MALBAC单细胞全基因组测序详细解析
硕士 毕业 单细胞测序 biomamba-概述说明以及解释

硕士毕业单细胞测序biomamba-概述说明以及解释1.引言1.1 概述概述单细胞测序技术是一种能够对单个细胞进行高通量基因组学研究的创新技术,它可以以高空间分辨率和高灵敏度探索生物体内单个细胞的基因表达和基因组变异等信息。
通过对单个细胞进行测序,我们可以深入了解细胞的多样性、特异性和动态特征,从而揭示细胞发育、功能调控以及疾病发生机制等重要生物学问题。
在过去的几十年里,基因组学研究主要集中在大规模样本的测序上,这种方法无法区分不同细胞间的异质性。
因此,细胞群体测序无法揭示细胞个体之间的差异和亚群分布。
而单细胞测序技术则能够克服这一限制,为我们提供了一种全新的研究细胞个体间异质性的方法。
单细胞测序技术的出现,为很多领域的研究提供了全新的机会。
在生物医学领域中,单细胞测序技术可以帮助我们更好地理解疾病的发生机制、提高疾病的早期诊断和预测,并为个性化医疗提供更多可能性。
此外,在生物科学研究中,单细胞测序技术可以进一步揭示生物系统的功能组成和调控网络,拓宽我们对生命的认知。
然而,单细胞测序技术也面临着一些挑战。
首先,单细胞测序技术的数据量庞大,分析和解读这些数据需要强大的计算和分析能力。
其次,在单细胞水平进行测序需要克服样本损失和串扰等技术挑战。
此外,单细胞测序技术在样本获取和准备方面也存在一定的困难。
综上所述,单细胞测序技术无疑是基因组学研究的重要突破之一,它为我们深入了解细胞个体间的差异提供了一种全新的视角。
随着技术的不断发展和成熟,单细胞测序技术在生物医学领域的应用前景将不可限量,为我们揭示更多关于生命奥秘的答案。
1.2文章结构1.2 文章结构本文主要围绕着单细胞测序技术展开,探讨其意义、应用、技术原理以及前景和挑战。
文章结构如下:引言部分将概述单细胞测序技术的背景和意义,介绍该技术在生物医学领域中的重要性和应用前景。
同时,引言部分还将对文章的结构和主要内容进行简要介绍,为读者提供整体的框架。
正文部分将首先阐述单细胞测序的意义和应用。
单细胞基因测序技术

单细胞基因测序技术单细胞基因测序技术是近年来发展迅速的生物技术领域中的热门研究方向。
它的出现使得科学家们能够深入了解单个细胞的基因组信息及其在生物体中的作用,为精准医学和生物学研究提供了重要的工具。
本文将对单细胞基因测序技术进行详细解读,从技术原理、应用领域到发展趋势进行全面分析。
一、单细胞基因测序技术的原理和方法1. 原理单细胞基因测序技术是一种能够对单个细胞进行基因组测序的方法。
其原理是通过分离单个细胞,并将其DNA进行放大扩增,然后进行测序分析。
由于单细胞存在于复杂的细胞组织中,因此在分离和扩增过程中需要克服一些技术难题,如单细胞损伤、污染等问题。
2. 方法目前常用的单细胞基因测序技术包括单细胞RNA测序(scRNA-seq)、单细胞DNA测序(scDNA-seq)等。
scRNA-seq能够分析单个细胞的转录组信息,揭示细胞类型和功能的差异;而scDNA-seq则能够对单个细胞的基因组进行测序,探究遗传变异的发生和影响。
二、单细胞基因测序技术的应用领域1. 癌症研究单细胞基因测序技术在癌症研究中具有重要价值。
通过对肿瘤细胞进行单细胞基因测序可以揭示肿瘤内部的遗传变异和克隆演化过程,有助于精准化治疗策略的制定。
2. 免疫学研究单细胞基因测序技术可以帮助科学家们深入了解免疫细胞的转录组和表观组学特征,从而揭示不同类型免疫细胞的功能和相互作用,为免疫相关疾病的治疗提供理论支持。
3. 胚胎学研究在胚胎发育过程中,单细胞基因测序技术可以追踪细胞的分化过程,揭示胚胎细胞谱系发展的规律和机制,有助于揭示胚胎发育的分子调控网络。
三、单细胞基因测序技术的发展趋势1. 技术趋势随着测序技术的不断进步和成本的不断降低,单细胞基因测序技术将更加快速、准确、经济,同时提高数据量和分辨率。
2. 数据分析趋势随着单细胞测序数据的不断增加,数据分析方法也在不断完善,包括单细胞测序数据的质控、批次效应的消除、细胞类型的鉴别等。
单细胞全转录组扩增总结

1、多重退火环状循环扩增技术MALBACMALBAC single cell whole genome amplification. A single cell is picked and lysed.First, genomic DNA of the single cell is melted into single-stranded DNA molecules at 94°C. MALBAC primers then anneal randomly to single-stranded DNA molecules at 0°C and ar e extended by a polymerase with displacement activity at elevated temperatures, creating semi-amplicons. In the following five temperature cycles, after the step of looping the full amplicons, single stranded amplicons and the genomic DNA are used as template to produce full amplicons and additional semi-amplicons, respectively. For full amplicons, the 3′ end is complementary to the sequence on the 5′ end. The two ends hybridize will form the looped DNA, which can efficiently preventsthe full amplicon from being used as template, therefore warrant a close-to-linear amplification. After the five cycles of linear preamplification, only the full amplicons can be exponentially amplified in the following PCR using the common 27-nucleotide sequence as the primer. PCR reaction will generate microgram level of DNA material for sequencing experiments.MALBAC覆盖率高、扩增均一,目前MALBAC试剂盒已经做到单管操作,经细胞裂解、MALBAC预扩增、指数式扩增三步,4小时内获得约2-4μg产物2、SMART-seq2TSO (5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′) Oligo-dT30VN (5′–AAGCAGTGGTATCAACGCAGAGTACT30VN-3′) ISPCR oligo (5′-AAGCAGTGGTATCAACGCAGAGT-3′)3、MDAMDA的扩增效率比MALBAC方法高,但各个片段的扩增倍数一致性则没有MALBAC 方法好。
【高中生物】单细胞测序,你为何如此令人痴迷

【高中生物】单细胞测序,你为何如此令人痴迷?最近几年,关于单细胞测序的报道日益增多。
事实上,单细胞测序是一个新兴的领域。
据了解,单细胞测序萌芽于2021年,是单细胞基因组学突飞猛进的一年。
谢晓亮教授哈佛大学课题组与北京大学BIOPIC李瑞强研究员小组合作,将创建的MALBAC技术应用于人类单个精子基因组的测序研究中。
12月10日,解放军总医院诞生了一对特殊的双胞胎?国内首例应用单细胞扩增技术(MALBAC)同时进行PGD/PGS阻断了遗传性耳聋的健康双胞胎。
单细胞测序分为单细胞转录组测序和单细胞基因组测序。
单细胞转录组测序分为:单细胞DGE、单细胞polyA测序、单细胞lncRNA测序。
单细胞基因组测序分为:单细胞外显子组测序和单细胞全基因组重测序。
单细胞发展的历史据了解,1990年,NormanIscove的课题组首次证实对单细胞进行转录组分析是可行的,他们用PCR技术实现了对cDNA分子的指数级扩增。
7月,来自斯坦福大学的StephenQuake在Cell上发表了一篇文章《Genome-wideSingle-CellAnalysisofRecombinationActivityandDeNovoMutationRatesinHumanSperm》,研究采用单细胞测序的方法,测定了来自一项研究的100个精子的重组率,发现了许多新的重组热点和与间接方法发现的相一致的比率。
同年,纽约冷泉港实验室的研究生TimourBaslan正利用单细胞技术来研究癌细胞。
1月《自然?方法学》(NatureMethods)上发表年度特别报道,将“单细胞测序”(Singledoutforsequencing)的应用列为度最重要的方法学进展。
基因组学前沿研讨会将单细胞组学单独列为一个单元,可见单细胞测序在当前基因组学前沿研究中的热度。
单细胞测序为何赢得众生命科学家们火热的心?因为即使来源相同的单个细胞,由于随机生物过程和环境扰动的原因,彼此在许多方面也存在差异,即细胞的异质性。
MALBAC单细胞扩增技术简介

单细胞测序新技术-MALBAC简介随着第二代测序平台的大规模普及,人类单倍型计划、千人基因组计划、癌症基因组计划、Meta-Hit计划等重大国际合作项目相继开展,将基因组研究日渐推向高潮。
然而,迄今为止使用的测序材料无一例外都是大量细胞的混合DNA样本。
一方面,在微生物生态学、癌症基因组、法医学、微量诊断、遗传印记等研究中,显然无法满足测序的mg级样品量需求;另一方面,细胞之间存在很大的异质性,对群体样品或混合样品进行研究得到的结果只是一群细胞中信号的平均值,或者只代表其中占优势数量的细胞信息。
而单细胞全基因组扩增技术为解决以上难题打开了一扇崭新的大门。
全基因组扩增(Whole Genome Amplification, WGA)技术是一种对全部基因组序列进行非选择性、均匀扩增的技术,其目的是在没有序列偏向性的前提下大幅增加DNA的总量。
常用的WGA技术主要分为两种类型:1.基于热循环以PCR为基础的WGA技术,如简并寡核苷酸引物PCR (Degenerate oligonucleotide primer PCR, DOP-PCR)、连接反应介导的PCR (ligation mediated PCR, LM-PCR)、扩增前引物延伸反应(Primer extension preamplification, PEP)等;2.基于等温反应不以PCR为基础的WGA技术,如多重置换扩增(Multiple displacement amplification, MDA) 和基于引物酶的全基因组扩增(Primase-based whole genome amplification, pWGA)。
上述技术可以对少量样品进行扩增,但对于极微量样品进行全基因组扩增时往往会产生非特异的扩增假象,影响实验结果。
为此,亿康基因(/)基于哈佛大学谢晓亮院士研究组研发的一项专利技术——多次退火环状循环扩增技术(Multiple Annealing and Looping Based Amplification Cycles,简称MALBAC)推出了单细胞全基因组/转录组测序服务,解决了基因组扩增对微量初始模板过大的扩增偏倚,使基因组测序的模板需求量从µg级降至单细胞水平。
人类单细胞基因组测序
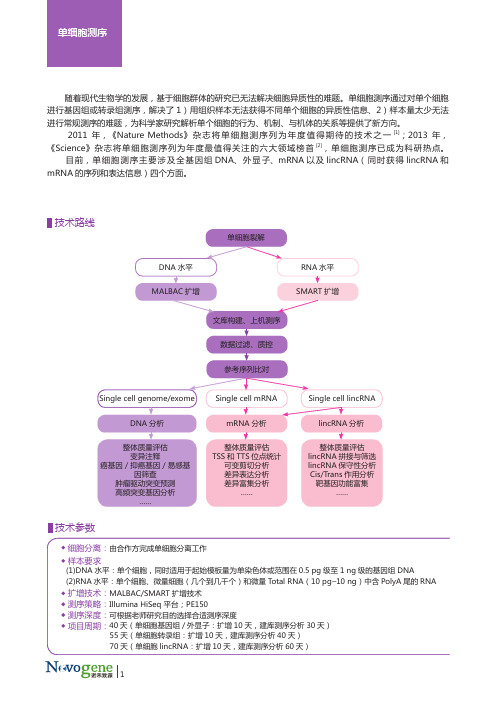
随着现代生物学的发展,基于细胞群体的研究已无法解决细胞异质性的难题。
单细胞测序通过对单个细胞进行基因组或转录组测序,解决了1)用组织样本无法获得不同单个细胞的异质性信息、2)样本量太少无法进行常规测序的难题,为科学家研究解析单个细胞的行为、机制、与机体的关系等提供了新方向。
2011 年,《Nature Methods》杂志将单细胞测序列为年度值得期待的技术之一[1];2013 年,《Science》杂志将单细胞测序列为年度最值得关注的六大领域榜首[2],单细胞测序已成为科研热点。
目前,单细胞测序主要涉及全基因组DNA、外显子、mRNA以及lincRNA(同时获得lincRNA和mRNA的序列和表达信息)四个方面。
技术路线技术参数产品优势最严格的数据指控标准诺禾建立了新一代高通量测序平台,包括10台Hiseq X 测序仪,10台Hiseq 2500,从样品检测到文库构建采用最严格的指控标准,从源头保证了数据的可靠性。
采用国际认可的全基因组扩增技术DNA 水平:诺禾致源采用多次退火环状循环扩增技术(MALBAC),MALBAC 扩增具有低偏倚性。
最新《Science》文章比较MALBAC 与MDA 扩增技术,发现MALBAC 扩增后的数据与混合建库的数据质量更为接近(如图1)[3]。
该技术应用于单精子测序,研究成果[4]已在《Science》杂志发表。
RNA 水平:诺禾致源采用SMARTer 扩增技术(美国Clontech 公司原装试剂盒),能够对单个细胞、微量细胞(几个到几千个)和微量Total RNA(10 pg-10 ng)中含PolyA 尾的RNA 全长进行准确而高效地扩增,扩增后的cDNA 片段适用于Illumina 建库测序平台,为单细胞样本的RNA-seq 分析提供了利器。
图2 SMARTer 扩增无3'和5'端偏好性定制化信息分析针对不同项目,除了进行标准信息分析外,通过和合作伙伴共同探讨,制定可行的个性化信息分析方案,整合各种主流软件,优化分析结果,以确保结果的准确性及创新性。
全基因组测序技术的原理与分析

全基因组测序技术的原理与分析近年来,全基因组测序技术成为了基因研究的主要手段之一,其在医学、农业、动植物基因遗传与演化等领域都得到了广泛的应用。
本文将围绕全基因组测序技术的原理和分析方法进行探讨。
一、全基因组测序技术的原理全基因组测序技术是指将包括人类、动物或植物在内的所有生物体的基因组中的所有DNA序列拍摄下来的过程。
通俗来讲,就是把所有的基因序列测出来。
全基因组测序技术的基本原理是DNA测序。
DNA测序是指通过化学或物理手段进行段扩增后测出DNA的碱基序列。
DNA测序技术的发展经历了多个阶段,从早期的Sanger测序法到最新的Next Generation Sequencing(NGS)技术。
下面将分别介绍这些技术的原理。
1、Sanger测序法Sanger测序法是最初的DNA测序技术,也称为链终止法或二进制测序法。
它是通过在PCR扩增过程中使用针对DNA模板的脱氧肌酸毒素(ddNTPs)来终止DNA链合成,再通过电泳分离产生不同长度的DNA片段,不断重复这个过程来得到DNA序列信息。
Sanger测序法可获得准确的序列信息,但需要大量的时间和财力。
因此,它在测序突变等小范围的DNA变化方面还有广泛应用。
2、Next Generation Sequencing(NGS)NGS技术是一系列基于核酸混合液的建立DNA大量复制,检测与测序的技术,包括Illumina Solexa、Roche 454、Ion Torrent PGM、Pacific Biosciences SMRT等。
NGS技术的原理是将DNA 片段规整至少数百份,将单个片段子剖成只有50-100碱基长度的小片段,多次抽取这些小片段进行测序。
NGS技术与Sanger技术相比较,具有更快的处理速度和较低的成本,且它可以同时检测大量的DNA序列。
但由于NGS技术测序错误率较高,因此对于数据的分析和解析也更加复杂。
二、全基因组测序技术的分析全基因组测序技术的数据分析和解读是后测序分析中一个非常关键的步骤。
单细胞测序技术-

受精卵可能只有一个是正常的
单细胞测序技术的优势与技术难题
用传统的随机引物PCR的方法来扩增,那么 常规的、用大量的DNA建库的方法,因为打断、 不同的扩增片段的扩增效率多少会有一些差异, 补平、加 A、加 这些扩增效率的差异会随着扩增循环数的增加呈 DNA 片段的损失,结果就是初始出指数放大的效果,其结果就是会发生严重的 优势: 表在单细 胞测序中,丢失大部分的起始 DNA 是不可接受的。 覆盖不均一,极少数区段的DNA 被大量扩增,测 分子,在上机的时候是被水冲走的,所以单细胞基因 1.测量基因表达水平更加精确; 单细胞测序要求几乎所有的原始基因组片段都得到 序后它深度非常深,但在大多数区段只有很低的 组扩增的方法还要有较高的扩增效率,至少要有上万 扩增,并且在后续的测序过程中被测序测到。这就 2.能检测到微量的基因表达子或罕见非编码 RNA 覆盖,甚至没有覆盖,那么我们就无法有效地判 倍到几十万倍的扩增效率,才能保证在全基因组测序 要求几乎所有的片段都会被得到扩增,而不只是少 的时候,大部分的片段都被测序测到。 断那些低扩增效率区段的基因序列的情况; 数片段得到有效扩增;
单细胞测序技术是指在单个细胞水平上对基因组进行扩增与测序的一项 新技术。 单细胞测序主要涉及单细胞基因组测序和转录组测序两方面,分别针对 单个细胞的DNA和RNA进行序列分析和比较,进而揭示基因组和转录组的变 化。
单细胞全基因组测序是对选定的目的细胞的全部基因组序列进行非选择
性、均匀扩增,随后利用外显子捕获技术进而高通量测序。 单细胞转录组测序是利用高通量测序技术进行cDNA测序,从而获取特 定器官或组织在某一状态下的几乎所有转录本
MDA方法是使用随机引物,让这些引物与基因组广泛结合,同时使用 一种特定的聚合酶,这种聚合酶能够置换与它自身附着在同一模板上的 DNA链片段,形成一种反复分支结构,扩增出大段的DNA。
全基因组扩增技术

MALBAC(multiple annealing and looping-based amplification cycles),即多次退火环状循环扩增技术,是"目前最先进的全基因组扩增技术"。
该技术于2012年12月刊登于《科学》杂志上(Science).
不同于以往的非线性或指数型扩增方法,MALBAC技术利用特殊引物,使得扩增子的结尾互补而成环,从而很大程度上防止了DNA的指数性扩增。
技术原理
技术特点
1. 起始模板量由微克级(μg)降低到皮克级(pg)。
2. 扩增均匀性远远好于其他扩增技术。
3. 90%以上位点可被成功扩增。
技术优势
1. 样本少,常规分析无法进行基因组或转录组分析。
2. 样本高度异质,细胞间存在重大差异。
相关产品
MALBAC技术单细胞基因组扩增试剂盒。
江苏亿康基因科技有限公司生产的单细胞基因组扩增试剂盒为目前全球范围内唯一以MALBAC技术为支持的基因组扩增实验试剂盒。
MALBAC技术和其它WGA技术相比较
表1 MALBAC技术和其它WGA技术相比较。
MALBAC单细胞全基因组测序详细解析

单细胞全基因组测序一直是生物学家梦想得到的结果。
但是其中必须解决的矛盾:1. 线性扩增。
如果用全基因组PCR扩增,因为PCR的扩增偏向性(bias),再加上PCR过程中较小的偏向性通过指数放大,其结果就会是严重的覆盖不均一,导致在许多地方只有很低的覆盖、甚至没有覆盖。
所以,保证模板被线性地扩增,是首要问题。
2. 全基因组覆盖。
常规的建库方法,因为补平、加A、接头连接效率的问题,起始DNA中有很大一部分会被浪费,没有形成有效文库分子(也就是两头都接好引物的DNA片段)。
在单细胞测序中,这是不可接受的。
3. 高扩增效率。
单细胞中的每个基因位置理论上都只有2个拷贝,而要从2个拷贝扩增到足够建立文库的DNA量,要经过许多次的扩增。
这就需要很高的扩增效率。
而一般的线性扩增很难有好的扩增效率谢晓亮教授创新的MALBAC方法(multiple annealing andlooping-based amplification cycles),一举解决了上述3个问题,达到:1. 线性扩增,2. 近乎全覆盖,3. 高扩增效率(高产量),并且最终可以用于检测单细胞的CNV。
原理:1. 第一步A. 用5’端有27个统一序列,而3’端是8个随机序列的引物,作为扩增引物。
用随机引物保证可以在模板链上的各处随机结合B. 0℃淬火,再65℃等温扩增,得到第一轮的复制的产物a. n个扩增产物(在图中标成蓝色),这些扩增产物都有在5’端有一个统一的引物b. 1个原来的模板(在图中标成黑色)C. 用有前链移开功能的聚合酶(ϕ29聚合酶)来进行扩增,这种酶的特点是会把酶前行方向上的前链从原来的模板链上进行解链、移开。
利用这种特点,可以让模板上的每个点在第一轮反应中,都有机会得到n个拷贝D.巧妙之处:a. 0℃淬火,在延伸开始之前,就把n个引物杂交到模板上b. ϕ29聚合酶可以把前面的链给推开,结合前面的淬火步骤,一次复制出n个扩增子2. 第2轮起的m轮扩增A. 每个循环a. 先0℃淬火,再65℃扩增b. 粘到第一轮所产生的扩增产物上的引物,又会产生一轮扩增。
malbac技术原理

malbac技术原理MALBAC技术,即多重位点扩增基因组单细胞测序技术,是一种单细胞测序技术,可以将单个细胞的基因组DNA进行扩增和测序,从而实现单细胞基因组测序的目的。
它的原理可以分为以下几个步骤。
第一步:单细胞分离MALBAC技术的第一步是将单个细胞分离出来,可以采用常规的离心分离、粘贴分离或者微流控芯片分离的方法。
分离出来的单细胞需要进行后续的扩增和测序。
第二步:整体细胞溶解和逆转录将单细胞放入含有整体细胞裂解缓冲液的96孔板中,同时加入逆转录酶和随机引物进行逆转录反应。
整体细胞裂解缓冲液和随机引物的作用是获得来自单细胞不同位点的DNA模板。
第三步:MARS-MALBAC扩增MARS-MALBAC是MALBAC技术的关键步骤,它可以扩增单个细胞的全基因组DNA。
MARS-MALBAC包括:首先,将逆转录反应得到的cDNA进行首轮线性扩增,并加入含有一定比例略微含有错误的引物;第二,将首轮扩增产物作为后续扩增的模板,进行数轮指数扩增,以产生大量cDNA;最后,进行线性扩增,得到适量的扩增产物。
第四步:测序最后,对MARS-MALBAC扩增产物进行测序,得到单细胞基因组DNA的序列信息。
这个过程中需要将扩增产物用PCR产物纯化试剂进行纯化,去除空间和杂质的DNA,并进行校验、定量、文库构建和测序。
综上所述,MALBAC技术的原理基于MARS-MALBAC扩增法,其主要思想在于通过多次扩增,产生足够的扩增产物,并引入略微有误的引物,减少扩增的偏好性,并避免某些基因组区域的扩增优先。
通过MALBAC技术,可以对单个细胞的基因组进行全基因组扩增和测序,为单细胞研究提供了有力的工具。
MALBAC

MALBAC®白金微量RNA扩增试剂盒【产品编号】KT110700724KT110700796【产品名称】通用名:MALBAC® 白金微量RNA扩增试剂盒英文名:MALBAC® Platinum Single Cell RNA Amplification Kit【包装规格】24测试/盒,96测试/盒【产品描述】MALBAC® 白金微量RNA扩增试剂盒能在5-6小时内,由1-2000个细胞或10 pg-20 ng经提取的真核生物总RNA为起始,特异性针对其中的mRNA进行逆转录,并以MALBAC® Template-switching专利技术在cDNA的3’端添加一段带有表达定量分子标签的接头序列,通过该接头序列进行后续的PCR扩增,获得全长cDNA扩增产物,有效避免了cDNA合成过程中的3’偏好性和基因组DNA与rRNA的污染,同时表达定量分子标签可辅助基因表达量计算,在完整扩增mRNA序列信息的同时保留链来源信息,对基因表达进行精确定量。
一般情况下,一个反应视投入量可以扩增出2-50 ng高质量全长双链cDNA。
本试剂盒可获得95%以上的逆转录与扩增成功率,全长cDNA扩增产物可无缝衔接主流测序平台,如Illumina、Ion torrent、PGM、Ion Proton测序仪,下机数据(2.5M Reads)可检测到90%以上的基因表达,基因表达一致性超过90%,扩增无明显偏倚,且需要的样本投入量更少。
【预期用途】本产品主要用于单细胞RNA-Seq前的样本扩增。
单细胞RNA-Seq是指通过对单个细胞的mRNA进行逆转录和PCR扩增,使用高通量测序手段,对单细胞中mRNA进行基因表达定量、功能富集、代谢通路等分析。
本产品可以解决传统RNA逆转录与扩增技术在早期胚胎发育、干细胞、癌症、免疫等研究领域中存在的样品量极低或细胞异质性的问题,是在单细胞水平研究基因表达强有力的工具,极大地拓展了RNA-Seq的应用范围:⚫极微量样品的表达分析(单个细胞)⚫胚胎早期发育研究⚫肿瘤细胞异质性研究⚫免疫细胞群研究⚫干细胞分化研究【产品原理】扩增原理图本产品可实现高效且均一的全长转录本扩增。
全基因组测序原理

全基因组测序原理全基因组测序,即Whole Genome Sequencing (WGS),是指将一个生物体的全部基因组进行高效、高通量的测序。
其原理基于二代测序技术,如Illumina测序技术,通过将待测DNA样本进行多次放大、断裂和现代测序技术的串接等处理,最终得到包含了整个基因组信息的DNA序列。
全基因组测序的步骤主要包括DNA提取、DNA文库构建、文库扩增、测序反应、质控和数据分析等环节。
首先,从生物体的细胞中提取DNA,并经过物理或化学方法断裂,得到以一定片段长度分布的DNA片段。
然后,将这些DNA片段连接到特定的测序适配体上,构建DNA文库。
接下来,通过PCR扩增文库中的DNA片段,使其数量增加到一个可测量的范围。
然后,将扩增后的文库进行测序反应。
在测序反应中,首先将文库DNA片段固定在测序模板上,然后利用特定的标记测序引物逐个加入碱基,发出荧光信号,记录测序信息。
在碱基加入的过程中,由于每次只添加一种碱基,所以可以通过检测不同荧光信号的强度来确定添加的碱基类型。
最后,通过测序仪读取和记录荧光信号,得到DNA片段的序列信息。
在全基因组测序过程中,质控是一个重要的环节。
通过质控可以检测文库的质量和纯度,以及测序数据的可靠性和准确性。
通常使用聚合酶链反应(PCR)和凝胶电泳等方法来检测文库的质量,并利用质控软件对测序数据进行分析和处理。
数据分析过程包括序列修饰、比对、SNP和Indel检测、基因注释等,通过这些步骤可以获得最终的基因组序列信息。
全基因组测序的应用广泛,可以用于研究基因组结构和功能,寻找遗传变异、突变和致病基因,以及进行进化分析、物种鉴定、种群遗传学研究等。
此外,全基因组测序还在医学领域中应用广泛,如个体基因组医学、癌症靶向治疗、药物个体化治疗等方面。
随着二代测序技术的不断发展和成熟,全基因组测序已经成为一种常用的高通量测序方法,为基因组学研究提供了强有力的工具。
全基因组测序的数据分析和生物学解读

全基因组测序的数据分析和生物学解读随着生物学的不断发展,全基因组测序已经成为了一项非常重要的技术。
基因组是细胞中存储着信息的重要组成部分,它所包含的信息能够指导生命体的生长、发育和适应环境的能力。
基因组测序就是通过对生物体DNA的高通量测序,获得它们的基因组序列信息。
全基因组测序的数据分析和生物学解读则是对产生的海量数据进行精细化处理和解读的步骤。
全基因组测序的数据分析步骤可以大致分为预处理、序列比对和变异鉴定三个部分。
预处理预处理是指对测序数据进行质量控制、去除污染和过滤低质量序列的过程。
前期质控可以通过FastQC等软件进行评估,检查数据中是否存在低质量序列、接头污染、含有接头的剪切等情况。
一旦存在这些情况,我们可以通过Trim Galore!、Fastp等软件进行过滤和去除。
而低质量序列过滤常常是基于读长、GC含量、质量分数等指标进行判断和筛选。
这些步骤都是为了保障后续分析的准确性。
序列比对序列比对是指将测序得到的reads进行比对,并确定它们在参考基因组上的位置。
由于基因组大小不一,测序技术的限制等原因,大多数应用都选择了将reads 比对到参考基因组(reference-based)上进行分析。
这个过程能够帮我们寻找到与参考序列对应的单条或多条读取序列,为后续进行基因注释、突变检测等分析提供依据。
变异鉴定变异鉴定是指利用序列比对的结果来查找基因组间的变异,并将它们分为基因缺失、突变、插入等。
常用的工具包括GATK、SAMtools、FreeBayes等。
这些工具可以有效地识别变异,比如SNP(单核苷酸多态性)和InDel(插入/删除),并进行标注、分类、统计和过滤等等。
数据分析过程蕴含着诸多的技术和细节,这里我们介绍了其中三个部分,旨在提供一个基本框架和流程。
全基因组测序的生物学意义意义非凡,它不仅可以帮助研究人员更好的理解生命的本质,还可以有助于开发新药物、治疗方法等等。
比如对于基因突变、癌症等人类疾病研究,全基因组测序都起着极为重要的作用。
不同单细胞全基因组扩增方法的比较及MALBAC在辅助生殖中
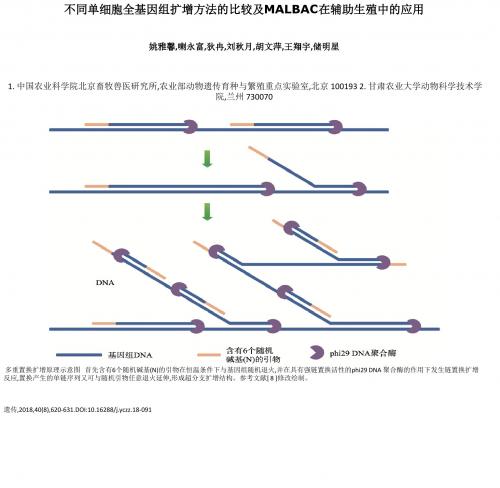
遗传,2018,40(8),620-631.DOI:10.16288/j.yczz.18-091
不同单细胞全基因组扩增方法的比较及MALBAC在辅助生殖中的应用
姚雅馨,喇永富,狄冉,刘秋月,胡文萍,王翔宇,储明星
1. 中国农业科学院北京畜牧兽医研究所,农业部动物遗传育物科学技术学 院,兰州 730070
多重置换扩增原理示意图 首先含有6个随机碱基(N)的引物在恒温条件下与基因组随机退火,并在具有强链置换活性的phi29 DNA 聚合酶的作用下发生链置换扩增 反应,置换产生的单链序列又可与随机引物任意退火延伸,形成超分支扩增结构。参考文献[ 8 ]修改绘制。
MALBAC

MALBAC®微量基因组快速扩增试剂盒说明书【产品编号】KT110700324/KT110700396【产品名称】通用名:MALBAC®微量基因组快速扩增试剂盒英文名:MALBAC®Single Cell DNA Quick-Amp Kit 【包装规格】24测试/盒,96测试/盒【预期用途】本产品可用于单细胞或其他微量样本中全基因组DNA 扩增,扩增产物可用于实时定量PCR 、高通量测序等技术平台。
也可用于多种下游实验:●基因点突变分析检测●单核苷酸多态性(SNP )基因分型●基因拷贝数(CNV )分析●微阵列比较基因组杂交(Array CGH )●SNP 芯片技术【检测原理】本产品采用MALBAC 专利技术(即多次退火环状循环扩增技术)对单细胞或稀有细胞样本进行全基因组高效、快速裂解和扩增;该技术无需提取核酸,利用独特的具有链置换活性的DNA 聚合酶进行准线性的全基因组扩增和指数式扩增,扩增均一性高,脱扣率较低,只需两步即可为下游分析提供充足的实验材料。
序号名称规格及数量储存条件24测试96测试1RAPE Mix 12µL ×1管48µL ×1管-20℃2Rapid Solution 108µL ×1管432µL ×1管3RWGA Enzyme Mix 48µL ×1管192µL ×1管4Rap-WGA Solution1440µL ×1管1440µL ×4管【储存条件及有效期】-20℃保存,避免反复冻融。
有效期:24个月。
【自备物品】1.试剂:无核酸酶水、1×PBS 缓冲液;2.仪器:微型离心机、涡旋混匀仪、紫外分光光度计、PCR 仪。
【产品特点】●单细胞经扩增后可获得2~5μg 的DNA 产物;●单管操作、2步完成、仅需2小时;●可从流式分选出的细胞或0.5pg 级DNA 中扩增出全基因组DNA ,且成功率达95%以上;●可在AT-GC富集区得到准确、高重复度的连续扩增结果;●全基因组覆盖度高,仅存在<10%的基因座丢失及等位基因丢失。
全基因组测序方法

全基因组测序方法
全基因组测序是一种高通量的DNA测序技术,其可以快速、精确地测定个体的全部基因组序列。
全基因组测序方法已经成为生命科学研究的重要工具之一,可用于研究生物多样性、基因组演化、疾病诊断和预防等领域。
全基因组测序方法主要分为两种:短读测序和长读测序。
短读测序技术使用高通量平台对DNA进行断片、连接、扩增和测序,产生数百万条短序列,然后通过计算机软件将这些序列拼接起来,得到全基因组序列。
常用的短读测序平台包括Illumina、Ion Torrent和SOLiD等。
长读测序技术则可以产生数千到数万个比短读更长的DNA序列,可以更准确地组装基因组序列。
目前,长读测序技术主要包括第三代测序技术(如PacBio和Oxford Nanopore)和高通量单细胞测序技术。
全基因组测序方法可以用于研究基因组结构和变异,包括单核苷酸多态性(SNP)、插入/缺失(Indel)、结构变异和基因重复等。
它还可以用于研究基因表达和调控,包括转录组测序和表观基因组测序等。
总之,全基因组测序方法已经成为现代生命科学研究的重要技术之一,为我们揭示了生命的奥秘,为人类健康和环境保护提供了巨大的帮助。
- 1 -。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
单细胞全基因组测序一直是生物学家梦想得到的结果。
但是其中必须解决的矛盾:
1. 线性扩增。
如果用全基因组PCR扩增,因为PCR的扩增偏向性(bias),再加上PCR过程中较小的偏向性通过指数放大,其结果就会是严重的覆盖不均一,导致在许多地方只有很低的覆盖、甚至没有覆盖。
所以,保证模板被线性地扩增,是首要问题。
2. 全基因组覆盖。
常规的建库方法,因为补平、加A、接头连接效率的问题,起始DNA中有很大一部分会被浪费,没有形成有效文库分子(也就是两头都接好引物的DNA片段)。
在单细胞测序中,这是不可接受的。
3. 高扩增效率。
单细胞中的每个基因位置理论上都只有2个拷贝,而要从2个拷贝扩增到足够建立文库的DNA量,要经过许多次的扩增。
这就需要很高的扩增效率。
而一般的线性扩增很难有好的扩增效率
谢晓亮教授创新的MALBAC方法(multiple annealing andlooping-based amplification cycles),一举解决了上述3个问题,达到:1. 线性扩增,2. 近乎全覆盖,3. 高扩增效率(高产量),并且最终可以用于检测单细胞的CNV。
原理:
1. 第一步
A. 用5’端有27个统一序列,而3’端是8个随机序列的引物,作为扩增引物。
用随机引物保证可以在模板链上的各处随机结合
B. 0℃淬火,再65℃等温扩增,得到第一轮的复制的产物
a. n个扩增产物(在图中标成蓝色),这些扩增产物都有在5’端有一个统一的引物
b. 1个原来的模板(在图中标成黑色)
C. 用有前链移开功能的聚合酶(ϕ29聚合酶)来进行扩增,这种酶的特点是会把酶前行方向上的前链从原来的模板链上进行解链、移开。
利用这种特点,可以让模板上的每个点在第一轮反应中,都有机会得到n个拷贝
D.巧妙之处:
a. 0℃淬火,在延伸开始之前,就把n个引物杂交到模板上
b. ϕ29聚合酶可以把前面的链给推开,结合前面的淬火步骤,一次复制出n个扩增子
2. 第2轮起的m轮扩增
A. 每个循环
a. 先0℃淬火,再65℃扩增
b. 粘到第一轮所产生的扩增产物上的引物,又会产生一轮扩增。
其产物5’端带有统一引物序列,3’端带有与统一引物序列,称为完整扩增产物
c. 粘到原始模板上的引物,也产生一轮扩增。
但是其产物是只在5’端带有统一引物序列,而在3’端没有统一引物序列,称为半扩增产物
d. 98℃解链
e. 58℃保温,让完整产物的两端发生链内杂交,复性后3’端的序列不再能与游离的互补引物发生杂交,这就阻止了指数扩增。
B. 重复A步骤中从a~e的5个步骤,共5次。
经过m个循环后,得到:
a. m* n2个“完整扩增产物”
b. (m+1)* n个“半扩增产物”
c. 1个原始模板
C. 巧妙之处:
a. 线性扩增
i. 其巧妙之处在于,每个循环的最后,加了一步58℃退火。
这一退火过程,让完整扩增产物的两端发生链内杂交。
这样3’端的序列就不能与新的游离引物发生杂交,也就不会引发起始于3’端的扩增,也就是避免了“完整扩增产物”的自我指数扩增。
ii. 如果发生以8个随机碱基为引物3’末端的指数扩增,3’末端的碱基序列不一致性就会导致扩增效率的Bias,再经过指数放大,就会变成明显的扩增效率Bias。
iii. 现在,还是8个随机碱基的引物在模板上随机地找结合位点,所有位点的被扩增机会大致均等。
iv. 完整产物的数量是m * n2,也就是说,扩增产物与m成正比,而不是与m2成正比,更不是与2m成正比。
也就是说扩增产物与扩增的次数成线性关系。
这达成了单细胞测序所要求的:线性扩增。
b. 全基因组覆盖
i. 再次利用ϕ29聚合酶的前链移开功能,在一个模板上扩增出多个扩增子。
这样从第1轮扩增开始,每个模板就会得到多个扩增子。
再加上以后5轮的扩增中,原始模板都有机会再次被扩增出多个扩增子。
这样本保证每个原始模板都产生n个扩增子。
建库时被漏掉一些扩增子,还是能保证大多数的基因组区域可以被建成库
c. 高扩增效率
i. 在扩增中,所得到的“完整扩增产物”的个数是m * n2个。
这个“n2”,还是利用了ϕ29聚合酶的“前链移开功能“,保证了扩增有较高的效率。
可以得到较多的扩增产物,以供下面的实验之用。
结果
1. Lorenz曲线
可以从上面的Lorenz曲线中看出来,MALBAC方法的覆盖均一性明显好于MDA方法,但还是弱于大批量细胞的测序结果。
蓝箭头和绿箭头,分别指出MALBAC方法和MDA方法没有测到序列部分占全基因组的比例。
2. CNV
3. A、B、C、D中的绿线是按Markov模型估算出来的CNV数
4. A、B、C,分别是3个单细胞MALBAC方法测序后的覆盖率。
5. D是大量细胞测序后的覆盖率。
6. E是MDA方法测序后的覆盖率。
7. 测序的深度都是平均25倍。
参考文献:
Chenghang Zong1, Sijia Lu1, Alec R. Chapman, X. Sunney Xie. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science 21 December 2012:Vol. 338 no. 6114 pp. 1622-1626
Rubicon Genomics公司推出了商业化的单细胞测序试剂盒,Rubicon并没有公布其技术细节,但是根据其公开的技术材料,基本与MALBAC技术相近:
∙都是通过扩增产物的末端自封闭来抑制指数扩增。
∙通过数轮线性扩增得到大量的扩增产物。