、参 须 ) 、 玄参、丹参、苦参、沙参 ( 包括南沙参、北沙参 ) ; 芍药 不分白芍、赤芍; 朴硝包括芒硝、玄明粉; 砒霜包括砒 石; 巴豆包括巴豆霜; 牙硝包括马牙硝、芒硝、玄明 粉; 官桂包括肉桂。
WANG X ,i ZHONG Gan sheng#, L IU Jia, OU L i na, CH EN Shao hong ( Schoo l of PreclinicalM edic ine, B eijing U niversity of Ch inese M edic ine, B eijing! 100029)
根据 珍珠囊补遗药性赋 [ 藻戟遂芫俱战草, 诸参辛芍叛藜芦。#以及十九畏歌诀 ∀ 硫黄原是火中 精, 朴硝一见便相争; 水银莫与砒霜见; 狼毒最怕密 陀僧; 巴豆性烈最为上, 偏与牵牛不顺情; 丁香莫与 郁金见; 牙硝难合京三棱; 川乌、草乌不顺犀; 人参最 怕五灵脂; 官桂善能调冷气, 若逢石脂便相欺; 大凡 修合看顺 逆, 炮爁炙 煿莫 相依 #。 结合 2010 年版
双虎肿痛宁 ! 参茸黑锡丸 ! 骨增生镇痛膏 安阳精制膏 ! 筋痛消酊 ! !
所含反药药对 Coup let m ed icinal of incom patib lem edicinal 生川乌、生草乌、半夏 ! ! ! 制附子、制半夏 ! ! ! ! ! 生川乌、生草乌、半夏 ! ! ! 生川乌、生草乌、白蔹、白及 制川乌、制草乌、浙贝母 ! !
Formulas containing couplet m edicinal of incompatible medicinal recorded in Instructions of China Pharmacopoeia App lication Chinese m ed icine Volum e (Edition 2005 )
金匮要略 甘遂半夏汤中甘遂与甘草同用, 附子粳 米汤中附子与半夏同用; 明代陈实功 外科正宗 海 藻玉壶汤、清代 医宗金鉴 消核散, 均以海藻与甘 草同用等。反药能否同用, 在历史上一直存在争议, 探讨反药配伍禁忌的实质, 在现代中医药研究领域 中已成为亟待解决的问题之一。
中华人民共和国药典临床用药须知 [ 3 ] (以下 简称 药典临床用药须知 ) 是 中华人民共和国药 典 (以下简称 中国药典 )的配套丛书之一, 由国家 药典委员会组织全国各学科的权威医药学专家, 根据 临床用药经验并结合国内外公认的资料编写而成, 有 助于指导我国医药工作人员准确了解和合理使用 中 国药典 中各类药品, 保证临床用药安全、有效。 药 典临床用药须知 分 ∀ 化学药和生物制品卷 #以及 ∀ 中 药卷 #2卷, 其中 药典临床用药须知 中药卷 ( 2005 年版 )是指导临床中、西医合理使用中成药的专业书 籍, 它不仅是临床医生的案头必备书, 也是中医药教 学、科研、中成药生产与管理工作不可或缺的参考书, 还是药品监管部门实施药品监督管理工作的重要依 据。因此现就 药典临床用药须知 中药卷 ( 2005 年版 )中涉及到反药配伍的中成药品种作一系统分 析, 期望对广大临床医师安全合理使用反药配伍提供 借鉴, 并为探讨反药的配伍本质提供研究思路。 1! 检索条件
第 V
34 o.l
卷第 1期 34 N o. 1
2011年 1月 Jan. 2011
!
北京中医药大学学报 ! Journa l of B eijing U niversity of T raditiona lCh inese M edic ine
27
药典临床用药须知 中药卷 ( 2005年版 )中含 反药药对成方制剂收载情况与分析*
中药配伍禁忌理论最早出现于 神农本草经 , 但此时并未记载药物配伍禁忌的具体内容。据文献 考证 [ 1- 2] , 北齐徐之才 药对 是最早著录具体药物 配伍禁忌的古 籍。后来随着 药物和方剂研 究的盛 行, 历代医家开始重视药物的配伍禁忌。虽然药物 配伍禁忌令临床医生对反药配伍使用望而却步, 但 古今使用反药配伍的例子屡见不鲜。如东汉张仲景
! 王茜, 女, 在读博士生 # 通信作者: 钟赣生, 男, 硕士, 教授, 博士生导师, 主要研究方向: 中药配伍禁忌本质的研究, E m ai:l zhonggansheng@ sohu. com * 国家重点基础研究发展计划 ( 973计划 ) 项目 ( N o. 2011CB505306)
28
A bstract: T he concepts of e ighteen incom pat ib le m ed icinal and nineteen m edic inal o f mu tual antagonism are the synonym for proh ibited com binat ion of Chinese m edic ina,l w h ich have been believed by physic ians o f all tim es. There w ere, how ever, literature and c lin ical reco rds abou t contrary m ed icinal used together in form ula designing, and the essentia l o f com binat ion of contrary m ed icina l has been a focus study. Instructions of China Pharm acop oeia App lication is one of assorted series of Ch ina N ational Pharm acopoeia and has be ing indispensable reference for Ch inese m ed ica l w orkers in reasonab le adm inistrat ion o f a ll types of m ed icina,l wh ich records the form u las containing coup let m ed icinal of incom patible m ed icina.l Instructions of Ch ina Pharm acop oeia App lication Chinese m ed icine V olum e (Ed ition 2005 ) records tota lly 1 423 paten t form ulas, am ong them there are 18 form u las conta in ing coup let m ed icina l o f incom pat ib le m edicina l including 9 contain ing coup let m edicina l of e ighteen incom patible m edic inal and 9 containing couplet m ed icina l o f nineteen m edic inal of m utua l antagon ism. T he frequency of couplet m edic inal o f contrary m ed icina l in above m entioned form ulas w as ana lysed, and the functions, dosage form s, usages and ingredients o f these form u las w ere summ arized in the paper a im ing at study ing the usage law s o f contrary m ed icinal com binat ion in clin ica l pract ice and gu id ing reasonable applica tion of e ighteen incom patible m edic inal and n ineteen m edicina l o f m utua l an tagon ism.