电解与极化作用
电解与极化作用小结

(A)都溶解
(B)Fe(s)不溶,Cd(s)溶解
(C)都不溶解
(D)Fe(s)溶解,Cd(s)不溶
答 (B) 设构成电池 Cd(s)|Cd2+||Fe2+|Fe(s)
则 电池反应为 Cd(s) + |Fe2+ = Cd2+ + Fe(s)
E
=
EO
−
RT 2F
ln
a(Cd 2+ ) a( Fe2+ )
解:
(1)
ϕ Cd 2+/ Cd
+
RT
F
ln
a Cd
2+
= −0.403 +
RT ln 0.01 = −0.4621V 2F
ϕ Cu 2+ / Cu
=ϕO Cu 2+ / Cu
+ RT F
ln
a Cu
2+
= 0.337 +
RT ln 0.02 = 0.2868 V 2F
仍不会有 H2(g)析出,问溶液的 pH 值应控制在多少为好? 已知 H2(g)在 Zn(s)上的超电势为 0.72V,并设此值与溶液浓度无关。 (设 γ±=1)已知: ϕ O (Zn2+/Zn)=-0.7628V .
解: φ(Zn2+/Zn)= ϕ O (Zn2+/Zn) -RT/2F×ln 1/a(Zn2+) = -0.8811 V
例题 12 298K, pO 下,以 Pt 为阴极,电解含 FeCl2(0.01mol·kg-1)和 CuCl2(0.02mol·kg
-1)的水溶液。若电解过程中不断搅拌,并设超电势可略去不计,已知ϕ O (Fe2+/ Fe)
10-电解与极化作用

阳,析出 阳,可逆 阳
3、极化曲线的测定
超电势或电极电势与电流密度之间的关系曲线称
为极化曲线,极化曲线的形状和变化规律反映了电化
学过程的动力学特征。
+
测定超电势的装置
如右图所示:
A
电极1为待测电极,
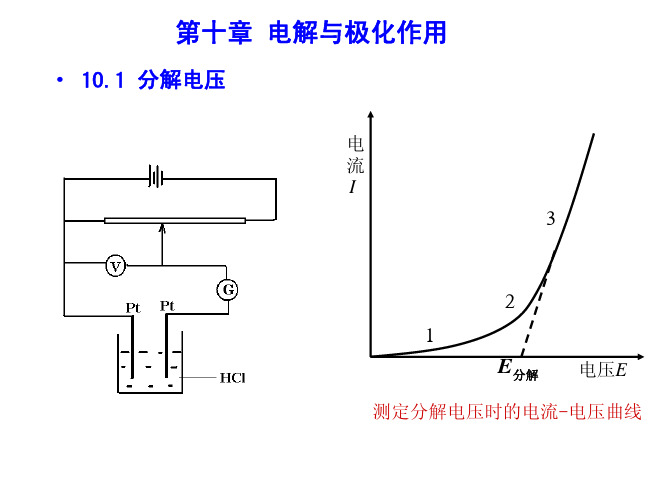
测定分解电压时的电流-电压曲线
二、分解电压的测定
当外压增至2-3段,氢 气和氯气的压力等于大
气压力,呈气泡逸出,反电
动势达极大值 Eb,max。
电
E外 Eb,max IR
流 I
再增加电压,使I 迅速增 加。将直线外延至I = 0 处,
得E(分解)值,这是使电解 池不断工作所必需外加的
最小电压,称为分解电压。
(2)电化学极化
以铜电极为例: 电极反应进行缓慢
作为阴极:则由外电源输入阴 极的电子来不及消耗,即溶液 中Cu2+不能马上与电极上的电 子结合,变成Cu,结果使阴极 表面积累了多于平衡状态的电 子,导致电极电势比平衡电极 电势更小。
-
- 电源 +
e-
+
e-
Cu
Cu
CuSO4
电解池
作为阳极:类似的,作为阳极时,会使阳极表面的电 子数目小于平衡状态的电子,导致电极电势比平衡电 极电势更大。
Ag ,Ag
-
RT F
ln
1 c,e
c,e c0
阴,不可逆 阴,可逆
c'
扩散层
在浓度梯度作用下(ce’ < c0)Ag+向 电极表面的迁移
阴极浓差极化的结果是阴极电极电势比可逆时变小。
(1)浓差极化
阳极: Ag Ag++e , v扩<v反,c0 < ce`
第十章电解与极化作用

(1)浓差极化——扩散过程的迟缓性而引起的极化。
浓差极化是在电流通过时,由于电极反应的反
应物或生成物迁向或迁离电极表面的缓慢而引起的
电极电势对其平衡值的偏离。
阴极:Ag++eAg,
v扩<v反,
m,<m=m
’ Ag
/ Ag
>
RT F
ln1 m' Nhomakorabea
’
即: 可 逆 > 不 可 逆 阴极极化的结果是阴 极电极电势变得更负。
η阳 j(电流密度)
E可逆 -ΔE不可逆
η阴
E可逆
电极电势
电解池中两电极的极化曲线
原电池与电解池极化的差别
当有电流通过电解池, 电解池的端电压大于平 衡电池电动势。
即:E = E平 +ηa +ηc
当有电流通过原电池, 原电池的端电压小于平 衡电池电动势。 即:E = E平 -ηa-ηc
影响超电势的因素
(1) 电流密度J 一般 , J越大 , 超电势越大。不同的物质,其 增大的规律不一样。 (2)电极材料及其表面状态 以氢电极为例:
J= 100 A/m2时,
若电极材料为Ag,η= 0.13V;
若电极材料为Pt(光滑),η= 0.16V;
若电极材料为Pt(镀有铂黑),η= 0.03V。
(3)温度 温度升高,超电势减小。 一般,每增高1度,超电势减小2mV。 除了以上因素外,电解质的性质、溶液中的杂 质对超电势均有影响。所以,超电势的重现性不好。 一般说来析出金属的超电势较小,而析出气体 (特别是氢、氧)的超电势较大。
§10.2 极化作用
§10.3 电解时电极上的竞争反应 §10.4 金属的电化学腐蚀、防腐与金属的钝化 §10.5 化学电源
物理化学(第五版傅献彩)第10_电解与极化作用

无电流
ϕ可逆
= ϕy Ag+ |Ag
−
RT F
ln
1 aAg+
有电流
ϕ不可逆
= ϕy Ag+ |Ag
−
RT F
ln
1 aAg+ , e
η阴
= ϕ可逆
− ϕ不可逆
=
RT F
ln aAg+ aAg+ , e
>0
aAg+ , e < aAg+ ϕ不可逆 < ϕ可逆
阳极上的情况类似,但 ϕ不可逆 > ϕ可逆
的金属先在阴极析出,这在电镀工业上很重要 例如,利用氢的超电势,控制溶液的pH,实
现镀 Zn,Sn,Ni,Cr 等
25
阴极上发生还原反应
发生还原 (1) 金属离子 的物质: (2) 氢离子 (中性水溶液 aH+ = 10−7 ) 判断在阴极上首先析出何种物质,应把各 种可能还原的物质的电极电势求出来(气 体要考虑超电势,金属可不考虑超电势)
2H+ + 2e- = H2
ϕ可逆
=ϕΟ H+ |H2
− RT 2F
ln
pH2 / p Ο a2
H+
= −0.059pH = −0.414V
ϕ不可逆 = ϕ可逆 −η = −0.414V − 0.584V = −0.998V
Zn2+ + 2e- = Zn
ϕ可逆
=ϕΟ Zn2+ |Zn
− RT 2F
1 ln
=−
RT 2F
ln
aH2 a2
H+
−ηH2
设 pH2 = p Ο
11章电解池与极化作用全解

可逆电池:电极反应是在电流趋于零的平衡条件 下进行的,此时的电极电势为可逆电 极电势或平衡电极电势。
实际上电池对外供电或进行电解时,都有一定的电流 通过电极,使电极反应在偏离平衡态下进行而成为不 可逆过程,导致电极电势也偏离平衡电极电势。现以 电解池为例讨论这种偏离现象产生的原因及在实际中 的意义。
由于化学反应本身的迟缓性而引起的的电极极化。
电极反应总是分若干步进行,若其中一步反应速
率较慢,需要较高的活化能。为了使电极反应顺利进
行所额外施加的电压称为称为活化超电势。
例:2H+ + e– H2
e
v反应 慢,阴极积累电子e- 2H+ + 2e– H2
电化学极化使阴极电势降低,使阳极电势升高
两种极化结果均使 阴极电势降低
7
§11.2 极化作用
1. 电极的极化
定义:电流通过电极时,电极电势偏离其平衡电极 电势的现象称为电极的极化。
阳极极化: E阳 E阳, 平 使 E阳 变大(正) 阴极极化: E阴 E阴, 平 使 E阴 变小(负)
离子扩散速率慢 浓差极化 极化产生的原因:
反应速率慢 电化学极化
8
(1) 浓差极化 由于电极表面附近薄液层的浓度和本体溶液的 浓度的差异所导致的电极极化。
所以标准氢电极中的 铂电极要镀上铂黑。
影响超电势的因素很多,如电极材料、电极表面状态、电
流密度、温度、电解质的性质、浓度及溶液中的杂质等。
14
Tafel 公式(Tafel’s equation)
早在1905年,Tafel 发现,对于一些常见的电 极反应,超电势与电流密度之间在一定范围内存 在如下的定量关系:
电解与极化作用

第九章 电解与极化作用前边讨论的电池与电极都是可逆的,那么应用能斯特公式来处理电化学体系时,它的前提就是该体系必须是处于热力学平衡态,但是对于一些现实的电化学过程来说一般都是不可逆过程,因此应用Nernst 公式研究电化学问题就具有很大的局限性。
事实上当原电池或电解池,只要有电流通过,就有极化作用发生,该过程就是不可逆过程。
研究不可逆电极反应及其规律性对电化学工业是十分重要的,所以我们要讨论不可逆电极过程。
在这一部分除了讨论电解池中的极化作用外,还要简单介绍一些电解在工业上的应用上及金属的防腐和化学电源等。
§9-1 电极的极化1、不可逆条件下的电极电势一个不可逆电池所具备的条件有两个:①电池反应在充电与放电时互为逆反应;②通过电池的电流I →0,即没有电流通过电池。
显然组成可逆电池的两个电极都是可逆电极,那么可逆电极的电极反应都是在可逆的条件下发生的。
这时电极所具有的电势就称为可逆电极电势。
可逆电极电势对许多电化学和热力学问题的解决是相当重要的。
但是在实践当中许多电化学过程,如进行电解和使用化学电源做电功时,并不是在可逆情况下进行的,也就是说要有电流通过电池或电解池,此时的电极反应就是不可逆的了,不可逆电极的电极电势用“I ϕ”表示,当然这个电极电势与可逆电极的电极电势r ϕ是不相同的,那么我们就把电极在有限电流通过时所表现的电极电位I ϕ与可逆电极电势产生偏差的现象叫做电极的极化。
偏差的大小(绝对值),称为“过电势”。
用“η”表示,||r I ηϕϕ=-,对于原电池,在可逆放电时,两电极的端电压是最大的,这个端电压就是电动势E ,它等于两个可逆电极的电位差。
()()()()r r r r E ϕϕϕϕ=-=-正阳阴负在不可逆条件下进行放电,两电极的端电压用E I 表示,它一定要小于原电池的电动势E ,E I <E ,E I =E-△E其电动势的降低主要是由于两个因素引起的,当有电流通过时, ①电池具有一定的内阻R 的消耗电位降IR ;②不可逆条件电极要产生极化,也会造成电动势下降,所以不可逆电池两电极的电位差通常就叫端电压。
物理化学课件6.3章电解与极化作用

实验材料
电解槽、电极、电源、电解质溶液等。
电解实验的设计与操作
实验步骤 1. 准备实验材料,配置电解质溶液。
2. 将电极插入电解槽中,连接电源。
电解实验的设计与操作
3. 观察并记录电极反应现象,测量电流和电压。 4. 分析实验数据,得出结论。
极化作用的实验研究方法
实验目的
通过实验研究,探究极化作用对电极反应的影响,理解极化作用的原理。
电解分离与提纯
总结词
电解分离和提纯是利用电解的原理将混 合物中的不同组分进行分离或提纯的方 法。
VS
详细描述
电解分离是通过电解过程中不同物质在电 极上的吸附、氧化还原反应等特性差异实 现分离。电解提纯则是利用电解过程将杂 质去除,实现物质的纯化。
05 极化作用的应用
电化学反应器
电解槽
利用电解原理进行物质转 化的设备,如氯碱工业中 的隔膜电解槽和电解水制 氢装置。
详细描述
电镀是将金属离子在电场作用下还原成金属并沉积在阴极表面,用于表面防护和装饰。电冶金则是利 用电解过程提取金属,从矿石或盐类等原料中分离和提纯金属。
电解制取气体
总结词
电解水是制取氢气和氧气的常用 方法,具有清洁、高效的特点。
详细描述
通过电解水可以将水分子分解成 氢气和氧气,分别在阴极和阳极 析出。电解水制取的气体可用于 燃料电池、医疗、潜水等领域。
电极反应的极化曲线
极化曲线是描述电极电势与电流密度之间关系的曲线,可以用来研究电极反应的动 力学过程和机理。
在极化曲线上,可以根据电流密度的大小来判断电极反应的速率快慢,以及电极电 势偏离可逆电势的程度。
通过测量不同温度下的极化曲线,可以研究电极反应的热力学性质和动力学过程。
电解与极化作用

物理化学论文电解与极化作用化工093班姓名:李寒萌学号:12 号电解与极化作用一、分解电压使电能转变成化学能的装置称为电解池。
当直流电通过电解质溶液,正离子向阴极迁移,负离子向阳极迁移,并分别在电极上起还原和氧化反应,从而获得还原产物和氧化产物。
若外加一电压在一个电池上,逐渐增加电压直至使电池中的化学反应发生逆转,这就是电解。
实验表明,对任一电解槽进行电解时,随着外加电压的改变,通过该电解槽的电流亦随之变化。
例如,使用两个铂电极电解HCl 溶液时,使用图9.1 的线路装置,改变可变电阻,记录电压表和电流表的读数,则可测量电解槽两端电位差与电流强度的关系曲线。
开始时,当外加电压很小时,几乎没有电流通过电解槽;电压增加,电流略有增加;当电流增加到某一点后,电流随电压增大而急剧上升,同时电极上有连续的气泡逸出。
在两电极上的反应可表示如下:阴极2H+(a H+)+2e.→H2(g, p)阳极2Cl-.(a Cl-)→Cl2(g, p)+2e. 图9.1 分解电压的测定装置当电极上有气泡逸出时,H2和Cl2的压力等于大气压力。
电解过程分析:当开始加外电压时,还没有H2和Cl2生成,它们的压力几乎为零,稍稍增大外压,电极表面上产生了少量的H2和Cl2,压力虽小,但却构成了一个原电池(自发地进行如下反应)(-) H2(p)→2H+ (a H+)+2e-(+) Cl2(g)+2e-→2Cl-(a Cl-)此时,电极上进行反应的方向正好与电解所进行的反应的方向相反。
它产生了一个与外加电压方向相反的反电动势E b。
由于电极上的产物扩散到溶液中了,需要通过极微小的电流使电极产物得到补充。
继续增大外加电压,电极上就有H2和Cl2继续产生并向溶液中扩散,因而电流也有少许增加,相当于图9.2 中I-E 曲线上的1-2段。
此时由于p H2和p Cl2不断增加,对应于外加电压的反电动势也不断增加,直至气体压力增至等于外界大气压力时,电极上就开始有气泡逸出,此时反电动势E b达到最大值E b, max将不再继续增加。
207-223 第十章电解与极化作用

为了不使 H2 析出,问溶液的 pH 值应控制在多少为好?
解:若 E(Zn2+|Zn)>E(H+|H2),则 Zn(S)析出而 H2 不能析出.
即: -0.763V+ 0.5916V lg10−5 >-0.05916V pH-0.75V 2
pH>2.72.
例 3 25°时,用 Zn 电极作为阴极,电解 a±=1 的 ZnSO4 水溶液。
( ) 阳极
H2O ⎯⎯→ 2H+
aH+
+
1 2
O2
(g
)
+
2e−
E阳,析出
=
E O2 H2O H+
+
RT 2F
ln
a2 H+
+O2
= 1.23
V+ RT 2F
ln (0.01)2
+ 0.5
V = 1.612
V
( ) E分解 = E阳,析出 − E阴,析出 = 1.612 + 0.17 V=1.782 V
(1)已知,水溶液为中性,则 Zn2+在 Zn 的平衡电极电势 Ee(Zn2+|Zn)及 H2 在 Zn 电 极上析出的平衡电极电势 Ee(H+|H2)各位多少?
(2)又知在某一电流密度下,H2 在 Zn 极上的超电势为 0.7 V,则 H2 在 Zn 上实际析出 的电势 EH2=?
(3) 若 Zn 在 Zn 电极上的超电势可忽略不计,则上述电解过程中在 Zn 极上优先析出的 是什么?
E 分解=E 可逆+△E 不可逆+IR 2.产生极化作用的原因主要有哪几种?原电池和电解池的极化现象有何不同? 答:产生极化作用的主要原因是电化学极化和浓差极化。电解时,电流密度愈大,超电 势愈大。外加电压也要增大,所消耗能量越多。原电池放电时,有电流在电极上通过,随着 电流密度增大。由于极化作用,正极比可逆电视愈来愈小,负极比可逆电势愈来愈大,原电 池的电动势逐渐减小,它所能作的电功逐渐减小。 3.什么叫超电势?它是怎样产生的?如何降低超电势的数值? 答:把某一电流密度下的电势 φ 与 不可逆 φ 可逆之间的差值称为超电势,超电势产生的原因 有,电化学极化和浓差极化,及电解过程中,在电极表面形成一层氧化膜或其他物质,从而 对电流的通过产生阻力(电阻超电势),在外加电压不大的情况下,把溶液剧烈搅动可以降 低浓差极化,但由于电极表面扩散层的存在,不可能把浓差极化完全除去。除此之外,还可 以加入去极化剂和减小体系的阴值 R 来减低超电势的值。 4.析出电势与电极的平衡电势有何不同?由于超电势的存在,使电解池阴、阳极的析出
第十章 电解与极化作用

电解与极化作用一、简答题1.什么叫极化作用?什么叫超电势?极化作用主要有几种?阴、阳极上由于超电势的存在其不可逆电极电势的变化有何规律?2.在电解过程中,阴、阳离子分别在阴、阳极析出的先后次序有何规律?3.电化腐蚀主要有哪些类型?在盛水的铁锅中,为什么在水周围比在水下的部分先生锈?4.以Pt 为电极电解Na 2SO 4水溶液,在两级的溶液中各加数滴石蕊试液,在电解的过程中两极区溶液的颜色有何变化?5.当电流通过下列电解池时,判断有哪些物质生成或消失,并写出反应式。
(1)碳为阳极,铁为阴极,溶液为氯化钠;(2)银为阳极,镀有氯化银的银为阴极,溶液为氯化钠;(3)两铂电极之间盛以硫酸钾溶液。
6.电解ZnCl 2水溶液,两极均用铂电极,电解反应如何?若均改用锌电极,结果又如何?两者的分解电压有何差异?二、计算题1.用金作阳极,镍作阴极,电解 1.0 mol·dm -3H 2SO 4溶液,求:分解电压为多少伏?(O 2在Ni 上的超电势η(H 2)=0.4V ,O 2在金上的超电势η(O 2)=0.53V ,在298K 时φø [O 2/H 2O ,H +]=1.229V)。
[答案:V (分解)=φ(阳)-φ(阴)=1.899 V ]2.用Pt 电极电解CuCl 2溶液,通过的电流为20A ,经过20min 后,问:(1).在阴极上能析出多少质量的Cu ?(2).在阳极上能析出多少体积的298K ,100kPa 下的Cl 2(g)?[答案:(1)m=0.2009kg ;(2).V(Cl 2)=0.0031m 3]3.298K 时电解含两种金属离子的盐溶液)1,01.0(12=⋅=±-γkg mol b FeCl 和)1,02.0(12=⋅=±-γkg mol b CuCl 。
若电解过程中不断搅拌溶液,超电势忽略不计。
试问:①何种金属首先析出?②当第二种金属析出时,第一种金属离子在溶液中的浓度为多少?[答案:① Cu 先析出;②2910214.42-⨯=+Cu a ]4. 在411CuSO kg mol -⋅及42102.0SO H kg mol -⋅的混合液中,使铜镀到Pt 电极上。
第十章电解与极化作用本章要求:1.了解分解电压的意义,要使电解池

第十章 电解与极化作用本章要求:1.了解分解电压的意义,要使电解池不断工作必须克服哪几种阻力?2.了解什么是极化现象,什么是超电势?极化作业有哪几种?如何降低极化作用?3.了解电解的一般过程及应用,特别是有关电解分离提纯方面的应用。
4.了解金属腐蚀的类型以防止金属腐蚀的常用方法。
电解池:使电能转变成化学能的装置当一个电池与外接电源反向对接时,只要外加电压大于该电池的电动势E ,电池中的反应逆向发生,原电池就要变成电解池,要使电解池继续正常工作,外加电压要比电池电动势E 大很多,这些额外的电能一部分用来克服电阻,一部分用来克服电极的极化作用极化作用:当电流通过电极时,电极电势偏离其平衡的现象,且该过程是步可逆过程。
§10.1 分解电压在电池上外加一个直流电源,并逐渐增加电压,使电池中的物质在电极上发生化学反应,称为电解。
如电解HCl 水溶液阴极: ).(()p g aH e H H 222→+-++阳极:).(p g Cl e Cl 222→--- 总反应: )()()(p P aq Cl H HCl 222+−−→−电解 分解装置P118图10.1,并绘制电流─电压曲线。
由P118图10. 电流─电压曲线可看出:① 当开始加外电压时,还没有)()(g g Cl H 22和生成,P=0 电路中几乎没有电流通过。
② 当稍增大外电压,电极表面有少量)()(g g Cl H 22和产生,其压力虽小,却构成了一个原电池,产生了与外加电压方向相反的反电动b E 由于压力很小,低于大气压力,产生气体不能离开电极自由逸出,而是扩散到溶液中消失,此时此时就需要通入极微小的电流使电极产物得到补充,相当于图1─2段。
③ 继续增大外电压,电极时上)()(g g Cl H 22和继续产生,当22cL H P P 和等于外界大气压力时,电极上开始有气泡逸出,此时反电动势b E 达到了最大值MAX b E 而不在继续增加,若此时继续增大外电压,则电流急曾,如图曲线2─3段直线部分。
10章 电解与极化作用

二、电极极化的原因 有电流通过电极时, 电极上会发生一系列过程(离子 的扩散、电极反应…), 每一步或多或少存在阻力, 要克服这些阻力需要一定的推动力, 反映在电极上表 现为电极电势的偏离. 根据极化产生的不同原因,通常把极化大致分为两类 :浓差极化和电化学极化。 (1)浓差极化 当有限电流通过电极时,由于 离子扩散的迟缓性导致电极表面与本体溶液离子浓
2H (aq) 2e H2 (g) i /A 1 阳极 2OH (aq) H 2 O(l) O 2 (g) 2e 2 对抗电解过程的具有反电动势的原电池: 阴极
(Pt) H2(g)|H2SO4(0.5mol· dm-3)|O2(g)(Pt)
1 O 2 E0 E /V 3
在缄性溶液中:
H2O Me e Me H OH
(b) H3O+和已被吸附电极表面的H原子反应生 成H2; 也为电化学脱附.
H3O Me - H e Me H2 H2O
④ 吸附在电极上的H原子化合为H2
Me H Me H 2Me H2
⑤ H2从电极上扩散到溶液内或形成气泡逸出.
2
2
2
0.894V
2
可见,由于氢超电势的存在,H2析出困难的多; 即使在 pH = 4 的溶液, H2析出也在Cd之后,此时:
H ,析 0.2366 H 0.7166 V
2 2
可见, 由于氢超电势存在, 使得活动次序在H 之 前的活泼金属能优先析出; 甚至Na+在汞阴极上也会 生成钠汞齐, 不会放出H2.
例2 电解 CdSO4(a± = 1) 水溶液, 氢在金属Cd 析出超电势为0.48V. 解: 在阴极上析出反应:
(整理)第10章电解与极化作用

第十章电解与极化作用一、本章主要内容§10.1 分解电压§10.2 极化作用§10.3 电解时电极上的反应§10.4 金属的电化学腐蚀与防腐§10.5 化学电源二、本章重点与难点1、分解电压的概念。
2、极化作用。
3、电解时电极上的反应。
4、金属的电化学腐蚀与防腐。
5、化学电源。
三、教学目的1、掌握电化动力学的一般原理;2、掌握电化学的基本理论和技能,为后续专业课的学习奠定坚实的理论基础。
四、教学要求1、了解分解电压的意义。
2、了解产生极化的原因,了解氢超电势在电解中的作用。
3、能计算一些简单的电解分离问题。
4、了解金属腐蚀的原因和各种防腐的方法。
5、了解化学电源的类型及应用。
五、授课时数8学时用Nernst 方程式处理电化学体系时,都有一个前提,即该体系需处于热力学平衡态。
所以用Nernst方程研究的问题具有很大的局限性。
一切实际的电化学过程都是不可逆过程。
对不可逆电极过程进行的研究,无论是在理论上或实际应用中,都有非常重要的意义。
因为要使电化学反应以一定的速度进行,无论是原电池的放电或是电解过程,在体系中总是有显著的电流通过。
因此,这些过程总是在远离平衡的状态下进行的。
研究不可逆电极反应及其规律对电化学工业有着十分重要的意义。
因为它直接涉及工艺流程、能量消耗、原料消耗等因素。
本章我们将讨论电解过程中在电极上进行的不可逆反应,从中得出不可逆电极过程的一些规律,将它们应用于电镀、电化学腐蚀、化学电源等方面。
§10.1 分解电压一、理论分解电压使某电解质溶液能连续不断发生电解反应时所必须外加的最小电压称为理论分解电压。
理论分解电压在数值上等于该电解池作为可逆电池时的可逆电动势:E (理论分解电压)=E (可逆)二、分解电压的测定若外加一电压在一个电池上,逐渐增加电压,使电池中的化学反应发生逆转,这就是电解。
当直流电通过电解质溶液时,正离子向阴极迁移,负离子向阳极迁移,并分别在电极上起还原和氧化反应,从而获得还原产物和氧化产物。
电解与极化作用

第九章电解与极化作用对于电解池,它是一个使电能转变成化学能的装置,是一个电化学的反应器,电解池在工作时是必须有电流通过的通常,一个电解池要连续正常工作,所加的电压要比电动势大得多,这些额外的电能,一部分用以克服电阻,一部分消耗在克服电极的极化作用上,如下列反应:Cu2+(aq) + 2Cl-(aq) = Cu(s) + Cl2(g)E 外CuCl 2石墨Cu −+E 外加电压:η++>IR E E 平衡外电解反应才能顺利进行,实验现象为:Cu 电极:Cu 2++ 2e →Cu(s)石墨电极:2Cl -→Cl 2(g) + 2e Cu 2++ 2Cl -→Cu(s)+ Cl 2(g)无论是原电池还是电解池,只要有电流通过,在电极上就有极化作用发生,电极极化为不可逆过程(1)电极的极化作用(2)金属腐蚀与防腐和电化学的应用(3)化学电源本章主要介绍三方面的内容:§9.1 分解电压1. 分解电压测定在电解一给定的电解液时,对电解池至少需要施加多少大的电压才能使电解顺利进行⎯⎯分解电压。
以铂电极电解0.1mol·dm-3的NaOH水溶液为例,说明分解电压的测定。
分解电压测定装置:VG 阳极阴极0.1m NaOH 溶液Pt 通过可变电阻逐渐增加外加电压,同时记录电流计的变化值,得到电流~电压曲线。
EI 曲线可分为三段:(1)1之前电压升高,电流变化不大→0,现象:在电极上没有气泡的产生。
12(2)1~2 电压升高,电流开始慢慢增大,在电极上有少许的气泡产生,并吸附在电极上。
(3)2~3. V ↑→I ↑↑在电极有大量的气泡逸出。
3从曲线外推得到E 分解=1.69V 。
E 分解=1.69V实验测定的分解电压要大于原电池:Pt, H2(pθ) | NaOH(0.1 mol·kg-1) | O2(pθ), Pt的电动势E可逆=1.229VE分解为实际分解电压,各种电解液的分解电压是通过实验测定E分解>E可逆2. 产生分解电压的原因在电解NaOH 水溶液时,电极反应:阴极:2H 2O + 2e = H 2+ 2OH -阳极:2OH -= ½O 2+ H 2O + 2e 阴极析出氢气,阳极析出氧气,并有部分H 2和O 2吸附在铂电极表面,结果组成了氢氧原电池:-Pt, H 2(p) | NaOH (0.1mol·kg -1)| O 2, Pt +E 外该原电池的极性正好与外加电源的极性相同,产生反电动势。
10-物化-下-第十章-电解与极化作用

根据Tafel 公式,η~lnj 为直线,j (<1) →0,η→―∞, 与事实不符。当j→0,η→0,φ不可逆→ φ可逆。 即电流密度很小时,氢超电势不符合Tafel 公式,而遵守 η=ωj 即η与j成正比。 电解时H 在阴极放电机理: 电解时 +在阴极放电机理:(p.125) 对氢超电势研究较多的原因: 对氢超电势研究较多的原因: (p.126)
a
M
,2
a a
M
Z+
,1 ,2
= 10
7
M
Z+
若以溶液中的残余量小于 10-7mol/l 作为判断是否分离彻底 的标准,只须计算出当溶液中某种金属离子的浓度从1~10-7 mol/l 变化时电极电势的差值,即可将浓度差值转化到电势的 差值来进行判断。 当 浓度为c1时,φ1=φө+(0.059/z)lgc1 ; 当 浓度为c2时,φ2=φө+(0.059/z)lgc2 ; 若 c1=10-7mol/l ,c2=1mol/l , ∆φ=φ2-φ1=(0.059/z)lg(c2/c1)= (0.059×7)/z 则:当 z=1 时, ∆φ=0.41 伏 当 = = 当 z=2 时, ∆φ=0.21 伏 = = 当 z=3 时, ∆φ=0.14 伏 = = 利用上述原理,也可使两种离子同时在阴极上析出而形成 合金,即调整两种离子的浓度,使其具有相等的析出电势。
练习题: 练习题 1. 298K和pө下,用Zn电极电解含Zn2+的水溶液。若要使 Zn2+的浓度降到10-7mol/kg时才允许H2(g)析出,问应如何控制 溶液的pH值?设H2 (g)在Zn (s)上的超电势为0.7V并假定此值与 溶液浓度无关;已知:φө(Zn2+/Zn)=-0.763V,假定各离子活 度系数均为1。 解: Zn2+的浓度为10-7mol/kg时 φ(Zn2+/Zn)=φө(Zn2+/Zn) +(RT/2F)lna(Zn2+) =-0.736+(RT/2F)ln10-7=-0.97V φ(Zn2+/Zn) =φ(H+/H2)析 =φ(H+/H2)平=φө (H+/H2)+(RT/2F)lna(H+ )2-η -0.97 =-0.05916pH-0.7 pH = 4.56
物理化学第十章 电解与极化作用
3、析出电势 :
ϕ阳,不可逆 = ϕ阳,析出 = ϕ阳,可逆 + η阳 ϕ阴,不可逆 = ϕ阴,析出 = ϕ阴,可逆 − η阴
三、极化曲线-超电势的测定 1、测定超电势的装置
2、电解池中两电极的极化曲线
j(电流密度)
阴极曲线
阳极曲线
E可逆+ΔE不可逆
E可逆
η阴
η阳
电
−ϕ
+ϕ
电解池中两电极的极化曲线
正极: 负极:
LiCoO 2 , LiNiO 2 , LiMn 2 O 2
石墨,焦炭
2
正极反应: L i C o O
+
充 + Z Z Z X L i C o O + x L i + YZ Z Z 1 -x 2 放 −
充 ZZZ X Li C 负极反应: C+xLi + xe YZZ Z x 放
总反应:
Ag + (a ) Ag ( s ) Ag + (a ) + e − → Ag ( s ) RT 没有电流通过时 : ϕ Ag + / Ag (可逆) = ϕ + + ln a Ag + Ag / Ag F RT θ 有电流通过时:ϕ Ag + / Ag (不可逆) =ϕ + + ln a’ + Ag / Ag Ag F 扩散速度小于电极反应速度,a’ + < a Ag +
3、原电池中两电极的极化曲线
η阳
j(电流密度)
E可逆 -ΔE不可
η阴
负 极 曲 线 E可逆
正 3;ϕ
电解池中两电极的极化曲
4、氢超电势
电解和极化作用

金属型电解质
在熔融状态下能导电的金 属氧化物,如氧化铝、氧 化铁等。
电解质的导电性
电离程度
电解质在水溶液中的导电能力与其电离程度成正 比,电离程度越大,导电能力越强。
带电粒子
电解质在水溶液中导电依赖于带电粒子的运动, 带电粒子越多,导电能力越强。
离子迁移率
带电粒子在电场作用下的迁移率决定了电解质的 导电能力,迁移率越大,导电能力越强。
对物质导电性的影响
01
极化作用可以改变物质内部的电子分布,从而影响物质的导电
性能。
对物质介电常数的影响
02
介电常数是表征物质电介质性能的重要参数,极化作用可以显
著改变物质的介电常数。
对物质光学性质的影响
03
由于极化作用可以改变物质内部电子云的分布,因此对物质的
光学性质如折射率、反射率等也有重要影响。
对未来研究的展望
随着对电解和极化作用认识的深入,未来研究将更加 关注复杂电化学体系的反应机制和动力学过程,以揭
示更广泛的规律和现象。
输标02入题
新型电化学电极材料和电解质的研发将为电解和极化 作用提供更多可能性,有助于提高电化学过程的效率 和稳定性。
01
03
随着可持续发展理念的深入人心,电解和极化作用在 可再生能源转换和储存、环境治理等方面的应用将得
当物质处于外电场中时,物质中 的电荷会受到电场力的作用,产 生相对位移,导致极化现象的产
生。
热运动的影响
在热运动的作用下,物质中的原子 或分子的电子云分布会发生不规则 的涨落,从而导致极化现象的产生。
晶体结构的作用
物质的晶体结构对其极化性质也有 重要影响,不同晶体结构的物质具 有不同的极化率。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
表面
本体
则 IR R
浓差= IR - R
阳= IR - R 阳 阴= R - IR 阴
结论:
①浓差极化使阴减小,阳增加 ②可通过加热、搅拌降低浓差(因电极表面有 扩散层存在,不可能完全消除浓差)。 ③极谱分析(利用滴汞电极上所形成的浓差 极化)。
的作用。
3. 能计算一些简单的电解分离问题。
前
言
如前所述,对于G < 0的自发反应,可 设计成原电池,产生电功,但对于G > 0的 反应,必须对系统作功,例如加入电功,反应 才能进行。电解反应即是其中一种。
使电能转化为化学能的装臵称为电解池。当一 个电池与外接电源反向对接时,只要外加的电压大 于该电池的电动势E,电池接受外界所提供的电能, 电池中的反应发生逆转,原电池就变成了电解池。
2C l C l2 (g) 2e
上述电解产物与溶液中的相应离子在阴极和阳极 上又构成氢电极与氯电极,从而形成如下电池:
Pt | H2 | HCl( 1mol dm3 ) | Cl2 | Pt
此电池的电动势与外加电压相反,称为反电动势。
下面分析一下电流-电压曲线:
当开始施加外电压时, 几乎无电流通过,阴、阳极 上无H2 (g) 和Cl2(g)放出。
结论:活化极化 极化 = IR - R
析出电势: 阳,析出=阳,R 阳 阴,析出=阴,R - 阴 最终结果:阳极更正, 阴极更负
分解电压: E 分解 阳,析出 - 阴,析出 =E 可逆 阳 阴
3.极化曲线—超电势的测定
超电势或电极电势与电流密度之间的关系曲线称为极化曲线, 极化曲线的形状和变化规律反映了电化学过程的动力学特征。 +
极超电势 阳 分别定义为:
阴 (可逆 不可逆 )阴
阳 (不可逆 可逆 )阳
1.浓差极化
原因:由于电解过程中电极附近溶液的浓度和本体 溶液(指离开电极较远,浓度均匀的溶液)浓度发 生了差别所致。
如:Ag+(aq.)│Ag(s)电极 (1)阴极 Ag+ + e- Ag Ag+接受电子生成Ag速度快,而Ag+从溶液中 向电极表面扩散慢。
解:E = 甘汞 - 不可逆 = 1.0685V 而 不可逆 = 可逆-ηH2 (2H+ +2e-=H2) 所以:ηH2 = E- 甘汞+ 可逆 = 1.0685-0.2802+RT/FlnαH+ = 0.7883+0.05916lg(0.265×0.1) =0.6950V
(1)电解池中两电极的极化曲线
Emax) /R ,R 指电解池电阻。
以下,我们由表列实验结果,来分析分解电压与原电池的 电动势(即理论分解电压,由能斯特方程算得值)的关系。
表:10.1 几种电解质溶液的分解电压(室温,铂电极)
电解质 HCl HNO3 H2SO4 NaOH CdSO4 NiCl2 浓度 c / mol ·dm-3 1 1 0.5 1 0.5 0.5 电解产物 H2 + Cl2 H2 + O 2 H2 + O 2 H2 + O 2 Cd + O2 Ni + Cl2 E分解 /V 1.31 1.69 1.67 1.69 2.03 1.85 E理论/ V 1.37 1.23 1.23 1.23 1.26 1.64
反电动势达极大值 Eb,max。
电 流 I
3
再增加电压,使I 迅速增 加。将直线外延至I = 0 处, 得E(分解)值,这是使电解
2 1
E分解
电压E
池不断工作所必需外加的 最小电压,称为分解电压。
测定分解电压时的电流-电压曲线
我们把外加电压等于分解电压时两极的电极电 势分别称为它们的析出电势。
若外加电压大于分解电压,则电流 I = ( V –
中的杂质等。
Tafel 公式(Tafel’s equation)
早在1905年,Tafel 发现,对于一些常见的电 极反应,超电势与电流密度之间在一定范围内存在 如下的定量关系:
j a b ln j
式中j是电流密度,[j]是j的单位,这样表示 使对数项中为纯数。a,b是常数。常数a是单位电流
电势,才使得镀Zn,Sn,Ni,Cr等工艺成为现实。
氢在几种电极上的超电势
从氢气在几种电极上的超电势,在石墨和汞等
材料上,超电势很大,而在金属Pt,特别是镀了铂
黑的铂电极上,超电势很小,所以标准氢电极中的 铂电极要镀上铂黑。 影响超电势的因素很多,如电极材料、电极表面 状态、电流密度、温度、电解质的性质、浓度及溶液
后,电流随电压的增加而急剧增加,如下图:
这时两极出现气泡。图中的D点所示电压,是 使电解质在两极继续不断进行分解所须最小外加电 压,称为分解电压。
在外加电压作用下,氢离子向阴极(负极) 运动,在阴极被还原为氢气。
2H 2e H 2 (g)
氯离子向阳极(正极)运动,在阳极被氧化为 氯气。
A
电 极 2 电 极 1
电位计
甘汞电极
例:298K时,用Pb(s)电极来电解H2SO4溶液(0.10mol· kg-1, γ ±=0.265),若在电解过程中把Pb阴极与另一摩尔甘汞电极相连 组成原电池,测得其电动势E=1.0685V,试求H2(g)在Pb上的超 电势。 (只考虑H2SO4的一级电离。已知φ甘汞=0.2802V)
RT 1 RT ln ln a Ag F a Ag F
a Ag
IR R 平衡状态
表面
a Ag
本体
(2)阳极 Ag Ag+ + e-
RT ln a Ag F a Ag a Ag
§10.1 分解电压
理论分解电压 使某电解质溶液能连续不断发生
电解时所必须外加的最小电压,在数值上等于该电
解池作为可逆电池时的可逆电动势。
E(理论分解 ) E(可逆)
电解: 在电池上若外加一个直流电源,并逐渐增加电 压直至使电池中的物质在电极上发生化学反应,这 就是电解过程。
下图是一个实验:在大气压力下,于1 mol·dm-3 的盐酸溶液中放入两个铂电极,电极两端的电压由伏 特计V测出,电流由安培计G测出,电位大小由可变电 阻R的位臵调节。当外加电压很小时,几乎没有电流, 电压增加,电流仍增加很小。但当电压增到某一定值
可逆 (阳),可逆 (阴)
在有电流通过时,随着电极上电流密度的增加, 电极实际分解电势值对平衡值的偏离也愈来愈大,这 种对可逆平衡电势的偏离称为电极的极化。
在某一电流密度下,实际发生电解的电极电势 不可逆 与可逆电极电势
可逆 之间的差值称为超电势。
为了使超电势都是正值,把阴极超电势 阴 和阳
实际上若使电解池连续正常工作,所加电压E外>>E。
如:电解饱和食盐水制H2、Cl2
, NaOH的氯碱
工业,E=1.3V, 槽电压为2030V,为的是克服 电阻和极化作用(在原电池放电与电解池电解时 ,都有一定量电流通过电极,电极平衡状态被破 坏,电极过程为不可逆的,这种电极电势偏离平 衡电极电势的现象称为电极的极化。 本章介绍电解池的极化作用、电解在工业上的应 用及金属的防腐和化学电源等。
2.电化学极化 电极反应总是分若干步进行,若其中一步反应
速率较慢,需要较高的活化能,为了使电极反应顺
利进行所额外施加的电压称为电化学超电势(亦称
为活化超电势),这种极化现象称为电化学极化。
如:氢电极 (1)阳极:H2 2H+ + 2er(电化学反应速率) < r(电子迁移速率),导致 阳极缺电子,即正电荷积累,致使IR(阳)升高 (2)阴极:2H+ + 2e- H2 r(电化学反应速率) < r(电子迁移速率),导致阴 极富余电子,即负电荷积累 ,致使IR(阴)降低
利用这种极化降低金属的电化腐蚀速度。
η阳 j(电流密度)
E可逆 -ΔE不可逆
η阴
E去极化作用: 加入某物质(去极化剂)较易在电极上反应,可使极 化减小或限制在一定程度内。
金属在电极上析出时超电势很小,通常可忽略不计。 而气体,特别是氢气和氧气,超电势值较大,不可忽略。
=阳,R 阳 阳,析出 =阴,R - 阴 阴,析出
1.阴极上的反应
电解时阴极上发生还原反应
发生还原的物质通常有(1)金属离子,(2)氢离子
(中性水溶液中 aH 107 )。
+
判断在阴极上首先析出何种物质,应把可能发
生还原物质的电极电势计算出来,同时考虑它的超
电势。电极电势最大的首先在阴极析出。
中间三行分解产物均是氢气与氧气,实质是电解水。
电解质 HNO3
浓度 c / mol ·dm-3 1
电解产物 H2 + O2
E分解 /V 1.69
E理论/ V 1.23
H2SO4
NaOH
0.5
1
H2 + O2
H2 + O2
1.67
1.69
1.23
1.23
由此可看出,用平滑铂电极时,实际的 E分解 常大于E理论 。 即使将溶液、导线和接触点的电阻降到可忽略不计的程度,分 解电压还是大于原电池电动势。这主要是由于析出电极电势偏 离平衡电极电势的原因。为了对每一个电极上的过程进行深入 的研究,应当研究电流密度与电极电势的关系。
平衡电极电势 E理论
析出电极电势
实际E分解
实际分解电压
要使电解池顺利地进行连续反应,除了克服作
为原电池时的可逆电动势外,还要克服由于极化在
阴、阳极上产生的超电势 (阴) 和 (阳) ,以及克服电