3.2水中无机污染物的迁移转化(3)
环境化学(戴树桂)课后习题参考答案

中科院研究生院硕士研究生入学考试《环境化学》考试大纲1.《环境化学》考试大纲适用于中国科学院研究生院环境科学专业硕士研究生入学考试。
《环境化学》是环境科学与工程类专业的重要基础课程,包括了环境化学研究的内容、特点和发展动向,主要环境污染物的类别和它们在环境各圈层中的迁移转化过程,典型污染物在环境各圈层中的归趋和效应。
本考试大纲侧重于环境污染化学,着重于各类有害物质在环境介质中的存在、行为、效应以及减少或消除其产生的理论和方法。
主要内容包括水环境化学、大气环境化学、土壤环境化学及化学物质的生物效应与生态效应等经典内容。
对其中有机污染物的归趋模式、金属离子的存在形态及生物效应、化学物质结构与毒性关系,污染物的生物效应和生物浓缩机制以及全球范围内的温室效应、酸雨、臭氧层破坏等环境问题需加以重点掌握。
要求考生掌握基本概念、基本原理和计算方法,并具备综合运用所学知识分析和解决实际环境问题的能力。
考试内容:环境化学发展及基本内容1.环境化学基本概念、发展动向、研究内容及热点问题水环境化学2.天然水的基本特征组成;3.水体无机污染物的迁移转化。
着重配合作用、氧化-还原作用、沉淀和溶解、水体颗粒物的吸附作用等基本原理及其实际应用。
4.水体有机污染物的迁移转化:环境行为与归趋模式。
着重分配作用、挥发作用、水解作用等典型机制与迁移转化模式。
5.水体的富营养化问题:水体富营养化;水体富营养化的机理;营养物质的来源;富营养化的影响因素;湖水的营养化程度;水体富营养化的危害及其防治对策。
大气环境化学6.大气中污染物的特征;7.大气中污染物的迁移和转化(温室效应;气相大气化学:光化学反应、光化学烟雾、臭氧层的形成与耗损化学;液相大气化学:酸沉降化学、大气中液相反应;大气颗粒物化学);8.重要的大气环境化学问题:光化学烟雾的定义、特征及形成条件;光化学烟雾主要参与物质;光化学烟雾形成机理;光化学烟雾危害及防治;光化学烟雾与硫酸型烟雾的对比;太阳和地球辐射间的能量平衡;温室气体;温室效应;全球变暖及防治对策;降水的化学组成;酸雨的形成;酸雨的影响因素;酸雨的危害及防治;大气平流层的组成;臭氧层的形成和耗损的化学反应;臭氧洞的危害;臭氧层破坏现状及防治。
环境化学第3.2章水环境化学水中无机污染物的溶解和沉淀课件

20
第三章/第二节/2.3 溶解和沉淀
2.3.4 碳酸盐 四、碳酸盐在开放体系的溶解度(二价金属)
[H2CO3*] = KHpCO2 [CO32-] = K1K2KHpCO2/[H+]2
pH>pK2(10.33) pK1<pH<pK2 (6.35~10.33) [Me2+] ≈ Ksp[H+]2/K1K2KHpCO2 pH<pK1(6.35)
第三章/第二节 水中无机污染物的迁移转化
2.3 溶解和沉淀
溶解/沉淀对迁移过程的影响
溶解/沉淀影响金属化合物溶解度,溶解度决定随水迁移能力 溶解度大,迁移能力大;溶解度小,迁移能力小
溶解/沉淀理论
溶解/沉淀受反应平衡和反应速率控制(化学热力学和动力学控制) 固-液平衡体系中,用溶度积来表征溶解度
第三章/第二节/2.3 溶解和沉淀
2.3.3 硫化物
二、金属硫化物的溶解度(以二价金属为例)
1. 金属硫化物的沉淀-溶解平衡
MeS (s) ⇌ Me2+ + S2-
[Me2+] = Ksp/[S2-]
2. H2S的电离平衡
H2S ⇌ H+ + HS- K1 = 8.9×10-8
HS- ⇌ H+ + S2-
= 2.532×10-3 mol/L
15
第三章/第二节/2.3 溶解和沉淀
2.3.4 碳酸盐
一、碳酸盐的沉淀-溶解平衡(以二价金属为例)
MeCO3 ⇌ Me2+ + CO32[Me2+] = Ksp/[CO32-] = Ksp/(CTα2)
H2CO3* ⇌ HCO3- + H+
水中无机污染物的迁移转化.

4r 2 4 3 = r 3
3 r
(cm2/g)
子受力不均匀(因为表面分子周围的分子数量不相等),因而产生了所谓的
表面能。 计算实例:某湖泊底泥ρ=2.65g/cm3,10%为直径D=1um(10-4cm)的颗 粒物,求面积S=100m2,深度h=0.2m的底泥中,所有直径D=1um(10-4cm)的 颗粒物比表面积?
G kC
G kC
用对数表示:
1 n
式中:K——分配系数
F型(Freundlich)等温式
1 lg G lg k lg C n
L型(Langmuir)等温式
G G C /( A C )
0
1 / G 1 / G ( A / G )(1 / C )
0 0
G0—单位表面上达到饱和时的最大吸附量 A—常数
(5)第二极小值絮凝; (6)聚合物粘结架桥絮凝;
(7)无机高分子的絮凝;
(8)絮团卷扫絮凝; (9)颗粒层吸附絮凝;
(10)生物絮凝。
水中胶体颗粒物聚集的基本原理和方式
1、概述
胶体颗粒的聚集亦可称为凝聚或絮凝。在讨论聚集的化学概念时,这两个 名词常交换使用。这里把由电介质促成的聚集称为凝聚,而由聚合物促成 的聚集称为絮凝。 胶体颗粒长期处于分散状态还是相互作用聚集结合成为更粗粒子,将决定 着水体中胶体颗粒及其上面的污染物的粒度分布变化规律,影响到其迁移
在水环境中,配合离子、有机离子、有机高分子和无机高 分子的专属吸附作用特别强烈。例如,简单的Al3+、Fe3+高 价离子并不能使胶体电荷因吸附而变号,但其水解产物却 可达到这种效果,这就是发生专属吸附的结果。 水合氧化物胶体对重金属离子有较强的专属吸附作用,这 种吸附作用发生在胶体双电层的Stern层中,被吸附的金属 离子进入Stern层后。不能被通常提取交换性阳离子的提取 剂提取,只能被亲和力更强的金属离子取代,或在强酸性 条件下解吸。
【环境化学】第3.2章 水环境化学——第二节 水中无机污染物的迁移转化

22
吸附等温线和等温式
吸附等温线: 在固定的温度下,当吸附达到平衡时, 颗粒物表面上的吸附量(G)与溶液中溶质平衡浓度 (C)之间的关系,可用吸附等温线来表示。
吸附等温线类型:
Henry型(H型) Freundlich型(F型) Langmuir型(L型)
腐植质分子与金属络合的机理★
Hum
COO OH
-
+[Fe(OH)(H2O)x-1]2+
低pH
Hum
COO O
Fe
OH (O) x-1
+ H+
在低pH时,从腐植质的酸性基团中置换出一个质子
Hum COOHO-+ [Fe(OH)(H2O)x-1]2+
高pH
[ Hum
COO O
Fe
OOHH(H2O)x-2]-+2H+
23
H型等温式为: G = kc
k: 分配系数;等温线为直线型
F型等温式为:G = kc(1/n)
1)k 是c=1的吸附量,大致表示 吸附能力的强弱; 2)1/n为斜率,表示吸附量随 浓度增长的强度; 3)该等温线不能给出饱和吸附 量。
L型等温式: G = G0c/(A+c)
G0—单位面积上达到饱和时的 最大吸附量; A—常数。
胶体表面的化学反应(见下页胶片)
25
胶体表面的化学反应
是氢氧化物和氧化物的典型行为 与pH值有关
在酸性介质中 M(OH)n (s) + H+ → M(OH)n-1(H2O)+(s) 粒子带净正电荷
在碱性介质中 M(OH)n (s) → MO(OH)n-1-(s) + H+ 粒子带净负电荷
3.2水中无机污染物的迁移转化 (4)

或者在两个羧基间螯合:
或者与一个羧基形成配合物:
在环境中对污染物的影响 与金属作用:许多研究表明:重金属在天然水体中主要以腐殖酸的 配合物形式存在。Matson等指出Cd、Ph和Cu在美洲的大湖(Great Lake)水中不存在游离离子,而是以腐殖酸配合物形式存在。表3—9 列出不同来源腐殖酸与金属的配合稳定常数,并可看出,Hg和Cu有较 强的配合能力,在淡水中有大于90%的Ca、Hg与腐殖酸配合,这点对 考虑重金属的水体污染具有很重要的意义。特别是Hg,许多阳离子如 Li+ 、Na+ 、Co2+ 、Mn2+ 、Ba2+ 、Zn2+ 、Mg2+ 、La3+ 、Fe3+ 、A13+ 、Ce3+ 、 Th4+,都不能臵换Hg。水体的pH、Eh等都影响腐殖酸和重金属配合作 用的稳定性。
K2 [ Zn ( NH [ ZnNH
2 3 3
Zn(NH3)22+
)2
2
]
3
2 . 1 10 ]
2
][ NH
K1、K2称为逐级生成常数(或逐级稳定常数),表示NH3加至中心Zn2+上是
一个逐步的过程。
积累稳定常数是指几个配位体加到中心金属离子过程的加和。例如,
Zn(NH3)22+的生成可用下面反应式表示:
五、配合作用
1、概述
污染物特别是重金属污染物,大部分以配合物形态存在于水体,其 迁移、转化及毒性等均与配合作用有密切关系。重金属容易形成配合 物的原因是重金属为过渡性元素,最外层为s轨道电子数目为2或1, 次外层为d轨道或f轨道电子,数目为1-9,为充满,则过渡金属元素 失去外层s轨道电子后,未充满的d轨道仍旧可以接受外来电子,形成 配合的络合物或者螯合物。 天然水体中有许多阳离子,其中某些阳离子是良好的配合物中心体, 某些阴离子则可作为配位体。 天然水体中重要的无机配位体有OH-、Cl-、CO32-、HCO3-、F-、S2 - 。它们易与硬酸进行配合。如OH - 在水溶液中将优先与某些作为中 心离子的硬酸结合(如Fe3+、Mn3+等),形成羧基配合离子或氢氧化物 沉淀,而S2-离子则更易和重金属如Hg2+、Ag+等形成多硫配合离子或 硫化物沉淀。
第二节、水中无机污染物的迁移转化

于晶格中离子的同晶替代造成的,例如硅氧四面体中的Si4+被
Al3+所取代,或者铝氢氧八面体中的Al3+被Mg2+所取代等,都会 产生这种永久负电荷。另一部分为可变电荷,主要随着环境pH
的改变而发生改变,原因是 Si-OH中的H+ 在碱性溶液中的离解。
Si-OH+OH-=Si-O-+H2O。
特征:这种吸附是一种可逆反应,能够迅速达到平衡。 不受温度影响,酸碱条件下均可进行,其交换吸附能力 与溶质的性质、浓度及吸附剂性质等有关。对于那些具 有可变电荷表面的胶体,当体系pH高时,也带负电荷并
n
以lgG对lgc作图可得一直线。lgk为截距,因此, k值是c=1时的吸附 1
n 量,它可以大致表示吸附能力的强弱。
浓度增长的强度。
该等温线不能给出饱和吸附量。 L型等温式为:G=G0c/(A+c)
为斜率,它表示吸附量随
式中:G0——单位表面上达到饱和时间的最大吸附量; A——常数。 G对c作图得到一条双曲线,其渐近线为G=G0,即当c→∞时,G→G0。 在等温式中A为吸附量达到时溶液的平衡浓度。 转化为:1/G = 1/G0 + (A/G0)(1/c)
1 以G
1 对 作图,同样得到一直线。 c
等温线在一定程度上反映了吸附剂与吸附物的特性,其形式在许
多情况下与实验所用溶质浓度区段有关。当溶质浓度甚低时,可能在 初始区段中呈现H型,当浓度较高时,曲线可能表现为F型,但统一起 来仍属于L型的不同区段。 影响吸附作用的因素有以下几种: 首先是溶液pH值对吸附作用的影响。在一般情况下,颗粒物对重金 属的吸附量随pH值升高而增大。当溶液pH超过某元素的临界pH值时, 则该元素在溶液中的水解、沉淀起主要作用。吸附量(G)与pH、平衡 浓度(C)之间的关系可用下式表示:G = A· C· 10BpH式中:A、B—常数。
第三章 水环境化学水中无机污染物的迁移转化汇总

20
1. 胶体颗粒凝聚的基本原理和方式
1) 带电胶粒稳定性的经典理论--DLVO理论 带电胶粒的两种相互作用力
双电层重叠时的静电排斥力 粒子间的长程范德华吸引力
DLVO理论认为,当吸引力占优势时,溶胶发生聚 沉; 当排斥力占优势,并大到足以阻碍胶粒由于 布朗运动而发生聚沉时,则胶体处于稳定状态。 颗粒在相互接近时两种力相互作用的总位能随相 隔距离的变化而变化: 总位能 VT=VR+VA 式中:VA——由范德华力所产生的位能; VR——由静电排斥力所产生的位能。
4
一 、 颗粒物与水之间的迁移
2、水环境中颗粒物的吸附作用
专属吸附是指吸附过程中,除了化学键的作
用外,尚有加强的憎水键和范德华力或氢键在 起作用。
专属吸附作用不但可使表面电荷改变符号, 而且可使离子化合物吸附在同号电荷的表面上。
5
表3-8水合氧化物对金属离子的专属吸附 与非专属吸附的区别
项目 非专属吸附 专属吸附 发生吸附的表面净电荷的符号 - -、0、+ 金属离子所起的作用 反离子 配位离子 吸附时发生的反应 阳离子交换 配位体交换 发生吸附时体系的PH值 >零电位点 任意值 吸附发生的位置 扩散层 内层 对表面电荷的影响 无 负电荷减少, 正电荷增多 注:本表摘自陈静生主编,1987。
(4)水体悬浮沉积物
悬浮沉积物是以矿物微粒,特别是粘土矿物 为核心骨架,有机物和金属水合氧化物结合在矿 物微粒表面上,成为各微粒间的粘附架桥物质, 把若干微粒组合成絮状聚集体(聚集体在水体中 的悬浮颗粒粒度一般在数十微米以下),经絮凝 成为较粗颗粒而沉积到水体底部。
(5)其他
3
一、 颗粒物与水之间的迁移
污染物迁移和转化规律研究及其对环境的影响

污染物迁移和转化规律研究及其对环境的影响随着人类社会的发展,工业、农业、交通等活动产生了大量污染物,它们对环境的影响日益严重。
其中,污染物迁移和转化规律的研究是解决环境污染问题的重要途径之一。
本文将从污染物的定义、污染物的迁移和转化规律以及污染物对环境的影响等方面进行探讨。
一、污染物的定义污染物是指有害物质或能够危害环境和生态系统的物质,主要包括有机物、无机物、微生物等。
它们的来源可以是生产、生活、交通等活动,也可以是自然界中本就存在的物质。
污染物对环境的影响主要表现为水、土和空气污染,其中水污染是最为严重的。
二、污染物的迁移和转化规律污染物的迁移和转化是指污染物经过环境因素的作用而改变其状态和位置的过程。
这个过程既受污染物自身特性的影响,也受环境因素(如水、气、土壤等)的影响。
具体来说,它包括以下几个方面:1. 水环境中的污染物迁移和转化水是污染物迁移和转化的主要载体之一。
水环境中的污染物主要通过水流、沉淀和沉积等方式向周围环境扩散。
同时,包括悬浮、溶解等方式使污染物转移。
根据化学反应的性质,污染物也会发生最终处理、降解、微生物氧化、光化学反应等变化以实现转化。
2. 土壤环境中的污染物迁移和转化土壤是自然环境中污染物迁移和转化的重要载体。
污染物首先通过雨水等渗入土壤,然后通过土壤孔隙和土壤结构向周围的空气或水中扩散。
在土壤环境中,污染物的迁移和转化受到土壤的物理、化学、生物等因素的影响。
污染物会通过吸附、分解等过程来实现转化。
3. 大气环境中的污染物迁移和转化大气是污染物中转的环境之一。
污染物通过气流和降水等途径进入大气环境,进而经过物理、化学、生物等过程实现迁移和转化。
在大气环境中,光化学反应、电化学反应等化学反应起到了重要的作用。
三、污染物对环境的影响污染物的迁移和转化不仅对环境自身造成了不利影响,还对人类健康产生了危害。
污染物进入人体,会导致多种疾病的发生,如呼吸系统疾病、胃肠疾病等。
此外,污染物还会对环境中的生态系统产生不利影响。
颗粒物与水之间的迁移水环境中颗粒物的吸附作用水环境中胶体

1.胶体颗粒凝聚的基本原理和方式
典型胶体的相互作用是以DLVO物理理论为 定量基础。
1.胶体颗粒凝聚的基本原理和方式
异体凝聚理论:适用于处理物质本性不同、粒径不等、
电荷符号不同、电位高低不等之类的分散体系。 异体凝聚理论的主要论点为:如果两个电荷符号相异的 胶体微粒接近时,吸引力总是占优势;如果两颗粒电荷符号 相同但电性强弱不等,则位能曲线上的能峰高度总是决定于 荷电较弱而电位较低的一方。因此,在异体凝聚时,只要其 中有一种胶体的稳定性甚低而电位达到临界状态,就可以发 生快速凝聚,而不论另一种胶体的电位高低如何。
天然水环境和水处理过程中所遇到的2)专属吸附凝聚; (3)胶体相互凝聚 (4)“边对面”絮凝;(5)第二极小值絮凝;(6)聚合物粘结架桥 絮凝;(7)无机高分子的絮凝; (8)絮团卷扫絮凝; (9)颗粒层吸附絮凝;(10)生物絮凝
第二节 水中无机污染物的迁移转化
G
影响吸附作用的因素
溶液pH值对吸附作用的影响。在一般情况下,颗 粒物对重金属的吸附量随pH值升高而增大。当溶
液pH超过某元素的临界pH值时,则该元素 在溶液中的水解、沉淀起主要作用。
影响吸附作用的因素
溶液pH值对吸附作用的影响。在一般情况下, 颗粒物对重金属的吸附量随pH值升高而增大。 颗粒物的粒度和浓度对重金属吸附量的影响。 颗粒物对重金属的吸附量随粒度增大而减少,并 且,当溶质浓度范围固定时,吸附量随颗粒物浓 度增大而减少。
二、水中颗粒物的聚集
胶体颗粒的聚集亦可称为凝聚或絮凝。在讨论
聚集的化学概念时,这两个名词时常交换使用。这里把
3.2水中无机污染物的迁移转化 (2)

数浓度图可看出,同价金属离子的各线均有相同的斜率,靠图右边斜
线代表的金属氢氧化物的溶解度大于靠左边的溶解度。根据此图大致 可查出各种金属离子在不同pH溶液中所能存在的最大饱和浓度。
可见众多金属随着溶液pH的降低,pC增加,即溶解度增加,这说明酸
性条件下,有利于金属氢氧化合物的溶解,而碱性条件有利于其形成 沉淀。
不过图3—11和式(3—43)所表征的关系,并不能充分反映出氧化 物或氢氧化物的溶解度,应该考虑这些固体还能与羟基金属离子配 合物[Me(OH)nz-n]处于平衡。如果考虑到羟基配合作用情况,可以 把金属氧化物或氢氧化物的溶解度(MeT)表征如下:
MeT [ Me ] [ Me (OH ) n
PbO(s) + 2H2O→Pb(OH)3- + H+
1g*Ks3 = –15.4
根据上式,Pb2+ 、PbOH+ 、Pb(OH)20 和Pb(OH)3 - 作为pH值函数的特 征线分别有斜率–2、–1、0和+1,把所有化合态都结合起来,可以得 到图3—12中包围着阴影区域的线。因此,[Pb(Ⅱ)T]在数值上可由下 式得出: [Pb(Ⅱ)T] = *Ks0[H+]2 + *Ks1[H+] + Ks2 + *Ks3[H+]-1 图3—12表明固体的氧化物和氢氧化物具有两性的特征。它们和 质子或羟基离子都发生反应,存在有一个pH值,在此pH值下溶解度为 最小值,在碱性或酸性更强的pH值区域内,溶解度都变得更大。
下面着重介绍金属氧化物、氢氧化物、硫化物、碳酸盐及多种成分 共存时的溶解—沉淀平衡问题。
1、氧化物和氢氧化物 : Al(OH)3 、 Fe(OH)3 、 Fe(OH)2 、 Hg(OH)2 、
第三章 第二节 水中无机污染物的迁移转化要点

常见的吸附等温线
G G
n是一个经验 值,不是由一个 过程控制,一般 适用于有机物 lgG
lgK
H型
G0/2
单分子吸附 适用于金属
L型 c
L型 1/c
0 A
当溶质浓度甚低时,可能在初始阶段呈现 H 型,当浓度较 高时,可能表现为 F 型,但统一起来仍属于 L 型的不同区段。
2、异体凝聚理论
(1)适用条件:适用于物质本性不同、粒径不等、电荷符
号不同、电位高低不等的分散体系。 (2)主要论点: A、电荷符号相异的胶体微粒接近时,吸引力总是占优势; B、电荷符号相同但电性强弱不等,则位能曲线上的能峰高 度总是决定于荷电较弱而电位较低的一方。
因此异体凝聚时,只要有一种胶体的稳定性甚低而电位
纯饱和溶液中 [S2-]= Ksp/ [H+]2 = 1.16×10-23 / 8.9×10-9 = 1.3×10-15mol/L 任意水体中 [S2-]= 1.16×10-23 / [H+]2 [Me2+] [S2-]=Ksp 因此,在 H2S 和硫化物均达到饱和的溶液中,溶液重金属离子
的饱和浓度为:
2、硫化物
H2S H++ HSK1 = 8.9×10-8 HSH++ S2K2 = 1.3×10-15 两者相加可得: H2S 2H+ + S2K12 = K1·K2 = 1.16×10-22
在饱和水溶液中,H2S 浓度总是保持在 0.1mol/L,因此可认
为饱和溶液中 H2S 分子浓度也保持在 0.1mol/L,得: [H+]2[S2-] =1.16×10-22×0.1 = 1.16×10-23 = Ksp´
环境化学教案 第三节水中无机污染物的迁移转化(氧化还原2)
由此反应可知,当有机物进入天然水后,水体中溶解氧的含量会迅速降低。如果有机物的数量非常多的话,可以使水体中溶解氧全部被消耗掉,甚至可以使水体转换成为沼泽。在水中,NH4+只有被氧化成NO3-后,氮才能被藻类利用。
例2求被大气氧所饱合的中性天然水的pE值。
解:该体系溶解氧起决定电势作用,溶解氧的氧化还原反应为:
pE=pE0+ [H+]
已知:pE0=20.75
pH=7 [H+]=10-7
将已知条件代入pE的表达式,得:pE=13.58。该体系pE值较高,是一个氧化性体系。
例3求微生物作用产生甲烷的中性厌氧水的pE值及溶解氧的分压
天然水是一个非常复杂的混合体系,其中存在着众多的氧化剂和还原剂。其中常见的氧化剂包括溶解氧、Mn(IV)、Fe(III)、和S(VI),常见的还原剂包括有机物、Mn(II)、Fe(II)、S(-II)。当我们要求得某种天然水的pE值时,首先需要确定哪种物质起决定电势作用,然后根据起决定电势作用物质的氧化还原反应,求得体系的pE值。一般的天然水体中起决定电势作用的物质是溶解氧,当有机物含量非常高时,则有机物起决定电势作用。铁和锰起决定电势作用的情况则比较少见。下面我们来看两种极端情况下体系的pE值。
pE=-4.13
将已知条件代入pE的表达式,得: =3.0×10-72atm。
由此可见该体系中溶解氧的分压非常低,一般水体中的溶解氧的分压均超过此值。由于天然水的pE值随水中溶解氧的减少而降低,因此表层水pE值较高,底层水pE值较低。
例3:从湖水中取出深层水,其PH=7.0,含溶解氧浓度0.32mg/L,请计算PE和Eh。(KH=1.26*10-8[mol/(L·Pa)]
污染物在水体中的迁移转化方式

污染物在水体中的迁移转化方式主要有以下三种途径:
(1)氧化-还原作用。
天然水体中有许多无机和有机氧化剂和还原剂,如溶解氧、Fe3+、Mn4+、Fe2+、S2-及有机化合物等,这些物质对污染物的转化起重要作用。
如环境中重金属在一定氧化-还原条件下,容易发生价态变化,结果是其化学性质改变,迁移能力也会发生改变。
水体中的氧化-还原类型、速率和平衡,在很大程度上决定了水中重要溶质和污染物的性质。
如在一个厌氧湖泊中,湖下层的元素以还原态存在:碳还原成CH4,氮还原成[*]等,而表层水由于可被大气中氧补充,成为氧化性介质,达到热力学平衡时,碳成为CO2,氮成为[*]。
显然这种变化对水生生物和水质影响很大。
(2)络合作用。
天然水体中有许多无机配位体,如OH-、Cl-[*]、[*]和有机配位体如氨基酸、腐殖酸,以及洗涤剂、农药、大分子环状化合物等,它们可以与水中的污染物,特别是重金属发生络合反应,改变其性质和存在状态,影响污染物在水体中发生、迁移、反应和生物效应。
(3)生物降解作用。
水体中的微生物,特别是底泥中的厌氧微生物,可以使一些污染物发生转化,如把无机汞转变为有机汞。
第二节 水无机污染物的迁移转化

第二节水中无机污染物的迁移转化水中无机污染物特别是重金属污染物进入水体,不能被生物降解,主要是通过沉淀-溶解、氧化-还原、配合作用、胶体形成、吸附-解吸等作用进行迁移转化。
一、颗粒物与水之间的迁移1、矿物颗粒物和黏土颗粒物常见矿物颗粒物为石英、长石、云母及黏土矿物等硅酸盐矿物,主要由物理作用形成。
2、金属水合氧化物:铝、铁、锰、硅等金属以无机高分子及溶胶等形态存在。
例:铝在岩土中是丰量元素,在水中浓度低,<0.1mg/L。
水解,主要形态是:Al3+Al(OH)2+Al2(OH)24+Al(OH)22+Al(OH)3+等铁水合氧化物:Fe3+Fe(OH)2+Fe(OH)2+ Fe2(OH)24+Fe(OH)3等硅酸聚合物:Si n O2n-m(OH)2m3、腐殖质是一种代负电的高分子弱电解质。
4、水体悬浮沉积物是以矿物微粒为核心骨架,有机物和金属水合氧化物结合在矿物微粒表面上,经絮凝成为较粗颗粒而沉积在底部。
5、其它藻类、细菌、病毒、表面活性剂、油滴等。
二、水环境中颗粒物的吸附作用1、表面吸附:胶体具有巨大的表面积和表面能;属物理吸附,胶体表面积越大,吸附越强。
2、离子吸附:由于胶体表面的电荷引力。
3、专属吸附:除了化学键以外,尚有加强的憎水键及范德华力或氢键起作用。
水锰矿对Co、Cu、Ni、K和Na离子的吸附及其随pH的变化图:对于碱金属离子,在低浓度时,体系pH在水锰矿ZPC以上时发生吸附。
表明其为离子吸附。
而Co 、Cu 、Ni 等在体系pH 在ZPC 处或小于时都能进行吸附,这表明不带电荷或带正电均能吸附过渡金属。
4、吸附理论――有效层流脱理论5、吸附方向和推动力6、吸附等温线和等温式(1) 等温吸附经验式――弗罗因德利希式Freundlich 型等温式为: G =kC 1/n两边取对数: log G =log k +1/nlog C,nkP P k n Γ=Γ--吸附量-吸附压力常数(2) 单分子层吸附理论――兰格缪尔吸附等温式单分子层吸附吸附剂表面是均匀被吸附的分子与其它同气体分子无作用力吸附是一个动态平衡φ被吸附质分子覆盖的吸附表面积覆盖率()=吸附剂的总表面积 Langmuir 型吸附等温线:G =G 0C /(A +C ) 1/G =1/G 0+(A /G 0)(1/C )G0------单位表面上达到饱和时间的最大吸附量; A-------常数(3)Henry 型吸附等温线为直线,等温式为: G =kCk------分配系数影响吸附作用的因素:(a) pH 值的影响:一般情况下,吸附量随pH 升高而增大。
水中无机污染物的迁移转化

二、水中颗粒物的聚集 聚集 分散
凝聚——利用电解质促成。
[SiO2] + Al(Ⅲ) → [AlO2- ] + Si(Ⅳ)
第二节 水中无机污染物的迁移转化
3、水环境中颗粒物的吸附作用 吸附:指溶液中的溶质在界面层浓度升高的 现象。 表面吸附:由于颗粒物具有巨大的比表面和 表面能,产生表面吸附;物理吸附。
第二节 水中无机污染物的迁移转化
离子交换吸附:胶体颗粒大部分带负电荷,容 易吸附各种阳离子;物理化学吸附。
例如:去离子水的制备。
第二节 水中无机污染物的迁移转化 专属吸附:有化学键、憎水键、范德华力、氢
键等作用。
pH 水锰矿对Co、Cu、Ni、 K和Na离子的吸附及其随pH的变化
第二节 水中无机污染物的迁移转化
表3-8 水合氧化物对金属离子的专属吸附与非专属吸附的区别 项 目 非专属吸附 反离子 阳离子交换 >零电位点 扩散层 无 专属吸附 -、0、+ 配位离子 配位体交换 任意值 内层 负电荷减少,正电 荷增加
临界pH 最大吸附量 (mg/g)
(b) 颗粒物的粒度和浓度的影响
第二节 水中无机污染物的迁移转化 (2) 氧化物表面吸附的配合模式:
由于表面离子配位不饱和,金属氧化物与水
配位,水发生离解吸附而生成羟基化表面。 ≡MeOH2+ ≡MeOH + H+
Ks a1 = {≡MeOH }[ H+] / {≡MeOH2+ }
发生吸附的表面净电荷的符号 金属离子所起的作用 吸附时所发生的反应 发生吸附时要求体系的pH值 吸附发生的位置 对表面电荷的影响
第二节 水中无机污染物的迁移转化
(1)吸附等温式和吸附等温线
3-2水中无机污染物的迁移转化

第二节 水中无机污染物的迁移转化一、 颗粒物与水之间的迁移1.水中颗粒物的类别(1)矿物微粒和粘土矿物 硅酸盐矿物(2)金属水合氧化物 Al 、Fe 、Mn 、Si 等(3)腐殖质 带负电荷的高分子弱电解质,多含有–COOH 、–OH 等(4)水体悬浮沉积物 胶体物质的聚集物,结构组成不固定(5)其他 藻类、细菌、病毒、表面活性剂或油滴。
2.水环境中颗粒物的吸附作用表面吸附—物理吸附,与胶体的比表面积有关。
离子交换吸附—物理化学吸附,胶体每吸附一部分阳离子,同时也放出等量其它阳离子。
可逆,不受温度影响,在酸碱条件下均可进行,交换吸附能力与溶质的性质、浓度及吸附剂性质等有关。
专属吸附—吸附过程中,除了受化学键作用外,尚有加强的憎水键、 范德华力或氢键等在起作用。
可使表面电荷改变符号或使离子化合物吸附在同号电荷的表面上;吸附作用发生在胶体双电层的Stern 层中,作用力较大。
配合离子、有机离子、 有机和无机高分子的专属吸附强烈;水合氧化物胶体对金属离子有较强的专属吸附。
(1)吸附等温线和等温式吸附是指溶液中的溶质在界面层浓度升高的现象。
在固定的温度下,当吸附达到平衡时,颗粒物表面上的吸附量(G )与溶液中溶质平衡浓度 (C) 之间的关系用吸附等温式表达。
H 型( Henry )等温式(直线型)式中:K ——分配系数F 型(Freundlich )等温式:用对数表示:Langmuir 型吸附等温线G =G 0C /(A +C )1/G =1/G 0+(A /G 0)(1/C ) G 0------单位表面上达到饱和时间的最大吸附量; A-------常数kC G =n kCG 1=C nk G lg 1lg lg +=(2)氧化物表面吸附的配合模式金属氧化物表面都含有≡MeOH 集团;把具体表面看作一种聚合酸,其大量羟基可发生表面配合反应。
在配合平衡过程中需将邻近集团的电荷影响考虑在内。
3.沉积物中重金属的释放1)盐浓度升高:碱金属和碱土金属离子可将吸附在颗粒物表面的重金属离子置换出来,重金属解吸的重要途径之一。
水体污染物的迁移转化

水体污染物的迁移转化摘要:水是人类生存所必须,因而水体遭到污染则人类生存的环境品质就会大大的受损。
本文探讨了水体污染物的概念,总结了一下目前世界上所发现的水体污染物的主要种类,并且总结了水体污染物迁移转化的过程和方法,从中得出对水体污染物处理的一些解决方法。
关键词:迁移转化水体污染物1.水体污染物的概念水体污染物是指造成水体水质、水中生物群落以及水体底泥质量恶化的各种有害物质(或能量)。
水体污染物从化学角度可分为无机有害物、无机有毒物、有机有害物、有机有毒物4类。
2.水体污染物的分类和介绍2.1 耗氧污染物在生活污水、食品加工和造纸等工业废水中,含有碳水化合物、蛋白质、油脂、木质素等有机物质。
这些物质以悬浮或溶解状态存在于污水中,可通过微生物的生物化学作用而分解。
在其分解过程中需要消耗氧气,因而被称为耗氧污染物。
这种污染物可造成水中溶解氧减少,影响鱼类和其他水生生物的生长。
水中溶解氧耗尽后,有机物进行厌氧分解,产生硫化氢、氨和硫醇等难闻气味,使水质进一步恶化。
2.2 植物营养物植物营养物主要指氮、磷等能刺激藻类及水草生长、干扰水质净化,使BOD5升高的物质。
水体中营养物质过量所造成的"富营养化"对于湖泊及流动缓慢的水体所造成的危害已成为水源保护的严重问题。
富营养化(eutrophication)是指在人类活动的影响下,生物所需的氮、磷等营养物质大量进入湖泊、河口、海湾等缓流水体,引起藻类及其他浮游生物迅速繁殖,水体溶解氧量下降,水质恶化,鱼类及其他生物大量死亡的现象。
在自然条件下,湖泊也会从贫营养状态过渡到富营养状态,沉积物不断增多,先变为沼泽,后变为陆地。
这种自然过程非常缓慢,常需几千年甚至上万年。
而人为排放含营养物质的工业废水和生活污水所引起的水体富营养化现象,可以在短期内出现。
植物营养物质的来源广、数量大,有生活污水(有机质、洗涤剂)、农业(化肥、农家肥)、工业废水、垃圾等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
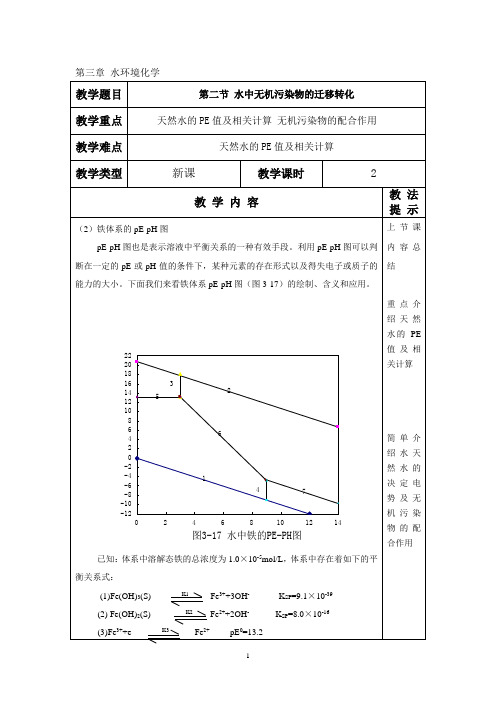
第三章:水环境化学——污染物存在形态第二节、水中无机污染物的迁移转化一、颗粒物与水之间的迁移、二、水中胶体颗粒物聚集的基本原理和方式 三、溶解和沉淀 四、氧化—还原1、概述2、天然环境中的氧化剂和还原剂3、氧化还反应概念回顾4、电子活度和氧化还原电位5、天然水体的pE-pH 关系图● 在氧化还原体系中,往往有H +或OH -离子参与转移,因此,pE 除了与氧化态和还原态浓度有关外,还受到体系pH 的影响,这种关系可以用pE-pH 图来表示。
该图显示了水中各形态的稳定范围及边界线。
● 由于水中可能存在物类状态繁多,于是会使这种图变得非常复杂。
例如一个金属,可以有不同的金属氧化态、羟基配合物、金属氢氧化物、金属碳酸盐、金属硫酸盐、金属硫化物等。
(1)水的氧化-还原限度绘制pE —pH 图时,必须考虑几个边界情况。
首先是水的氧化还原反应限定图中的区域边界。
选作水氧化限度的边界条件是1.0130×105Pa 的氧分压,水还原限度的边界条件是1.0130×105Pa 的氢分压(此时P H2=1,P O2=1),这些条件可获得把水的稳定边界与pH 联系起来方程。
天然水中本身可能发生的氧化还原反应分别是: 水的还原限度(还原反应):221H e H ↔++ pE 0=0.00 pE = pE 0 – lg((P H2)1/2/[H +])pE = –pH水的氧化限度(氧化反应):O H e H O 222141↔+++ pE 0 = +20.75]}[lg{4120++=H po pE pEpE=20.75—pH(2)pE—PH图假定溶液中溶解性铁的最大浓度为1.0×10-7mol/L,没有考虑Fe(OH)2+及FeCO3等形态的生成,根据上面的讨论,Fe的pE—pH图必须落在水的氧化还原限度内。
下面将根据各组分间的平衡方程把pE—pH的边界逐一推导。
①Fe(OH)3(s)和Fe(OH)2(s)的边界。
Fe{OH}3(s)和Fe(OH)2(s)的平衡方程为:Fe(OH)3(s)+H+ + e→Fe(OH)2(s)+H2O lgK = 4.62 ]][[1e HK+=,所以 pE =4.62–pH以pH对pE作图可得图3—17中的斜线①,斜线上方为Fe(OH)3(s)稳定区。
斜线下方为Fe(OH)2(s)稳定区。
②Fe(OH)2(s)和FeOH+的边界。
根据平衡方程:Fe(OH)2(s)+H+→FeOH++ H2O lgK = 4.6可得这两种形态的边界条件:pH = 4.6–lg[FeOH+]将[FeOH+]=1.0×10-7mol/L代人,得:pH= 11.6故可画出一条平行pE轴的直线,如图3—17中②所示,表明与pE无关。
直线左边为FeOH+稳定区,直线右边为Fe(OH)2(s)稳定区。
③Fe(OH)3(s)与Fe2+的边界。
根据平衡方程:Fe(OH)3(s) +3H+ +e→Fe2+ + 3H2O lgK=17.9可得这二种形态的边界条件:pE=17.9–3pH–lg[Fe2+]将[Fe2+]以1.0×10-7mol/L代入,得:pE=24.9–3pH得到一条斜率为–3的直线,如图3—17中③所示。
斜线上方为Fe(OH)3(s)稳定区,斜线下方为Fe(OH)2(s)稳定区。
④Fe(OH)3(s)与FeOH+的边界。
根据平衡方程:Fe(OH)3(s)+2H+ +e→FeOH++2H2O lgK=9.25将[FeOH+]以1.0×10-7mol/L代入,得:pE=16.25-2pH得到一条斜率为–2的直线,如图3—17中④所示。
斜线上方为Fe(OH)3(s)稳定区,下方为FeOH+稳定区。
⑤Fe3+与Fe2+的边界。
根据平衡方程:Fe3++e →Fe2+ lgK = 13.1可得: ][][lg 1.1323+++=Fe Fe pE 边界条件为[Fe 3+]=[Fe 2+],则:pE=13.1因此,可绘出一条垂直于纵轴平行于pH 轴的直线,如图3-17中⑤所示。
表明与pH 无关。
当pE>13.1时,[Fe 3+]>[Fe 2+];当pE<13.1时,[Fe 3+]<[Fe 2+] 。
⑥Fe 3+与FeOH 2+的边界。
根据平衡方程:Fe 3+ + H 2O →FeOH 2+ + H + lgK = –2.4K=[FeOH 2+][H +]/[Fe 3+]边界条件为[FeOH 2+]=[Fe 3+],则:pH=2.4故可画出一条平行于pE 的直线,如图3—17中⑥所示。
表明与pE 无关,直线左边为Fe 3+稳定区,右边为FeOH 2+稳定区。
⑦Fe 2+与FeOH +的边界。
根据平衡方程:Fe 2++ H 2O →FeOH ++ H +lgK= –8.6K=[FeOH +][H +]/[Fe 2+]边界条件为[FeOH +]=[Fe 2+],则: pH=8.6同样得到一条平行于pE 的直线,如图3—17中⑦所示。
直线左边为Fe 2+稳定区,右边为FeOH +稳定区。
⑧Fe 2+与FeOH 2+的边界。
根据平衡方程:Fe 2++H 2O →FeOH 2++H ++e lgK=–15.5可得:pH Fe FeOH pE -+=++][][lg 5.1522边界条件为:[FeOH 2+]=[ Fe 2+],则:pE = 15.5–pH得到一条斜线,如图3-17中⑧所示。
此斜线上方为FeOH 2+稳定区,下方为Fe 2+稳定区。
⑨FeOH 2+与Fe(OH)3(s)边界。
根据平衡方程:Fe(OH)3(s)+2H +→FeOH 2++2H 2O ,lgK=2.4K=[FeOH 2+]/[H +]2将[FeOH 2+]以1.0×10-7mol/L 代入,得:pH=4.7可得一平行于pE 的直线,如图3-17中⑧所示。
表明与pE 无关。
当pH>4.7时,Fe(OH)3(s)将陆续析出。
化介质),Fe3+是主要的;在低酸度的氧化介质中,固体Fe(OH)3(s)是主要的存在形态,最后在碱性的还原介质中,具有低的H+活度及高的电子活度,固体的Fe(OH)2是稳定的。
注意:在通常的水体PH范围内(约5—9),Fc(OH)3或Fe2+是主要的稳定形态。
6.天然水的pE和决定电位天然水中含有许多无机及有机氧化剂和还原剂。
水中主要的氧化剂有溶解氧、Fe(Ⅲ)、Mn(Ⅳ)和S(Ⅵ),其作用后本身依次转变为H2O、Fe(Ⅱ)、Mn(Ⅱ)、和S(–Ⅱ)。
水中主要还原剂有种类繁多的有机化合物、Fe(Ⅱ)、Mn(Ⅱ)和S(–Ⅱ),在还原物质的过程中,有机物本身的氧化产物是非常复杂的。
由于天然水是一个复杂的氧化还原混合体系,其pE应是介于其中各个单体系的电位之间,而且接近于含量较高的单体系的电位。
若某个单体系的含量比其他体系高得多,则此时该单体系电位几乎等于混合复杂体系的pE,称之为“决定电位”。
在一般天然水环境中,溶解氧是“决定电位”物质,而在有机物累积的厌氧环境中,有机物是“决定电位”物质,介于二者之间者,则其“决定电位”为溶解氧体系和有机物体系的结合。
从这个概念出发,可以计算天然水中的pE。
若水中PO2=0.21×105Pa,以[H+]=1.0×10-7mol/L代入OHeHO222141↔+++ pE0 =+20.75,则:pE = 20.75 + lg{( PO2/1.013×105)0.25×[H+]}=20.75 + lg[(0.21×105/1.013×105)1/4×1.0×10-7]=13.58说明这是一种好氧的水,这种水存在夺取电子的倾向。
若是有机物丰富的厌氧水,例如一个由微生物作用产生CH4及CO2的厌氧水,假定P CO2=PCH4和pH=7.00,其相关的半反应为:OHCHeHCO242418181+↔+++ pE0=2.87125.04125.020/][lgCHCOpHppEpE+⋅+==2.87+lg[H+]=–4.13这个数值并没有超过水在pH=7.00时还原极限–7.00,说明这是一还原环境,有提供电子的倾向。
从上面计算可以看到,天然水的pE随水中溶解氧的减少而降低,因而表层水呈氧化性环境,深层水及底泥呈还原性环境,同时天然水的pE 随其pH 减少而增大。
7、无机氮化物的氧化-还原转化水中氮主要以NH 4+或NO 3-形态存在,在某些条件下,也可以有中间氧化态NO 2-。
像许多水中的氧化—还原反应那样,氮体系的转化反应是微生物的催化作用形成的。
下面讨论中性天然水的pE 变化对无机氮形态浓度的影响。
假设总氮浓度为 1.00×10-4mol/L ,水体pH=7.00。
(1)在较低的pE 值时(pE<5),NH 4+是主要形态。
在这个pE 范周内,NH 4+的浓度对数则可表示为:lg[NH 4+]=–4.00➢ lg[NO 2-]—pE 的关系可以根据含有NO 2-及NH 4+的半反应求得:O H NH e H NO 24231613461+↔++++- pE 0=15.14 在pH=7.00时就可表达为:614612][][lg82.5+-+=NH NO pE以[NH 4+]=1.00×10-4代入,就可得到lg[NO 2-]与pE 相关方程式:lg[NO 2-]=–38.92+6pE➢ 在NH 4+是主要形态并有1.00×10-4mol/L 浓度时,lg[NO 3-]—pE 的关系为:O H NH e H NO 24383814581+↔++++- pE 0=14.90 814813][][lg15.6+-+=NH NO pE (在pH=7.00)lg[NO 3-]=–53.20+8pE(2)在一个狭窄的pE 范围内,约pE=6.5左右,NO 2-是主要形态。
➢ 在这个pE 范围内,NO 2-的浓度对数根据方程给出:lg[NO 2-]=–4.00 用[NO 2-]=1.00×10-4代人式(O H NH e H NO 24231613461+↔++++- pE 0=15.14)中,得到:614614][)1000.1(lg82.5+-⨯+=NH pE ,因此lg[NH 4+]=30.92–6pE➢ 在NO 2-占优势的范围内,lg[NO 3-]的方程式可从下面的处理中得到:O H NO e H NO 223212121+↔++-+- pE 0=14.15pE NO 230.18]lg[3+-=- (当[NO 2-]=1.00×10-4mol/L 时)(3)当pE>7,溶液中氮的形态主要为NO 3-,此时:lg[NO 3-]= –4.00➢ lg[NO 2-]的方程式也可在pE>7时获得,将[NO 3-]=1.00×10-4mol/L 代入(O H NH e H NO 24231613461+↔++++- pE 0=15.14),得: 212214][)1000.1(lg15.7--⨯+=NO pE ,因此,lg[NO 2-]=10.30–2pE➢ 依次类似,代入式(O H NH e H NO 24383814581+↔++++- pE 0 =14.90)给出在NO 3-占统治区的lg[NH 4+]的方程式:814814][)1000.1(lg15.6+-⨯+=NH pE因此:lg[NH 4+]=45.20–8pE至此,绘制水中氮系统的对数浓度图所需要的全部方程式均已求得。