TALEN和CAS9技术移码敲除测序峰图分析方法说明-仅供参考
新一代TALEN对比cas9的优势

新一代TALEN对比cas9的优势上海斯丹赛生物技术有限公司●新一代TALEN操作比cas9更加简单容易● TALEN比cas9更加精确● TALEN相比cas9总体而言,投入成本更低,更便宜①新一代TALEN操作比cas9更加简单容易做cas9需要对载体酶切,胶回收,而TALEN可以实现一步连接。
新一代TALEN连接1.设计序列,选择模块2.加样,进行连接4-5h完成Cas9系统质粒构建sgRNA设计和oligo合成引物退火形成带粘性末端的双链片段线性化表达载体的制备(酶切、胶回收)双链oligo片段连接入表达载体②TALEN比cas9更加精确很多的文章和报道都说明了cas9的脱靶问题。
No evidence for TALEN off-targetingDing Q, Lee YK, Schaefer EA, et al. A TALEN genome-editing system for generating human stem cell-based disease models [J]. Cell Stem Cell, 2013, 12(2):238-51.本文结合全基因组测序法,在6个细胞系中分别对设计的TALEN靶序列在基因组上可能的10万个脱靶位点处进行了检测,均未发现任何脱靶效应;对外显子区进行测序比对,未发现TALEN存在脱靶效应。
TALEN 无明显脱靶效应TALEN 对基因进行有效编辑时未检测到任何的脱靶突变Liu H, Chen Y, Niu Y, et al. TALEN-Mediated Gene Mutagenesis in Rhesus and Cynomolgus Monkeys[J]. Cell stem cell, 2014.在这项新研究中,研究人员通过采用显微注射法将MECP2靶向TALEN质粒导入猴受精卵中,成功对MECP2进行了有效基因编辑,且未检测到任何的脱靶突变。
TALEN靶向基因操作技术

TALEN靶向基因操作技术技术介绍:TALE 技术(Transcription activator–like effectors)是一种崭新的分子生物学工具。
科学家发现,来自植物细菌Xanthomonassp.的TAL蛋白的核酸结合域的氨基酸序列与其靶位点的核酸序列有恒定的对应关系。
利用TAL的序列模块,可组装成特异结合任意DNA序列的模块化蛋白,从而达到靶向操作内源性基因的目的。
目前TALE技术主要有两种应用:1)TALEN(transcription activator-like (TAL) effector nucleases)技术构建针对任意特定核酸靶序列的重组核酸酶,在特异的位点打断目标基因DNA,进而在该位点进行DNA操作,如Knock-out、Knock-in 或点突变。
它克服了常规的ZFN方法不能识别任意目标基因序列,以及识别序列经常受上下游序列影响等问题,而具有ZFN相等或更好的活性,使基因操作变得更加简单、方便。
2)TALEA(transcription activator-like (TAL) effector activator)技术,针对基因启动子上游任意特定DNA序列构建转录激活因子,可提高特异内源基因的表达水平,而不需要购买或克隆cDNA。
TALE技术已经成功应用到了细胞、植物、酵母、斑马鱼及大、小鼠等各类研究对象,日益成为功能强大的实验室工具,使得过去无法逾越的项目成为可能。
技术特点:1.无基因序列、细胞、物种限制。
2.TAL的核酸识别单元与A、G、C、T有恒定的对应关系。
实验设计简单准确、实验周期短、成本低。
3.成功率几乎可达100%。
4.毒性低、脱靶情况少。
5.克服了常规的ZFN方法不能识别任意目标基因序列,以及识别序列经常受上下游序列影响等问题,而具有ZFN相等或更好的活性。
TALE构建与应用:1.TALE靶点识别模块构建TAL的核酸识别单位为重复34个恒定氨基酸序列,其中的12、13位点双连氨基酸与A、G、C、T有恒定的对应关系,即NG识别T,HD识别C,NI识别A,NN识别G。
基因编辑简介(Cas9,Talen,ZFN)

TALEN基因编辑技术前言转录激活样效应因子核酸酶(transcription activator-like effector nuclease, TALEN)技术与锌指核酸酶(Zinc-finger nuclease, ZFN)技术组成了一大类强有力的基因组编辑工具,这一大类技术的发展重新划定了生物学研究的边界。
这些嵌合核酸酶由两部分组成——一个可编码的序列特异性DNA结合模块与一个非特异性的DNA切割结构域。
通过诱导DNA双链断裂(DNA double-strand break)来刺激容易出错的非同源末端连接或在特定基因所在的位置进行的同源定向修复,TALEN和ZFN能够完成一系列遗传学编辑修饰操作。
成簇规律间隔短回文重复(clustered regulatoryinterspaced short palindromic repeat, CRISPR)技术是最新出现的一种基因组编辑工具,它能够完成RNA导向的DNA识别及编辑。
与其它基因组编辑工具相比,CRISPR技术更易于操作,具有更强的可扩展性。
本文将以上述三种技术为例,介绍并探讨新一代位点特异性基因组工程技术的生物学原理、未来发展趋势,及其在遗传学研究领域的作用和潜在的医学应用前景。
一、TALEN技术TAL效应因子(TAL effector, TALE)最初是在一种名为黄单胞菌(Xanthomonas sp.)的植物病原体中作为一种细菌感染植物的侵袭策略而被发现的。
这些TALE通过细菌 III类分泌系统(bacterial type III secretion system)被注入植物细胞中,通过靶定效应因子特异性的基因启动子来调节转录,来促进细菌的集落形成。
由于TALE具有序列特异性结合能力,研究者通过将FokI核酸酶与一段人造TALE连接起来,形成了一类具有特异性基因组编辑功能的强大工具,即TALEN。
近年来, TALEN已广泛应用于酵母、动植物细胞等细胞水平基因组改造,以及拟南芥、果蝇、斑马鱼及小鼠等各类模式研究系统。
CRISPR与Talen基因组编辑技术对比分析

CRISPR与Talen基因组编辑技术对比分析基因组编辑技术一直是生物学和医学领域的研究热点。
近年来,CRISPR和Talen成为最受关注的两种基因组编辑工具。
本文将对CRISPR和Talen进行对比分析,从技术原理、效率、精确性、应用范围等方面综合评价它们在基因组编辑中的优缺点。
首先,我们来探讨这两种技术的原理。
CRISPR(聚合酶鏈反應辅助人工制造的双链RNA)技术利用CRISPR-Cas9系统进行基因组编辑,其中Cas9是一种核酸内切酶,能够识别特定DNA序列并实现DNA切割。
CRISPR系统通过引导RNA(gRNA)与Cas9结合,使其精确识别目标DNA序列,从而实现靶向编辑。
与之相比,Talen(转录激活样基因分子)技术使用转录激活样基因分子(TAL)蛋白质和DNA结合结构域构建。
Tal蛋白质通过与DNA靶位点结合,识别与目标DNA序列互补的核酸,并通过特定核酸串联域(repeats)和变异核酸接头(TAL effectorDNA-binding domain)实现DNA的编辑。
Talen的特点是能够精确定位到任意DNA序列,并且可以进行多点编辑,但其设计和构建过程较为复杂。
在效率方面,CRISPR技术相对于Talen具有更高的编辑效率。
由于CRISPR-Cas9系统利用了高度特异的RNA-DNA序列互补配对,因此能够更容易地找到并切割目标DNA序列。
相比之下,Talen需要通过设计和构建Tal蛋白质与特定DNA序列结合,这一过程较为繁琐。
因此,CRISPR在实现基因组编辑时具有更高的效率和灵活性。
精确性是基因组编辑技术的关键指标之一。
在这方面,CRISPR技术存在一定的局限性。
由于CRISPR-Cas9系统一般通过非同义突变来引导编辑,存在一定的脱靶效应,可能导致编辑到非目标位点的DNA序列。
虽然研究人员已经通过改进gRNA设计和Cas9蛋白质的改造来减少脱靶问题,但在实际应用中仍存在一定的风险。
TALEN和CRISPRCas9技术介导的靶向基因破坏和同源修复的开题报告

TALEN和CRISPRCas9技术介导的靶向基因破坏和同源修复的开题报告标题:TALEN和CRISPRCas9技术介导的靶向基因破坏和同源修复摘要:TALEN和CRISPRCas9技术是当前最常用的靶向基因编辑技术。
这两种方法都能够高效地切割DNA双链,引发靶向基因的破坏、转录调控或同源修复。
其中,靶向基因破坏和同源修复是应用最广泛的功能。
本文主要介绍TALEN和CRISPRCas9技术在靶向基因破坏和同源修复方面的原理、方法和应用。
在靶向基因破坏方面,我们详细分析了TALEN和CRISPRCas9技术介导的靶向基因破坏前端子的选择、设计和优化;在同源修复方面,我们阐述了TALEN和CRISPRCas9介导的同源修复的两种不同策略:基于质粒介导的同源修复和基于核酸修饰介导的同源修复。
关键词:TALEN;CRISPRCas9;靶向基因破坏;同源修复正文:一、TALEN和CRISPRCas9技术介绍TALEN技术是由转录激活样核苷酸修饰(Transcription activator-like effector nucleases)蛋白和双链DNA切割酶FokI蛋白组成的。
TALEN蛋白能够识别靶向DNA上的特定序列,由于携带着特殊的重复、变异结构域,因此它具有高选择性和高亲和力。
组合使用FokI蛋白产生两个股份的断裂,实现基因定点打靶和破坏。
CRISPRCas9技术是目前最为受欢迎和广泛应用的靶向基因编辑技术。
它由靶向RNA(guide RNA, gRNA)和具有核酸内切酶活性的Cas9蛋白组成。
gRNA指示Cas9精确定位到靶向基因上,并通过三个核酸酶活性位点之一引发DNA双链断裂,完成基因编辑。
二、TALEN和CRISPRCas9介导的靶向基因破坏1、TALEN和CRISPRCas9的优缺点TALEN和CRISPRCas9均有精准、高效的优点,但TALEN有更高的精度,因为TALEN识别靶向DNA序列更为精确;CRISPRCas9技术能够进行基因组广泛的编辑,因为它的gRNA能够非常灵活地修饰,从而获得不同的精度和选通性,并且gRNA片段长度更短,更容易与目标序列结合。
基因编辑技术6

CRISPR/Cas9的发现和发展
1987年,Ishino等研究大肠杆菌iap 基因的序列和功能时发现,在该基因的3’
端非翻译区存在一段高度保守的的重复序列,这些重复序列被长度为32nt 的非重复序列间隔开。 在2002 年将其命名为CRISPR (clustered regularly interspaced short palindromic repeats)。
Cas (CRISPR-associated genes)
(20~50 bp)
2007 年,Barrangou 等首次利用实验证明了嗜热链球菌的CRISPR
系统直接参与细菌对噬菌体的适应性免疫反应。
24
产脓链球菌CRISPR/Cas9 干扰噬菌体或外源质粒入侵示意图
CRISPR/Cas的三种类别
CRISPR/Cas9在PAM (5’-NGG) 上游 3bp进行切割。
实验的关键步骤是设计一对20bp左 右(不包括NGG在内的)与基因目标 区域完全互补的oligo插入载体元件 中。
设计sgRNA
36
基点 PAM:NGG
• PAM要在CDS区搜索; • PAM上下游20nt左右; • 对于复杂基因组,避免同源位点,以减少Off-Target效应。
16bp)分别进行TALE识别模块的构建。
TALENs基因打靶过程 9
TALEN基因敲除步骤
10
TALENs靶点识别模块构建
TALENs表达质粒构建
TALENs质粒对共转入
目标基因敲除突变体筛选
11
4种RVD单元
单元组装
12
第一轮 第二轮
第三轮
TALENs表达质粒构建
左侧半位点TALE模块
唯尚立德内部资料-TALEN CRISPR培训

Cas9-工作原理
Nature Biotech. 2012:30, 836–838
TALEN、Cas9/gRNA-神奇的基因剪刀
Double Strand Breaks (DSB) Repair(DNA双链断裂) 细胞内的DNA损失修复方式:
Double strand breaks (DSB)
Template
唯尚立德的大规模TALE制备技术
优势: 不同于传统的酶切连接方法,我们的方法利用在固相表面实现快速合成,实现 快速、高效大规模制备TALE, 一天可以合成上百条TALE Ref: Zhao Wang et al, An Integrated Chip for the High-throughput Manufacture of TAL Effectors. Angew. Chem. Int. Ed. 2012 (Accepted)
利用Cas9-gRNA-体外细胞水平敲除基因
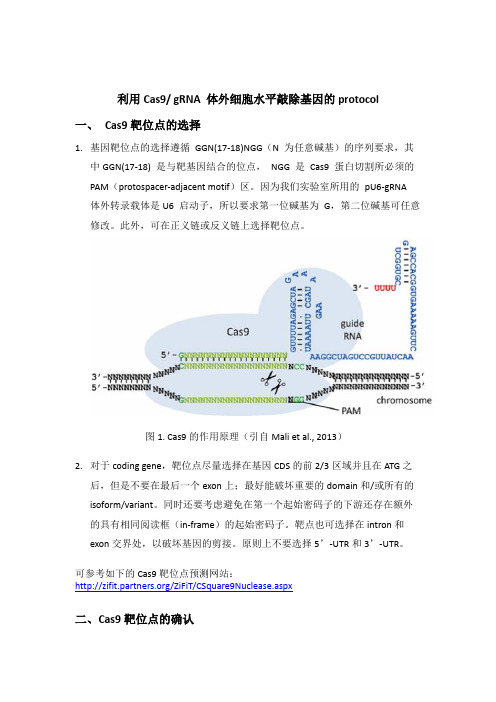
利用Cas9/ gRNA 体外细胞水平敲除基因的protocol一、Cas9靶位点的选择1.基因靶位点的选择遵循GGN(17-18)NGG(N 为任意碱基)的序列要求,其中GGN(17-18) 是与靶基因结合的位点,NGG 是Cas9 蛋白切割所必须的PAM(protospacer-adjacent motif)区。
因为我们实验室所用的pU6-gRNA 体外转录载体是U6 启动子,所以要求第一位碱基为G,第二位碱基可任意修改。
此外,可在正义链或反义链上选择靶位点。
图1. Cas9的作用原理(引自Mali et al., 2013)2.对于coding gene,靶位点尽量选择在基因CDS的前2/3区域并且在ATG之后,但是不要在最后一个exon上;最好能破坏重要的domain和/或所有的isoform/variant。
同时还要考虑避免在第一个起始密码子的下游还存在额外的具有相同阅读框(in-frame)的起始密码子。
靶点也可选择在intron和exon交界处,以破坏基因的剪接。
原则上不要选择5’-UTR和3’-UTR。
可参考如下的Cas9靶位点预测网站:/ZiFiT/CSquare9Nuclease.aspx二、Cas9靶位点的确认1. 确认靶点在基因组中的唯一性:靶位点序列在Ensembl/NCBI网站进行blast,确认是基因组中的单一位点。
2.PCR扩增靶点序列:从准备用于打靶的样品中PCR扩增靶点及附近的序列。
在靶位点周围设计引物,使其距离靶位点两侧都大于100 bp,并且PCR扩增产物最好不要超过500 bp,且为单一条带。
3.检测酶切效率:如果计划采用酶切法检测靶点的突变情况,则需要对上述PCR产物进行酶切验证。
要求尽量酶切完全,并且电泳后能明显与原条带区分开。
4.测序确认靶点序列:将PCR产物直接送去测序(不需要TA克隆)。
如果实测序列跟靶点的预测序列有出入,原则上应根据实测结果设计gRNA序列;但是,如果实测序列不再符合靶点选择的原则(例如,PAM区不是NGG,或5’端不是GG),则应重新选择靶点。
CRISPR及Cas9基因编辑技术具体步骤及方法

CRISPR/Cas9基因编辑技术具体步骤及方法CRISPR/Cas9 是一种能够对基因组的特定位点进行精确编辑的技术。
其原理是核酸内切酶Cas9蛋白通过导向性RNA(guide RNA, gRNA)识别特定基因组位点并对双链DNA进行切割,细胞随之利用非同源末端连接(Non homologous End Joining,NHEJ)或者同源重组(Homologous Recombination, HR)方式对切割位点进行修复,实现DNA水平基因敲除或精确编辑。
CRISPR基因敲除利用CRISPR / Cas9 进行单基因敲除目前研究最透彻、应用最广泛的II 型-CRISPR/Cas9 系统由两部分组成:1. 单链的guide RNA(single-guide RNA,sgRNA)2. 有核酸内切酶活性的Cas9 蛋白CRISPR/Cas9 系统利用sgRNA 来识别靶基因DNA,并引导Cas9 核酸内切酶剪切DNA(图1)。
当基因组发生双链DNA 断裂后,细胞通过非同源性末端接合(Non-homologous end joining, NHEJ) 将断裂接合,在此过程中,将随机引入N 个碱基的缺失或增加,若N 非3 的倍数,则目的基因发生移码突变,实表1 CRISPR/Cas9 基因敲除与RNAi 比较CRISPR过表达利用CRISPR / Cas9 进行单基因过表达通过修饰CRISPR/Cas9 系统中的一些元件,形成一种蛋白复合物-协同激活介质(SAM),可实现对多数细胞内源基因的特异性激活。
该系统灵活方便,为研究基因功能提供了极为便利的工具。
CRISPR-SAM 系统由三部分组成:1. 失去核酸酶活性的dCas9(deactivated Cas9)-VP64 融合蛋白2. 含2 个MS2 RNA adapter 的sgRNA3. MS2-P65-HSF1 激活辅助蛋白CRISPR-SAM 系统中的MS2-P65-HSF1 激活辅助蛋白就是SAM,全称为SynergisticActivation Mediator( 协同激活调节器),这也就是CRISPR-SAM 的命名由来。
手把手教你学会CRISPRCas9基因敲除技术(需要挑选成对的靶点一般在正义链和反义链上分。。。

⼿把⼿教你学会CRISPRCas9基因敲除技术(需要挑选成对的靶点⼀般在正义链和反义链上分。
h ttp:///a/214091994_177233(需要挑选成对的靶点⼀般在正义链和反义链上分别挑选靶点配对)C RISPR/Cas 是进⾏基因编辑的强⼤⼯具,可以对基因进⾏定点的精确编辑。
在向导 RNA(guide RNA, gRNA)和 Cas9 蛋⽩的参与下,待编辑的细胞基因组 DNA 将被看作病毒或外源 DNA,被精确剪切。
⼀、寻找⽬的基因的靶标使⽤在线设计⽹站 CRISPR direct,如需直接复制⽹址,可在⽣物学霸后台对话框回复 direct即可。
靶点挑选要点:1. 基因敲除靶点应设计在起始密码⼦附近(包括起始密码⼦)或者起始密码⼦下游的外显⼦范围内。
2. 不同Cas9/gRNA 靶点在基因敲除效率上有较⼤差异,因此同时设计构建2~3 个靶点的基因敲除载体再从中选出敲减效果较佳的靶点。
3. N1-N20NGG 靠近 PAM 的碱基对靶点的特异性很重要,前 7~12 个碱基的错配对 Cas9 切割效率影响较⼩。
设计好的靶点序列应在基因库中进⾏ BLAST 检测。
4. Cas9Nicknase 需要挑选成对的靶点。
⼀般在正义链和反义链上分别挑选相距 20~30bp 的靶点配对。
多对靶点的敲除效率常有较⼤差异。
由于基因敲除实验时间长,在正式对⽬的细胞进⾏敲除前对靶点进⾏验证和挑选⾮常必要。
⼆、插⼊⽚段设计插⼊寡核苷酸序列设计(必须 PAGE 纯化寡核苷酸):正向序列5’ACACCGNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTT3’反向序列3’TGTGGCNNNNNNNNNNNNNNNNNNNCAAAATCTCGATCTTTATCGTTCAATTTTATTCCGATCAGGCAA5’插⼊⽚段的合成1. ⽤⽔将寡核苷酸稀释为 100 µM。
基因敲除技术,talen技术

•
•
•
•
基因同源重组法敲除靶基因的步骤
•
•
有嵌合体得到基因敲除的纯合体小鼠
条件性基因敲除法
• 利用Cre/LoxP 和来自酵母的FLP—frt 系统可以研究特定组织器官 或特 定细胞中靶基因灭活所导致的表型[7]。通过常规基因打靶在基 因组的靶位点上 装上两个同向排列的1oxP,并以此两侧装接上loxP 的(“loxP floxed”)ES 细 胞产生“loxPfloxed”小鼠,然后,通过 将“loxP floxed”小鼠与Cre 转基因鼠 杂交(也可以其他方式向小 鼠中引入Cre 重组酶),产生靶基因发生特定方式(如 特定的组织特 异性)修饰的条件性突变小鼠。在“loxP floxed”小鼠,虽然靶基 因的两侧已各装上了一个loxP,但靶基因并没有发生其他的变化,故 “1oxP noxed”小鼠表型仍同野生型的一样。但当它与Cre 转基因 小鼠杂交时,产生的子代中将同时带有“loxP floxed”靶基因和Cre 基因。Cre 基因表达产生的Cre 重组酶就会介导靶基因两侧的1oxP 间发生切除反应,结果将一个loxP 和靶基因 切除。这样,靶基因的 修饰(切除)是以Cre 的表达为前提的。Cre 的表达特性决 定了靶基 因的修饰(切除)持性:即Cre 在哪一种组织细胞中表达,靶基因的修 饰 (切除)就发生在哪种组织细胞;而Cre 的表达水平将影响靶基因 在此种组织细胞中进行修饰的效率。所以只要控制Cre 的表达特异性 和表达水平就可实现对小鼠中靶基因修饰的特异性和程度[9,10]。
TALEN技术
• 表达一个重组核酸酶, 在靶点识别结构域的 作用下,识别靶点核 酸序列,核酸酶发挥 内切酶活性,打断目 标基因,完成基因敲 除的过程。
TALEN基因敲除基本流程
TALEN靶向基因敲除技术

TALEN靶向基因敲除TALENs(transcription activator-like (TAL) effector nucleases),中文名是转录激活因子样效应物核酸酶,TALENs是一种可靶向修饰特异DNA序列的酶,它借助于一种由植物病原菌黄单胞菌(Xanthomonas)自然分泌的天然蛋白即激活因子样效应物(TAL effectors, TALEs)---的功能:该蛋白能够识别特异性DNA碱基对。
人们可以设计一串合适的TALEs来识别和结合到任何特定序列,如果再附加一个在特定位点切断DNA双链的核酸酶,就生成了TALENs。
TAL效应核酸酶可与DNA结合并在特异位点对DNA链进行切割,从而利用这种TALEN就可以在细胞基因组中引入新的遗传物质。
相对锌指核酸酶(zinc-finger nuclease, ZFN)而言,TALEN能够靶向更长的基因序列,而且也更容易构建。
但是直到现在,人们一直都没有一种低成本的而且公开能够获得的方法来快速地产生大量的TALENs。
TALEN 靶向基因敲除技术是一种崭新的分子生物学工具,现已应用于植物、细胞、酵母、斑马鱼及大、小鼠等各类研究对象。
研究发现,Xanthomonas TAL蛋白核酸结合域的氨基酸序列与其靶位点的核酸序列有较恒定的对应关系。
研究者们利用来自Xanthomonas TAL的序列模块,构建针对任意核酸靶序列的重组核酸酶,在特异的位点打断目标基因,敲除该基因功能。
成功解决了常规的ZFN方法不能识别任意目标基因序列,以及识别序列经常受上下游序列影响识别特性的问题,使基因敲除变得简单方便。
技术特点:·无基因序列、细胞、物种限制。
·实验周期短。
·整个实验简单准确,成本低。
·成功率可达90%以上。
·毒性低、脱靶情况少。
·TAL的核酸识别单元与A、G、C、T有恒定的对应关系。
·靶序列识别模块不受上下游影响,特异识别并打断基因组中的任意靶序列。
基因编辑技术的使用教程及实践项目介绍

基因编辑技术的使用教程及实践项目介绍基因编辑技术是一种可以精确修改生物体基因组的工具,可以帮助我们了解基因在生物体发育和疾病中的作用。
本文将介绍基因编辑技术的使用教程以及一些实践项目,旨在帮助读者更好地理解和应用这一技术。
首先,我们需要了解基因编辑的主要工具,包括CRISPR-Cas9和TALEN。
CRISPR-Cas9是一种基于细菌天然防御机制的技术,可以通过对基因组进行切割和修复来实现基因编辑。
TALEN(转录活化剂效应核酸酶)是一种由转录因子结合结构域和核酸外切酶融合而成的人工核酸内切酶,能够精确切割DNA序列。
接下来,让我们以CRISPR-Cas9为例介绍基因编辑的步骤。
首先,选择目标基因组中需要编辑的位点,设计引物和gRNA(RNA导向的DNA切割)来指导Cas9酶的识别和切割。
然后,通过合成或转染的方式将gRNA和Cas9酶导入到细胞中。
Cas9酶通过与gRNA结合来找到目标基因组的特定位点,并将其切割。
在细胞的自身修复过程中,可以通过提供外源的DNA片段来插入或删除特定的基因序列,实现基因组的精确编辑。
基因编辑技术不仅可以在细胞水平上进行,也可以在整个生物体中进行。
现在让我们来介绍一些基因编辑的实践项目,以帮助读者更好地应用这一技术。
其中一个实践项目是利用基因编辑技术来研究人类疾病。
通过编辑特定的基因序列,研究人员可以模拟和研究许多常见疾病的发生机制,例如癌症、先天性疾病等。
这些研究有助于寻找新的治疗方法和药物,以改善人类的健康。
另一个实践项目是利用基因编辑技术来改良农作物。
基因编辑技术可以帮助农业科学家精确改变农作物的基因组,以提高产量、抵抗病虫害、改善抗逆性等。
例如,通过编辑水稻基因组,可以使其对抗部分病虫害更加有效。
这种方法可以减少农药的使用量,降低环境污染和农民的成本,为粮食安全和可持续农业发展做出贡献。
除了上述项目,基因编辑技术还可以应用于保护濒危物种、改变生物的特征以及生产药物等方面。
如何应对基因编辑实验中的偏倚序列问题

如何应对基因编辑实验中的偏倚序列问题基因编辑是一种重要的生物技术,可以用于改变生物体的基因组。
然而,在进行基因编辑实验时,我们必须面对一个关键问题,那就是偏倚序列问题。
偏倚序列是指在基因编辑过程中,某些特定的DNA序列在编辑中发生偏向性过度修复的现象。
这种偏倚会导致编辑结果的不准确性,从而影响实验的可靠性和可重复性。
因此,如何应对基因编辑实验中的偏倚序列问题是极为重要的。
首先,要解决偏倚序列问题,我们需要了解其产生的原因。
偏倚序列问题的产生往往与基因编辑技术中所使用的核酸酶有关。
常见的基因编辑技术包括CRISPR-Cas9和TALEN。
这些技术使用特定的核酸酶来识别和剪切靶基因的DNA。
然而,由于核酸酶的特异性不完全,它们不仅会剪切目标基因的DNA,还会剪切与目标序列相似的非目标序列。
这些非目标序列被剪切后,会引发细胞自身的DNA修复机制,从而导致偏倚序列的产生。
为了应对偏倚序列问题,我们可以采取以下几种方法。
首先,我们可以选择合适的基因编辑工具。
不同的基因编辑工具对偏倚序列的产生有不同的程度的影响。
研究表明,CRISPR-Cas9技术相比于TALEN技术更容易产生偏倚序列。
因此,在实验设计中,我们可以根据具体情况选择合适的基因编辑工具,以减少偏倚序列的产生。
其次,我们可以优化基因编辑实验的条件。
偏倚序列的产生很大程度上与核酸酶的特异性有关。
因此,我们可以调整核酸酶的浓度和反应条件,以增加其特异性。
此外,还可以通过优化引物设计和反应体系来减少偏倚序列的产生。
例如,选择合适的引物和放大条件,可以在一定程度上降低非目标序列的扩增,从而减少偏倚序列的发生。
除了调整实验条件,我们还可以利用新近开发的工具来解决偏倚序列问题。
肥大细胞启动子结合酶(dCas9)和基因组编辑活化和抑制系统(CRISPRa和CRISPRi)是近年来新兴的基因编辑技术工具。
与传统的CRISPR-Cas9技术不同,dCas9不具有切割功能,但可以识别和结合到目标靶基因的DNA上。
棉花团队系统总结基因编辑脱靶效应

棉花团队系统总结基因编辑脱靶效应基因组编辑(Genome Editing, GE)技术是当前生命科学研究的前沿领域,近年来基因组编辑技术发展迅猛,出现了多种不同的基因组编辑方法包括锌指核酸酶(ZFN)技术,转录激活样效应因子核酸酶(TALEN)技术和串联间隔短回文重复序列(CRISPR/Cas)等系统,其中以CRISPR/Cas9系统最为便捷高效,应用也最广泛。
目前CRISPR/Cas9系统已用于治愈人类遗传病、实现细胞个性化治疗、开发新药、作物遗传改良等领域。
但CRISPR技术存在的脱靶效应(Off-target effect)依旧是影响CRISPR技术能否广泛应用的主要限制因素,如何正确评估、检测脱靶效应,并提出相应的策略降低脱靶效应是当前基因编辑研究领域的重要研究方向。
为什么要关注基因编辑脱靶问题?CRISPR/Cas9系统在基因组编辑中显示出巨大的潜力,但其脱靶可能给宿主生物带来严重的问题。
脱靶可以导致染色体重排,在不完全匹配的基因组处造成损伤,并限制了GE在人类医学研究上的应用。
除干扰染色体稳定性外,脱靶效应还可能导致功能基因活性的丧失,从而导致各种生理或信号异常(图1)。
在植物中,CRISPR/Cas9已在超过30种植物物种和数百个基因实现成功编辑。
与人类的基因疗法和临床研究不同,植物研究不受伦理学问题的影响,并且对GE的脱靶效应的耐受性可能更高。
虽然对于植物脱靶效应可以通过育种过程表型筛选来消除脱靶突变或自发突变,但对科学问题的研究依然带来很大的不确定性。
因此非常有必要对基因组编辑中存在的脱靶问题进行系统分析,为未来更加高效、精准地利用基因编辑技术提供理论依据及解决方案。
图1. CRISPR/Cas9系统的脱靶效应基因编辑脱靶效应是如何产生的?Cas9与DNA之间的相互作用首先是通过PAM位点的识别和结合来介导的,同时crRNA和潜在靶标编辑序列之间的结合进行定向反应,形成稳定的R-循环,进而激活酶切活性使PAM近端DNA断裂。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.为什么TALEN和CAS9技术移码敲除项目需通过测序判定基因型?
TALEN和CAS9技术是通过特异识别和核酸酶切割,使DNA双链断开,机体启动DNA损伤修复机制,在非同源末端连接修复过程中,碱基的随机增减造成目标基因功能缺失。
这一修复过程通常会产生1-30个左右的碱基缺失,PCR后凝胶电泳无法将碱基缺失链和wt链区分开,所以只能通过测序判断。
2.如何判断TALEN和CAS9技术移码敲除项目的基因型?
a. 野生型或基因敲除的纯合子基因型的判定:
1)如下图所示,只有单峰的为野生型或基因敲除的纯合子。
2)将只有单峰的序列文件与wt序列比对,可判定野生型或基因敲除的纯合子基因型(网站或生物软件比对均可).
如下图的对比结果为野生型:
如下图的比对结果为缺失一个C的纯合子
b.杂合子基因型的判定:
1).如下图所示,测序图谱中出现叠峰的为杂合子
若将叠峰的序列文件直接与wt序列比对,叠峰后的序列会对应不上(因为叠峰的序列只显示了信号强一点的那个碱基,实际上每个叠峰对应两个碱基),如下图所示:
所以不能通过直接比对来分析,需通过峰图分析,在峰图中峰的颜色与碱基对应关系如下:A:绿色
T:红色
C:蓝色
G:黑色
2)将wt的峰图与杂合子的峰图用软件同时打开,从出现叠峰的位置开始分析(杂合子的一条链是wt链,一条链是缺失链):
上图第一个峰图是野生型的序列,与第二个峰图叠峰开始对应的序列为:
Gttaact ccgagcagcaaagaaatgatgtccc
上图第二个峰图是杂合子的测序峰图,从叠峰位置开始的序列为:
Gttaact ccgagcagcaaagaaatgatgtccc(wt链)
Ccgagcagcaaagaaatgatgtcccaagcctt(缺失链)
由此可判定为该杂合子的基因型为缺失gttaact(-7)。