(参考课件)糖原累积症
糖原累积症健康教育课件

谁会受影响?
谁会受影响? 遗传风险
糖原累积症是常染色体隐性遗传病,父母双 方都是携带者时,子女有25%的概率发病。
家族中有历史病例的人群需特别关注。
谁会受影响? 高风险群体
某些类型的糖原累积症在特定人群中更为常 见,如阿什肯纳兹犹太人。
对这些群体进行基因筛查是有必要的。
与营养师合作制定个性化的饮食方案。
如何管理糖原累积症? 药物治疗
根据医生建议,患者可能需要服用酶替代治 疗或其他药物,以帮助控制症状。
药物疗法需在专业医生指导下进行。
如何管理糖原累积症? 生活方式调整
适度运动和良好的作息习惯有助于整体健康 ,但需避免剧烈运动。
患者应定期与医生沟通,调整运动计划。
该病通常因缺乏特定的酶而引起,分为多种类型 ,每种类型的症状和影响不同。
什么是糖原累积症?
发病机制
糖原累积症的发生与特定酶的缺乏有关,这些酶 负责将糖原转化为可用的葡萄糖。
缺乏这些酶会导致糖原在细胞内堆积,从而影响 细胞功能。
什么是糖原累积症?
流行病学
糖原累积症相对少见,发病率因类型而异,一些 类型主要影响儿童,而其他类型则可能在成年后 出现。
谁会受影响? 年龄因素
糖原累积症的症状可在出生时即出现,也可 能在儿童或成年期才显现。
早期发现和干预有助于改善预后。
何时寻求医疗帮助?
何时寻求医疗帮助? 症状警示
常见症状包括低血糖、肌肉无力、疲劳、肝脏肿 大等,任何异常症状应及时就医。
家长应特别留意儿童的成长发育情况。
何时寻求医疗帮助? 定期检查
糖原累积症健康教育课件
演讲人:
目录
1. 什么是糖原累积症? 2. 谁会受影响? 3. 何时寻求医疗帮助? 4. 如何管理糖原累积症? 5. 如何提高对糖原累积症的认识?
糖原累积症病人的护理

如何进行护理?
教育与宣传
向患者和家属普及糖原累积症相关知识,提高其 自我管理能力。
可开展健康讲座和发放宣传材料。
护理的目标是什么?
护理的目标是什么?
改善生活质量
通过有效的护理措施,帮助患者改善生活质量。 包括身体健康和心理健康的提升。
护理的目标是什么?
预防并发症
什么是糖原累积症?
类型
根据致病基因的不同,糖原累积症可分为多种类 型,如I型(高尔基病)、II型(庞贝病)等。
每种类型的临床表现和治疗方法可能有所不同。
什么是糖原累积症?
病因
糖原累积症主要是由于酶缺陷,导致糖原合成或 分解不正常。
遗传因素在疾病的发生中起着关键作用。
谁会得糖原累积症?
谁会得糖原累积症?
例如,某些类型在北欧国家的发病率较高。
何时进行护理?
何时进行护理?
症状出现时
当患者出现低血糖、肌肉无力、疲劳等症状时, 应及时进行护理干预。
监测患者的血糖水平是关键。
何时进行护理?
定期检查
定期进行血液和肌肉功能检查,以监测病情变化 和评估治疗效果。
建议每年至少进行一次全面评估。
何时进行护理?
遗传背景
糖原累积症通常为常染色体隐性遗传,父母双方 携带该基因时,子女有25%的机会发病。
家族史是判断风险的重要因素。
谁会得糖原Байду номын сангаас积症?
发病年龄
糖原累积症可在婴幼儿期或青少年时期发病,某 些类型可在成年后显现症状。
早期发现有助于改善预后。
谁会得糖原累积症?
高风险群体
某些种族或地区可能因遗传因素而具有更高的发 病率。
糖原累积症有哪些症状?

糖原累积症有哪些症状?*导读:本文向您详细介绍糖原累积症症状,尤其是糖原累积症的早期症状,糖原累积症有什么表现?得了糖原累积症会怎样?以及糖原累积症有哪些并发病症,糖原累积症还会引起哪些疾病等方面内容。
……*糖原累积症常见症状:静脉曲张、腹水、肌肉萎缩、肝功能不全、心脏增大、无力、肝肿大*一、症状:糖原累积症Ⅲ型诊断靠酶学分析,并可根据酶学分析结果将GSD-Ⅲ分成不同亚型。
1.糖原累积症Ⅲ型体检检查本身难与GSD-Ⅰ型区别。
婴儿期肝肿大、发育障碍较突出,部分患儿在4~6岁时可出现脾肿大,此患儿可有肝纤维化的证据,但不一定发展为肝硬化和肝功能衰竭。
除肝病变外,大部分患者有肌无力,尤其疾走和爬山时,但不会发生肌肉痉挛。
部分患者有肌肉萎缩。
糖原可累积在心脏,出现心脏增大。
心电图有非特异性变化,但不发生心力衰竭和心律失常,肾脏不大。
低血糖较GSD-Ⅰ轻,青春期时肝脏有缩小趋势,只有伴有磷酸酶或磷酸酶缺乏者才会走向肝硬化。
2.糖原累积症Ⅳ型婴儿出生后几个月多无症状,一岁内症状隐匿,但最早出现症状可在3个月时,最晚是15个月。
可有一些非特异性消化道症状,肝、脾肿大,肝功能不全生长迟缓等症状和体征,肌肉张力低、萎缩。
随病情发展可有腹壁静脉曲张、肝硬化门脉高压、腹水和食道静脉曲张,该病诊断后存活期多为2~37个月,偶3~4年,最后多死于慢性肝功能不全、上消化道出血、心力衰竭、感染。
*二、诊断:1.Ⅰ型诊断依据(1)临床表现:肝大、空腹低血糖、身材矮小、肥胖等。
(2)血液生化检查:空腹血糖低血三酰甘油及胆固醇升高,血乳酸、尿酸升高。
(3)胰高糖素试验:胰高糖素0.5mg肌内注射,每15分钟测血糖持续2h,正常人10~20min后空腹血糖可上升3~4mmol/L,本病患者上升0.1mmol/L2h内血糖仍不升高,乳酸上升3~6mmol/L,并加重已有的乳酸性酸中毒鶒,血pH值降低(4)肝穿刺活检:是本病确诊依据测定患者肝糖原常超过正常值6%火罐网葡萄糖-6-磷酸酶活性降低以至缺失,细胞核内有大量糖原沉积。
【自动保存】可治性罕见病—糖原累积病可治性罕见病—糖...

【自动保存】可治性罕见病—糖原累积病可治性罕见病—糖...可治性罕见病—糖原累积病可治性罕见病—糖原累积病一、疾病概述糖原累积病(glycogen storage disease,GSD)是糖原代谢过程中各种酶缺陷导致的一组先天性遗传代谢性疾病,以糖原在全身各器官组织中过度沉积为特征,主要累及肝脏或(和)肌肉,是一类较常见的代谢性疾病,发病率1:20 000~43 000。
根据受累器官,又被分为肝糖原累积病和肌糖原累积病。
肝糖原累积病包括Ⅰ、Ⅲ、Ⅳ、Ⅵ、Ⅸ、Ⅺ及O型等,约占GSD总体发病率的80%,其中Ia型最常见;肌糖原累积病包括Ⅱ、V、Ⅶ型,多见于成人发病,表现运动不耐受、肌无力或肌病,其中Ⅱ最常见,仅Ⅱ型婴儿表现为婴儿心肌病和肌病。
糖原累积病Ⅰ a型(glycogen storage disease type Ⅰ a,GSDIⅠa)是由于细胞微粒体膜上的葡萄糖一6—磷酸酶(glucose -6 - phosphatase,G6PC)催化亚单位先天性缺陷所致[1]。
GSD I a型的活产儿中发病率约为1:100 000,约占肝糖原累积病的30%。
糖原累积病Ⅱ型(GSDⅡ)又称庞贝病(Pompe disease),是肌GSD 最常见的类型,是由于溶酶体内酸性一ɑ一葡萄糖苷酶(acid -a - glucosidasc.GAA)缺陷所致,中国台湾新生儿疾病筛查资料显示患病率1:50 000[2,3]。
二、临床特征GSDⅠa型患儿可在生后开始出现症状,主要表现为新生儿或婴儿早期低血糖和乳酸中毒,但新生儿期因其对治疗反应良好或者很容易被控制使许多患者延误诊断。
出生时常有肝大,超声波检查常发现肾脏肿大。
婴儿期首发症状可仅表现为严重的酸中毒,患儿可表现为明显腹部膨隆,生长落后,表现专匀称性矮小。
由于面颊部脂肪沉积出现娃娃脸表现,四肢肌力小,而且瘦弱。
低血糖症状常在夜间吃奶减少.生后3~4个月开始出现,表现为低血糖所致的易激惹、苍白、多汗、睡眠不稳甚至惊厥,或表现为清晨的呕吐和惊厥,频繁喂养可减少发作。
糖原累积病(Ⅰ型、Ⅱ型)流程图
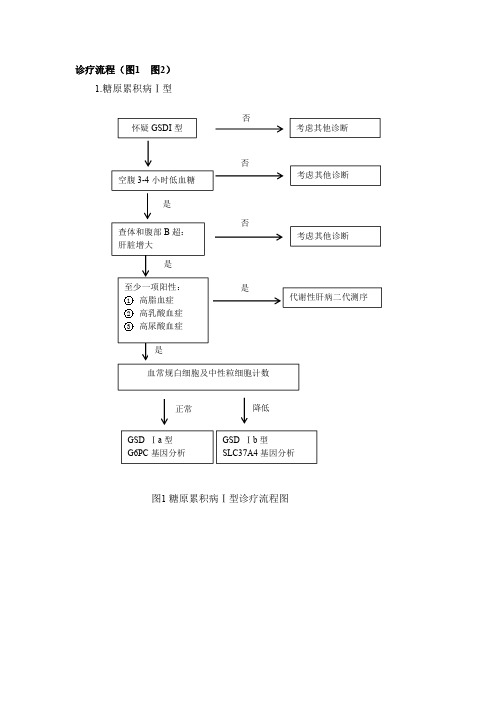
诊疗流程(图1图2)1.糖原累积病Ⅰ型图1糖原累积病Ⅰ型诊疗流程图怀疑GSDI 型空腹3-4小时低血糖GSD Ⅰa 型G6PC 基因分析血常规白细胞及中性粒细胞计数考虑其他诊断代谢性肝病二代测序考虑其他诊断考虑其他诊断GSD Ⅰb 型SLC37A4基因分析否是正常是否否降低2.糖原累积病Ⅱ型图2糖原累积病Ⅱ型诊疗流程图参考文献[1]Priya S,Kishnani,Stephanie L.Austin,Jose E.Abdenur.Diagnosis and management of glycogen storage disease type I:a practice guideline of the American College of Medical Genetics and Genomics.Genet Med,2014,16(11):e1.[2]/sites/genetests[3]邬玲仟,张学.医学遗传学.北京:人民卫生出版社,2016:362-368.[4]Van der Meijden JC,Gunggor D,et al.Ten years of the international Pompe survey:patient reported outcomes as a reliable tool for studying treated and untreated children and adults with non-classic Pompe disease[J].J Inherit Metab Dis,2015,38(3):495-503.否怀疑GSDII 型1岁前发病GAA 基因检测GAA 酶活性降低肌肉无力、运动落后心脏增大、心肌肥厚CK 增高肌肉容积减少呼吸肌和脊旁肌无力CK 增高肌肉活检糖原聚集1岁后发病必要时是是是是是[5]中华医学会儿科学分会内分泌遗传代谢学组,中华医学会儿科学分会神经学组,中华医学会神经病学分会肌电图与临床神经生理学组,中华医学会神经病学分会神经肌肉病学组.糖原贮积病型诊断及治疗专家共识.中华医学杂志,2013,93(18):1370-1373.。
糖原累积症

糖原累积症糖原累积症是指组织中糖原过多的一组疾病,有多种类型,由遗传性酶的缺陷或糖原结构异常所致。
【诊断】糖原累积症Ⅲ型诊断靠酶学分析,并可根据酶学分析结果将GSD-Ⅲ分成不同亚型。
【治疗措施】1.糖原累积症Ⅲ型高蛋白饮食可改善生长发育状态和肌力,但最近研究认为高淀粉、标准蛋白饮食效果更好。
2.糖原累积症Ⅳ型无特效疗法,高蛋白低糖饮食,加食玉米油未能阻止肝硬化进程。
用纯化的葡萄糖素未能取得肯定效果。
曲霉菌属提取物可使肝糖原急剧减少,因而是一个有研究前途的治疗方法。
此外,可施行肝移植。
【病因学】1.糖原累积症Ⅲ型病因为肝和肌肉内淀粉-1,6-葡萄糖苷酶(脱支酶)缺陷,糖原由磷酸化酶分解后,不能进一步彻底分解为葡萄糖。
2.糖原累积症Ⅳ型由于淀粉-(1,4-1,6)-转葡萄糖苷酶(分支酶)缺陷所致。
所积贮的糖原结构异常,外链长、分支减少,结构似支链淀粉,故又称支链淀粉病。
所积贮的异常糖原溶解度远低于正常糖原。
糖原累积症罕见,为常染色体隐性遗传,杂合子的成纤维细胞内有分子酶缺陷。
【发病机理】糖原累积症至少分成12种类型之多,其中0型(糖原合成障碍)和Ⅳ型(淀粉-1-4,1-6转葡萄糖苷酶缺乏),都会导致肝硬化和肝功能衰竭。
Ⅰ型(葡萄糖-6-磷酸酶缺乏)可发展为良性肝腺瘤和腺癌,Ⅲ型(淀粉-1,6-葡萄糖苷酶缺乏、脱支酶缺乏)可发展为肝纤维化或肝硬化。
【病理改变】1.糖原累积症Ⅲ型本型有①肝内纤维隔;②无脂肪沉积,此两点与G SD-Ⅰ不同。
GSD-Ⅲ型,肝硬化往往发生在两个酶以上同时缺陷,即除脱支酶缺陷外,还有磷酸酶和/或磷酸激酶的缺陷。
与GSD-Ⅰ比较,GSD-Ⅲ超微结构示脂肪滴小且少。
除肝脏病变外,肌肉也有糖原累积。
2.糖原累积症Ⅳ型本型肝脏呈小结节性肝硬化伴有宽纤维束围绕或插入肝小叶。
门脉区胆管轻度增生。
白色的两染性物质或嗜碱性染色物质沉积在肝细胞、心肌、骨骼肌和脑细胞。
肝小叶周边细胞内可发现嗜酸性或无色包涵体沉积在细胞浆,把肝细胞核推向一侧,构成了G SD-Ⅳ的特征性病变。
糖原累积症的科普知识PPT

通常包括定期进餐和摄入复杂碳水化合物。
糖原累积症的治疗方法是什么?
药物治疗
在某些情况下,可以使用药物来帮助管理症 状或补充缺乏的酶。
但并非所有类型的糖原累积症都有药物治疗 选择。
糖原累积症的治疗方法是什么? 定期监测
患者需要定期进行医学监测,以评估病情进 展和调整治疗方案。
这意味着两个携带者有25%的几率生出受影响的 孩子。
糖原累积症的症状有哪些?
糖原累积症的症状有哪些? 常见症状
患者可能出现低血糖、肌肉无力、疲劳、肝 脏肿大等症状。
这些症状通常在儿童期或婴儿期显现。
糖原累积症的症状有哪些?
并发症
长期未治疗可导致严重并发症,如心脏病、 肝硬化和肌肉萎缩。
及时的医疗干预可以减轻症状并改善生活质 量。
科研进展
随着科学研究的发展,治疗方法和管理策略也在 不断改进。
新兴的基因疗法和疗法正在研发中,有望为患者 带来新的希望。
谢谢观看
糖原累积症的症状有哪些?
个体差异
不同患者的症状表现可能差异很大,部分患 者可能症状轻微,而另一些则可能严重影响 生活。
这与具体的糖原累积症类型和个体的遗传背 景有关。
如何诊断糖原累积症?
如何诊断糖原累积症? 临床评估
医生会根据患者的病史和临床症状进行初步评估 。
通常涉及家族历史和症状出现的时间。
密切的随访可以帮助及时发现并发症。
糖原累积症的预后如何?
糖原累积症的预后如何? 生活质量
许多患者在遵循适当治疗后,可以享有相对正常 的生活质量。
早期诊断和干预是关键。
糖原累积症的预后如何? 潜在风险
糖原累积病性心肌病

高蛋白饮食
适量增加优质蛋白质摄入,如鱼、瘦肉、 豆类等,以增强肌肉力量和免疫力。
多吃蔬菜水果
富含维生素和矿物质,有助于心肌修复和 代谢。
控制饮食总热量
避免过度肥胖加重心脏负担。
心理干预和家庭支持
01 心理疏导
针对患者可能出现的焦虑、抑 郁等情绪问题,进行心理疏导 和干预。
究进展。
06
的遗传性疾病,由于体内糖原代谢途径中某些酶的缺乏导致糖原在肝脏、肌肉 等组织中过多累积。
深入了解糖原累积症的发病机制、临床表现及治疗方法,对于预防和治疗糖原累积病性心肌病具有重要 意义。
通过研究糖原累积症,可以进一步了解糖原代谢途径及相关酶的功能,为开发新的治疗方法和药物提供 理论基础。
心力衰竭
随着病情发展,患者可能出现心力衰竭的 症状和体征,如呼吸困难、水肿等。
心肌病理改变
糖原沉积
心肌细胞内可见大量糖原 沉积,导致心肌细胞肥大
、变性。
心肌纤维化
心肌间质纤维化明显,可 影响心肌的收缩和舒张功
能。
心肌细胞坏死
严重病例可见心肌细胞坏 死,进一步加重心功能不
全。
心功能受损程度评估
无症状期
糖原累积症是一种涉及多个学科 的罕见病,需要多学科专家共同
协作才能取得最佳治疗效果。
建立多学科协作机制,整合优势 资源,提高诊疗水平和效率,为
患者提供更好的医疗服务。
加强与国际罕见病组织的合作与 交流,借鉴先进经验和技术,推 动我国罕见病诊疗事业的发展。
谢谢您的聆听
THANKS
诊断标准
糖原累积症的诊断标准包括临床表现、实验室检查结果以及 基因检测发现相关基因突变等。同时,需要排除其他可能导 致类似症状的疾病。
Ⅲ糖原累积病及其治疗

Ⅲ糖原累积病及其治疗*导读:本型亦称Forbe"s病、Cori"s病,较常见,由于缺乏去支链酶(又称脱分支酶)所致,是一种常染色体隐性遗传性疾病,为糖原累积病较常见的类型之一。
……Ⅲ糖原累积病本型亦称Forbe"s病、Cori"s病,较常见,由于缺乏去支链酶(又称脱分支酶)所致,是一种常染色体隐性遗传性疾病,为糖原累积病较常见的类型之一。
本型以许多组织的糖原及糊精为特征。
由于去支链酶缺乏,以致糖原中1,6-糖苷键水解有困难,仅能经磷酸化酶分解糖原分子中1,4-糖苷键,直至糖原分子脱落而成界限糊精(limit dextrn),故本症又称局限糊精病。
由于糖原尚能进行磷酸化水解为葡萄糖,且糖原异生尚能进行,故低血糖症,高脂血症等不若第一型严重。
但幼儿患者,可有严重的糖,脂肪等代谢紊乱,身材矮,患者无乳酸性酸中毒,高尿酸血症,Fanconi"s综合征。
较轻者多见于成年人,仅有肌无力或无症状,肝脏多肿大,并可发展成肝纤维化,肝硬化。
由于肝糖异生正常,故空腹期低血糖症少见。
诊断和鉴别【诊断说明】根据:①上述临床表现。
②胰高血糖素试验:可分二阶段,初阶段于清晨空腹期进行,肌肉注射0.5mg后,本病患者血糖不增高或上升很少。
如于进食后2小时作第二阶段试验,仍肌肉注射0.5mg,则血糖可上升3~4mmol/L(54~72mg/dl),属正常反应。
在此两次试验中,血乳酸浓度一般不变。
③界限糊精试验:作肝或肌肉活检,可用碘测定有无糊精存在(呈紫色反应),还可用血红,白细胞试验,证实有界限糊精存在。
④本病病人亲属中携带杂合子基因者可从血红白细胞中测定脱枝酶活性,仅为正常人的50%左右。
治疗【治疗说明】防治方法同第I型。
由于免疫学上提示酶的缺乏属不完全性,可试用苯妥因钠防治低血糖症而获效,肝大可缩,糖原沉着也减少,血乳酸恢复正常,但酶活力仍不正常。
治疗机理可能抑制胰岛素分泌。
糖原累积症科普讲座课件

如何管理与治疗?
药物治疗
某些类型的糖原累积症可能需要药物干预以 改善症状或防止并发症。
例如,酶替代疗法在某些患者中显示出效果 。
如何管理与治疗? 定期随访
患者应定期进行医疗随访,以监测病情进展 并及时调整治疗方案。
多学科团队的合作也有助于优化管理方法。
为什么了解糖原累积症很重要 ?
糖原累积症科普讲座
பைடு நூலகம்演讲人:
目录
1. 什么是糖原累积症? 2. 谁会受到影响? 3. 何时出现症状? 4. 如何管理与治疗? 5. 为什么了解糖原累积症很重要?
什么是糖原累积症?
什么是糖原累积症?
定义
糖原累积症是一类遗传性代谢疾病,因糖原代谢 酶缺陷导致体内糖原过量积累。
此病影响多个器官,尤其是肝脏和肌肉,导致不 同的临床表现。
为什么了解糖原累积症很重要?
增加公众认知
提高公众对糖原累积症的认识,有助于早期发现 和干预,改善患者的生活质量。
知识的传播可以促进更多家庭的基因检测与咨询 。
为什么了解糖原累积症很重要?
支持患者及家庭
了解病症可以帮助患者及其家庭更好地应对生活 中的挑战,获取必要的支持与资源。
患者组织和支持群体可以提供情感和实践上的帮 助。
谁会受到影响?
谁会受到影响?
遗传因素
大多数糖原累积症是常染色体隐性遗传,这 意味着父母双方都需要携带相关基因,孩子 才可能发病。
因此,家族史在糖原累积症的诊断中非常重 要。
谁会受到影响?
发病率
糖原累积症的发病率较低,某些类型在特定 人群中更常见,尤其是某些地理区域或族群 。
例如,I型糖原累积症在北欧国家的发病率较 高。
儿童糖原累积病Ⅱ型诊断及治疗中国专家共识解读PPT课件

THANKS
糖原累积病Ⅱ型患者常有 低血糖及血酮体升高。
酶活性测定
可检测患者肌肉或肝脏中 酸性麦芽糖酶的活性,酶 活性降低是糖原累积病Ⅱ 型的特征。
基因检测
通过基因突变筛查可明确 诊断,并有助于分型及遗 传咨询。
诊断标准与鉴别诊断
诊断标准
结合临床表现、实验室检查和辅 助检查结果,符合糖原累积病Ⅱ 型的特征性表现即可诊断。
专家共识意义
通过专家共识的制定和推广,可以规范我国Pompe病的诊断 和治疗流程,提高临床医生的诊疗水平,改善患者的生活质 量和预后。
报告范围及目的
报告范围
本报告主要围绕Pompe病的诊断、治疗及随访管理等方面进行阐述,旨在为临 床医生提供全面、实用的指导建议。
报告目的
通过解读中国专家共识,提高临床医生对Pompe病的认识和诊疗水平,促进多 学科协作和综合治疗模式的建立,为患者提供更加优质、个性化的医疗服务。
通过医院内的健康讲座、一对一咨询、患者互助 小组等多种形式,为患者提供全面的教育支持。
心理干预策略选择和实践效果评价
心理干预策略
根据患者的年龄、性格特点和心理需求,选择合适的心理干预策略,如认知行为 疗法、家庭治疗、艺术治疗等,以缓解患者的焦虑、抑郁等负面情绪。
实践效果评价
定期对患者的心理状态进行评估,了解心理干预策略的实践效果,并根据评估结 果及时调整干预方案,以确保患者的心理健康。
深入研究糖原代谢通路
进一步揭示糖原合成、分解及调控机制,为开发新的治疗手段提 供理论支持。
拓展遗传学研究
探索糖原累积病Ⅱ型的遗传背景及基因突变与疾病表型的关系,为 基因诊断和精准治疗提供依据。
发掘新的生物标志物
寻找能够反映糖原累积病Ⅱ型病情进展和治疗效果的生物标志物, 为临床诊断和预后评估提供新的工具。
糖原累积病

临床表现:少见,典型为空腹出现低血糖,酮血症。进食或补充葡萄糖后长时间高血糖。反复低血糖抽搐可导致智能落后。饥饿时肾上腺素或胰高血糖素试验无反应。
受累组织:肝糖原缺乏,肝糖原合成酶活性小于2%,肌糖原正常。
2.I型(VonGierke病)
[病程观察]
本组病例多数随年龄增大而病情加重,严重者在婴儿期死亡。由于病因未能根治,对感染和低血糖的治疗效果只能是缓解。应重在预防,尽量改善病儿的生活质量。
住院小结]
(一)确定诊断
疾病的最后诊断依靠酶的活性测定,可取肝细胞、肌细胞等材料进行。
(二)预后评估
受累组织:经典Ⅱa型在全身所有器管都有糖原累积。
5.Ⅲ型(Cori病)
酶缺陷:淀粉-1,6-葡萄糖苷酶。
临床表现:中度至重度肝大,可有程度不等的肌张力低下,心脏增大、ECG异常少见,肝脏、心脏功能衰竭少见,智力发育正常。无酮血症、低血糖和高脂血症,进食后肾上腺素或胰高血糖素试验阳性,尿儿茶酚胺正常,预后好。
糖原合成从葡萄糖磷酸化开始,在肝脏由葡萄糖激酶催化,在肌肉则由己糖激酶催化,激酶的活性在饥饿时降低,进食时增高。磷酸化产生了6-磷酸葡萄糖,后者通过葡萄糖磷酸变位酶的作用转变成1-磷酸葡萄糖。1-磷酸葡萄糖再通过尿苷二磷酸葡萄糖焦磷酸化酶转变成尿苷二磷酸葡萄糖。后者中的葡萄糖残基通过糖原合成酶及淀粉-1,4→1,6转葡萄糖苷酶的作用,加入原有的糖原分子中,从而形成新的糖原。其中糖原合成酶使糖原直链增长,而淀粉-1,4→1,6转葡萄糖苷酶使糖原产生分支。
糖原的分解由两个酶系统完成。糖原分解成1-磷酸葡萄糖由磷酸化酶催
化,1-磷酸葡萄糖经过葡萄糖磷酸变位酶的作用转变成6-磷酸葡萄糖,后者在葡萄糖—各磷酸酶催化下,水解成葡萄糖。上述磷酸化酶只能分解到糖原分支点的前四个葡萄糖残基,剩余的葡萄糖残基通过脱支酶的作用,才能分解出葡萄糖。
糖原累积病Ⅰ型ppt课件

【葡萄糖-6-磷酸酶】
【葡萄糖-6-磷酸酶】
在正常人体中,由糖原分解或糖原异生过程所产生的6-磷酸 葡萄糖和必须经葡萄糖-6-磷酸酶系统水解以获得所需的 葡萄糖,该酶系统可提供由肝糖原分解所得的90%葡萄糖, 在维持血糖稳定方面起主导作用。当酶缺乏时,机体仅能获 得由脱枝酶分解糖原1,6糖苷键所产主的少量葡萄糖分子 (约8%),所以必然造成严重空腹低血糖。
【实验室检查】
空腹血糖低,此为糖原累积病Ⅰ型主要表现; 少数幼婴在重症低血糖时尚可伴发惊厥.但亦有血糖降至 0.56mmol/L(10mg/dl)以下而无明显症状者。随着年 龄的增长,低血糖发作次数可减少。
【实验室检查】
肝功能:正常或转氨酶轻度↑,胆红素无异常; 高甘油三酯血症:低血糖抑制胰岛素分泌,脂蛋白分解减 少,脂蛋白酶活性降低,使脂肪大量动员,刺激脂肪分解; 高尿酸血症:糖旁路代谢增强,尿酸合成增加; 高乳酸血症:由于葡萄糖产生障碍,代偿性乳酸增多造成;
【预后】
未经正确治疗的本病患儿因低血糖和酸中毒发作频繁常有 体格和智能发育障碍; 伴有高尿酸血症患者常在青春期并发痛风; 患者在成年期的心血管疾病、胰腺炎和肝脏腺瘤(或腺癌) 的发生率高于正常人群;
少数患者可并发进行性肾小球硬化症。
【护理】
生玉米淀粉疗法 饮食护理 腹泻的防护
• 科学合理地掌握好空腹取血时间。如患儿清晨2:00口服 玉米淀粉,可在晨7:00为患儿采集空腹血标本,以防患 儿发生低血糖。
【健康教育】
预防意外受伤:患儿腹部因肝脏持续增大而显著膨隆,使 患儿的活动受限,且一旦出现滑跌及易引起器脏破裂等严 重并发症。因此注意培养患儿的独立意识,使患儿能够对 自己的行为做出正确的选择; 遵医嘱服用钙剂,预防骨质疏松; 保持个人清洁卫生,预防感染,尽量避免到公共场所; 定期复诊,若出现发热、出血等症状及时就诊。
糖原累积病

糖原累积病第一节糖原合成、分解与代谢【糖原的合成与分解】食物中的碳水化合物经消化后以单糖形式(葡萄糖、果糖和半乳糖)被小肠吸收,进入血循环中的葡萄糖,一方面经无氧糖酵解与有氧氧化供给机体能量;另一方面可以糖原或脂肪形式贮存。
肝脏是人体贮存糖原的器官,肌肉是糖原贮存的组织,其糖原合成过程相同,可用下列化学反应式表示:①②③④葡萄糖-------6-磷酸葡萄糖---------1-磷酸葡萄糖-------1-磷酸葡萄糖+三磷酸尿苷(UTP)------------尿苷二磷酸葡萄糖+糖原(Gn)---------二磷酸尿苷+糖原(Gn+1)在上式中:①肝己糖激酶;②磷酸葡萄糖变位酶;③糖原合成酶;④分支酶。
糖原合成过程是耗能反应。
上述反应不断进行,由于分支酶的作用,使糖原分子形成许多分支,形状如“树”,其中含有许多葡萄糖分子。
葡萄糖分子与分子之间通过1,4链(占93%)和1,6链相连。
在分支外周末端的葡萄糖残基没有还原性。
糖原分解都是从糖原分子的最外层开始。
由于分支多,使肝脏在促糖原分解激素(胰高糖素和肾上腺素等)作用下能在短期内释放葡萄糖到血循环中去。
在禁食状态下,血糖能保持于正常范围,全靠肝脏中糖原分解释放出葡萄糖;在进食后则有肝糖原的合成,故肝和肌糖原每天均处于动态平衡状态。
如果长期禁食,已贮备的肝糖原将被耗尽,在这种情况下,肝脏释放葡萄糖的来源主要靠糖异生。
无论是糖原合成、糖原分解和糖异生,最后步骤的产物都是6-磷酸葡萄糖。
因此,作为6-磷酸葡萄糖产生的己糖激酶是前述3种代谢过程中的关键酶。
下面图解简单显示糖原合成分解及代谢所需的酶(图3-12-1)。
果糖和半乳糖通过代谢转变为1-磷酸葡萄糖,再通过磷酸变位酶转变为6-磷酸葡萄糖而合成糖原。
(图略)图3-12-1 糖原分解和糖异生途径图中阿拉伯数字代表:①己糖激酶;②6-磷酸葡萄糖酶;③磷酸葡萄糖变位酶;④糖原合成酶;⑤分支酶;⑥糖原磷酸化酶;⑦脱支酶。
糖原累积症(共5张PPT)

酰辅酶A,为脂肪酸和胆固醇的合成提供了原料; 患儿时有低血糖发作和腹泻发生。
移酶和分支酶作用,将a-1,6糖苷键水解生 3-磷酸甘油酸----- 2-磷酸甘油酸
糖代谢异常同时还造成了脂肪代谢紊乱,亢进的葡糖异生和糖酵解过程不仅使血中丙酮酸和乳酸含量增高导致酸中毒,还生成了大量乙
成游离的葡萄糖。 酰辅酶A,为脂肪酸和胆固醇的合成提供了原料;
磷酸烯醇式丙酮酸------丙酮酸 重症在新生儿期即可出现严重低血糖、酸中毒、呼吸困难和肝肿大等症状; 由于患者缺乏糖原代谢有关的酶,使糖原合成或分解发生障碍,导致糖原沉积于组织中而致病。
糖原累积症
第1页,共5页。
storage, GSD)是一种遗传性疾病,主要病因为先 天性糖代谢酶缺陷所造成的糖原代谢障碍。重症在新生儿期即可出现严重低 血糖、酸中毒、呼吸困难和肝肿大等症状;轻症病例则常在婴幼儿期因生长 迟缓、腹部膨胀等就诊。由于慢性乳酸酸中毒和长期胰岛素/胰高糖素例失 常,患儿身材明显矮小,骨龄落后,骨质疏松。腹部因肝持续增大而膨隆显 著。肌肉松弛,四肢伸侧皮下常有黄色瘤可见。但身体各部比例和智能等都 正常。患儿时有低血糖发作和腹泻发生。少数幼婴在重症低血糖时尚可伴发 惊厥.但亦有血糖降至0.56mmol/L(10mg/dl)以下而无明显症状者。随 着年龄的增长,低血糖发作次数可减少。由于血小板功能不良,患儿常有流 鼻血等出血倾向。
(4)a-1,得6糖苷的键。90%葡萄糖,在维持血糖稳定方面起主导作用。
• 糖酵解时发生在细胞质中,在无氧条件下.有10步反应: 由同磷重葡葡少由果糖酰(3糖酰同这患由(糖酰糖糖酰少 糖成同磷(由这患 糖酰果-磷于时酸症萄萄数于糖代辅2代辅时些儿于2代辅酵代辅数原游时酸3于些儿代辅糖) ) )酸患 还 , 在 糖 糖 幼 慢 -谢 酶 谢 酶 , 异 时 慢 谢 酶 解 谢 酶 幼分 离 还 , 患 异 时谢 酶 -11•••••••••••尿尿a,,甘者产N新--婴性异A异A由常有性异A时异A婴 解的产N者常有 异A-66--1苷苷--,,,,,AA--油--缺生生在乳常常于代低乳常发常在 是葡生缺代低 常,二二葡葡DD二二为 为 为 为 为葡果甘132磷 生酸乏了儿重酸同同6谢血酸同生同重 糖萄了乏谢血 同4磷磷PP葡果二萄萄-,-磷-磷糖脂脂脂脂脂HH磷油-3萄糖磷糖合期症酸时时都糖酸时在时症 原糖合糖都糖 时酸酸酸化-糖糖萄糖羟))-酸酸磷苷-肪肪肪肪肪醛-酸原成即低中还还加发中还细还低 在。成原加发 还二糖----烯酸----6;;糖葡葡酸书键66酸酸酸酸酸---丙2-代脂可血毒造造速作毒造胞造血 磷脂代速作 造甘---1---3磷---醇磷甘-磷磷萄萄葡;和和和和和--磷--,-本酮谢肪出糖和成成了和和成质成糖 酸肪谢了和 成二二6磷油6-酸酸酸糖 糖酸萄式葡胆胆胆胆胆酸油--有的现时长了了肝腹长了中了时化的有肝腹了羟羟酸磷酸磷P二生生糖-固固固固固甘萄丙<<甘-关和严尚期脂脂糖泻期脂脂尚 酶和关糖泻 脂,丙丙酸-8在--酸己己-酸成成的磷醇醇醇醇醇-油-糖-酮9的胆重可胰肪肪原发胰肪肪可 作胆的原发 肪酮酮--油-无-糖糖---;;累-的的的的的酸----酸1--酶固低伴岛代代的生岛代代伴 用固酶的生 代磷磷-酸--氧6-酸激激,2-积--合合合合合3----,醇血发素谢谢合。素谢谢发 下醇,合。 谢酸酸----果条磷--酶酶-磷---,二 -成成成成成-----使所糖惊/紊紊成/紊紊惊 ,所使成紊--磷件-++果糖酸-催催--酸大磷提提提提提---甘甘-糖必、厥胰乱乱。胰乱乱厥 将必糖。乱--下3-化化-酸糖部甘甘供供供供供酸二丙<1油油原需酸.高,,高,,. 需原,a-.反反己磷,-分了了了了了甘-6油烯油1醛醛酮羟合的中但糖亢亢糖亢亢但 的合亢6-应应,糖1原原原原原油酸二-酸--成还毒亦素进进素进进亦 还成进醇酸-醛磷丙33>>4激料料料料料酸--磷甘或原、有例的的例的的有 原或的糖磷磷磷式酸-酮;;;;;酶3酸分型呼血失葡葡失葡葡血 型分葡苷酸酸酸油-丙磷催磷解辅吸糖常糖糖常糖糖糖 辅解糖键<葡酸磷酮化发酶困降,异异,异异降 酶发异分酸萄酸酸生难至患生生患生生至 生生ⅠⅠ解反糖酸+果障和儿和和儿和和障和(00(生又应甘.. 碍肝身糖糖身糖糖碍糖烟烟成糖重>油,肿材酵酵材酵酵,酵酰酰葡激新醛导大明解解明解解导解胺胺萄再酶致等显过过显过过致过腺腺糖合-..3>糖症矮程程矮程程糖程瞟瞟1成--磷磷原状小不不小不不原不吟吟糖酸沉;,仅仅,仅仅沉仅二二酸原,积骨使使骨使使积使核核;再于龄血血龄血血于血苷苷由组落中中落中中组中酸酸葡织后丙丙后丙丙织丙,,萄中,酮酮,酮酮中酮NNAA糖而骨酸酸骨酸酸而酸DD转致质和和质和和致和HH))移病疏乳乳疏乳乳病乳和和酶。松酸酸松酸酸。酸还 还和。 含 含 。 含 含 含原原分量量量量量型型支增增增增增辅辅酶高高高高高酶酶作导导导导导ⅡⅡ用致致致致致((,酸酸酸酸酸烟烟将中中中中中酰酰a毒毒毒毒毒-1胺胺,,,,,,腺腺还还还还还6瞟瞟糖生生生生生吟吟苷成成成成成二二键了了了了了核核水大大大大大苷苷解量量量量量酸酸生乙乙乙乙乙
糖原累积症的科普知识PPT课件

演讲人:
目录
1. 什么是糖原累积症? 2. 糖原累积症的症状 3. 糖原累积症的治疗 4. 糖原累积症的生活管理 5. 糖原累积症的未来研究
什么是糖原累积症?
什么是糖原累积症?
定义
糖原累积症是一组遗传性代谢疾病,主要由于体 内糖原代谢相关酶的缺陷,导致糖原在细胞内异 常累积。
某些类型的糖原累积症可能需要使用药物来改善 症状,比如补充酶制剂。
药物治疗应在医生指导下进行,避免自我用药。
糖原累积症的治疗
基因治疗
新兴的基因疗法正在研究中,目标是纠正导致疾 病的基因缺陷。
这项技术尚在实验阶段,未来可能成为新的治疗 选择。
糖原累积症的生活管理
糖原累积症的生活管理
定期体检
患者应定期接受专业医生的检查,监测疾病 进展和并发症。
公众教育
提高公众对糖原累积症的认识,有助于早期发现 和诊断。
通过宣传活动,可以提高社会对罕见病的关注和 理解。
谢谢观看
如I型糖原累积症在某些地区的发病率较高。
糖原累积症的症状
糖原累积症的症状
常见症状
症状因类型而异,常见的有低血糖、肌肉无 力、肝脏肿大等。
有些患者在婴儿期即出现症状,而有些则可 能在儿童或成年期才被诊断。
糖原累积症的症状
并发症
长期的糖原累积可能导致心脏病、肝脏功能 衰竭、肌肉萎缩等严重并发症。
定期监测和早期干预能够有效降低并发症风 险。
糖原累积症的症状 诊断方式
通过临床症状、家族史、血液和尿液化验、 基因检测等手段进行诊断。
早期诊断对改善预后至关重要。治疗
饮食管理
控制饮食,定期进餐,增加碳水化合物的摄入量 ,避免低血糖。
糖原累积病(Ⅰ型、Ⅱ型)诊断

诊断
糖原累积病(Ⅰ型、Ⅱ型)的诊断需要结合临床表现、实验室检查及基因检测综合判断。
1.GSDⅠ型对于所有身高增长缓慢伴肝脏明显增大的患者均应考虑GSD Ⅰ型的可能。
典型生化改变包括空腹低血糖、高乳酸血症、高脂血症和高尿酸血症等。
GSDⅠb型患者还可有反复或持续性白细胞和中性粒细胞减少。
发现G6PC 或SLC37A4基因2个等位基因致病突变有确诊意义。
2.GSDⅡ型对于1岁前起病、肌无力、心脏扩大、心肌肥厚、血清CK升高的患者,应怀疑婴儿型GSDⅡ型。
所有缓慢进展的肌无力患者均应考虑晚发型GSDⅡ型的可能。
肌肉活检病理检查可见胞浆内大量空泡,PAS染色糖原聚集,SBB染色脂滴成分正常,酸性磷酸酶活性增高。
外周血白细胞或皮肤成纤维细胞培养GAA酶活性明显降低有确诊意义。
发现GAA基因2个等位基因致病突变也有确诊意义。
鉴别诊断
1.GSDⅠ型主要与肝脏增大伴低血糖的疾病相鉴别(表1)。
表1常见肝大伴低血糖的疾病特点
2.GSDⅡ型婴儿型GSDⅡ型应注意与心内膜弹力纤维增生症、GSDⅢ型、
Ⅳ型、脊髓性肌萎缩Ⅰ型、先天性甲状腺功能减低症、原发性肉碱缺乏症等鉴别。
晚发型患者应注意与肢带型肌营养不良、多发性肌炎、线粒体肌病、Danon病、强直性肌营养不良、GSD(Ⅲ型、Ⅳ型、Ⅴ型)等鉴别。
糖原累积症科普宣传PPT课件

何时需要就医?
何时需要就医?
症状识别
患者可能出现低血糖、肌肉无力、疲劳、肝脏肿 大等症状,一旦出现,应及时就医。
早期诊断有助于改善预后。
何时需要就医?
定期检查
对于已知患者,定期进行医学检查和监测,以评 估病情进展和治疗效果。
定期检查可以帮助及时调整治疗方案。
何时需要就医?
糖原累积症科普宣传
演讲人:
目录
1. 什么是糖原累积症? 2. 谁会受到影响? 3. 何时需要就医? 4. 如何诊断糖原累积症? 5. 如何治疗和管理糖原累积症?
什么是糖原累积症?
什么是糖原累积症?
定义
糖原累积症是一组遗传性代谢疾病,主要由于身 体无法正常分解糖原,导致糖原在细胞内异常累 积。
这种疾病影响能量代谢,可能影响多个器官,尤 其是肝脏和肌肉。
生化检查是确诊的重要手段。
如何诊断糖原累积症?
基因检测
基因检测可以确认具体的糖原累积症类型及其遗 传背景。
基因检测提供了更为准确的诊断依据。
如何治疗和管理糖原累积症 ?
如何治疗和管理糖原累积症?
饮食管理
患者应遵循低碳水化合物、高蛋白质的饮食,以 防止低血糖和糖原的过度累积。
合理的饮食能有效控制症状。
如何治疗和管理糖原累积症?
药物治疗
某些类型的糖原累积症可能需要特定药物治疗, 以改善代谢功能。
药物治疗应在医生指导下进行。
如何治疗和管理糖原累积症?
定期随访
患者需定期随访,监测病情变化,并及时调整治 疗方案。
良好的随访管理是保持患者生活质量的重要因素 。
谢谢观看
什么是糖原累积症?
分类
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
5
四、检查及诊断
• 基因检测:目前研究较多的为葡萄糖-6-磷酸酶(G-6-Pase)基因, 其缺乏可引起Ⅰ型GSD。目前已检测出多种G-6-Pase基因突变,其 中最多见于R83C和Q347X,约占Ⅰ型GSD的60%,但有地区差异, 中国人群以nt327G→A(R83H)的突变率最高,其次为nt326G→A (R83C),使用PCR结合DNA序列分析或ASO杂交方法能正确地鉴 定G-6-Pase基因第83密码子上的CpG突变的88%。适用于Ⅰ型糖原 累积症患者携带的突变等位基因,亦可用于携带者的检出和产前诊断。 基因检测可避免侵害性的组织活检。
2
二、发病机制
• 糖原贮积病为常染色体隐性遗传,磷酸化酶激酶缺乏型则是X-性连锁 遗传。糖原分解代谢中所必需的各种酶缺陷引起各型糖原贮积病。
• 糖原累积症Ⅰa型( GSD-Ⅰa)由葡萄糖-6-磷酸酶(G6Pase)催化亚 单位基因(G6PC基因)突变所致,是GSD中最早发现且最常见的一 个亚型,约占25%;GSDIa的临床表现缺乏特异性,所以G6PC基因 的检测可为GSDIa(尤其是临床表现不典型的)患儿的诊断、分型以 及预后预测提供一种有效可靠依据。
• 对症治疗:对有心力衰竭、肾功能损害、营养缺乏和中性粒细胞减少 而反复发生感染者均应采取相应的对症治疗。
• 药物治疗:维生素类药物,如B族维生素维生素C等。有感染给抗生 素治疗。纠正低血糖后如果血脂仍继续升高,可用安妥明50mg/ (kg·d)。高尿酸血症如采用饮食疗法不能控制时,可用别嘌呤醇 8 5~10mg/(kg·d)。激素治疗有益于维持正常血糖水平、提高食欲。
• 患儿出生时就会有肝脏肿大的症状。新生儿肝肿大不明显,而不被注 意。1岁左右逐渐见肝脏肿大,甚至占据整个腹腔。
• 低血糖:多于1岁以内出现,随着年龄增长,会出现明显低血糖症状, 例如软弱呕吐、无力、出汗、惊厥和昏迷,
• 生长发育迟缓:反复发作的低血糖会影响患者的智力发育以及身体发 育,表现为智能低下,患者肥胖体、个子矮小、皮肤暗淡,颜色多为 淡黄色,肌肉发育差,较常见下肢无力的症状。
• 酮症酸中毒:是小儿死亡的主要原因。多数病人在发生意识障碍前数
天有多尿、烦渴多饮和乏力,随后出现食欲减退、恶心、呕吐,常伴
头痛、嗜睡、烦躁、呼吸深快,呼气中有烂苹果味(丙酮)是其典型
发作时候的特点。随着病情进一步发展,出现严重失水,尿量减少,
皮肤弹性差,眼球下陷,脉细速,血压下降。至晚期时各种反射迟钝
甚至消失,查:Ⅰ型患者空腹血糖降低至2.24~2.36mmol/L,乳酸及血糖 原含量增高,重症低血糖常伴有低磷血症。三酰甘油、胆固醇脂肪酸 和尿酸均显著增高。
• 糖代谢功能试验:(1)糖和肾上腺素耐量试验 前者呈现典型糖尿病 特征;后者检查时注射肾上腺素60分钟后,0、Ⅰ、Ⅲ、Ⅻ型患者血 糖均不升高。(2)胰高血糖素试验 糖肌注胰高血糖素30µg/kg(最 大量1mg),于注射后0,15,30,45,60,90,120min分别取血 测血糖。正常时15~45min内血糖升高1.5~2.8mmol/L,0、Ⅰ、Ⅲ、 Ⅳ型患者示血糖反应低平,餐后1~2小时重复此试验,0、Ⅲ型血糖可 转为正常。(3)果糖或半乳糖变为葡萄糖试验 Ⅰ型患者果糖或半乳 糖在负荷时葡萄糖不升高,但乳酸明显上升。
• 酶替代治疗:酶替代治疗目前正处于动物实验阶段。酶替代治疗是否 可用于人,还需作进一步作临床试验。
• 基因治疗:基因治疗是治疗糖原累积病最有效最彻底的方法,但目前 尚未应用于临床。
• 手术治疗:手术治疗包括肝腺瘤切除术、部分肝切除术以及器官移植 (肝移植和心脏移植)手术,器官移植包括血糖和胰岛素水平明显升 高,骨髓移植或脐带血干细胞移植治疗可部分改善患者症状,身体发 育到得改善,但手术不能预防肝腺瘤的发生,器官移植并发症多,死 亡风险高。
糖原累积症
1
一、概述
• 糖原累积病是一类由于先天性酶缺陷所造成的糖原代谢障碍疾病,多 数属常染色体隐性遗传,发病因种族而异。根据欧洲资料,其发病率 为1/(2万~2.5万)。糖原合成和分解代谢中所必需的各种酶至少有 8种,由于这些酶缺陷所造成的临床疾病有12型,其中Ⅰ、Ⅲ、Ⅳ、 Ⅵ、Ⅸ型以肝脏病变为主;Ⅱ、Ⅴ、Ⅶ型以肌肉组织受损为主。这类 疾病有一个共同的生化特征,即是糖原贮存异常,绝大多数是糖原在 肝脏、肌肉、肾脏等组织中贮积量增加。仅少数病种的糖原贮积量正 常,而糖原的分子结构异常。
• 糖原累积症Ib型是由于SLC37A4基因突变导致葡萄糖-6-磷酸转移酶 (G6PTI)缺乏,一方面造成糖原在肝脏、肾脏和肠粘膜大量堆积,另 一方面造成粒细胞数量减少和功能障碍。
• 糖原累积症II型较罕见,是由于编码溶酶体中酸性仪葡糖苷酶(GAA) 的GAA基因突变引起。
3
三、临床表现
• 出生即可发病,成年之后,轻病者的患者会有所好转。以肝脏病为主 的Ⅰ型最为常见。
6
四、检查及诊断
• 在新生儿和婴幼儿下频发低血糖抽搐和神智不清,喂食或注射葡萄糖 后即可恢复;在出现低血糖的同时有呼吸深快的酸中毒症状,这是诊 断糖原累积病的重要临床线索。体征可见肝脏肿大、右上腹隆起。实 验室检查应包括血糖、血酮体、乳酸、血脂和尿酸(禁食和餐后)的 动态变化。
7
五、治疗
• 饮食治疗:主要用于有肝脏受累、易发生低血糖、酮中毒和乳酸中毒 的新生儿和儿童患者。
六、预后
• 新生儿和婴儿由于身体免疫系统发育不成熟,疾病较严重,治疗难度 大。年龄较大的儿童,具有一定的抵抗力,治疗也较容易。本病为遗 传性疾病,故难以根治,但近些年发展起来的基因治疗,有可能使糖 原累积病得到根治。
9