克氏综合征诊疗新进展
克氏综合征患者性激素水平分析

合 征 (Ks)患 者 的 应 用价 值 。方 法 染 色体 核 型 分 析 KS患 者 22例 为 实 验 组 ,正 常核 型 男 性 不 育 患者 2O例 为 对 照 组 。所 有 患 者
均为 雄 性 激 素 治 疗前 进 行 精 液 分 析及 血 清 性 激 素 水 平 检 测 。 结 果 实 验 组 FSH 和 LH 两 项 指 标 同 时 增 高 占 100 ,对 照 组 占
lating horm one (FSH )and luteinizing horm one (I H ) for rapid screening m ethod to diagnose Klinefelter syndrom e (K S).M ethods 42 infertile m ales were studied by retrospective analysis.There w ere 22 out of them with KS as the experim ental group and 20 with the m ale infertile of 46,XY karyotype as control group.Before the treatm ent of androgen horm one the sem en sam ples w ere analyzed according to the current W orld Health Organization Laboratory M anual(W orld Health Organization,1999)and the sex hormones
2330
· 临 床 研 究 ·
重庆 医学 2O1O年 9月 第 39卷 第 17期
2024CAH-X综合征研究进展(全文)

CAH-X综合征研究进展(全文)摘要CAH-X综合征是指先天性肾上腺皮质增生症(CAH)患者中,合并肌腱蛋白X(TNX)缺陷而出现埃勒斯-当洛综合征表型的特殊亚群,占CAH 患者的10%~15%。
TNX缺陷可导致一系列结缔组织症状,包括全身性关节活动过度、皮肤过度伸展、反复关节脱位、慢性疼痛、心脏缺陷等,严重影响患者生存质量。
CAH-X 综合征的遗传学病因是CYP21A2和TNXB基因的连续性缺陷,由于致病基因的复杂性,其分子诊断充满挑战。
现对CAH-X综合征研究进展进行综述,以提高临床医师对于这一新发现疾病的认识。
关键词先天性肾上腺皮质增生症;CAH-X综合征;埃勒斯-当洛综合征;TNXB 基因先天性肾上腺皮质增生症(congenital adrenocortical hyperplasia,CAH)是一类肾上腺类固醇合成酶缺乏的常染色体隐性遗传病,由CYP21A2基因缺陷所致的21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)是CAH中占比约95%的主要类型,以肾上腺皮质功能不全和高雄激素血症为临床特征[1]。
埃勒斯-当洛综合征(Ehlers-Danlos syndrome,EDS)是一组异质性遗传性结缔组织病,以全身性关节活动过度、皮肤过度伸展和组织脆性为特点[2]。
一部分EDS是由于肌腱蛋白X(tenascin-X,TNX)缺陷所致,编码TNX的TNXB 基因与编码21-羟化酶的CYP21A2基因紧密连锁。
现已发现一种CYP21A2及TNXB基因的连续性缺陷,可同时引起21-OHD和EDS表型,称为CAH-X综合征。
自该综合征于2013年被命名以来,全球范围内累计报道的患者数量已近200例,但鲜有中文文献报道。
近年研究显示,CAH-X综合征在CAH患者中占比10%~15%[3-8]。
为提高临床医师对于这一新发现疾病的认识,现就CAH-X综合征研究进展进行综述。
克氏综合征是什么病

克氏综合征是什么病很多人对克氏综合症并不是特别的了解,这是一种遗传疾病,是因为性感色体异常而导致患者睾丸功能衰减引起的病变,患者常常会出现第二性征异常,会引起生精障碍,导致性功能减退,甚至会引起智力低下,得了这种疾病,早期治疗意义非凡,通过早期的性激素方面的治疗,能够达到很好的治疗效果,当然这种病症本身就会终身不育,即使用药也无法生育,但是通过一定的药物治疗,能够促进第二性征的发育,还能够改善患者的性功能。
★1、第二性征发育不良病人在出生时和儿童期与正常人没有什么差别,只是到了青春期,才显露出一些病态,外观是男性,但睾丸小,阴茎可有一定程度的发育,但也较正常人小。
第二性征也有不同程度的发育,有的有少许阴毛及胡须或无,喉结小或无,发音尖或女性声音。
★2、“女性化”特征身材较高大(同其兄长高),骨骼较细,四肢相对较长,测量身体下部从耻骨到足底得出较长于身体上部的距离。
皮肤较白,因皮下脂肪丰富,故皮肤好如女性,臀部较为宽大也宛如女性,大约一半的患者乳腺呈“女性化”特征,部分病人有男性乳房增生。
★3、不能生育如结婚,有的无性交能力,有的性功能差,多数患者阴茎能勃起,也可射精,但精液中无精子或精子量极少,因故称无精症,占97%大多数患者不能生育。
化验检查卵泡刺激素明显增高,黄体生成素偏高或增高,睾酮偏低。
★4、1/4患者智力发育迟缓此病患者性格和行为也有异常表现,约1/4患者智力发育迟缓,胆怯,生活意志被动以及薄弱,有依赖性,感情不稳定,情绪多变,性格和行为有个体差异不同程度的表现为女性化倾向。
患者男性第二性征发育差,有女性化表现,如无胡须,体毛少,阴毛分布如女性,阴茎龟头小等,约25%的患者有乳房发育。
★5、本病患者的睾丸小而硬组织学检查可见睾丸曲细精管纤维化和透明样变,管腔闭塞,无精子发生,间质细胞增生或聚集成团,且功能低下,睾酮生成减慢,血睾酮浓度低,对外源性促性腺激素(hMG)刺激反应低,而患者的血浆及尿中黄体生成素及促卵泡激素升高,黄体生成素分泌多,将刺激睾丸间质细胞,使雌二醇增高,雌二醇/睾酮比值上升,从而使病人的乳房发育呈女性型乳房。
克氏综合征诊断与治疗进展

!造庄凼塾盘壶垫塑生兰旦筮堑鲞筮三翅』g丛垫!!望丛型:丛坐h至业:!竺!:堑:盟Q:兰克氏综合征诊断与治疗进展李江源[中图分类号]R442.8[文献标识码]Adoi:10.3969/j.issn.1001-9057.2009.03.006[关键词】克氏综合征克氏综合征为HarryKlinefeher于1942年首先报告,后来发现这些患者的染色体核型为47,XXY。
20世纪70年代大规模的新生儿细胞遗传学研究确定47,XXY核型的发病率为1/500新生儿,在男性群体中的患病率为1/600。
克氏综合征患者约10%进行了产前细胞遗传学检杏,在出生前明确诊断;约25%的患者在青春期或成年期诊断,约75%的患者在他们的一生中始终没有得到诊断【1J。
在一些特殊人群[如精神病、行为异常和(或)学习困难]中,47,XXY核型的发病率比普通人群高4—6倍。
一、致病原因产生47,XXY核型的原因是配子在减数分裂或合子在有丝分裂时发生了错误,出现性染色体不分离的XY或XX配子,它们与正常的x或Y配子结合即产生非整倍体合子。
在47,XXY核型患者中,精子第一次减数分裂性染色体不分离的几率为53%,卵子不分离的几率为34%,卵子在第二次减数分裂不分离占9%,合子有丝分裂错误占3%。
早年的研究已证明,卵子分裂错误与母亲年龄有关,卵f第一次减数分裂错误母亲的平均年龄为32岁,第■次减数分裂错误母亲的平均年龄为27岁。
当时没有发现精子分裂错误作者单位:100853北京,解放军总医院内分泌科・161・・综述与讲座・与父亲年龄有关。
近年来应用更先进的荧光原位杂交(FISH)方法分析精子,发现每10000个精子中有1—10个异常性染色体精子。
有人研究17例克氏综合征患者的父亲,年龄33~54岁,XY二体型精子的百分率与父亲年龄直线相关‘2】。
二、临床表现1.不同发育阶段的表现:婴儿期可能有出生体重低,头围小,阴茎小和隐睾,发牛隐睾的几率比正常新生儿高3倍。
克氏综合征认识新进展

67克氏综合征(Klinefelter Syndrome )的主要核型是47,XXY ,发病率约为150/10万,是男性中最常见的性染色体异常疾病。
克氏征自1942年被首次报道以来,随着对其认识的不断深入,其伴随的一系列临床特征陆续被认知和描述,涉及遗传学、流行病学、儿科学、内分泌学、心血管病学、精神病学、泌尿科学等学科。
一、遗传学克氏征表型的遗传学机制主要存在额外的性染色体。
这种遗传异常也见于家养和野生动物[1]。
有研究报道其遗传表型可能与存在于X 染色体上未被灭活的额外基因有关。
在这些基因中,唯一被阐明影响克氏征表型的是位于拟常染色体(pseudoautosomal )p1区的矮生高同源框基因(SHOX )。
SHOX 能促进患者骨骼生长[2]。
SHOX 的转录目标是脑利钠肽和成纤维细胞生长因子受体[3, 4],这将有助于我们对克氏征临床表型的理解。
雄激素受体中CGA 基因的重复数量也与克氏征表型有关[5],如影响患者身高、血细胞比容等。
然而来源于父母的额外X 染色体是否会影响克氏征表型值得进一步研究,因为在X 染色体中有超过10%的基因在睾丸中表达[6]。
二、流行病学(一)患病率最初,克氏征被认为“极为罕见”。
直到在新生儿中大规模的染色体分析后,“真的”患病率才确定。
克氏征的患病率在增长,而且在不同的人群中患病率也可能存在差异[7]。
丹麦学者通过产前诊断发现新生男婴的患病率为152/10万[8];格鲁吉亚在36124例新生婴儿干燥血清DNA 的筛查中患病率为158/10万[9];澳大利亚最近报道的患病率为223/10万[10];亚洲来自韩国Jo 等一项回顾性研究显示患病率为22/9387[11],而我国尚未见相关报道。
(二)诊断率克氏征的诊断严重滞后。
丹麦通过调查发现只有约25%患者被诊断,而这些病例中又仅有10%的患者是在青春期前确诊[8]。
来自英国的同类研究也报道了相似的诊断率,大约为每年100/525[12]。
《克氏综合症简介及其家庭指南》_已获美国授权共享

克兰费尔特综合症简介:XXY男性及其家庭指南作者:罗伯特博克美国国立卫生研究院全国儿童健康与人类发展部研究报告原稿:1993年8月最后更新:2000年8月15日目录什么是克兰费尔特综合症--------------------------------------------------------------------------3染色体和克氏综合症------------------------------------------------------------------------------------3原因--------------------------------------------------------------------------------------------------------4诊断--------------------------------------------------------------------------------------------------------4如何告诉家人、朋友和XXY男生-----------------------------------------------------------------5童年--------------------------------------------------------------------------------------------------------6语言问题的早期检测-------------------------------------------------------------------------------------6检测语言问题的指导方针---------------------------------------------------------------------------7课堂上的XXY男孩-------------------------------------------------------------------------------------7寻求法律帮助-----------------------------------------------------------------------------------------------7教学技巧-----------------------------------------------------------------------------------------------8青春期---------------------------------------------------------------------------------------------------------9睾酮治疗-----------------------------------------------------------------------------------------------10性染色体变异-----------------------------------------------------------------------------------------------10性倾向-----------------------------------------------------------------------------------------------11不育---------------------------------------------------------------------------------------------------------11健康的思考-----------------------------------------------------------------------------------------------12成年---------------------------------------------------------------------------------------------------------12有关机构-----------------------------------------------------------------------------------------------13什么是克兰费尔特综合症1942年,波士顿麻省总医院的哈里克氏博士及其同事发表了一篇有关9个男性的一些特殊临床症状的报告。
克氏综合症的诊治新进展

克氏综合症的诊治新进展摘要】克氏综合征是人类常见的性染色体疾病,也是男子无精症最常见的病因,以“小而硬的睾丸、不育和雄激素缺乏状态”为临床特征。
在青春期严重影响身体和生殖系统发育,既往认为是不能生育的疾病,随着显微外科技术在男科的应用,部分患者可在睾丸中获得精子,联合卵胞浆内单精子注射技术可以获得生育能力。
【关键词】克氏综合征;无精症;micro-TESE;卵胞浆内单精子注射【中图分类号】R698+.2 【文献标识码】A 【文章编号】1007-8231(2015)16-0003-02克莱恩费尔特综合症(Klinefelter syndrome,KS),简称克氏综合征,最早在1942年由Klinefelter提出,是人类常见的性染色体疾病,也是男性无精子症最常见的病因[1]。
其在男性人群的发病率为0.1%~0.2%,其中只有1/4的病例被诊断,约80%的病例为染色体数目畸变47,XXY,剩余20%包括46,XY/47,XXY的嵌合体,更多的X染色体(如48,XXXY;49,XXXXY)。
KS患者可出现青春期性征发育异常、身体机构异常、智力低下等问题,同时又因为睾丸发育障碍导致无精症发生,对其生育权提出了挑战。
如何更好的促进青春期身体发育,同时获得生育能力,目前在男科界是比较有争议的话题,该文就以上问题作一综述。
1.遗传病理与其他性染色体数目异常综合征一样,生殖细胞减数分裂过程中染色体不分离是克氏综合征的遗传病理,导致性染色体二体生殖子产生。
额外的X染色体导致精子细胞发育障碍,位于X染色体上逃避X失活的基因剂量效应可能是遗传病理之一。
染色体不分离可以发生在卵原细胞,也可以发生在精原细胞,各约占50%,精原细胞的染色体不分离只限于减数分裂I期,生成XY生殖子后与正常卵子受精得到XXY合子,约3/4的卵细胞染色体不分离发生在MⅠ期,1/4的卵细胞不分离发生在MⅡ期。
2.临床表现Klinefelter综合征常以“小而硬的睾丸、不育和雄激素缺乏状态”为临床特征。
克氏综合症是什么病,有哪些症状?

克氏综合症是什么病,有哪些症状?很多人对克氏综合症并不了解,其实它是一种先天性的疾病,很多时候是因为染色体异常所引起的,所以我们需要通过克氏综合征患者,所表现出的一些症状去判断这种疾病,只有及时的了解发现,才能够及时进行干预和治疗,比如最常见的症状是病人的乳房发育成女性型的乳房,还有外观虽然是男性,但是睾丸比较小,而且有一些患者有男性乳房增生,阴茎能勃起,可以射精,但是精液中无精子等等这些情况,而这些症状是需要通过全面检查去了解的。
★1、病人的乳房发育呈女性型乳房本病患者的睾丸小而硬,组织学检查可见睾丸曲细精管纤维化和透明样变,管腔闭塞,无精子发生,间质细胞增生或聚集成团,且功能低下,睾酮生成减慢,血睾酮浓度低,对外源性促性腺激素(hMG)刺激反应低,而患者的血浆及尿中黄体生成素及促卵泡激素升高,黄体生成素分泌多,将刺激睾丸间质细胞,使雌二醇增高,雌二醇/睾酮比值上升,从而使病人的乳房发育呈女性型乳房。
★2、外观是男性,但睾丸小病人在出生时和儿童期与正常人没有什么差别,只是到了青春期,才显露出一些病态,外观是男性,但睾丸小,阴茎可有一定程度的发育,但也较正常人小。
第二性征也有不同程度的发育,有的有少许阴毛及胡须或无,喉结小或无,发音尖或女性声音。
身材较高大(同其兄长高),骨骼较细,四肢相对较长,测量身体下部从耻骨到足底得出较长于身体上部的距离。
★3、皮肤较白,因皮下脂肪丰富皮肤较白,因皮下脂肪丰富,故皮肤好如女性,臀部较为宽大也宛如女性,大约一半的患者乳腺呈"女性化"特征,部分病人有男性乳房增生。
如结婚,有的无性交能力,有的性功能差,多数患者阴茎能勃起,也可射精,但精液中无精子或精子量极少,因故称无精症,占97%大多数患者不能生育。
化验检查卵泡刺激素明显增高,黄体生成素偏高或增高,睾酮偏低。
此病患者性格和行为也有异常表现,约1/4患者智力发育迟缓,胆怯,生活意志被动以及薄弱,有依赖性,感情不稳定,情绪多变,性格和行为有个体差异不同程度的表现为女性化倾向。
克氏综合症信息指南与深入研究

The KS StoryKlinefelter’s SyndromeAN INFORMATIVE GUIDE & FURTHER STUDYCompiler: Iain W McKinlay 47,XXYForeword by: Prof Milton Diamond Ph.D.本书是一种常见的遗传性疾病克氏综合征的介绍,为克氏综合征患者、家长、家属、伴侣和普通公众提供信息,同时为医疗健康等专业人士提供参考资料。
KS故事克氏综合征信息指南和深入研究瓷娃娃罕见病关爱中心中国克氏互助会2013年4月16日本书由瓷娃娃罕见病翻译志愿者刘宇超、赵丽伶、周末、石大雨、田蕊、黄栩芾、彭洋、张新明、寒江、孟晶晶翻译,孟晶晶校对、统筹,感谢他们的辛勤和卓越工作,希望本书对中国的克氏综合征病友群体和希望了解该疾病的人士有所裨益。
P1:KS故事你并不孤单信息指南和深入研究KS故事是克氏综合症(KS,细精管发育不全综合征)12年研究进展巅峰的产物,同时也体现了研究工作中的情感历程。
本书的初衷是作为支持KS研究的团体,医学专家以及研究小组的一种沟通渠道,并有重要章节专门介绍深层次的研究,其中涵盖了广泛的支持演说,网络部分,相关精选书籍,研究论文,医学期刊和科学杂志。
除此之外,这本书还涉及到KS的其它许多方面,给我们展示了KS对患病个体以及关爱他们的人的影响。
KS故事的编者是一个本身患有克氏综合征(47.xxy型)的患者,他受到英国乃至世界上各个领域的科学家们的帮助和支持,包括生物学,细胞遗传学,内分泌学,神经学,性学。
为了平衡大量枯燥的数据,书中穿插了一些有趣的连环画和卡通图作为弥补。
唯一对读者的要求就是请您丢弃固有的思维定势,用一种积极开放的心态来阅读。
KS日记与KS共存敏感,恐惧,力量,触摸,原谅,爱…….计划的第二本书被命名为KS日记,它将向读者呈现那些每天都在与克氏综合征战斗的患者们的童年,青年和成年后的生活经历,了解他们的生活。
Mounier-Kuhn综合征病例报告1例
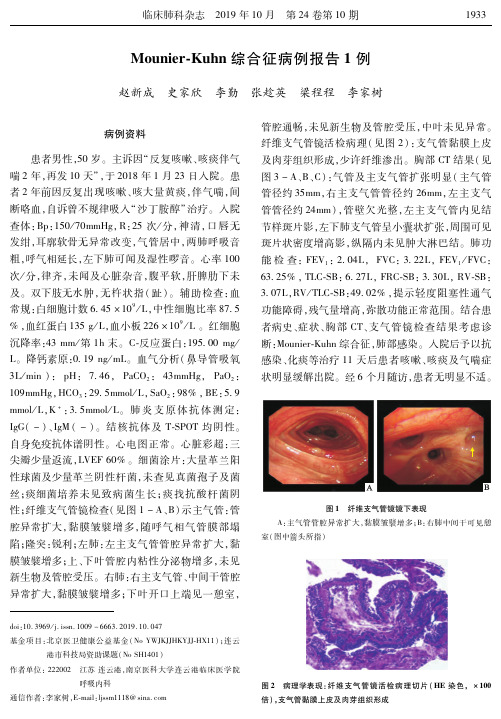
图 2 病理学表现:纤维支气管镜活检病理切片(HE染色, ×100 倍),支气管黏膜上皮及肉芽组织形成
1934ห้องสมุดไป่ตู้
临床肺科杂志 2019年 10月 第 24卷第 10期
图 3 CT影像学表现 A:胸部 CT冠状位示气管及左、右主支气管扩张,憩室形成;B:胸部 CT见主气管扩张明显,最大直径 3462mm;C:胸部 CT见左、右支气管 扩张明显,最大直径分别为 3405mm、2808mm
图 1 纤维支气管镜镜下表现 A:主气管管腔异常扩大,黏膜皱襞增多;B:右肺中间干可见憩 室(图中箭头所指)
doi:10.3969/j.issn.1009-6663.2019.10.047 基 金 项 目:北 京 医 卫 健 康 公 益 基 金 (NoYWJKJJHKYJJHX11);连 云
港市科技局资助课题(NoSH1401) 作者单位:222002 江苏 连云港,南京医科大学连云港临床医学院
讨 论
MounierKuhn综合征(MounierKuhnsyndrome, MKS)又 称 巨 气 管 支 气 管 症 (Tracheobronchomega ly),是一种罕见的病因未明的慢性气道疾病。张艺 等[1]初 步 估 算 MKS的 发 病 率 在 05% ~16%, MKS的男女性别比为 85:1,起病年龄中位数为 30 ~50岁。MKS的组织病理学特征[2]是气管、主支气 管的弹性纤维和平滑肌层变薄、萎缩,引起管腔特征 性的扩大 以 及 呼 气 末 期 气 管 过 度 塌 陷。 Krustins[3] 通过系统地回顾过去 25年发表的 128个 MKS病例 获得如下临床特征:支气管扩张,气管憩室病和气管 支气管运动障碍是普遍存在的合并症(分别占比为 492%,336%和 289%);咳嗽,呼吸困难和反复 呼吸道感染(分别占比为 711%,516%和 508%) 是最常见的主诉。值得注意的是本文报道的该患者 2年前即有反复咳嗽、咳痰及气喘等症状,曾于当地 基层医院诊断为 “哮喘”,不规律予以 “沙丁胺醇” 吸入治疗,未及时行胸部 CT、气管镜、肺功能等检查 明确诊断,导致患者延迟至今才得以确诊。 Himalstein和 Gallagher[4]通过对 100例男性患 者进行尸检,获取了气管和主支气管的直径数据值, 基于这项研究结果,若成人气管、右主支气管和左主 支气管的直径分别超过 正 常 直 径 上 限 值 (分 别 为 30mm、24mm和 23mm)即有确诊意义。MKS的胸部 CT特征性表现及诊断标准[5]为:男性患者:气管横 径 >25mm,气管矢状径 >27mm,右主支气管直径 > 211mm,左主支气管直径 >184mm;女性患者:气 管横径 >21mm,气管矢状径 >23mm,右主支气管直 径 >198mm,左主支气管直径 >174mm。MKS可
克氏综合征

苏 杭
KS定义及流行病学
克氏综合征(klinefelter syndrome,KS)由 HarryF. Klinefelter于1942年首次描述,又称 为原发性小睾丸症或生精小管发育不良,是男 性中最常见的性染色体异常疾病。 据统计,KS在新生儿中发病率约为1/500~ 1/800。KS在男性不育患者中约占3%,在无 精子症患者中约占13%。
儿童期的克氏患儿与正常同龄儿相比,除了精原细胞数量 减少外,睾酮、FSH、LH、E2、AMH等激素水平无明显 差异;
睾丸的主要组织学变化发生在青春期,如曲细精管纤维化 和透明样变,管腔闭塞,无/少精子发生,支持细胞变性、 间质细胞增生等。
KS 与认知心理
近年对KS 患者的认知及心理方面研究较多。KS 患者 临床表型多样,但其在认知方面仍存在一些特征性表 现: 智力水平处于平均及以下,与适应、规划及反应抑制 等相关的执行能力较差,患者的语言表达能力最易受 到严重影响,包括早期的语言发展迟滞、音标及语法 学习困难及拼写阅读能力低下等。 与同等教育水平的对照组相比,KS 患者的视觉及操 作能力未受明显影响。 一项丹麦的研究表明与一般人群相比,克氏患者因精 神障碍入院治疗的几率是一般人群的6.5 倍,病因包 括抑郁、焦虑、精神分裂、自闭症、多动症、注意力 涣散等。
KS 产前筛查
KS 被确诊的三个主要时期是出生前、儿童期和青春 期,不同时期患者均需行染色体核型分析 (karyotyping)确诊。 早孕期胎儿颈项透明层厚度增加、孕妇血清中游离 β⁃HCG 及妊娠相关血浆蛋白A(PAPP⁃A)水平升高 以及中孕期孕妇血清α⁃甲胎蛋白(AFP)、HCG 及 游离雌三醇(uE3)等水平升高并非诊断KS 的特异 指标,KS 的产前诊断需要行羊水穿刺进行胎儿染色 体检查。 是否对所有胎儿进行KS 染色体筛查存在伦理争议, 如美国、瑞士、以色列及加拿大的文献报道指出产前 筛查出克氏胎儿孕妇的妊娠终止率> 70%。
邢台地区89例克氏征实验室诊断论文

邢台地区89例克氏征的实验室诊断与分析[摘要] 目的了解kiinefelter综合征的病因,预后和防治。
方法对247例无精症患者进行精液分析、生殖激素测定、染色体检查、睾丸活检。
结果染色体数目增加89例。
结论克林非氏综合症因其损害的不可逆性给治疗带来困难,应积极预防和早期治疗。
[关键词] kiinefelter综合征;病因;防治[中图分类号] r730.43[文献标识码] b[文章编号] 1005-0515(2011)-07-009-01the laboratory diagnose that 89 example kiinefelter collect synthetically analyseshu yizhen,zheng bo,guan lijun, zhou junxia, zhao lingling ( the xingtai dysgenesia dysgenesisprofessional training hospital of hebeixingtai,hebei054009)[abstract] objective to study the kiinefelter syndrome the cause of disease, the prognosis and the prevention. methods carries on the seminal fluid analysis, the reproduction hormone determination, the chromosome inspection, the testicle biopsy to247example not essence sickness patient. results chromosome number picks out89 examples. conclusion kiinefelter syndrome its harm’s irreversibility brings the difficulty to the treatment, should prevent positively withthe early time treats.[keywords] kiinefelter syndrome;cause ofdisease;prevents and controlskiinefeiter综合征是一种先天性疾病,是染色体异常引起的。
非嵌合Klinefelter综合征患者年龄、血清生殖激素与精液量关系研究

非嵌合 克氏症患者精液量下 降支 持克 氏症患者雄激 素缺乏理论 。
K ie h r 合 征 , l f e综 ne 又称 克 氏症 , 一 种 常 见 性 是 染色 体 数 目异 常 引 起 的 多 种 表 型 异 常 综 合 征 ] 。
利 用有 刻度 的锥 形 量筒 测定 。
123 染 色 体检 查 ..
摘 要: 目的 探讨非嵌 合克氏症患者 年龄 、 血清生殖 激素与精液量 的关系 。方法 分析 7 3例非嵌 合克 氏症 患
者标本 , 进行常规精液 分析 , 采用放射 免疫分析法检 测血清生 殖激素 。结果 非嵌合 克 氏症 患者精 液量 降低 ( P<
0 O ) 年龄与精液量呈 显著负相 关 ( =一0 3 7 P< .5 , 液量 与 F H、 H、 .1, r .8 , 0 0 ) 精 S L T之间无 相关性 ( P>0 0 ) 而 与 .5 , L/ H T比率之 间呈显著负相关 ( =一0 3 , r .3 P<0 0 ) .5 。结论
小 于 5 。血 清 生殖 激 素参 考 值 :S . % F H0 8~1 i/ 5ml d
mlL 0 4-6 8nI /n , . , H . . 1U r T 2 6~1 . g n l 3 2 n /  ̄。
13 统计 学 方法 .
采 用 S S 30统计 软 件 , P S1 . 数据
1 2 研 究方 法 .
2 1 精 子检 出率 .
在 7 3例 非 嵌 合克 氏症 患者 中 ,
经 过样 本 离 心后 , 现 2例精 液 中有精 子 , 发 年龄 分别 是2 8和 2 9岁 , 2 7 % 。生 育 组 精 液 量 为 ( .0 占 .4 3 4
±1 4 ) l克 氏症 组 为 ( .5±1 0 ) lP< . 1 .2 m , 16 .4 m , 0 0 。
产前诊断克氏综合征一例

07 - 04. 月经周期规则,月经量中等,颜色正常,无血块、无痛 对克氏综合征早期筛查的有效方法。本报告患者产前检查
经。28 岁结婚,爱人体健。妊娠 1 次,生产 0 次,无流产、早 均正常,仅 B 超显示胎儿颈部皮肤厚度 0. 33cm,略大于正常
产、手术产、死产,无节育、绝育。无子女。
( 小于 0. 3cm) 胎儿颈部皮肤厚度。处于优生考虑,医精管 纤维化和透明样变,管腔闭塞,无精子发生,间质细胞增生或
[J]. 中外医学研究,2009,7( 5) : 91. [2]魏瑷,杨孜. 胎儿颈部透明带厚度检测在产前筛查中的应用 [J].
中国优生与遗传杂志,2006,14( 7) : 53 - 54. 收稿日期: 2010 - 11 - 12
肤较白,因皮下脂肪丰富,故皮肤细腻如女性,臀部较为宽大 也宛如女性,大约一半的患者乳腺呈“女性化”特征,部分病
胞遗传学报告: 克氏综合征,核型 47,XXY。患者要求行引产 人有男性乳房增生。成年后有的无性交能力,有的性功能
术。
差,多数患者阴茎能勃起,也可射精,但精液中无精子或精子
既往史: 否认肝炎、结核、传染病史,否认高血压、糖尿病 量极少,97% 的患者不能生育。性格和行为也有异常表现,
笔者遇孕 21 周羊水细胞培养经染色体核型分析确诊克 氏征[1]1 例,核型 47,XXY。
源性促性腺激素( hMG) 刺激反应低,而患者的血浆及尿中黄 体生成素及促卵泡激素升高,黄体生成素分泌多,将刺激睾
临床资料
丸间质细胞,使雌二醇增高,雌二醇 / 睾酮比值上升,从而使 病人的乳房发育呈女性型乳房。
家族史: 父母体健,有一弟健康,未婚。否认家族性遗传 其进行羊水染色体检查,从而避免了此患儿的出生。本病例